

Abstract
Objective:
Recurring issues in clinical trial design may bias results toward the null, yielding findings inconclusive for treatment effects. This study evaluated for powering bias among high-impact critical care trials and the associated risk of masking clinically important treatment effects.
Design, Settings, and Patients:
Secondary analysis of multicenter randomized trials of critically ill adults in which mortality was the main endpoint. Trials were eligible for inclusion if published between 2008–2018 in leading journals. Analyses evaluated for accuracy of estimated control group mortality, adaptive sample size strategy, plausibility of predicted treatment effect, and results relative to the minimal clinically important difference. The main outcome was the mortality risk difference at the study-specific follow-up interval.
Interventions:
None.
Measurements and Main Results:
Of 101 included trials, 12 met statistical significance for their main endpoint, five for increased intervention-associated mortality. Most trials (77.3%) overestimated control group mortality in power calculations (observed minus predicted difference: −6.7% ± 9.8%; p<0.01). Due to this misestimation of control group mortality, in 14 trials the intervention would have had to prevent at least half of all deaths to achieve the hypothesized treatment effect. Seven trials prespecified adaptive sample size strategies that might have mitigated this issue. The observed risk difference for mortality fell within 5% of predicted in 20 trials, of which 16 did not reach statistical significance. Half (47.0%) of trials were powered for an absolute risk reduction ≥10%, but this effect size was observed in only three trials with a statistically significant treatment benefit. Most trials (67.3%) could not exclude clinically important treatment benefit or harm.
Conclusions:
The design of most high-impact critical care trials biases results toward the null by overestimating control group mortality and powering for unrealistic treatment effects. Clinically important treatment effects often cannot be excluded.
Keywords: clinical trials as topic, bias, minimal clinically important difference, sample size, critical care
INTRODUCTION
Lack of statistical significance of clinical trial results may not exclude clinically important findings (1–3). “Negative” randomized controlled trials (RCTs)—those that do not demonstrate a statistically significant treatment benefit—may indeed reflect an ineffective treatment on average for the study population in some instances. Yet, “negative” RCTs also may be the product of design choices that mask important treatment effects within the population of study.
RCTs examining treatments for critically ill patients afford an ideal case study for design challenges. Phenotypic heterogeneity (4–7), practice variation (8, 9), and secular trends in mortality from critical illness (10) may contribute to inaccurate prediction of control group mortality and treatment effect size, which in turn threaten statistical power. The complexity of critical illnesses and ubiquity of competing risks often preclude adoption of endpoints other than all-cause mortality. Trials may power for overly optimistic treatment effects that do not reflect smaller but clinically important differences or expend substantial resources to include many participating centers and accrue large sample sizes within a reasonable time. Neither approach is ideal.
This study evaluated multicenter critical care RCTs published in leading medical journals to determine recurring power and sample size estimation issues that contribute to “negative” trial results, and to assess whether such trials reliably exclude clinically important differences in survival. The main hypothesis was that two recurring issues—control group mortality misestimation and overly optimistic predicted treatment effects—bias trial results toward the null. This study also evaluated how often trial results fail to exclude potentially clinically important treatment effects.
METHODS
Eligibility Criteria for Trials
Studies eligible for inclusion were multicenter RCTs of critically ill adults in which mortality was the main endpoint. For inclusion, the publication must have appeared between January 2008 and December 2018 in one of seven high-impact general medical or specialty journals: New England Journal of Medicine, JAMA, Lancet, American Journal of Respiratory and Critical Care Medicine, Lancet Respiratory Medicine, Intensive Care Medicine, or Critical Care Medicine. Journals were selected by considering impact factor and content relevance in effort to identify trials most influential to future trial design and clinical practice. Trials were excluded if not designed as superiority trials or if patient-level randomization was not employed.
Data Extraction
Tables of contents for each journal issue were screened independently by two study physicians and a PubMed search conducted to identify articles for inclusion (Supplement). Results from all search strategies were combined with discordance resolved by an independent third reviewer to create the final study list.
Data were extracted in duplicate, blinded fashion. Discordant entries were resolved by an independent third reviewer. Data sources included the main trial publication, accompanying online data and protocol supplements, published trial protocols, and online trial registration sites.
Main Outcome
The main outcome for this study was the unadjusted risk difference in mortality (treatment group minus control group) at each study-specific interval for primary follow-up.
Determination of Minimal Clinically Important Difference
The minimal clinically important difference (MCID) was defined from the clinician’s perspective as the smallest treatment effect required to change routine clinical practice for a comparable patient (11). A 5% absolute risk difference and 20% relative risk difference in mortality were selected a priori as the MCID based on existing literature (12–17) and concern that smaller differences might be viewed as unlikely to change clinical practice. Other MCID thresholds were considered in secondary analyses (Supplement).
Statistical Analysis
Predicted control group mortality and predicted risk difference were obtained directly from each trial’s reported sample size calculation. Observed risk difference was calculated using observed enrollment and mortality extracted from each trial; corresponding 95% confidence intervals (CI) were computed from extracted data.
A paired t-test was used to determine whether the average difference in observed versus predicted control group mortality among trials was significantly different from zero, with observed and expected mortality handled as paired observations for each trial.
To evaluate impact of control group mortality misestimation on statistical power, the predicted absolute risk reduction was assessed as a fraction of the true observed control group risk of death. The association between control group mortality and statistical power was evaluated graphically for representative sample sizes and relative risk.
Conclusiveness of trial results for important treatment effects was evaluated according to the approach of Kaul and Diamond (18). Trials were evaluated for whether the risk difference 95% CI included the MCID. Trials also were evaluated for whether the risk difference 95% CI included the trial’s own predicted treatment effect. Bayesian models were developed via the Markov Chain Monte Carlo method with noninformative priors, 1000 burn-in iterations, 50,000 iterations and thinning rate of five. Model results were used to calculate the posterior probability of observing the proposed MCID on the absolute and relative risk scales. Posterior probabilities for treatment effect of each individual trial were reported. Other treatment effect thresholds for clinical importance were considered in secondary analyses (Supplement).
Frequentist hypothesis testing applied a two-sided alpha threshold of 0.05 without adjustment for multiple comparisons. Analyses were conducted using SAS 9.4 and PASS 14.
RESULTS
Characteristics of Included Trials
Of 657 unique publications identified on initial screen, 101 multicenter superiority trials with patient-level randomization and mortality as the main endpoint were included (Table 1, Figure S1). A complete list of included trials is provided in the online supplement. Twelve trials (11.9%) met the statistical significance threshold for the primary endpoint, five of which demonstrated increased mortality with the intervention.
Table 1.
Characteristics of included trials
| Characteristic | Value (n = 101) |
|---|---|
| Journal, no. (%) | |
| New England Journal of Medicine | 48 (47.5%) |
| JAMA | 18 (17.8%) |
| Critical Care Medicine | 13 (12.9%) |
| American Journal of Respiratory and Critical Care Medicine | 6 (5.9%) |
| Intensive Care Medicine | 6 (5.9%) |
| Lancet | 6 (5.9%) |
| Lancet Respiratory Medicine | 4 (4.0%) |
| Main source of funding, no. (%)1 | |
| Government outside US | 54 (53.5%) |
| Industry | 23 (22.8%) |
| Non-profit organization | 11 (10.9%) |
| US government | 8 (7.9%) |
| Local institution | 5 (5.0%) |
| Geographic region(s) in which trial conducted, no. (%) | |
| Europe | 75 (74.3%) |
| US | 27 (26.7%) |
| Canada | 23 (22.8%) |
| Australia and/or New Zealand | 19 (18.8%) |
| Asia | 22 (21.8%) |
| Central or South America | 10 (9.9%) |
| Africa | 6 (5.9%) |
| Topic of trial, no. (%) | |
| Sepsis or infection | 34 (33.7%) |
| Respiratory | 21 (20.8%) |
| Cardiovascular | 19 (18.8%) |
| General critical care | 14 (13.9%) |
| Renal | 5 (5.0%) |
| Trauma | 4 (4.0%) |
| Other2 | 4 (4.0%) |
| Trial design, no. (%) | |
| Two-arm parallel group | 84 (83.2%) |
| Three-arm parallel group3 | 8 (7.9%) |
| Factorial 2×24 | 9 (8.9%) |
| Trial stopped early, no. (%)5 | 35 (34.7%) |
| Sample size in final analysis, median (IQR) | 843 (411–1588) |
| Main endpoint, no. (%) | |
| ≤ 7-day mortality | 3 (3.0%) |
| 28-day or 30-day mortality | 63 (62.4%) |
| 60-day or 90-day mortality | 23 (22.8%) |
| ≥ 180-day mortality | 3 (3.0%) |
| In-hospital mortality without specified interval | 9 (8.9%) |
| P-value ≤ 0·05 for primary endpoint, no. (%) | 12 (11.9%) |
| Significant benefit with intervention | 7 (6.9%) |
| Significant harm with intervention | 5 (5.0%) |
For studies with multiple funding sources, the primary funding source was considered governmental whenever the government was a trial sponsor regardless of other funding sources reported.
For all analyses, the five three-arm parallel group trials were handled as two-arm trials. One trial merged two groups in the main analysis. Two trials stopped enrollment in one arm early and reported as the main result a two-group comparison. Two trials explicitly stated the a priori main analysis consisted of two-group comparison without consideration for the third study arm.
Eight trials of factorial 2×2 design, reported in four publications, specified pairwise comparison of both factors in the main analysis. One trial of factorial 2×2 design specified comparing only one factor in the main analysis.
Reasons for trials stopping early included futility (n = 17), harm (n = 10), poor enrollment (n = 6), sponsor termination (n = 1), and drug/device withdrawal from market (n = 1).
Sample Size
The median (IQR) sample size for analysis of the main endpoint was 843 (411–1588) patients. The smallest sample size was 62 patients, and the largest was 20,127 patients. Seven trial protocols (6.9%) incorporated an adaptive sample size design that permitted increasing enrollment targets if prespecified criteria were met (19–24).
Control Group Mortality
Reported power calculations contained sufficient data to ascertain predicted control group mortality in 97 of 101 trials. Median (IQR) predicted control group mortality was 40% (30–45%).
Power calculations significantly overestimated control group mortality (observed: 33.9 ± 18.0%; predicted: 40.6 ± 17.5%; mean difference −6.7 ± 9.8%; p < 0.01). Seventy-five trials (77.3%) overestimated control group mortality, whereas only 22 trials (22.7%) underestimated control group mortality.
The distribution of control group mortality misestimation is displayed graphically in Figure 1.
Figure 1. Waterfall plot of control group mortality misestimation.
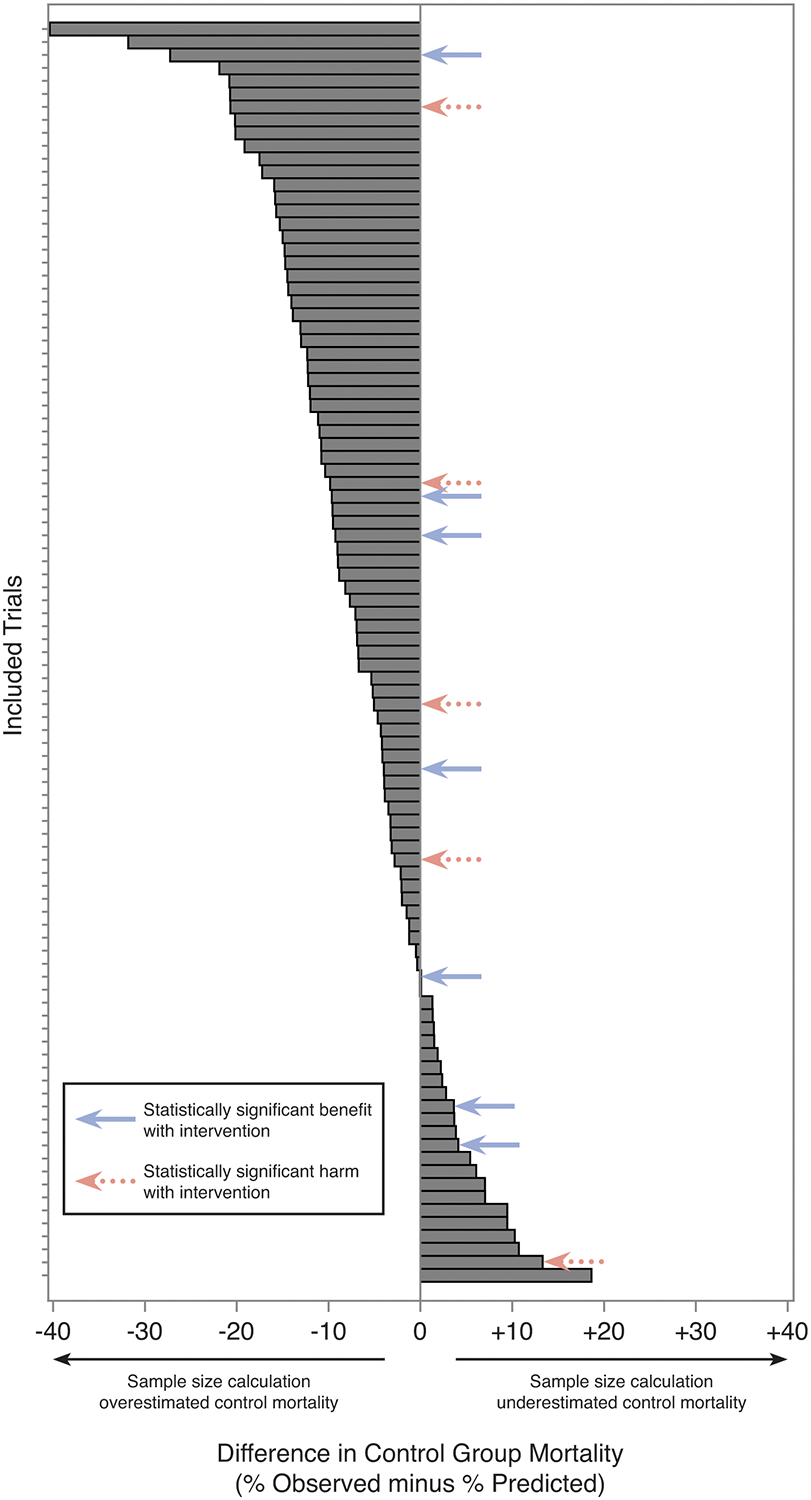
Absolute difference in control group mortality, observed minus expected. Each bar represents an individual trial.
Predicted Risk Difference
On the absolute risk scale, trials were powered to detect a median (IQR) mortality risk difference of 9.0% (6.0–12.0%), corresponding to an NNT of 11 (8–17) patients. Only nineteen trials (19.0%) were powered to detect a risk difference of 5% or less (NNT = 20) (Figure 2). Large predicted effect sizes were common across all included journals and disease areas (Figures S2–S3).
Figure 2. Butterfly plot of predicted and observed mortality risk difference.
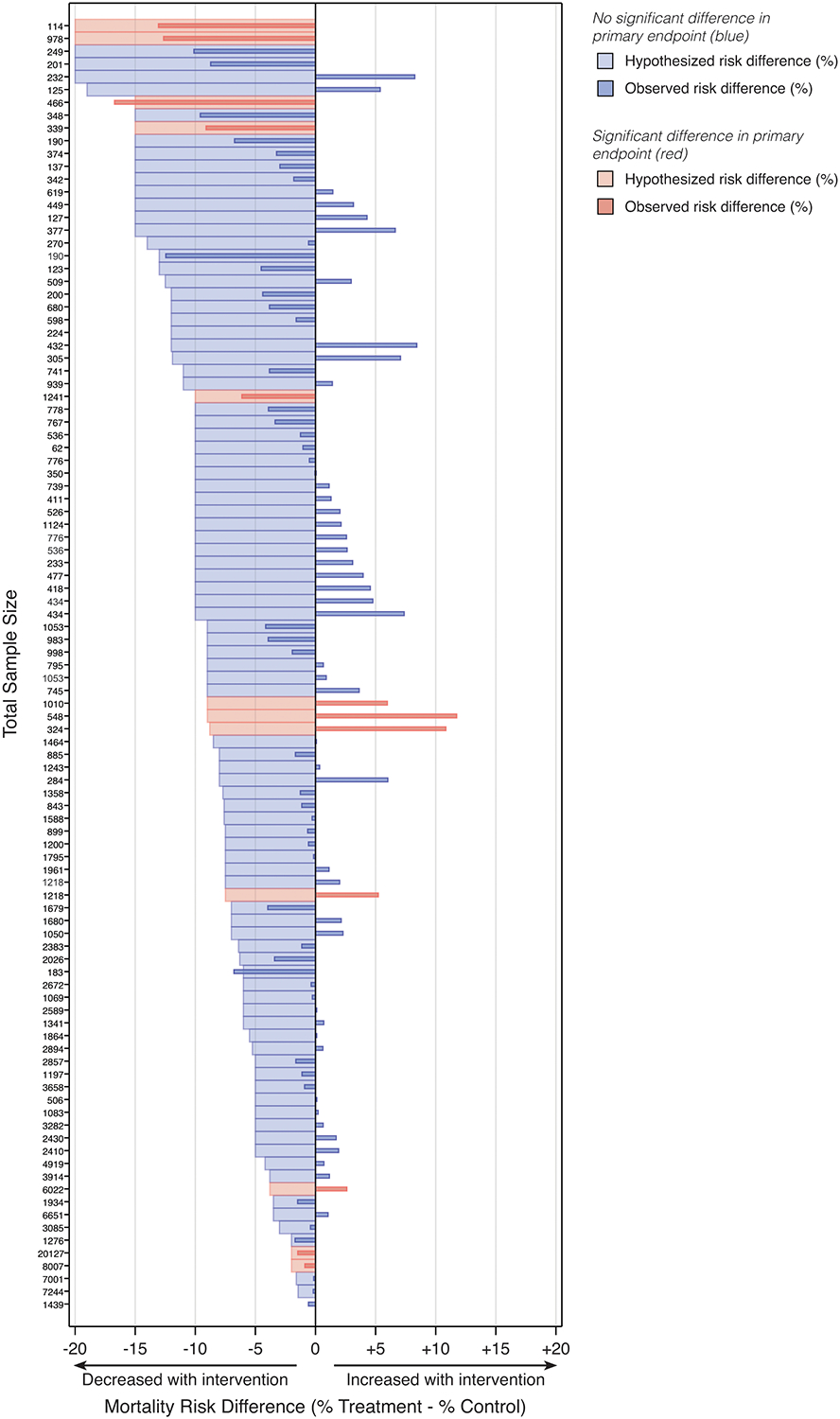
Data are sorted by sample size within each study type. One trial (sample size 1,439 patients) contained insufficient information in its reported power and sample size determination to identify predicted risk difference.
On the relative risk scale, trials were powered to detect a median (IQR) relative risk reduction of 23% (20–30%). The hypothesized relative risk reduction was at least one-third of all deaths in nineteen trials and at least half of all deaths in five trials.
Influence of Control Mortality Misestimation on Predicted Treatment Effect
The hypothesized absolute risk reduction in mortality, as a percent of the true observed baseline (control group) risk in the trial, was median (IQR) 28.7% (20.1–42.5%). To achieve the hypothesized absolute risk reduction, in 14 trials the intervention would have had to prevent at least half of all deaths given the observed control group mortality, an exceedingly unlikely treatment effect. In two trials, the hypothesized absolute risk reduction could not have been achieved even if the intervention prevented all deaths.
The relationship between control group mortality, risk ratio, sample size, and statistical power is shown in Figure 3.
Figure 3. Impact of control group mortality misestimation, hypothesized risk ratio, and sample size on statistical power.
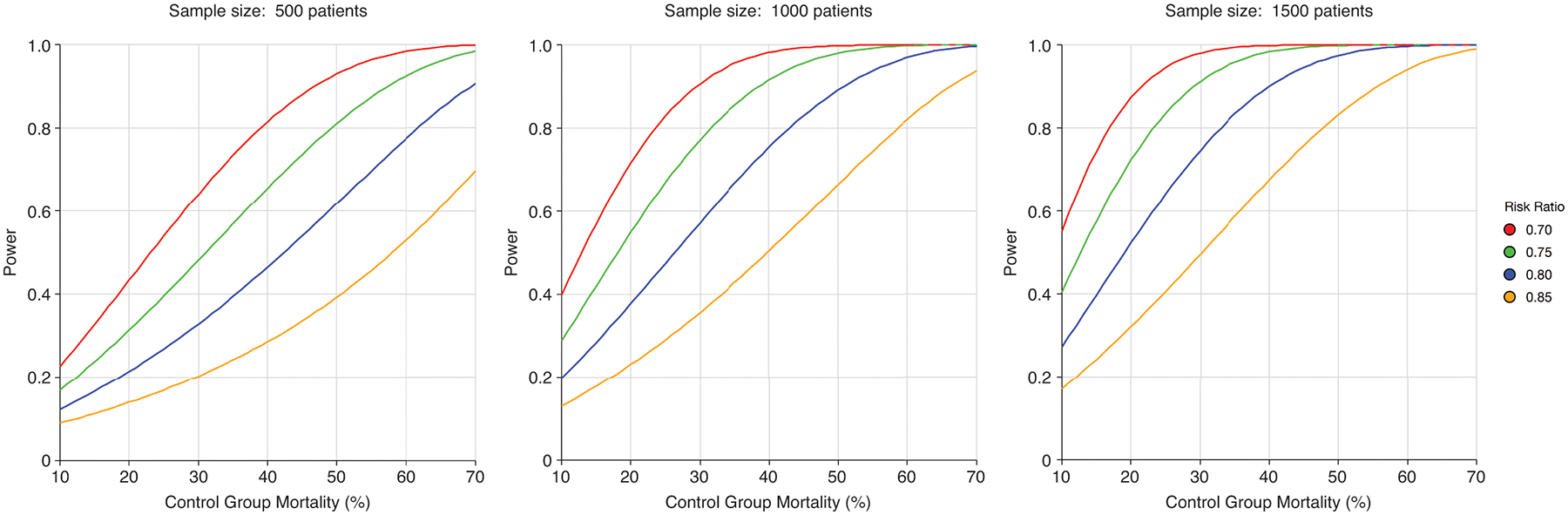
For a given sample size and risk ratio (also known as relative risk), power decreases with lower control group mortality. Overestimation of control group mortality in sample size calculations decreases power (1 – ß) and therefore increases probability of a “false negative” trial result (ß). The impact of lower control group mortality on diminishing power is exacerbated by smaller sample size.
Observed Risk Difference
The median (IQR) observed risk difference (treatment minus control) in mortality was −0.2% (−1.7% to 2.0%), and ranged from −16.7% to 11.7% (Figure 2).
Among trials observing a statistically significant treatment benefit, the median (IQR) observed risk difference was −9.1% (−13.1% to −1.5%), corresponding to an NNT of 11 (8 to 68) patients to prevent one death.
In only five trials (5.0% of included trials) was an absolute risk reduction of 10% or greater observed (NNT ≤ 10) (25–29), and two of these trials did not reach statistical significance (26, 27). The sample size for four of the five trials with this large observed treatment benefit fell within the smallest third of included trials, with corresponding wide confidence intervals, raising doubts about the large magnitude of treatment effect (Figure 4).
Figure 4. Trial results according to clinically important difference in mortality on absolute and relative scales.
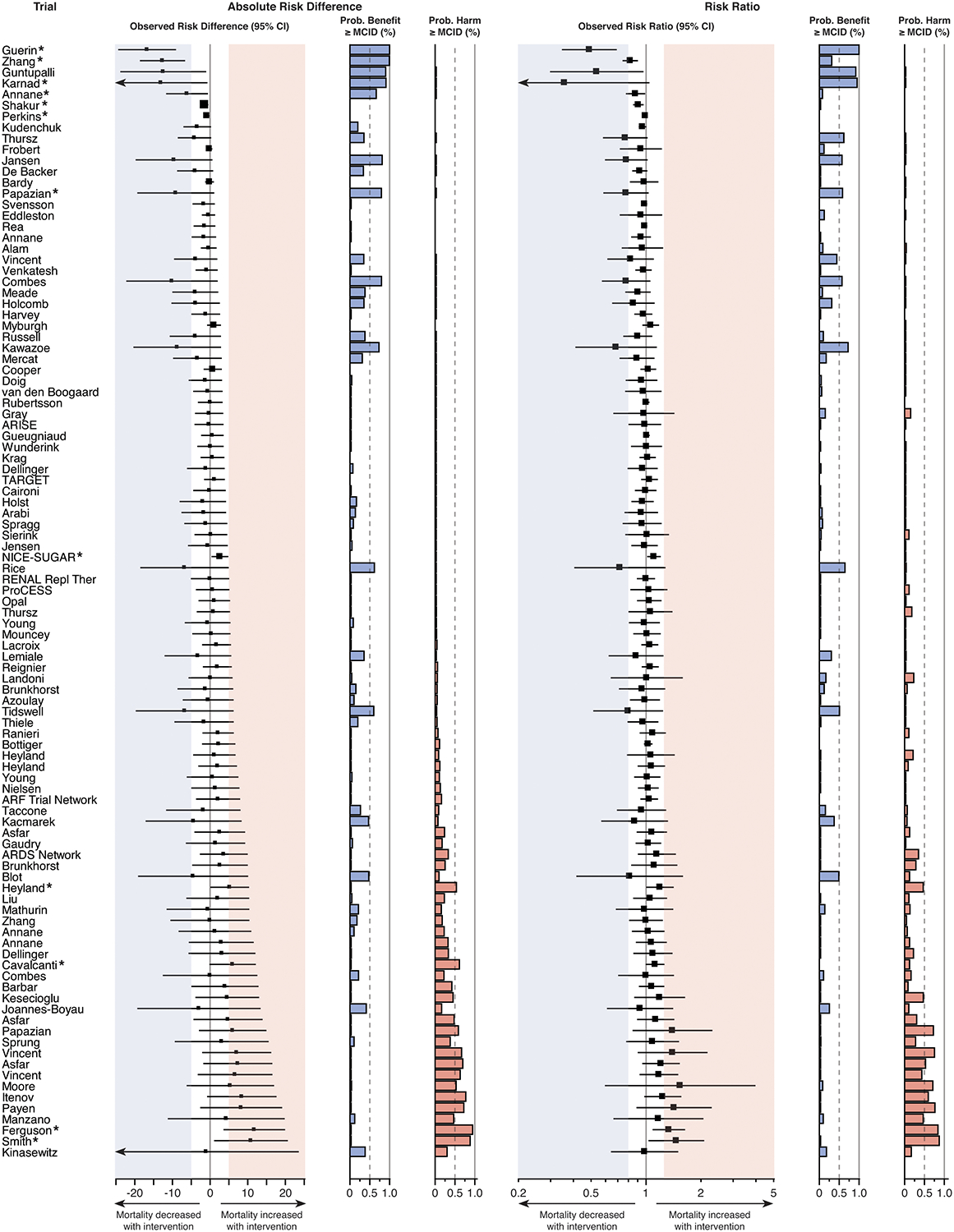
Several trials that did not achieve statistical significance in conventional frequentist analysis nevertheless failed to exclude clinically important benefit or harm for the intervention studied. The prespecified threshold used for MCID was a 5% absolute risk difference (number needed to treat = 20) or 20% relative risk difference (risk ratio ≤ 0.8 or ≥ 1.2) for either benefit or harm with treatment. Thresholds are indicated by the shaded areas: blue for benefit and red for harm. Forest plots indicate the effect estimate and 95% confidence interval for absolute risk difference (left) and risk ratio (right) for each trial. The size of each square corresponds to the trial sample size relative to other included trials. The probability of a clinically important treatment effect (benefit or harm), given the trial results, was calculated for both the absolute risk difference and risk ratio using Bayesian statistics. MCID = minimal clinically important difference. * denotes statistical significance according to the trial’s main analysis.
Clinically Important Difference in Treatment Effect
The observed risk difference for mortality fell within ± 5% of the predicted treatment effect in 20 of 100 trials with requisite data. Yet, 16 of these 20 trials did not achieve statistical significance for the primary endpoint (Figure 2). Larger sample size was strongly correlated with a smaller difference in observed versus hypothesized treatment effect (Pearson r = −0.42, 95% CI −0.57 to −0.24; p < 0.01).
“No benefit” trials:
In 21 of the 94 trials (22.3%) without a statistically significant benefit, the 95% CI for risk difference included the effect size the trial was designed to detect. In 44 of these trials (46.8%), the 95% CI included a 5% absolute risk reduction in death (Figure 4), the a priori MCID selected for this analysis.
“No harm” trials:
In 49 of the 96 trials (51.0%) without a statistically significant worsening of mortality with the intervention, the 95% CI included a 5% absolute risk increase in death, the MCID (Figure 4). Overlap with other risk difference thresholds for benefit and harm are presented in the Supplement (Tables S6–S8).
Inconclusive frequentist results for important treatment effects:
Most trials (68 of 101 included trials; 67.3%) failed to exclude a clinically important treatment benefit or harm, assessed via the treatment effect estimate 95% CI crossing the 5% absolute risk difference threshold for either benefit or harm without achieving statistical significance.
Inconclusive Bayesian results for important treatment effects:
The intervention of study was more probable than not (posterior probability > 0.5) to decrease risk of death by at least the 5% MCID (treatment benefit) in eleven trials, six of which did not reach statistical significance (Figure 4). The intervention of study was more probable than not to increase risk of death by at least the 5% MCID (treatment harm) in eleven trials, seven of which did not reach statistical significance. Results grouped by journal and disease area are presented in Figures S4–S5. Results for other treatment effect thresholds and the posterior probability calculated for each trial, as both absolute risk difference and risk ratio, are presented in Figure 4 and Figures S6–S9.
DISCUSSION
The main findings of this study are that multicenter RCTs of critical illness routinely overestimate control group mortality, power for overly optimistic treatment effects, and fail to provide conclusive results for clinically important differences in survival. These issues collectively may be referred to as powering bias.
Relevance for Clinicians
These findings have important implications for clinicians seeking to practice evidence-based medicine. Particularly in critical care, widespread adoption of a given treatment often hinges on results of one or two trials (30–33). When one large-scale trial does not find a statistically significant difference for main endpoints, the common interpretation is often conclusive, that the therapy is ineffective, thereby dissuading use in clinical practice. If trial results confidently exclude the MCID and other sources of bias are not identified, this interpretation may be reasonable. However, the present study indicates most multicenter critical care trials power for improbably large effect sizes, an issue compounded by misestimation of control group mortality, biasing results toward the null regardless of true treatment effect. In such trials, the correct interpretation may be that data are inconclusive due to powering bias.
Learning to assess for powering bias therefore is an essential skill for the astute clinician weighing evidence to guide management decisions. Classic teaching on how to assess trials for bias (34, 35) does not routinely include these potential sources, related to power and sample size calculations, which occur often and may bias results toward the null.
The present study brings to light two simple questions to ask when reading a primary trial report to ascertain the threat of powering bias. One, did the control group mortality predicted in the sample size calculation overestimate the actual control group mortality observed in the trial? Two, did the treatment effect size for which the trial was powered fail to include what the reader deems a minimal clinically important difference that would change his/her practice? If the answer to either of these questions is yes, then powering bias may be present. Particularly when the treatment effect 95% confidence interval includes the MCID, a “negative” trial might better be described as inconclusive. Regardless, interpretation and implications for clinical practice always should be weighed in context of the totality of trial results (beneficial and harmful effects), existing literature, and current understanding of pathophysiology.
Relevance for Trialists
The present study also highlights several issues to consider in trial planning. Phenotypic (4, 6, 36) and practice heterogeneity (37) inherent in critical care are not easily mitigated even with rigorous trial eligibility criteria and protocol standardization of care. Coupled with secular trends in mortality (10), such heterogeneity likely accounts for the systematic overestimation of control group mortality found here.
Powering for overly optimistic treatment effects is common in critical care trials. Nearly half (47%) of trials in this study powered for an absolute mortality reduction of 10% or greater; yet, this treatment effect was seen in only five trials, four of which had modest sample sizes (114–466 patients) with wide 95% CIs, suggesting the true effect might be smaller. Thus, trials designed to detect a mortality risk difference exceeding 10% may bias toward a “negative” result and should require compelling justification to go forward.
Several factors may contribute to improbably large predicted treatment effects, including: (i) need to balance sample size against budget constraints and trial feasibility; (ii) publication bias in existing literature; and (iii) large effect sizes in early-phase trials, when observed, may tend to overestimate treatment effects but also are likeliest to garner support for advancing to large-scale trials (38–43). Because not all factors reflect overly optimistic views of treatment effect per se (“optimism bias” (38)), the term “delta inflation” has been proposed to refer more broadly to improbably large predicted effect estimates regardless of rationale (13).
Targeting large sample sizes to detect risk differences approaching the MCID often requires adding trial sites or extending the enrollment period, which may introduce additional phenotypic and practice heterogeneity to the trial. With large sample size, costs may become prohibitive or reduce the number of studies that can be supported by a given funding body.
All-cause mortality remains the most widely accepted critical care endpoint for phase III trials in part because validated disease-specific or intermediate surrogate endpoints are lacking for most critical illnesses (44). Yet, mortality may not always be the appropriate main endpoint (45), particularly when lower baseline risk of death is anticipated, an issue best evidenced by considering relative risk reduction. For example, a trial powered to detect a 5% absolute risk reduction with control group mortality of 15% would require that the treatment prevent one-third of all deaths to achieve statistical significance, an improbable treatment effect observed in only three trials in this study. Powering for an absolute risk reduction less than 5% to maintain the predicted relative risk reduction within a plausible range may be appropriate in select instances. Simply enrolling patients with higher risk of death may not improve trial performance if doing so dilutes disease- and target-specific attributable risk, i.e. the proportion of deaths due to the particular disease of interest and mechanism being targeted (46–48).
Addressing Powering Bias in Trial Design and Reporting
Two underutilized strategies for mitigating powering bias are adaptive trial design and MCID-based reporting.
Adaptive sample size strategies can overcome misestimation of control group mortality (49) but were employed by only 6.9% of multicenter trials in this analysis. Adaptive trial designs of any kind require flexibility from funding and regulatory agencies to account for and respond to increases in sample size, and due care must be taken to avoid introducing bias (50, 51).
Trial analysis plans should include assessment of clinical importance relative to a prespecified MCID to facilitate interpretation for clinical practice. We favor a two-pronged strategy (Figure 4) (18). First, report where the effect estimate 95% CI falls relative to the MCID (52, 53). If the 95% CI includes the MCID, clinical importance cannot be excluded, and the trial may be considered inconclusive (54). Second, calculate the Bayesian posterior probability of treatment effect greater than or equal to the MCID (55). By assuming a noninformative prior, the computed posterior probability is effectively determined by trial data alone, an approach more familiar to readers than specifying various informative Bayesian priors. The resultant statistic is intuitive for clinical practice: the probability of a clinically important treatment effect based on trial results. The MCID should be prespecified in the trial protocol, and both benefit and harm should be evaluated (56). Incorporating MCID into trial design may help establish benchmarks to persuade funding agencies to promote smarter and more clinically meaningful trials (57).
Limitations
This study is not the first to identify issues around powering bias (12, 13, 58). However, it does confirm its persistence in recent critical care clinical trials. This study builds on existing literature in part by evaluating the problem according to an MCID and by using Bayesian statistics to quantify the posterior probability of achieving the MCID. Importantly, it also reframes the issue of powering bias as an essential consideration not just for trialists but also for clinicians to weigh in interpreting the evidence.
This study included only multicenter RCTs published in seven high-impact journals, criteria intended to select trials most influential to scientific direction, future trial design, and clinical practice. Whether powering bias is similarly widespread in smaller trials published in lower-impact journals could be important because such trials often have outsized influence in meta-analyses. To facilitate comparison between trials, only trials with mortality as the main endpoint were included. This study did not restrict eligible trials to intensive care unit-based interventions only but instead included the more broadly defined population of critically ill adults, potentially increasing heterogeneity among included trials.
Other strategies for improving trial design, including prognostic and predictive enrichment, were not explored in this analysis.
This analysis prespecified a 5% absolute risk difference and 20% relative risk difference as the MCID for all studies, but MCID may depend on the risk profile of the associated intervention and extent of long-term morbidity among survivors. MCID is a difficult concept to apply to mortality when characterized from the individual patient’s perspective. Instead, it was defined for this analysis using a clinical perspective, as an effect size sufficient to change clinical practice, an approach similar to that proposed by professional societies for use in oncology trials (57). To our knowledge, no validated mortality MCID exists for the critical illnesses studied.
The Bayesian posterior probability of achieving the MCID was modeled using a noninformative prior distribution so that the posterior probability would be determined entirely by study data as in a frequentist analysis; however, in practice a trial reporting Bayesian results might incorporate informative prior distributions based on preliminary data.
CONCLUSION
Most multicenter RCTs of critical illness bias toward the null by overestimating control group mortality and powering for overly optimistic treatment effects. As a result of this powering bias, trials rarely provide conclusive results on smaller but clinically important treatment effects. Including assessment of trial results relative to the MCID would greatly improve clinical interpretation. Incorporating adaptive designs in future trials may mitigate the impact of misestimation in power and sample size calculations.
Supplementary Material
Acknowledgements:
The authors thank Dr. B. Taylor Thompson, Massachusetts General Hospital, for his thoughtful feedback on the draft manuscript. The authors also thank Dr. Quixuan Chen, Columbia University Mailman School of Public Health, for her expert guidance with the Bayesian analysis.
Copyright form disclosure: Dr. Montesi’s institution received funding from United Therapeutics and she receives funding from the Francis Family Foundation and the Scleroderma Foundation. Dr. Brodie’s institution received funding from ALung Technologies, Baxter, Xenios, BREETHE, and Hemovent. Dr. Beitler’s institution received funding from National Heart, Lung, and Blood Institute (NHLBI); he received funding from Hamilton Medical; and he received support for article research from National Institutes of Health (NIH). The remaining authors have disclosed that they do not have any potential conflicts of interest.
Funding: U.S. National Heart, Lung, and Blood Institute (K23-HL133489, R21-HL145506). The NHLBI had no role in the design or conduct of the study, the collection, analysis, or interpretation of the data, the preparation, review, or approval of the manuscript, or the decision to submit the manuscript for publication.
Footnotes
Disclosures: Dr. Brodie reports fees to his university from ALung Technologies, personal fees from Baxter, and anticipated fees from BREETHE and Xenios, as well as an unpaid association with Hemovent outside the submitted work. Dr. Montesi has received research funding through her institution from Merck, United Therapeutics, and Promedior outside the scope of this work, royalties from Wolters Kluwer for contributions to UpToDate, and is supported by grants from the Francis Family Foundation, the Scleroderma Foundation, and the U.S. National Institutes of Health. Dr. Beitler reports speaking fees from Hamilton Medical and consulting fees from Sedana Medical outside the scope of this work and receives research funding from the U.S National Institutes of Health. All other authors report nothing to disclose.
REFERENCES
- 1.Significance of significant. N Engl J Med 1968; 278:1232–1233. [DOI] [PubMed] [Google Scholar]
- 2.Pocock SJ, Stone GW: The primary outcome fails - what next? N Engl J Med 2016; 375:861–870. [DOI] [PubMed] [Google Scholar]
- 3.Greenland S: Nonsignificance plus high power does not imply support for the null over the alternative. Ann Epidemiol 2012; 22:364–368. [DOI] [PubMed] [Google Scholar]
- 4.Calfee CS, Delucchi K, Parsons PE et al. : Subphenotypes in acute respiratory distress syndrome: latent class analysis of data from two randomised controlled trials. The Lancet Respiratory medicine 2014; 2:611–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seymour CW, Kennedy JN, Wang S et al. : Derivation, validation, and potential treatment implications of novel clinical phenotypes for sepsis. JAMA 2019; 321:2003–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adrie C, Adib-Conquy M, Laurent I et al. : Successful cardiopulmonary resuscitation after cardiac arrest as a “sepsis-like” syndrome. Circulation 2002; 106:562–568. [DOI] [PubMed] [Google Scholar]
- 7.Reynolds HR, Hochman JS: Cardiogenic shock: current concepts and improving outcomes. Circulation 2008; 117:686–697. [DOI] [PubMed] [Google Scholar]
- 8.Garland A, Shaman Z, Baron J et al. : Physician-attributable differences in intensive care unit costs: a single-center study. Am J Respir Crit Care Med 2006; 174:1206–1210. [DOI] [PubMed] [Google Scholar]
- 9.Bellani G, Laffey JG, Pham T et al. : Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA 2016; 315:788–800. [DOI] [PubMed] [Google Scholar]
- 10.Stevenson EK, Rubenstein AR, Radin GT et al. : Two decades of mortality trends among patients with severe sepsis: a comparative meta-analysis. Crit Care Med 2014; 42:625–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.du Bois RM, Weycker D, Albera C et al. : Six-minute-walk test in idiopathic pulmonary fibrosis: test validation and minimal clinically important difference. Am J Respir Crit Care Med 2011; 183:1231–1237. [DOI] [PubMed] [Google Scholar]
- 12.Harhay MO, Wagner J, Ratcliffe SJ et al. : Outcomes and statistical power in adult critical care randomized trials. Am J Respir Crit Care Med 2014; 189:1469–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aberegg SK, Richards DR, O’Brien JM: Delta inflation: a bias in the design of randomized controlled trials in critical care medicine. Crit Care 2010; 14:R77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim WB, Worley B, Holmes J et al. : Minimal clinically important differences for measures of treatment efficacy in Stevens-Johnson syndrome and toxic epidermal necrolysis. J Am Acad Dermatol 2018; 79:1150–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Target Investigators, ANZICS Clinical Trials Group, Chapman M et al. : Energy-dense versus routine enteral nutrition in the critically ill. N Engl J Med 2018; 379:1823–1834. [DOI] [PubMed] [Google Scholar]
- 16.TARGET Investigators, Australian and New Zealand Intensive Care Society Clinical Trials Group: Statistical analysis plan for the augmented versus routine approach to giving energy trial (TARGET). Crit Care Resusc 2018; 20:15–21. [PubMed] [Google Scholar]
- 17.Ridgeon EE, Bellomo R, Aberegg SK et al. : Effect sizes in ongoing randomized controlled critical care trials. Crit Care 2017; 21:132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaul S, Diamond GA: Trial and error. How to avoid commonly encountered limitations of published clinical trials. J Am Coll Cardiol 2010; 55:415–427. [DOI] [PubMed] [Google Scholar]
- 19.Brunkhorst FM, Engel C, Bloos F et al. : Intensive insulin therapy and pentastarch resuscitation in severe sepsis. N Engl J Med 2008; 358:125–139. [DOI] [PubMed] [Google Scholar]
- 20.Caironi P, Tognoni G, Masson S et al. : Albumin replacement in patients with severe sepsis or septic shock. N Engl J Med 2014; 370:1412–1421. [DOI] [PubMed] [Google Scholar]
- 21.Writing Group for the Alveolar Recruitment for Acute Respiratory Distress Syndrome Trial Investigators, Cavalcanti AB, Suzumura EA et al. : Effect of lung recruitment and titrated positive end-expiratory pressure (PEEP) vs low PEEP on mortality in patients with acute respiratory distress syndrome: a randomized clinical trial. JAMA 2017; 318:1335–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De Backer D, Biston P, Devriendt J et al. : Comparison of dopamine and norepinephrine in the treatment of shock. N Engl J Med 2010; 362:779–789. [DOI] [PubMed] [Google Scholar]
- 23.Holcomb JB, Tilley BC, Baraniuk S et al. : Transfusion of plasma, platelets, and red blood cells in a 1:1:1 vs a 1:1:2 ratio and mortality in patients with severe trauma: the PROPPR randomized clinical trial. JAMA 2015; 313:471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ranieri VM, Thompson BT, Barie PS et al. : Drotrecogin alfa (activated) in adults with septic shock. N Engl J Med 2012; 366:2055–2064. [DOI] [PubMed] [Google Scholar]
- 25.Guerin C, Reignier J, Richard JC et al. : Prone positioning in severe acute respiratory distress syndrome. N Engl J Med 2013; 368:2159–2168. [DOI] [PubMed] [Google Scholar]
- 26.Combes A, Hajage D, Capellier G et al. : Extracorporeal membrane oxygenation for severe acute respiratory distress syndrome. N Engl J Med 2018; 378:1965–1975. [DOI] [PubMed] [Google Scholar]
- 27.Guntupalli K, Dean N, Morris PE et al. : A phase 2 randomized, double-blind, placebo-controlled study of the safety and efficacy of talactoferrin in patients with severe sepsis. Crit Care Med 2013; 41:706–716. [DOI] [PubMed] [Google Scholar]
- 28.Zhang Q, Li C, Shao F et al. : Efficacy and safety of combination therapy of shenfu injection and postresuscitation bundle in patients with return of spontaneous circulation after in-hospital cardiac arrest: a randomized, assessor-blinded, controlled trial. Crit Care Med 2017; 45:1587–1595. [DOI] [PubMed] [Google Scholar]
- 29.Karnad DR, Bhadade R, Verma PK et al. : Intravenous administration of ulinastatin (human urinary trypsin inhibitor) in severe sepsis: a multicenter randomized controlled study. Intensive Care Med 2014; 40:830–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van den Berghe G, Wouters P, Weekers F et al. : Intensive insulin therapy in critically ill patients. N Engl J Med 2001; 345:1359–1367. [DOI] [PubMed] [Google Scholar]
- 31.NICE SUGAR Study Investigators, Finfer S, Chittock DR et al. : Intensive versus conventional glucose control in critically ill patients. N Engl J Med 2009; 360:1283–1297. [DOI] [PubMed] [Google Scholar]
- 32.Rivers E, Nguyen B, Havstad S et al. : Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med 2001; 345:1368–1377. [DOI] [PubMed] [Google Scholar]
- 33.Investigators ProCESS, Yealy DM Kellum JA et al. : A randomized trial of protocol-based care for early septic shock. N Engl J Med 2014; 370:1683–1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Anthon CT, Granholm A, Perner A et al. : Overall bias and sample sizes were unchanged in ICU trials over time: a meta-epidemiological study. J Clin Epidemiol 2019; 113:189–199. [DOI] [PubMed] [Google Scholar]
- 35.Cochrane Handbook for Systematic Reviews of Interventions, version 5.1.0; http://handbook-5-1.cochrane.org.
- 36.Hochman JS: Cardiogenic shock complicating acute myocardial infarction: expanding the paradigm. Circulation 2003; 107:2998–3002. [DOI] [PubMed] [Google Scholar]
- 37.Grissom CK, Hirshberg EL, Dickerson JB et al. : Fluid management with a simplified conservative protocol for the acute respiratory distress syndrome. Crit Care Med 2015; 43:288–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chalmers I, Matthews R: What are the implications of optimism bias in clinical research? Lancet 2006; 367:449–450. [DOI] [PubMed] [Google Scholar]
- 39.Mann H, Djulbegovic B: Choosing a control intervention for a randomised clinical trial. BMC Med Res Methodol 2003; 3:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chan AW, Hrobjartsson A, Haahr MT et al. : Empirical evidence for selective reporting of outcomes in randomized trials: comparison of protocols to published articles. JAMA 2004; 291:2457–2465. [DOI] [PubMed] [Google Scholar]
- 41.Bassler D, Briel M, Montori VM et al. : Stopping randomized trials early for benefit and estimation of treatment effects: systematic review and meta-regression analysis. JAMA 2010; 303:1180–1187. [DOI] [PubMed] [Google Scholar]
- 42.Ioannidis JP: Contradicted and initially stronger effects in highly cited clinical research. JAMA 2005; 294:218–228. [DOI] [PubMed] [Google Scholar]
- 43.de Grooth HJ, Parienti JJ, Postema J et al. : Positive outcomes, mortality rates, and publication bias in septic shock trials. Intensive Care Med 2018; 44:1584–1585. [DOI] [PubMed] [Google Scholar]
- 44.Spragg RG, Bernard GR, Checkley W et al. : Beyond mortality: future clinical research in acute lung injury. Am J Respir Crit Care Med 2010; 181:1121–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ospina-Tascon GA, Buchele GL, Vincent JL: Multicenter, randomized, controlled trials evaluating mortality in intensive care: doomed to fail? Crit Care Med 2008; 36:1311–1322. [DOI] [PubMed] [Google Scholar]
- 46.Bekaert M, Timsit JF, Vansteelandt S et al. : Attributable mortality of ventilator-associated pneumonia: a reappraisal using causal analysis. Am J Respir Crit Care Med 2011; 184:1133–1139. [DOI] [PubMed] [Google Scholar]
- 47.Shankar-Hari M, Harrison DA, Rowan KM et al. : Estimating attributable fraction of mortality from sepsis to inform clinical trials. J Crit Care 2018; 45:33–39. [DOI] [PubMed] [Google Scholar]
- 48.Girbes ARJ, de Grooth HJ: Time to stop randomized and large pragmatic trials for intensive care medicine syndromes: the case of sepsis and acute respiratory distress syndrome. Journal of thoracic disease 2020; 12:S101–S109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Elsasser A, Regnstrom J, Vetter T et al. : Adaptive clinical trial designs for European marketing authorization: a survey of scientific advice letters from the European Medicines Agency. Trials 2014; 15:383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bhatt DL, Mehta C: Adaptive designs for clinical trials. N Engl J Med 2016; 375:65–74. [DOI] [PubMed] [Google Scholar]
- 51.van der Graaf R, Roes KC, van Delden JJ: Adaptive trials in clinical research: scientific and ethical issues to consider. JAMA 2012; 307:2379–2380. [DOI] [PubMed] [Google Scholar]
- 52.Sterne JA, Davey Smith G: Sifting the evidence-what’s wrong with significance tests? BMJ 2001; 322:226–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gardner MJ, Altman DG: Confidence intervals rather than P values: estimation rather than hypothesis testing. Br Med J (Clin Res Ed) 1986; 292:746–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sackett DL: The principles behond the tactics of performing therapeutic trials In: Clinical Epiidemiology: How to Do Clinical Practice Research. Edited by Haynes RB, Sackett DL, Guyatt GH, Tugwell P. Philadelphia, PA: Lippincott Williams & Wilkins; 2006: 173–243. [Google Scholar]
- 55.Diamond GA, Kaul S: Prior convictions: Bayesian approaches to the analysis and interpretation of clinical megatrials. J Am Coll Cardiol 2004; 43:1929–1939. [DOI] [PubMed] [Google Scholar]
- 56.Lewis RJ, Angus DC: Time for clinicians to embrace their inner Bayesian?: Reanalysis of results of a clinical trial of extracorporeal membrane oxygenation. JAMA 2018; 320:2208–2210. [DOI] [PubMed] [Google Scholar]
- 57.Ellis LM, Bernstein DS, Voest EE et al. : American Society of Clinical Oncology perspective: raising the bar for clinical trials by defining clinically meaningful outcomes. J Clin Oncol 2014; 32:1277–1280. [DOI] [PubMed] [Google Scholar]
- 58.Moher D, Dulberg CS, Wells GA: Statistical power, sample size, and their reporting in randomized controlled trials. JAMA 1994; 272:122–124. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.