

Abstract
Parkinson’s disease (PD) is a progressive neurodegenerative disorder that is a major public health concern due in part to prevalence, debilitating symptoms, and links to environmental exposures. Much research has focused on environmental factors that may lead to dopaminergic neurotoxicity that occurs in PD. In the study of neuronal uptake and neurotoxicity, critical species differences have been observed. For example, neuromelanin is a molecule formed in part by the breakdown products of dopamine metabolism, along with lipid and protein components. Interestingly, human catecholaminergic neurons contain readily detectable amounts of neuromelanin, while rodent models form far lower levels of neuromelanin that is barely detectable. This discrepancy is potentially an important translational weakness. Recently, we showed that neuromelanin formation modulates heterocyclic aromatic amine (HAA)-induced neurotoxicity in cellular models. HAAs are dietary toxins that have primarily been studied as carcinogens, with emergent literature on selective neurotoxicity. The goal of the present study was to identify whether mitochondria in neuromelanin forming cells may be especially sensitive to HAAs. Here, we exposed galactose-supplemented SH-SY5Y cells to HAAs and tested mitochondrial function and mitophagy. The ectopic formation of neuromelanin was found to increase mitochondrial oxidative stress, decrease membrane potential, increase mitochondrial bioenergetic impairments, and impair mitophagy relative to HAA-treated cells that do not form neuromelanin. These results suggest that neuromelanin has a critical role in HAA toxicity and adverse effects on mitochondria. The data also further cement the need to conduct both mechanistic and risk assessment studies on PD-relevant neurotoxicity in models that form neuromelanin.
Keywords: Parkinson’s disease, heterocyclic aromatic amine, neuromelanin, mitophagy
INTRODUCTION
Parkinson’s disease (PD) is a progressive and chronic movement disorder characterized by gradual loss of dopaminergic neurons from the midbrain region, called the substantia nigra pars compacta (SNpc) (Jackson-Lewis et al., 2012; Jenner, 2007; Kowal et al., 2013; Lee and Trojanowski, 2006). In the US, at least 2 out of 100 people over the age of 60 have been diagnosed with PD, making it the second most common neurodegenerative disease (Moore et al., 2005). Currently, there is no therapeutic cure for PD, and patients rely on symptomatic relief-targeted strategies. Although etiopathogenesis of PD remains unclear, it is believed that gene-environment interactions may contribute to the disease etiology (Kanthasamy et al., 2008; Keeney et al., 2009; Lee and Trojanowski, 2006). In this context, environmental factors such as, pesticides, heavy metals, tobacco, coffee, NSAIDs, and alcohol have been shown to modulate disease risk (Campdelacreu, 2014). In addition, dietary habits, including meat consumption, have been associated with PD (Anderson et al., 1999; Johnson et al., 1999; Logroscino et al., 1996). Epidemiological studies were performed to investigate the effect of different diets on PD; however, no conclusive relationship can be established as these studies majorly considered the natural components of food and not processed diet (reviewed in (Ascherio and Schwarzschild, 2016)). Hence, our lab and others investigated the effect of byproducts produced during preparation of food on dopaminergic function and neuropathology. Heterocyclic aromatic amines (HAAs) are produced during high temperature cooking of meat (Skog et al., 1998). HAAs have been widely investigated in the cancer field due to their genotoxic properties (Skog et al., 1998; Turesky, 2007). However, limited evidence is available demonstrating their role in neurodegenerative diseases (Agim and Cannon, 2015). Preliminary studies demonstrated the negative effect of direct HAA infusion on striatal dopamine (DA) levels (Ichinose et al., 1988; Kojima et al., 1990), but there were limited relevant in vivo studies performed until recently by our research group. Our studies have revealed that various HAAs cause dose-dependent cell death specifically to dopaminergic neurons derived from rat embryos (Cruz-Hernandez et al., 2018; Griggs et al., 2014). The study revealed that 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP), 2-amino-3,4-dimethylimidazo[4,5-f]quinoline (MeIQ), 1-methyl-9H-pyrido[3,4-b]indole (harmane) and 2-amino-9H-pyrido[2,3-b]indole (AαC) showed significantly higher toxicity than other HAAs (Cruz-Hernandez et al., 2018). Another study in male Sprague-Dawley rats showed that PhIP causes selective dopaminergic neurotoxicity when administered systemically (Agim and Cannon, 2018).
Importantly, current models used to assess HAA neurotoxicity have significant translational weaknesses (these weaknesses likely apply to the assessment of other dopaminergic neurotoxins/toxicants). In this context, one key weakness in typical cellular and animal models is far lower formation of neuromelanin versus that which occurs in human nigral DA neurons (Aimi and McGeer, 1996; Hasegawa, 2010). Histological evidence reveals that neuromelanin, a polymeric dark pigment of 5,6-dihydroxyindole monomers, is produced mainly in the substantia nigra (SN) and locus coeruleus (LC) regions of the brain (Charkoudian and Franz, 2006). These regions contain high proportions of catecholaminergic neurons which, upon exposure to parkinsonian factors, degenerate, leading to disease pathology (Davie, 2008). In dopaminergic neurons, the excess DA oxidizes to a reactive species called DOPA-o-quinone (Matsunaga et al., 2002; Zucca et al., 2017), which undergoes spontaneous autoxidation steps to generate neuromelanin (Schroeder et al., 2015). Furthermore, tyrosinase, a copper-containing metalloprotein, may oxidize DA to form melanin pigments, probably through DOPA-o-quinone (Miranda et al., 1984). The exact role of neuromelanin remains unclear to date. Initial investigations suggested a neuroprotective role of neuromelanin due to its ability to bind different metals, especially iron, forming stable complexes and blocking neurotoxic effects that could take place through redox reactions (Zecca et al., 1994; Zecca et al., 2001). Nevertheless, increasing evidence indicates that neurons with high neuromelanin content are more susceptible to death (Gibb, 1992; Hasegawa, 2010; Hirsch et al., 1988; Mann and Yates, 1983). Interestingly, postmortem analysis of PD brains reveals loss of neuromelanin concentrations by 60%, suggesting significant loss of neuromelanin-expressing nigral neurons (Zecca et al., 2002). Neuromelanin can promote neurotoxicity by impairing the ubiquitin-proteasomal system (Shamoto-Nagai et al., 2004), causing pro-oxidant effects on mitochondria (Shamoto-Nagai et al., 2006), and by inducing activated microglia-mediated neuroinflammation (Zhang et al., 2011). However, these studies fail to explain the modes of neuromelanin-mediated toxicity that can relate to environmental toxicant-mediated PD pathogenesis.
Emergent data suggest that neuromelanin may be critical in mediating HAA-induced neurotoxicity. We previously showed that immortalized human SH-SY5Y cells, a widely used PD in vitro model, supplemented with galactose and ectopically forming neuromelanin are significantly more sensitive to HAAs compared to wild type cells (Lawana et al., 2020). Similar to other known parkinsonian toxicants (Karlsson et al., 2009; Langston et al., 1999; Lindquist et al., 1988; Ostergren et al., 2007), we demonstrated that HAAs bind with neuromelanin (Lawana et al., 2020). We also showed that neuromelanin formation increases intracellular accumulation of PhIP in neuronal cells compared to cells without neuromelanin. Thus, models that lack neuromelanin likely underestimate brain accumulation and neurotoxicity. While our first report points to the role of neuromelanin in modulating intracellular dose and potency, the underlying mechanisms of HAA-mediated neurotoxicity in neuromelanin forming cells remain unknown. Thus, in this study, we aimed to assess the molecular mechanisms, including mitochondrial and autophagolysosomal system functions, in galactose-supplemented SH-SY5Y cells. In addition to providing a new approach for investigating toxicants in a more translational PD model, the present study also identifies mechanisms of mitophagy that are triggered in cells in response to toxicant exposure, turning on the cytotoxic mechanisms and loss of neurons.
MATERIALS AND METHODS
Cell Culture, Ectopic Neuromelanin Formation, and Treatments:
Immortalized human SH-SY5Y neuroblastoma cells, a commonly used model for toxicant-based PD (Singh et al., 2018), were stably supplemented with 0.18% D-galactose (Teknova, Hollister, CA, CAT# G050) without glucose for higher oxidative phosphorylation dependency to mimic in vivo DA neuronal systems (de Rus Jacquet et al., 2017). The cells were obtained from ATCC and cultured in RPMI-1640 growth media (Sigma-Aldrich, CAT# R1382), containing 15% NuSerum (Corning Inc., Corning, NY, CAT# 355500), 1% Penicillin/Streptomycin (Invitrogen, Carlsbad, CA, CAT# 15140–122), 1% Glutamine (Invitrogen, CAT# 3505–061), 0.01% w/v sodium pyruvate (Sigma-Aldrich, CAT# P5280), 0.37% w/v sodium bicarbonate (Sigma-Aldrich, CAT# S5761) and 1% 1 M HEPES (Corning Inc., CAT#61–034RM). To form neuromelanin in vitro, galactose-supplemented SH-SY5Y cells were transfected with tyrosinase (TYR) constructs as detailed in our recent paper (Lawana et al., 2020). In brief, p3xFLAG-TYR (3 ng/million cells; human tyrosinase, plasmid # 32780, Addgene, Watertown, MA) was transfected into SH-SY5Y cells using the Lipofectamine™ 3000 transfection kit (Invitrogen, CAT# L3000008) following the manufacturer’s instructions. The 1435 pSG5L Flag HA empty vector (3 ng/million cells; EV, plasmid # 10791, Addgene, Watertown, MA) was used as a transfection control. Forty-eight hours after transfection, both EV and TYR cells were exposed to 10 μM PhIP or 10 μM harmane (HAR) for 1 to 48 h, as detailed in each procedure. The doses were chosen based on our previous dose-dependent study (Lawana et al., 2020). Post-treatment cells were analyzed for various biochemical markers as follows.
MitoSOX Assays:
Cells were plated in 96-well plates and transfected with EV or TYR constructs followed by treatment with HAAs (PhIP or HAR) for various time intervals. The generation of mitochondrial superoxide was analyzed using a previously shown technique with MitoSOX (Lawana et al., 2017). Briefly, post-treatment cells were incubated with 5 μM MitoSOX™ Red Mitochondrial Superoxide Indicator dye (Invitrogen, CAT# L3000008) for 20 min, followed by two washes with HBSS buffer per the manufacturer’s protocol. Change in mitochondrial superoxide generation was quantified using a fluorescence microplate reader with excitation and emission wavelengths of 510 nm and 580 nm, respectively. The values were converted to % of EV control group.
Mitochondrial Membrane Potential:
The mitochondrial membrane integrity was measured using a lipophilic JC-1 dye (Invitrogen, CAT# T3168) as described by Ghosh et al. (Ghosh et al., 2010). In brief, post-treatment cells were exposed to JC-1 (2 μg/mL) dye. After a 30 min incubation, cells were washed with PBS and analysis was performed by measuring red fluorescence with excitation at 590 nm over a 600 nm emission and green fluorescence with excitation at 485 nm over a 535 nm emission. JC-1 dye accumulates within healthy mitochondria forming red aggregates; however, the dye remains as a green monomer in unhealthy mitochondria. The mitochondrial membrane potential (MMP) was determined by calculating the ratio of the intensity of red fluorescence (intact MMP) to green fluorescence (impaired mitochondrial membrane).
Measurement of Oxygen Consumption Rate:
Mitochondrial oxidative phosphorylation via oxygen consumption rate (OCR) measurements were performed using the Seahorse XFp Extracellular Flux analyzer (Agilent, Santa Clara, CA), as described previously (Pike Winer and Wu, 2014). Briefly, galactose-supplemented SH-SY5Y cells (10,000/well) were plated in XFp polystyrene cell culture plates (Seahorse Bioscience, North Billerica) and transfected with TYR or EV followed by treatment with 10 μM PhIP or 10 μM HAR for 24 h. Sensor cartridges were hydrated overnight in a non-CO2 incubator. Post-treatment, cells were washed with pre-warmed bicarbonate-free, XF cell assay medium containing 10 mM glucose, 1 mM sodium pyruvate and 2 mM L-glutamine. Cell plates were then incubated at 37 °C in a non-CO2 incubator for 1 h prior to the start of the assay. Meanwhile, the sensor cartridge was loaded with the following treatments (final concentrations indicated): oligomycin (ATP synthase inhibitor, 1 μM), carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP, potent uncoupler of mitochondrial oxidative phosphorylation, 1 μM) and rotenone (Mitochondrial Complex I inhibitor, 0.5 μM) + antimycin A (Mitochondrial Complex III inhibitor, 0.5 μM) in the respective ports. The instrument took 3 baseline measurements, referred to as baseline OCR, followed by sequential addition of mito-stressors as indicated above. OCR was expressed as pmoles/min and normalized to mg protein. Basal respiration, proton leak, maximal respiration, spare respiratory capacity, non-mitochondrial respiration, and ATP production were calculated using the Seahorse mito-stress test. The data were collected and plugged in the Microsoft Excel Template provided by the instrument manufactures; the produced values were then plotted on GraphPad Prism to generate data shown here.
Western Blotting:
Whole cell lysates were prepared using modified RIPA buffer as described previously (Panicker et al., 2015). To analyze the intracellular translocation, mitochondrial fractions were extracted using an extraction technique described by Dennis and Bennett (2003). In brief, cells were washed with PBS, centrifuged and stored at < −80 °C. The cell pellets were homogenized on ice in buffer (10 mM Tris-HCl and 0.3 M sucrose, with 1X protease-phosphatase inhibitor cocktail, pH 7.0), followed by centrifugation for 1 h at 10,000 × g at 4 °C. The supernatant (cytoplasmic fraction) was discarded and the pellet was suspended in ice-cold buffer (10 mM Tris-HCl, 1% Triton X-100, 150 mM NaCl, with 1X protease-phosphatase inhibitor cocktail, pH 7.4), sonicated on ice and centrifuged for 30 min at 10,000 × g at 4°C. The supernatant was collected and stored at < −80 °C (mitochondrial fraction). Protein amount was estimated using Bradford reagent and equal amounts of protein were resolved on a 7.5–15% gradient SDS-polyacrylamide gel. Resolved proteins were transferred to nitrocellulose membrane and membranes were blocked for 1 h by incubating with 2% Bovine Serum Albumin (BSA) blocking buffer, followed by incubation with specific primary antibodies overnight at 4 °C. Anti-LAMP1 rabbit mAb (1:1000, CAT# 9091), anti-SQSTM1/p62 (D5L7G) mouse mAb (1:2000, CAT# 88588), anti-LC3B rabbit mAb (1:800, CAT# 3868), anti-Parkin mouse mAb (1:1000, CAT# 4211) and anti-optineurin (1:1000, CAT# 70928) were purchased from Cell Signaling Technology, Danvers, MA. Anti-actin antibodies were purchased from MilliporeSigma, Burlington, MA (1:3000, CAT# MAB1501). Membranes were washed and incubated with specific fluorescent-labelled secondary antibodies from LI-COR Biosciences, Lincoln, NE [IRD Dye 680CW anti-mouse (1:10,000; CAT# 926–68072), donkey IRD Dye 680CW anti-chicken (1:10,000; CAT# 925–68075), donkey IRD Dye 800CW anti-rabbit (1:10,000; CAT# 926–32213)]. The membrane containing antibody-bound protein was scanned using an Odyssey IR Imaging System (LI-COR Biosciences, Lincoln, NE) and data were analyzed using Odyssey 2.0 software.
Immunocytochemistry and Confocal Imaging:
Immunostaining was done with galactose-supplemented SH-SY5Y cells (Panicker et al., 2015). In brief, cells were plated on glass coverslips, transfected with TYR or EV and treated with 10 μM HAR for 24 h. Post-treatment, cells were fixed with 4% paraformaldehyde for 30 minutes followed by 3 washes with PBS and blocked with 2% BSA solution containing 0.5% Triton-X-100 and 0.05% Tween 20. After blocking, cells were incubated with MitoTracker™ Red (Invitrogen, CAT# M22425) for 20 min. The cells were then incubated with anti-LC3B primary antibody (1:300; Cell Signaling Technology, Danvers, MA; CAT# 3868) at 4 °C overnight and then had a 90 min incubation with secondary antibodies (Donkey anti-rabbit Alexa Fluor® 488). Lastly, cells were exposed to Hoechst 33342 (10 μg/mL) for 10 min to stain nuclei. After each step, cells were washed with 1X PBS thrice. Coverslips were mounted on glass slides using Fluoroshield™ mounting medium (Sigma-Aldrich, CAT# F6182) and were allowed to dry overnight in the dark. Confocal imaging was performed to visualize mitochondria and LC3B-tagged puncta colocalization. High resolution images were obtained using a Nikon A1R inverse scanning confocal microscope (Nikon Instruments, USA) and images were acquired with a 60X oil-immersion objective. Prior to acquisition, microscope and detector settings were optimized to avoid saturation (Syeda et al, 2020). Mitochondrial morphological assessments were done using ImageJ software (NIH open access) with the ‘Mitophagy’ plug-in (Dagda et al., 2008) In brief, the mitophagy plug-in was opened along with ImageJ and the desired image was uploaded to ImageJ. The ‘polygon selection tool’ from the macro region of interest (i.e. individual cell) was selected. Then, using a macro, the RGB image was then split into individual channels (blue, green, and red), followed by closing the blue and green channels, extracting the red channel to grayscale and photographic inversion of the pixels. The macro then measured mitochondrial morphology (circularity and area/perimeter), mitochondrial content (percentage of cytosolic area occupied by mitochondria) and cellular area and perimeter. The macro also provided output of the total mitochondrial particles (red channel). Here, cells were individually observed for (i) total mitochondrial particles (mitochondrial content) and (ii) circularity. The data obtained were plotted in GraphPad Prism to a obtain histograph.
Acridine Orange Assay:
Activity of intracellular acidic sacs can be viewed by acridine orange (AO) dye, which tends to accumulate in acidic vacuoles and produce red fluorescence. EV and TYR transfected SH-SY5Y cells in 96-well plates were treated with HAR. Post treatment, the cells were incubated with 5 μg/mL AO solution for 20 min at 37 °C, followed by washing with PBS (Moriyama et al., 1982). Using a SpectroMax™ plate reader, we estimated red fluorescence at excitation/emission of 584/612 nm and green fluorescence at excitation/emission of 485/520 nm, respectively. The fluorescence reading ratio of green:red was calculated and plotted. An increase in the ratio may indicate elevated lysosomal permeability and/or decreased number of intact acidic vacuoles including lysosomes (Singh et al., 2018).
Electron Microscopy:
Electron microscopic images were obtained at the Life Science Microscopy Facility (LSMF), Purdue University. For Transmission Electron Microscopy (TEM) images of SH-SY5Y cells, the LSMF standard protocol was followed. In brief, SH-SY5Y neuroblastoma cells were fixed with 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer 48 h after transfection with EV or TYR constructs and treatment with HAR as above. Cells were post-fixed in 1% osmium tetroxide buffer containing 0.8% potassium ferricyanide, and stained in 1% aqueous uranyl acetate. Cells were then dehydrated with a graded series of acetonitrile and embedded in EMbed-812 resin. Thin sections (80 nm) were cut on a Leica EM UC6 ultramicrotome (Leica Camera, Wetzlar, Germany) and stained with 2% uranyl acetate and lead citrate. Cell sections were mounted on TEM grids and imaged using a Gatan Orius side mount CCD camera on an FEI/Philips CM-100 transmission electron microscope operating at 40 kV.
LysoTracker™ Assay:
The analysis of lysosomal membrane permeability was performed using LysoTracker™ red dye purchased from Invitrogen (CAT# L7528) following the manufacturer’s protocol. In brief, SH-SY5Y cells were grown in a 96-well plate and were transfected and treated as above. Following treatment, cells were incubated with 100 nM LysoTracker™ dye for 30 min. Post-incubation, cells were washed with PBS multiple times and were analyzed using a SpectraMax fluorescence plate reader at excitation/emission of 445 nm/505 nm. A decrease in fluorescence correlates with reduced number of lysosomes.
Statistical Analyses:
Results are expressed as mean ± standard error of the mean (SEM). All data were analyzed with Prism 8.0 software (GraphPad, La Jolla, CA). Most data contained multiple variables and were analyzed using two-way ANOVA, followed by post-hoc Sidak’s test to compare means of different treatment groups. Students’ t-tests were used for comparing two groups. For all analyses, differences with p < 0.05 were considered statistically significant. GraphPad Prism 8 was also utilized to generate all curves and graphs.
RESULTS
Intracellular neuromelanin increases HAA-induced mitochondrial oxidative stress and decreases mitochondrial membrane potential
Galactose-supplemented human SH-SY5Y neuroblastoma cells were transfected with TYR constructs to ectopically form neuromelanin in vitro, as described previously (Lawana et al., 2020). In a recent study, we showed that 10 μM harmane or PhIP can induce cell death and promote aberrant reactive oxygen species (ROS) generation in SH-SY5Y cells, which is more severe in the presence of intracellular neuromelanin (Lawana et al., 2020). 48 h after TYR or empty vector (EV) transfection, cells were treated with 10 μM PhIP or harmane for 1, 6, 9, 12, 18, 24, 36, or 48 h. Post-treatment, cells were analyzed for mitochondrial superoxide levels using MitoSOX™ Red dye and mitochondrial membrane potential (MMP) using cationic JC-1 dye. EV SH-SY5Y cells showed significant increases in mitochondrial superoxide generation by 38% at 24 h (p<0.05) leading up to 82% at 48 h (p<0.0001) after 10 μM PhIP exposure (Figure 1A). Further, TYR SH-SY5Y cells showed a significant increase as early as 18 h (53% higher than control, p<0.05) with a subsequent increase up to 160% greater than the respective EV control at 48 h (p<0.0001) post treatment with 10 μM PhIP. Interestingly, HAA treated TYR-mediated neuromelanin forming SH-SY5Y cells showed significantly higher (p<0.0001) mitochondrial superoxide levels compared to cells without neuromelanin (EV-transfected) after PhIP exposure. In a similar way, EV SH-SY5Y cells showed a 39%, 61%, 63% and 140% increase, and TYR SH-SY5Y cells showed a 75%, 119%, 166% and 223% increase in MitoSOX levels after 10 μM harmane exposure for 18, 24, 36 and 48 h, respectively, compare to baseline control (Figure 1B). Notably, neuromelanin forming cells showed higher toxicity compared to cells without neuromelanin in response to harmane treatment.
Figure 1: HAAs increase mitochondrial superoxide and reduce mitochondrial membrane potential production in a time-dependent manner in neuromelanin forming SH-SY5Y cells.
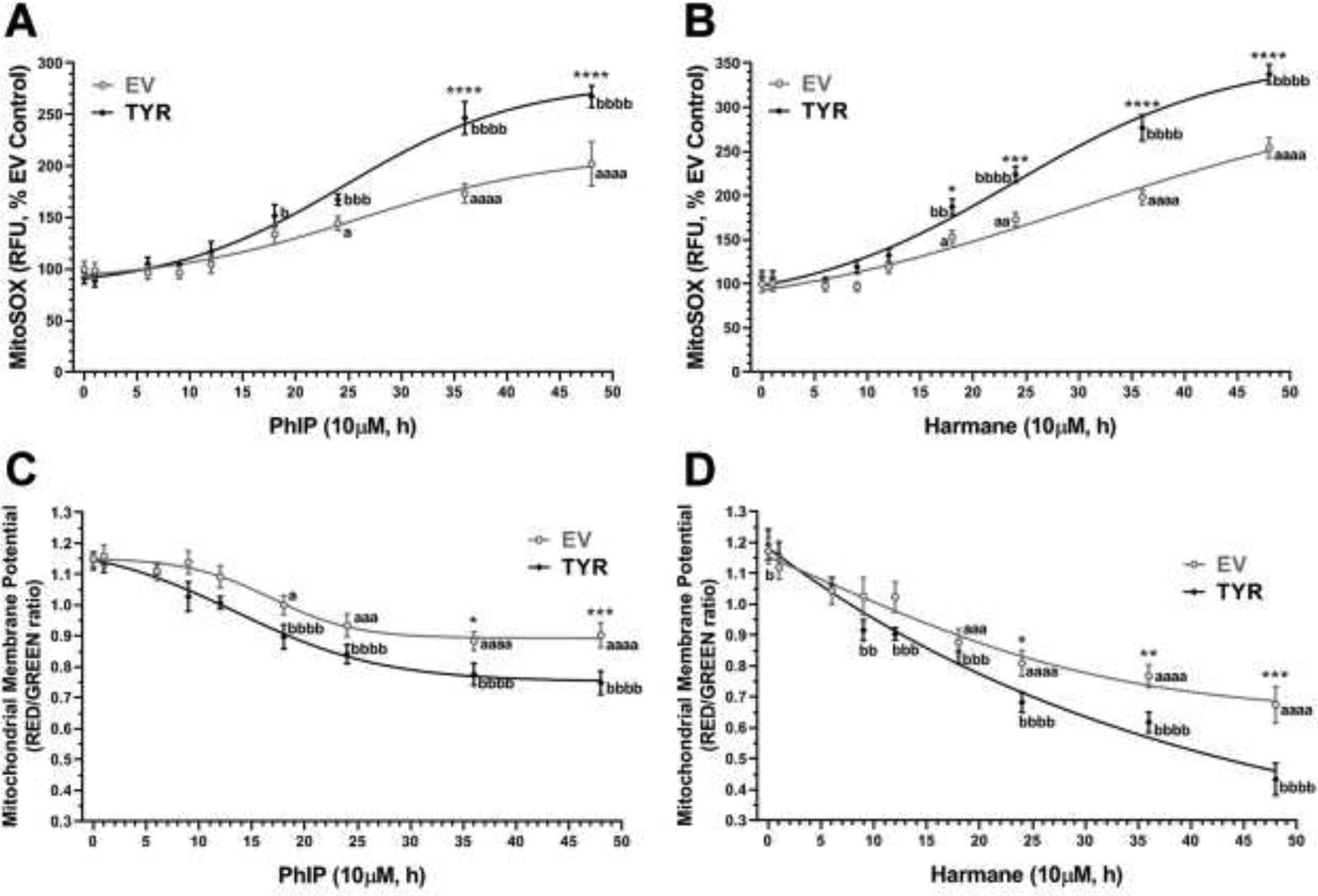
Galactose-supplemented SH-SY5Y neuroblastoma cells were transfected with EV or TYR. After 48h of transfection, cells were treated with either 10 μM HAR or 10 μM PhIP in a time-dependent manner from 1 h to 48 h. A, B. Mitochondrial superoxide (MitoSOX) levels analyzed for each time-point post-treatment with PhIP or HAR, respectively. C, D. Time-dependent changes in mitochondrial membrane potential (Δψm) post-treatment with PhIP or HAR, respectively. The data are represented as mean ± SEM for n = 6, analyzed by two-way ANOVA followed by Sidak’s post-hoc test. ap<0.05, aaap<0.001 and aaaap<0.0001 compared EV non-treated control group; bp<0.05, bbp<0.01, bbbp<0.001 and bbbbp<0.0001 compared TYR non-treated control group; while, *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001 demonstrate significant difference between EV versus TYR-transfected SH-SY5Y cells at given concentration.
Next, we analyzed MMP in EV and TYR SH-SY5Y cells exposed to PhIP or harmane. MMP was found to be depressed by HAA treatment. PhIP (10 μM) exposure decreased MMP by 18%, 22%, 26% and 24%, in EV SH-SY5Y neuroblastoma cells; whereas, decreases up to 28%, 30%, 33% and 36% were detected in TYR SH-SY5Y cells, at 18, 24, 36 and 48 h (Figure 1C). Likewise, 10 μM harmane treatment depleted MMP by 30–61% from 18 to 48 h in EV SH-SY5Y cells. While in TYR SH-SY5Y cells MMP reduced by 28% as early as 9 h after harmane treatment leading up to 90% at 48 h post harmane exposure (Figure 1D). Interestingly, our data demonstrated that neuromelanin-forming SH-SY5Y cells were more susceptible to HAA-induced damage to mitochondrial membrane integrity and caused 18–31% greater reduction (p<0.05) in MMP compared to cells without neuromelanin.
Neuromelanin exacerbates HAA-mediated mitochondrial bioenergetic deficits
To further assess the role of TYR-mediated neuromelanin formation on mitochondrial health in the presence of neurotoxic HAAs, we used a Seahorse XFp Analyzer. Using the Seahorse XFp Cell MitoStress test reagents as described in the Methods section, we determined the various aspects of mitochondrial respiration: the electron transport chain and proton translocation. Here, we treated SH-SY5Y cells (with or without neuromelanin) with 10 μM harmane (Figure 2) or 10 μM PhIP (Figure 3) for 24 h and subjected these cells to the MitoStress assay. Figure 2A represents the time versus OCR plot showing real-time changes in OCR levels in EV and TYR transfected SH-SY5Y cells treated with harmane with sequential additions of mitochondrial stressors (noted with doted lines). The bar graphs in Figure 2B–I show the different parameters of mitochondrial bioenergetics. As shown in Figure 2B, we noted a variation in baseline OCR levels among the four groups, EV-Control, EV-HAR, TYR-Control, and TYR-HAR. These data demonstrate that 10 μM harmane can reduce baseline mitochondrial respiration by 24–31% in cells without neuromelanin (p<0.001) and 66–78% in cells with neuromelanin formation (p<0.0001). Interestingly, the neuromelanin forming cells also showed a slight, insignificant decrease in baseline OCR. The baseline data, an indirect parameter implying decreased oxidative phosphorylation (OXPHOS) (Singh et al., 2018) reiterates that harmane may cause substantive mitochondrial damage after 24 h of treatment which is exaggerated by neuromelanin, suggesting that harmane may reduce OXPHOS. Notably, neuromelanin alone did not reduce basal respiration but HAR treatment to neuromelanin forming cells showed dramatic reduction by up to 72% compare to neuromelanin control cells (p<0.0001).
Figure 2. Neuromelanin forming SH-SY5Y cells are more susceptible to harmane-mediated mitochondrial stress compare to cells without neuromelanin.
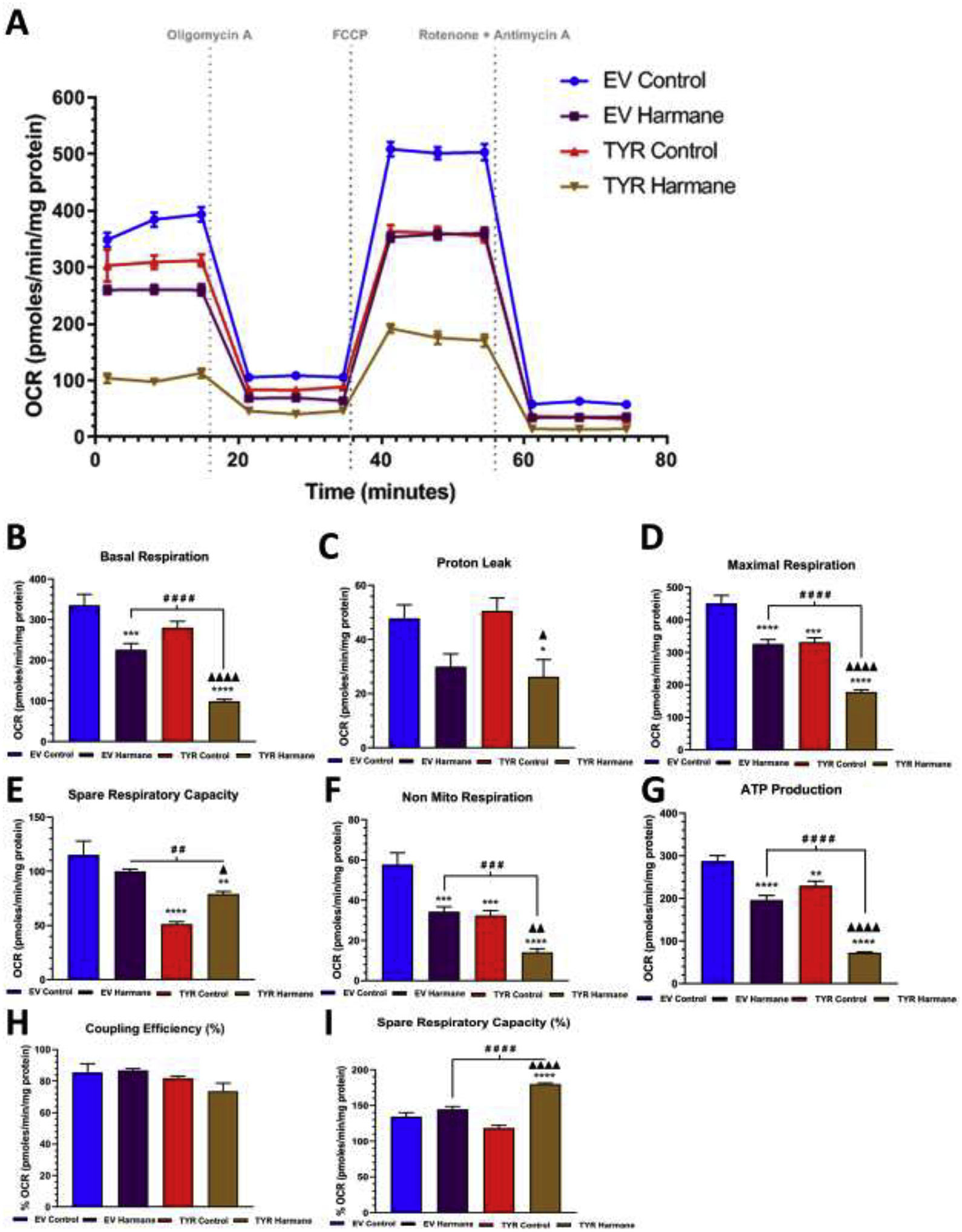
A. A schematic representation of the mitochondrial bioenergetics profile in relation to oxygen consumption rate levels, in both EV and TYR SH-SY5Y neuroblastoma cells treated with or without HAR (10 μM) for 24 h. Post treatment, the change in the Oxygen consumption rate (OCR) levels were determined using a Seahorse XFp analyzer. B Basal respiration, C proton leak, D maximum respiration, E spare respiratory capacity, F non-mitochondrial respiration, G ATP production, H coupling capacity (%) and I spare respiratory capacity (%) were calculated by measuring change in OCR levels post sequential addition of oligomycin (1 μM), FCCP (1 μM), and Rotenone (0.5 μM) +Antimycin A (0.5 μM), respectively, as indicated by arrows in Figure 2A. Results are representative of at least 4 independent experiments performed in replicates of four. Data are expressed as mean ± S.E.M. *p<0.05, *p<0.01, ***p<0.001 and ****p<0.0001 vs EV Control group; # #p<0.01, # # #p<0.001 and # # # #p<0.0001 vs EV HAR-treated cells; Δp<0.05, Δ Δ Δp<0.001 and Δ Δ Δ Δp<0.0001 vs TYR control cells.
Figure 3. Neuromelanin forming SH-SY5Y cells are more susceptible to PhIP-mediated mitochondrial stress compare to cells without neuromelanin.
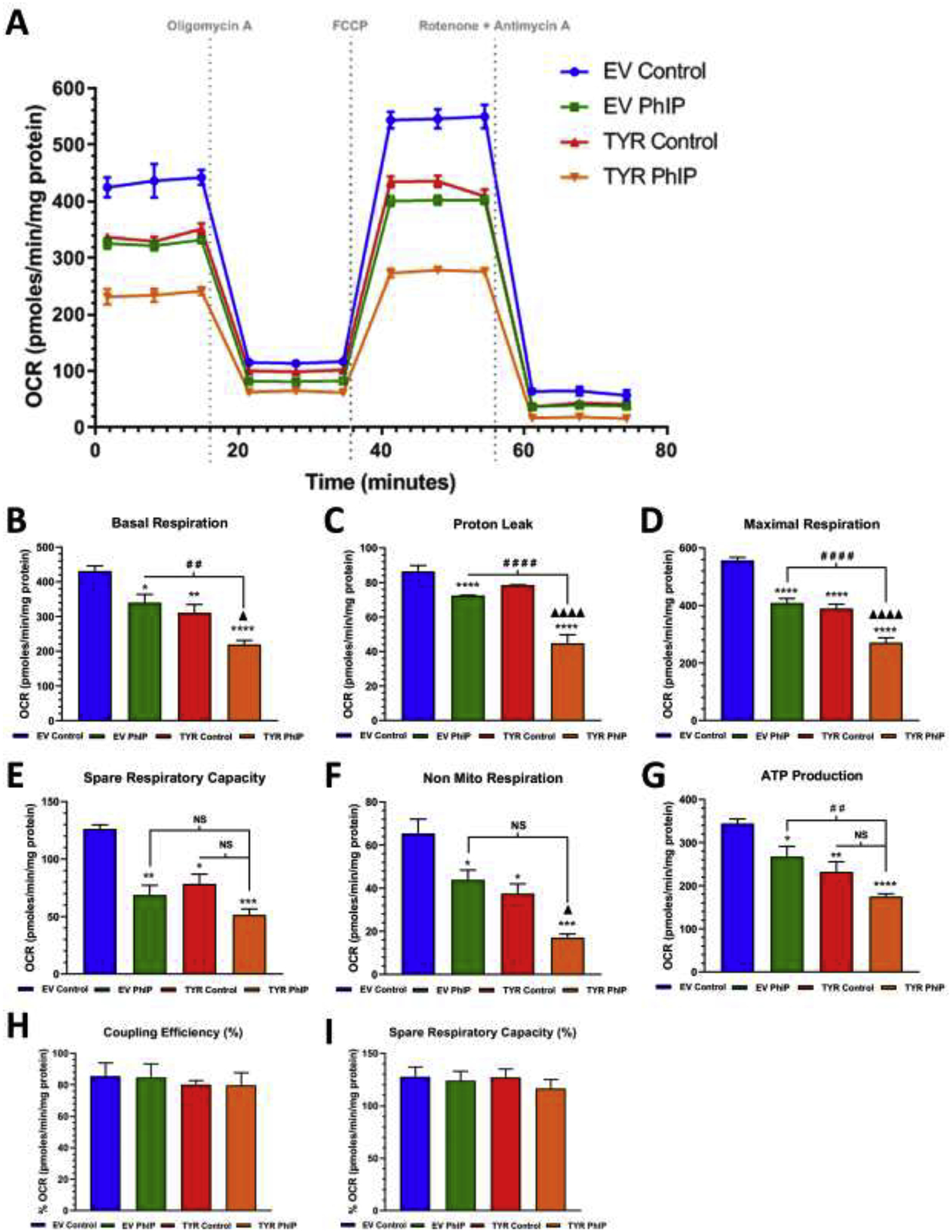
A. A schematic representation of the mitochondrial bioenergetics profile in relation to oxygen consumption rate levels, in both EV and TYR SH-SY5Y neuroblastoma cells treated with or without PhIP (10 μM) for 24 h. Post treatment, the change in the oxygen consumption rate (OCR) levels were determined using a Seahorse XFp analyzer. B Basal respiration, C proton leak, D maximum respiration, E spare respiratory capacity, F non-mitochondrial respiration, G ATP production, H coupling capacity (%) and I spare respiratory capacity (%) were calculated by measuring change in OCR levels post sequential addition of oligomycin (1 μM), FCCP (1 μM), and Rotenone (0.5 μM) +Antimycin A (0.5 μM), respectively, as indicated by arrows in the Figure 3A. Results are representative of at least 4 independent experiments performed in replicates of four. Data are expressed as mean ± S.E.M. *p<0.05, *p<0.01, ***p<0.001 and ****p<0.0001 vs EV Control group; # #p<0.01, # # #p<0.001 and # # # #p<0.0001 vs EV PhIP-treated cells; Δp<0.05 and Δ Δ Δ Δp<0.0001 vs TYR control cells.
With reduced OXPHOS, harmane also lowered ATP production by 20–27% in cells without neuromelanin and 75–82% in cells with neuromelanin compare to EV control group (p<0.0001), respectively (Figure 2G). While cells producing neuromelanin without treatment (TYR Control) showed 18–23% reduction in ATP production compare to EV control (p<0.01), the HAR treatment to neuromelanin forming cells showed 62–66% reduction in ATP production compared to neuromelanin control cells (p<0.0001). Further, direct comparison between harmane groups show that neuromelanin formation results in a more severe impairment of ATP production (p<0.0001, Figure 2G). The cellular maximal respiration capacity was determined by treating with FCCP, an uncoupler of the electron transport chain (ETC), causing an ATP synthase-independent proton gradient increase across the mitochondrial inner membrane to the matrix and elevates oxygen consumption (Schneider et al, 2011). As shown in Figure 2D, maximal respiration was suppressed by neuromelanin formation by 22% (p<0.0001) and harmane-treated EV SH-SY5Y cells showed up to a 25% drop (p<0.001) compared to the EV SH-SY5Y control cells. Interestingly, harmane treatment to TYR SH-SY5Y showed as much as a 56%, 33% and 32% reduction in maximal respiration compared to EV control (p<0.0001), TYR control (p<0.0001) and EV HAR (p<0.0001) groups, respectively.
Other parameters of mitochondrial respiration including proton leak (Figure 2C), non-mitochondrial respiration (Figure 2F) and coupling efficacy (Figure 2H) were also assessed. Specifically, we noted 52% and 57% reduction in proton leak in HAR-treated neuromelanin-forming SH-SY5Y cells compared to EV control (p<0.05) and TYR control groups (p<0.05), respectively. Notably, no difference was noted in proton leak values in HAR-treated cells with or without neuromelanin (Figure 2C). Lastly, we noted 44% loss in non-mitochondrial respiration in neuromelanin control cells compare to EV control cells (p<0.001). Intriguingly, we noted 74%, 55% and 52% decrease in non-mitochondrial respiration in TYR cells treated with HAR compared to EV control (p<0.0001), EV HAR (p<0.001) and TYR control groups (p<0.01). Together, the data from the MitoStress assay further illustrates the adverse effects of HAR on mitochondrial integrity, which was significantly exacerbated in the presence of neuromelanin.
Figure 3 demonstrated that PhIP exposure also produced bioenergetic deficits. Here, PhIP exposure in EV SH-SY5Y cells depleted baseline respiration rate by 15% (p<0.05), which was further reduced by 44% in TYR SH-SY5Y compare to EV control cells (p<0.0001) (Figure 3A). Further, similar to HAR, PhIP exposure also reduced mitochondrial, maximal respiration (Figure 3D), spare respiratory capacity (Figure 3E), non-mitochondrial respiration (Figure 3F), and ATP production (Figure 3G) in cells without neuromelanin, which was significantly reduced in cells forming neuromelanin. No notable change was reported in percentage of coupling efficacy (Figure 3H).
Based on results from Figures 1–3, harmane exposure produced greater mitochondrial insults in comparison to PhIP (± neuromelanin). Hence, we tested for additional markers of HAA-induced mitochondrial structural damage in neuromelanin-forming SH-SY5Y cells treated with 10 μM HAR (24 h) and stained with MitoTracker Red (Figure 4). Qualitative analyses of immunocytochemical images showed changes in mitochondrial morphology in EV HAR cells (Figure 4C) compared to EV control cells (Figure 4A). We also noted impaired mitochondrial morphology in TYR control cells (Figure 4B), which appeared to deteriorate even further in TYR HAR cells (Figure 4D). ImageJ-based quantification of mitochondrial morphology revealed no change in overall mitochondrial content per cell (Figure 4E). However, our quantitative assessment demonstrated that mitochondria in harmane-treated cells exhibited greater (~2.2-fold) fission-related circularity (p<0.05) which is further increased to 4.1-fold in the presence of neuromelanin (p<0.0001) compared to EV control cells (Figure 4F).
Figure 4. Harmane modulates mitochondrial morphology in neuromelanin forming SH-SY5Y cells.
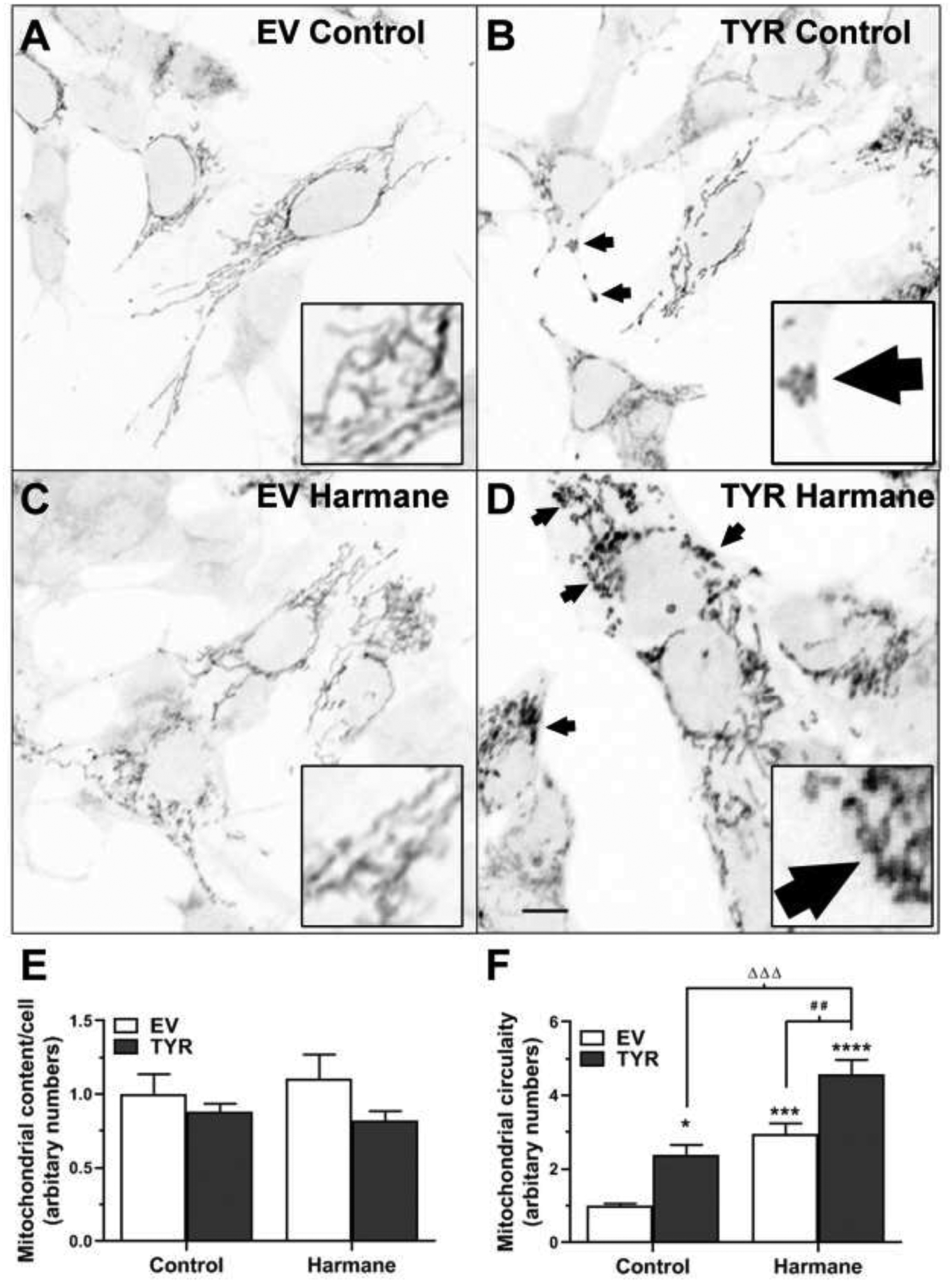
A-D. MitoTracker staining shows mitochondrial morphology in EV-transfected control (A), EV-transfected HAR treated (C), TYR-transfected control (B) and TYR-transfected HAR-treated (D) SH-SY5Y cells. Scale bar, 20 μm. E. Total mitochondrial content and F. mitochondrial circularity (indicative of mitochondrial fragmentation) was determined using imageJ and values were converted to arbitrary numbers (representative examples shown in with arrows and 3x insets). Data expressed as mean ± S.E.M. *p<0.05, ***p<0.001 and ****p<0.0001 vs EV control group; # #p<0.01 vs EV HAR-treated cells; and Δ Δ Δp<0.001 vs EV HAR-treated cells.
Harmane-mediated autophagolysosomal system dysfunction in cells is exacerbated by neuromelanin formation
Lysosomal destabilization has been shown to contribute to neurodegeneration (Ghavami et al., 2014; Micsenyi et al., 2013; Wu et al., 2015a). Therefore, we assessed common protein markers of the autophagolysosomal system, such as Lysosomal-associated membrane protein 1 (LAMP1), p62, and LC3B (Figure 5A). Our immunoblotting assessment revealed that SH-SY5Y cells without neuromelanin formation exposed to harmane had no significant change in LAMP1, a lysosomal marker, expression. However, SH-SY5Y cells forming neuromelanin showed a 71–78% decrease in LAMP1 protein post 24 h exposure to 10 μM HAR (Figure 5A). Alternately, we noticed that harmane treatment increased the protein expression of p62 and LC3B II, markers for autophagy, in SH-SY5Y cells with or without neuromelanin formation, however, the increase in neuromelanin forming SH-SY5Y cells was much greater (p<0.001 compare to EV SH-SY5Y treated with HAR).
Figure 5. Harmane induces autophagy in neuromelanin forming SH-SY5Y cells.
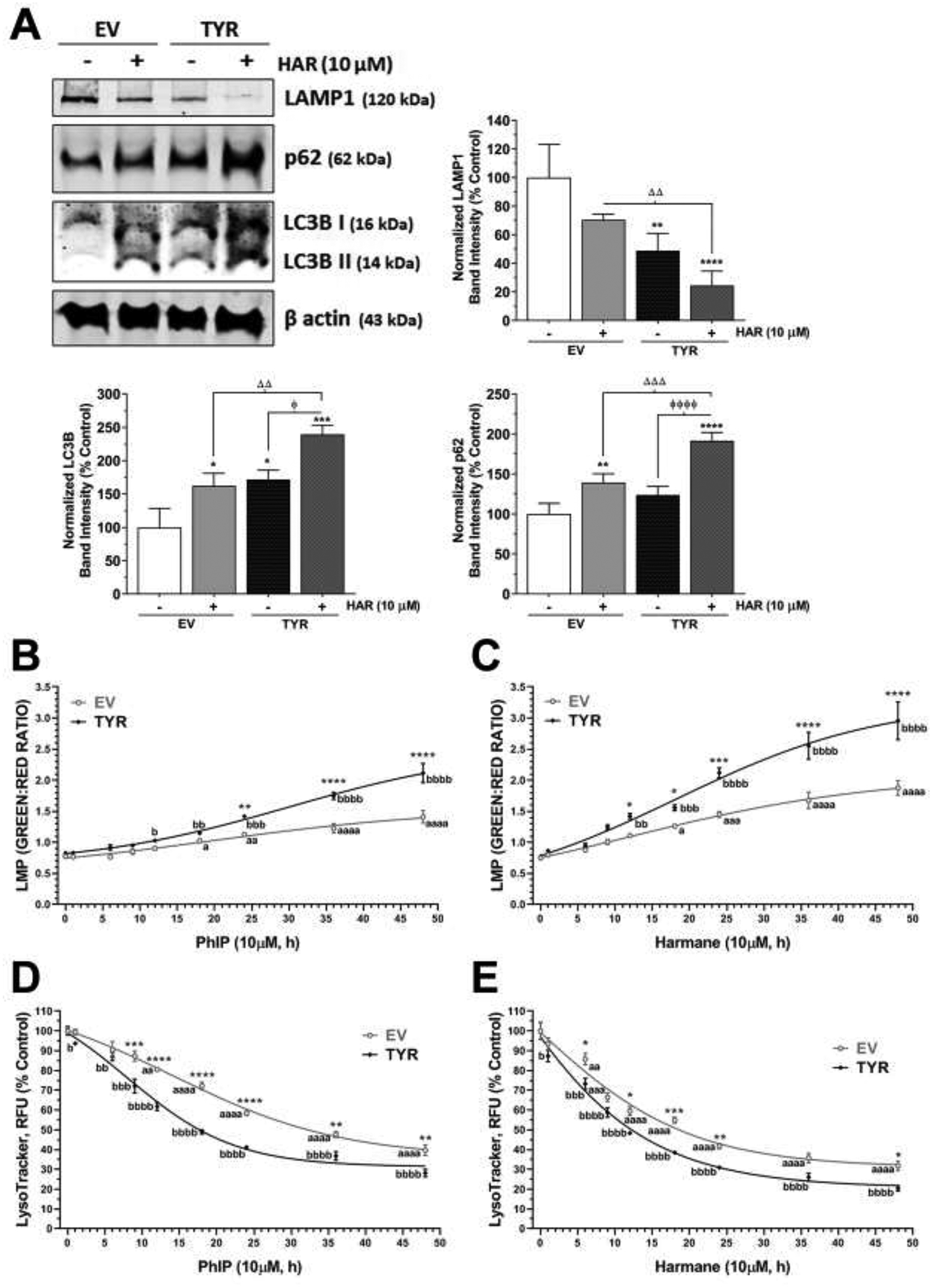
The galactose supplemented SH-SY5Y neuroblastoma cells were transfected with EV or TYR. After 48 h of transfection, cells were treated with 10 μM HAR or 10 μM PhIP for various time-points. A, B. The acridine orange assay was performed to assess lysosomal membrane permeability at various time-points from 1 h and 48 h post HAR and PhIP exposure, respectively. C, D. The LysoTracker™ green assay was performed to assess cellular lysosomal content at various time-points from 1 h and 48 h post HAR and PhIP exposure, respectively. ap<0.05, aaap<0.001 and aaaap<0.0001 compared EV non-treated control group; bp<0.05, bbp<0.01, bbbp<0.001 and bbbbp<0.0001 compared TYR non-treated control group; while, *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001 demonstrate significant difference between EV versus TYR-transfected SH-SY5Y cells at given concentration. E. Representative immunoblot showing expression of LAMP1, p62 and LC3B in SH-SY5Y cells transfected with EV or TYR constructs for 48 h followed by HAR (10 μM, 24 h) treatment. β actin was used as a loading control. F. The bar histogram represents the normalized densitometry of respective proteins. The data are represented as mean ± SEM, n = 3–4. *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001 vs EV control group; ɸp<0.05 and ɸ ɸ ɸp<0.05 vs TYR control cells; and Δ Δp<0.01 and Δ Δ Δp<0.001 vs EV HAR-treated cells.
Because of these changes in autophagolysosomal proteins, we utilized the acridine orange (AO) method, where the dye acridine fluoresces red in healthy lysosomes and switches to green in cells with perturbed lysosomal function. Hence, increases in green:red ratio may indicate a reduction in lysosomal acidity due to damaged lysosomal membrane and increased lysosomal membrane permeabilization. A marked increase in green fluorescence was evident with HAA exposure and this effect was augmented in cells forming neuromelanin, indicative of lysosomal destabilization (Figure 5B,C). In particular, we noted that EV SH-SY5Y cells treated with 10 μM HAR showed about 1.8-fold increase in green:red AO ratio, while HAR exposure to the TYR SH-SY5Y cells showed up to 4.2-fold increase in the ratio at 48 h time-point compare to control (Figure 5C). Likewise, PhIP (10 μM, 48 h) treatment showed 1.3-fold and 2.6-fold increases in green:red AO ratio in EV and TYR SH-SY5Y cells, respectively (Figure 5B).
We also used the fluorescent dye LysoTracker® Red, which exhibits a pH-dependent increase in fluorescence intensity upon acidification as compared with LysoSensor probes, to determine lysosome function. As shown in Figure 5D,E we observed that both harmane and PhIP induced loss of fluorescence intensity starting at 6–12h (p<0.05) and a persistent reduction was observed during the remaining treatment period up to 48h (p<0.0001) in EV SH-SY5Y cells. Moreover, cells forming neuromelanin and treated with HAAs demonstrated a more significant reduction in fluorescence intensity. This data, along with the acridine orange data indicates dysfunctional lysosomal function.
Mitophagic impairments induced by harmane are exacerbated by neuromelanin
Mitophagy is the selective clearance of damaged mitochondria through autophagy. Typically, damaged mitochondria undergo PINK1-parkin interaction to initiate the mitophagy cascade by polyubiquitination. Then, optineurin (OPTN) is recruited by this multi-protein complex on the mitochondrial outer membrane, which, along with p62, helps in anchoring the PINK1-parkin-ubiquitin labelled damaged mitochondria to the LC3B-asssociated autophagic membrane, ultimately leading to formation of mitophagic vacuoles (Weil et al, 2018).
After demonstrating alterations in autophagic markers from HAR treatment, we performed western blotting on mitophagy markers using whole cell lysates or mitochondrial fractions. First, our whole cell immunoblots showed that harmane increased OPTN protein levels by 20–29% (p<0.05) in EV SH-SY5Y cells. Furthermore, harmane exposure increased OPTN levels by 62–72% (p<0.0001) in TYR SH-SY5Y cells compare to EV control cells (Figure 6A). Next, our immunoblotting showed the opposite trend for parkin expression in whole cell lysates. As shown in Figure 6A, HAR treatment (10 μM, 24 h) caused a 24–30% (p<0.01) decrease and a 48–57% (p<0.0001) decrease in parkin expression in EV and TYR SH-SY5Y cells, respectively, compared to EV SH-SY5Y cells. Our immunoblot demonstrated no significant change in OPTN and parkin protein expressions between EV and TYR non-treated SH-SY5Y cells, demonstrating no effect of neuromelanin alone on expression of these mitophagic markers.
Figure 6. Neuromelanin modulates mitophagy in harmane-exposed SH-SY5Y cells.
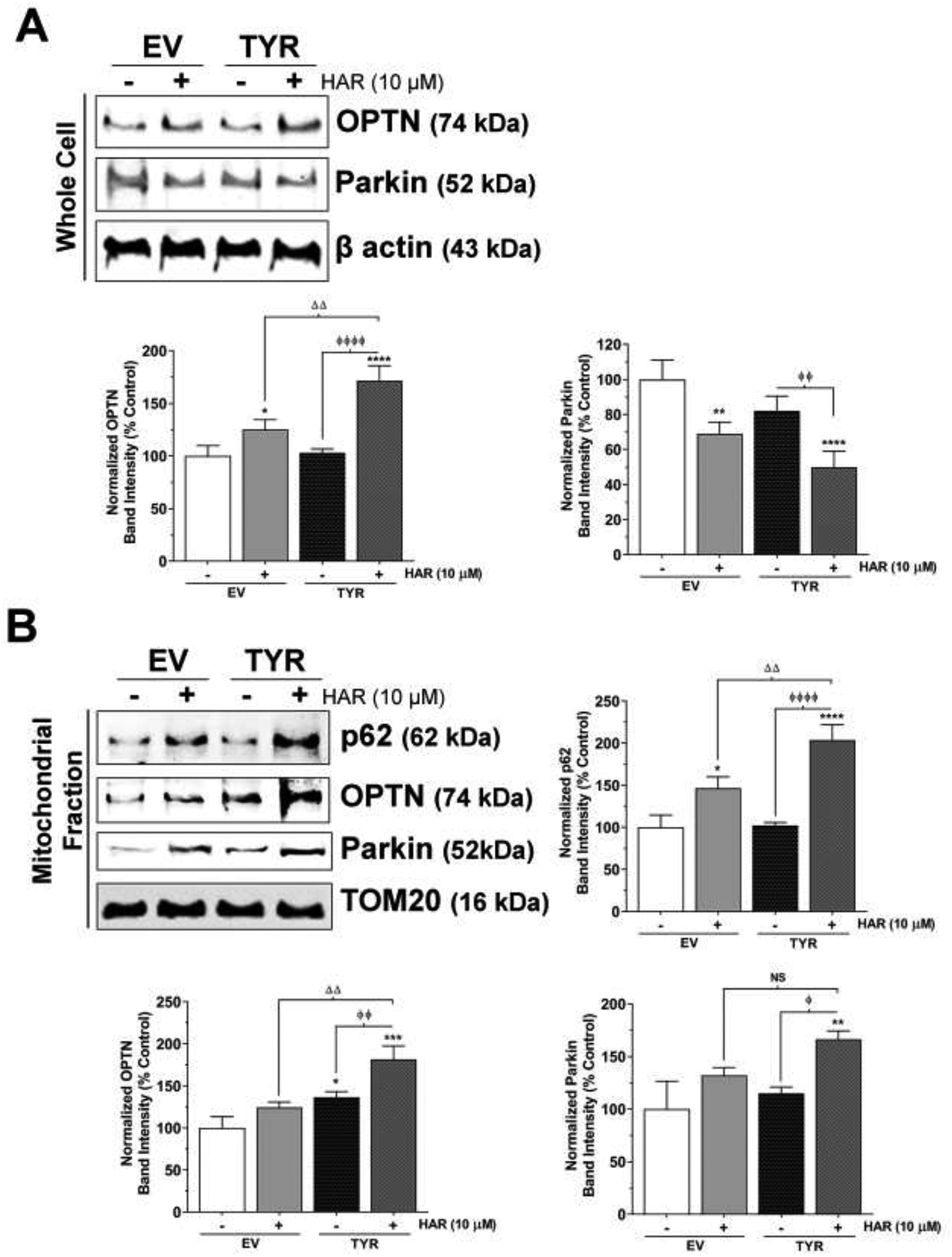
SH-SY5Y cells were transfected with TYR to express melanin in vitro, while EV transfected cells were used as control cells. EV and TYR SH-SY5Y cells were treated with 10 μM HAR for 24 h. A. Representative immunoblot showing expression of OPTN and parkin in whole cell lysates of EV or TYR-transfected HAR-treated cells. β actin was used as a loading control. B. The bar histogram represents the normalized densitometry of respective proteins. The data are represented as mean ± SEM, n = 4. C. Representative immunoblot showing expression of p62, OPTN and parkin in mitochondrial fraction of EV or HAR-transfected HAR-treated cells. TOM20 was used as a loading control. D. The bar histogram represents the normalized densitometry of respective proteins. The data are represented as mean ± SEM, n = 3. *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001 vs EV Control group; ɸp<0.05 and ɸ ɸ ɸp<0.05 vs TYR Control cells; and Δ Δp<0.01 and Δ Δ Δp<0.001 vs EV HAR-treated cells. NS= not significant.
We then analyzed the protein levels of autophagy/mitophagy markers in mitochondrial extracts. Our data showed no changes in p62 and parkin expressions between EV control and TYR control groups, while OPTN levels were increased by 24% in TYR control group compared to EV control group (p<0.05). Further, an increase of 48% in p62 expression (p<0.05) was observed in the absences of changes to OPTN and parkin expression in EV SH-SY5Y cells treated with HAR compared to the EV control group (Figure 6B). Similarly, we noted that HAR treatment to TYR SH-SY5Y cells showed a further increase in p62, OPTN, and parkin expression by 92–104% (p<0.0001), 75–83% (p<0.001) and 61–66% (p<0.01), respectively, compared to the EV SH-SY5Y control group. While compared to the neuromelanin control group, HAR treatment to neuromelanin forming cells caused 99–108% (p<0.0001), 49–56% (p<0.01) and 60–66% (p<0.05) increases in protein expression of p62, OPTN, and parkin, respectively. TOM20 was used as loading control for mitochondrial lysates and to normalize the protein expressions represented in Figure 6B.
Further, we assessed mitophagy by staining cells with a mitochondrial stain, MitoTracker Red, and an autophagic marker, LC3B (green). Figure 7A shows the representative images of immunofluorescence confocal microscopic images. Similar to immunoblotting data, immunofluorescence images show an increase in LC3B autophagy vacuoles (AVs) in harmane-treated EV SH-SY5Y cells, which was further increased in HAR-treated TYR SH-SY5Y cells. Interestingly, our data demonstrate increased colocalization of MitoTracker Red and LC3B in TYR SH-SY5Y cells treated with HAR (10 μM, 24 h) (Bottom row, Figure 7A). To further demonstrate the colocalization, we performed Z-stack imaging using a confocal microscope to create 3-dimensional images. The X-axis and Y-axis thin-line images from marked points are shown at the bottom and on the right of the merged confocal images, respectively. These images show no notable overlap of red (MitoTracker) and green (LC3B) pixels in control TYR SH-SY5Y cells, however, the colocalization of these two signals is apparent in HAR-treated TYR SH-SY5Y cells (Figure 7B).
Figure 7. Immunocytochemical images of increased mitophagy in HAR-treated SH-SY5Y cells forming neuromelanin.
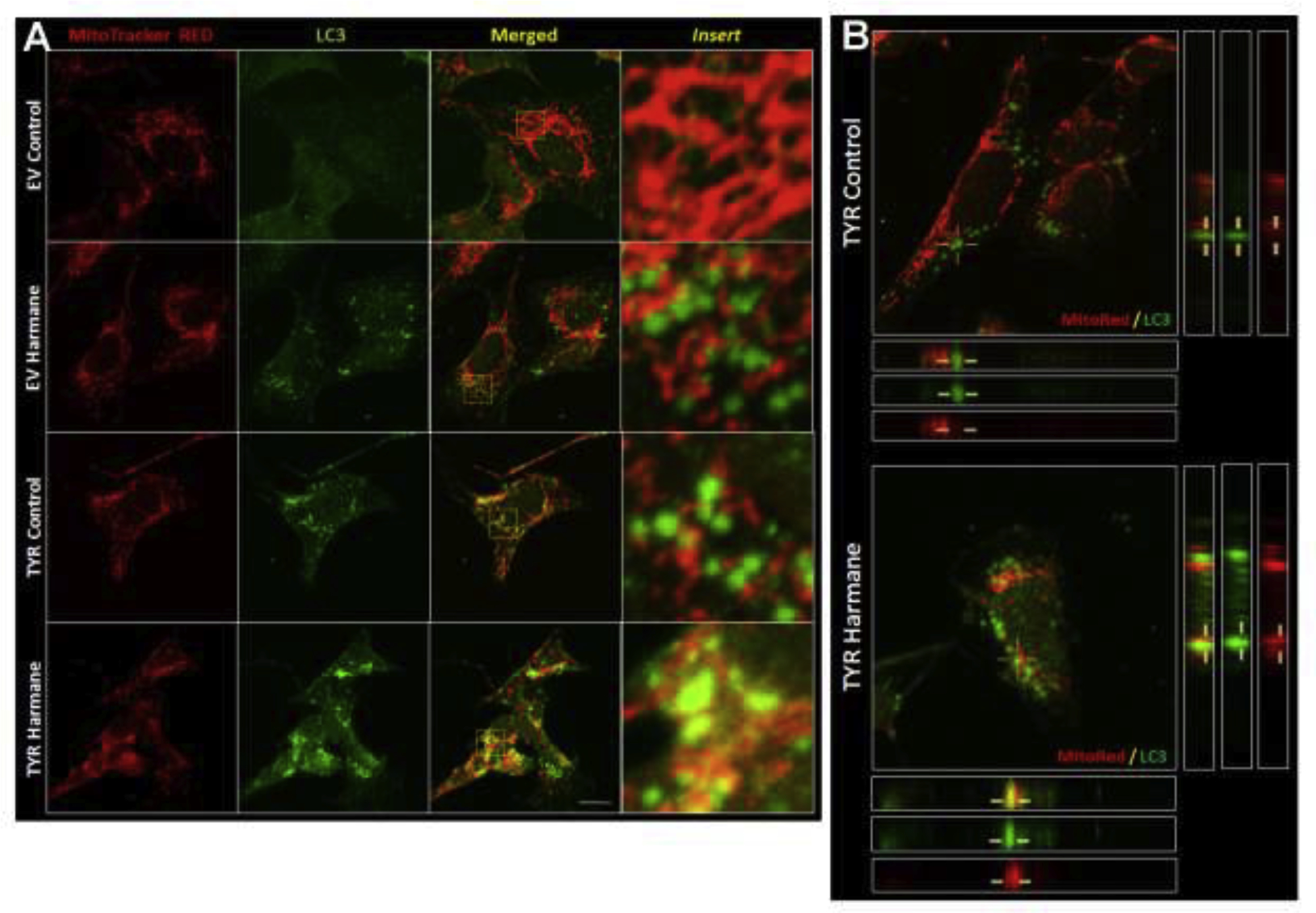
A. Representative immunofluorescence images of EV and TYR-transfected cells SH-SY5Y cells with or without HAR (10 μM, 24 h) treatment stained with MitoTracker (red) and LC3 (green). The third column shows merged images and the fourth column shows enlarged images from respective merged images. B. Transverse sections of Z-stacks of representative confocal images from TYR-transfected SH-SY5Y cells with or without HAR (10 μM, 24 h) treatment. The yellow lines represent the LC3-tagged vacuoles. Scale bar = 30μm.
Transmission electron microscopic analysis shows increased accumulation of autophagosomes and mitophagosomes in HAA-treated cells forming neuromelanin
We recently used TEM assessment to show the presence of neuromelanin-like pigments in TYR SH-SY5Y cells (Lawana et al., 2020). Using a similar approach, we further demonstrated the presence of autophagosomes and mitophagosomes in neuromelanin-containing SH-SY5Y cells after HAR treatment (10 μM, 24 h) (Figure 8). The representative images of control EV SH-SY5Y neuroblastoma cells shown in Figure 8A,B demonstrate healthy mitochondria at two different magnifications. A lack of laboratory access and staff shortages did not permit EM analysis of all groups (EV or TYR treated ± harmane). However, the presence of AVs by EM analysis in TYR transfected cells treated with harmane provides further support of the morphological and biochemical evidence of mitophagic alterations observed in Figures 4–7. Here, Figure 8C,D show the presence of multilamellar (onion skin-like) structures indicative of accumulated AVs (arrows). Further, we also observed the presence of multilayer AVs containing damaged mitochondria, as noted by a swollen mitochondrial matrix and irregular mitochondrial cristae, as highlighted by white arrows in Figure 8E,F,G,H at different magnifications denoted by scale bars in the left bottom corners.
Figure 8. Transmission electron microscopic images of mitophagy.
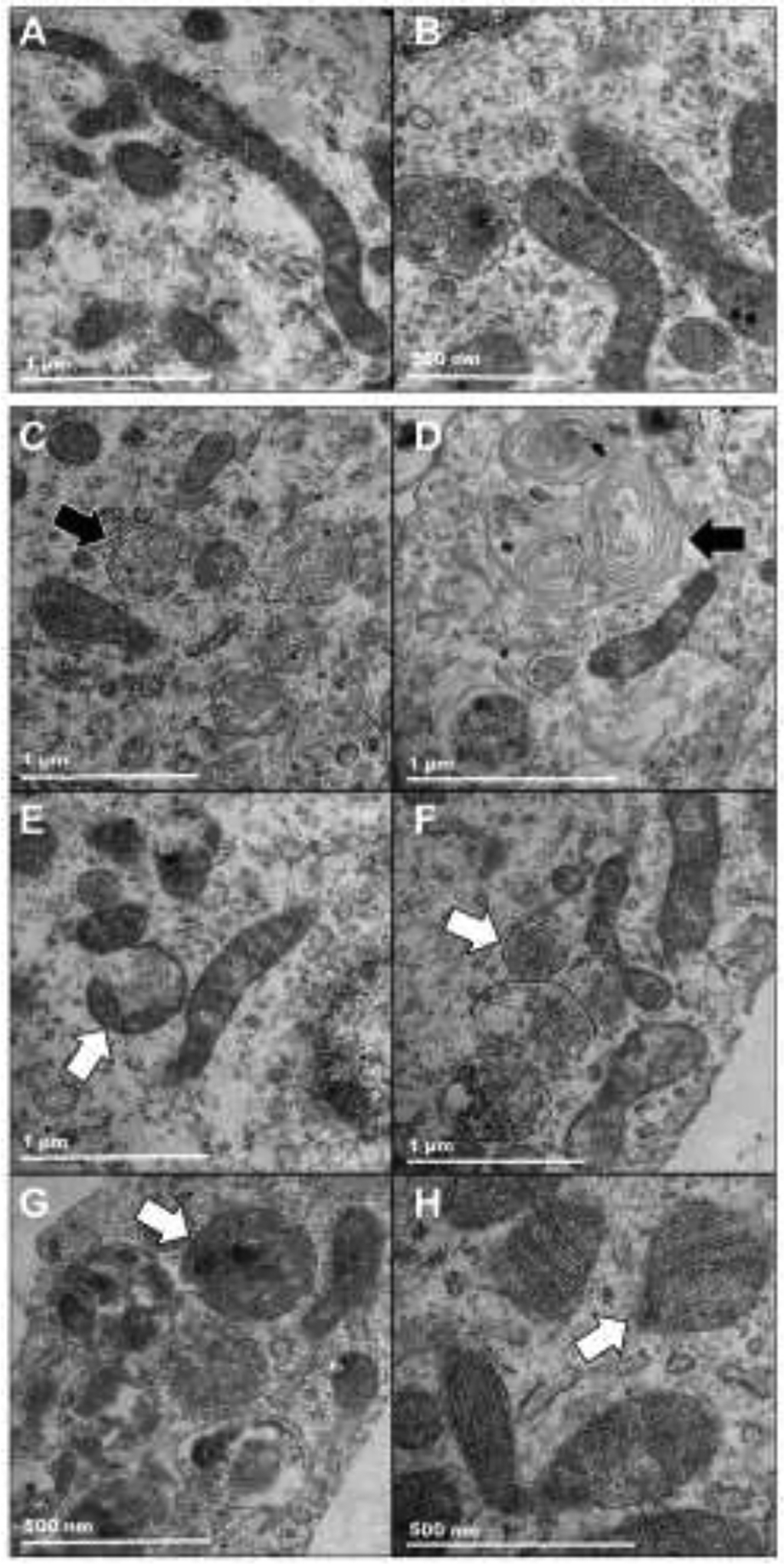
SH-SY5Y neuroblastoma cells were transfected with empty-vector (EV) or tyrosinase (TYR) constructs and 48 h post-transfection. EV-transfected control cells and TYR-transfected HAR-treated cells were fixed for TEM and images were captured with a FEI/Philips CM-100 transmission electron microscope at different magnifications. A,B. Representative images of EV-transfected control cells show healthy mitochondria of EV SH-SY5Y cells. C-H. Representative TEM images of TYR SH-SY5Y cells treated with HAR (10 μM, 24 h). Black arrows represent multilayered autophagic vacuoles. White arrows represent autophagic vacuoles containing damaged mitochondria (mitophagic vacuoles). The scale bars denote 1 μm in A-F; and 500 nm G,H.
DISCUSSION
Commonly used in vitro and in vivo PD models lack detectable levels of neuromelanin. Thus, there has been limited evidence demonstrating mechanistic roles of neuromelanin in modulating PD-relevant neurotoxicity. Recently we showed that ectopic expression of the human TYR gene in SH-SY5Y neuroblastoma cells dramatically increases neuromelanin formation in vitro. We also demonstrated that neuronal cells forming neuromelanin demonstrate higher sensitivity to neurotoxic HAAs by elevating ROS generation (Lawana et al., 2020). The goal of the present study was to determine if mitochondrial function is especially sensitive to interactions between HAAs and neuromelanin formation. Our study showed that several aspects of mitochondrial function are targeted by HAA exposure and that neuromelanin formation is a critical modulator of either the presence or magnitude of mitochondrial toxicity. These data are important because they identify the mitochondria as potentially important targets and suggest mechanisms that should be experimentally examined relative to how neuromelanin may increase neurotoxicity and also underscore the need to study dopaminergic neurotoxicity in models that more accurately replicate human neurobiology.
Our data show that two HAAs with high neurotoxic potential, PhIP and HAR, (i) increased the cellular levels of mitochondrial superoxide, (ii) reduced MMP, (iii) increased lysosomal membrane permeabilization and, (iv) reduced lysosomal acidity, in a time-dependent manner in EV SH-SY5Y neuroblastoma cells. Our data further demonstrated that both PhIP and HAR negatively altered mitochondrial bioenergetics. This included a decrease in basal respiration, ATP synthesis, and maximal capacity. This indicates that HAAs are decreasing functional mitochondria, potentially through inhibition of specific complexes of the electron transport chain or through opening of the mitochondrial permeability transition pore, although both of these hypotheses require further research to determine if they are a mechanism of HAA toxicity. However, harmane-mediated mitochondrial degeneration was much greater compared to PhIP at 10 μM and 24 h treatment.
Hence, we further investigated the effect of harmane on mitochondrial morphology, autophagy, and mitophagy. In this context, our data showed that harmane (i) reduced healthy mitochondria by increasing mitochondrial circularity, indicating aberrantly high mitochondrial fission, (ii) increased AV accumulation as shown by elevated expression of LC3B and p62 as well as higher LC3B-positive puncta, and (iii) increased mitophagy, indicated by higher translocation of parkin, p62, and OPTN to mitochondria and increased colocalization of LC3B and TOMM20, a mitochondrial marker. Collectively, these data illustrate that harmane induced mitochondrial dysfunction, leading to elevated mitophagy and potential impairments to autophagic flux. Determination of flux-specific affects requires full autophagic pathway analyses. Previous research showed that derivatives of harmane also caused mitochondrial dysfunction and a lesion in the substantia nigra when injected into the substantia nigra; however, this research did not test potential mitochondrial dysfunction caused by harmane (Albores et al., 1990; Neafsey et al., 1995).
Next, we demonstrated the effect of neuromelanin on the aforementioned HAA-mediated neurotoxic mechanisms using TYR SH-SY5Y neuroblastoma cells. Our results revealed that ectopic TYR expression-dependent neuromelanin formation markedly increased mitochondrial superoxide generation and MMP depolarization at later time points, starting 24 h after PhIP or harmane exposure. In addition, neuromelanin formation also worsened the effect of HAAs on mitochondrial function and mitochondrial morphology, demonstrating deteriorated mitochondrial overall health, including further reduced ATP production. We also noted that neuromelanin formation increased the harmane-mediated autophagic flux impairment by further elevating the expression of LC3B II and p62 as well as increasing LC3B-tagged puncta. Analysis of the mitochondrial fraction showed increased accumulation of p62, parkin, and OPTN in harmane-treated neuromelanin-forming cells, which was further confirmed by increased colocalization of AVs and Mitotracker dye. Lastly, our TEM assessment further bolstered our mitophagy findings by showing aberrant presence of AVs and mitophagic vacuoles. Altogether, our data suggest that harmane promotes neurotoxicity by causing mitochondrial and autophagolysosomal dysfunction, leading to defective mitophagy. The presence of intracellularly formed neuromelanin worsens these effects, which may contribute to higher cell death induced by HAR treatment. Based on our previous study (Lawana et al., 2020), it is likely that neuromelanin-mediated exaggerated neurotoxic effects could be due to greater retention of HAAs compared to cells without neuromelanin. We have shown that harmane and PhIP both cause mitochondrial toxicity, which is enhanced by the presence of neuromelanin. This is especially important in light of the fact that harmane is increased in both blood and cerebral spinal fluid of patients with PD (Kuhn et al., 1995a; Kuhn et al., 1996; Kuhn et al., 1995b; Louis et al., 2014). Therefore, the presence of neuromelanin indicates that dopaminergic neurons of the substantia nigra could have increased susceptibility to HAA toxicity due to the presence of neuromelanin. Furthermore, traditional models systems, such as rats and mice, could be downplaying the amount of toxicity that could be occurring after exposure to specific neurotoxins.
Highlights:
Tyrosinase was utilized to form neuromelanin in SY5Y cells
Neuromelanin increases heterocyclic aromatic amine-induced mitochondrial toxicity
Interactions between neuromelanin and heterocyclic aromatic amines impair mitophagy
Neuromelanin formation is critical to translationally study Parkinson’s disease
ACKNOWLEDGMENTS
We would like to thank the Life Science Microscopy Facility (LSMF) at Purdue University for assistance with electron microscopy. We specifically thank the facility directors Dr. Christopher J. Gilpin, Robert Seiler and Laurie Mueller, for providing training, sample preparation, and utilization of electron microscopes.
FUNDING
This work was supported by the National Institute of Environmental Health Sciences at the National Institutes of Health (R01ES025750 to J.R.C.); the Ralph W. and Grace M. Showalter Research Trust (to J.R.C.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Portions of this work were presented at a presentation entitled, “Interactions between neuroinflammation and mitophagy in Parkinson’s disease models” in the Immune dysregulation as a primary mechanism of early neurotoxicity – relevance to disease session of the 17th meeting of the International Neurotoxicology Association, convened September 29 – October 3, 2019 in Düsseldorf, Germany, entitled: “Translational Neurotoxicology”.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
REFERENCES
- Agim ZS, and Cannon JR (2018). Alterations in the nigrostriatal dopamine system after acute systemic PhIP exposure. Toxicology letters 287, 31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agim ZS, and Cannon JR (2015). Dietary Factors in the Etiology of Parkinson’s Disease. Biomed Res Int doi: Artn 672838 10.1155/2015/672838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aimi Y, and McGeer PL (1996). Lack of toxicity of human neuromelanin to rat brain dopaminergic neurons. Parkinsonism & related disorders 2(2), 69–74. [DOI] [PubMed] [Google Scholar]
- Albores R, Neafsey EJ, Drucker G, Fields JZ, and Collins MA (1990). Mitochondrial respiratory inhibition by N-methylated beta-carboline derivatives structurally resembling N-methyl-4-phenylpyridine. Proceedings of the National Academy of Sciences of the United States of America 87(23), 9368–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson C, Checkoway H, Franklin GM, Beresford S, Smith-Weller T, and Swanson PD (1999). Dietary factors in Parkinson’s disease: the role of food groups and specific foods. Mov. Disord 14(1), 21–7. [DOI] [PubMed] [Google Scholar]
- Ascherio A, and Schwarzschild MA (2016). The epidemiology of Parkinson’s disease: risk factors and prevention. Lancet Neurol 15(12), 1255–1270. [DOI] [PubMed] [Google Scholar]
- Campdelacreu J (2014). Parkinson’s disease and Alzheimer disease: environmental risk factors. Neurología (English Edition) 29(9), 541–549. [DOI] [PubMed] [Google Scholar]
- Charkoudian LK, and Franz KJ (2006). Fe(III)-coordination properties of neuromelanin components: 5,6-dihydroxyindole and 5,6-dihydroxyindole-2-carboxylic acid. Inorganic chemistry 45(9), 3657–64. [DOI] [PubMed] [Google Scholar]
- Cruz-Hernandez A, Agim ZS, Montenegro PC, McCabe GP, Rochet JC, and Cannon JR (2018). Selective dopaminergic neurotoxicity of three heterocyclic amine subclasses in primary rat midbrain neurons. Neurotoxicology 65, 68–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagda RK, Zhu J, Kulich SM, and Chu CT (2008). Mitochondrially localized ERK2 regulates mitophagy and autophagic cell stress: implications for Parkinson’s disease. Autophagy 4(6), 770–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davie CA (2008). A review of Parkinson’s disease. Br Med Bull 86, 109–27. [DOI] [PubMed] [Google Scholar]
- de Rus Jacquet A, Timmers M, Ma SY, Thieme A, McCabe GP, Vest JHC, Lila MA, and Rochet JC (2017). Lumbee traditional medicine: Neuroprotective activities of medicinal plants used to treat Parkinson’s disease-related symptoms. J Ethnopharmacol 206, 408–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Chandran K, Kalivendi SV, Joseph J, Antholine WE, Hillard CJ, Kanthasamy A, and Kalyanaraman B (2010). Neuroprotection by a mitochondria-targeted drug in a Parkinson’s disease model. Free Radic Biol Med 49(11), 1674–84 (Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibb WRG (1992). Melanin, Tyrosine-Hydroxylase, Calbindin and Substance-P in the Human Midbrain and Substantia-Nigra in Relation to Nigrostriatal Projections and Differential Neuronal Susceptibility in Parkinsons-Disease. Brain Res 581(2), 283–291. [DOI] [PubMed] [Google Scholar]
- Griggs AM, Agim ZS, Mishra VR, Tambe MA, Director-Myska AE, Turteltaub KW, McCabe GP, Rochet JC, and Cannon JR (2014). 2-Amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) is selectively toxic to primary dopaminergic neurons in vitro. Toxicological sciences : an official journal of the Society of Toxicology 140(1), 179–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa T (2010). Tyrosinase-Expressing Neuronal Cell Line as in Vitro Model of Parkinson’s Disease. Int J Mol Sci 11(3), 1082–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch E, Graybiel AM, and Agid YA (1988). Melanized Dopaminergic-Neurons Are Differentially Susceptible to Degeneration in Parkinsons-Disease. Nature 334(6180), 345–348. [DOI] [PubMed] [Google Scholar]
- Ichinose H, Ozaki N, Nakahara D, Naoi M, Wakabayashi K, Sugimura T, and Nagatsu T (1988). Effects of Heterocyclic Amines in Food on Dopamine Metabolism in Nigro-Striatal Dopaminergic-Neurons. Biochem Pharmacol 37(17), 3289–3295. [DOI] [PubMed] [Google Scholar]
- Jackson-Lewis V, Blesa J, and Przedborski S (2012). Animal models of Parkinson’s disease. Parkinsonism & related disorders 18 Suppl 1, S183–5. [DOI] [PubMed] [Google Scholar]
- Jenner P (2007). Oxidative stress and Parkinson’s disease. Handbook of clinical neurology 83, 507–20. [DOI] [PubMed] [Google Scholar]
- Johnson CC, Gorell JM, Rybicki BA, Sanders K, and Peterson EL (1999). Adult nutrient intake as a risk factor for Parkinson’s disease. Int. J. Epidemiol 28(6), 1102–9. [DOI] [PubMed] [Google Scholar]
- Kanthasamy AG, Kitazawa M, Yang Y, Anantharam V, and Kanthasamy A (2008). Environmental neurotoxin dieldrin induces apoptosis via caspase-3-dependent proteolytic activation of protein kinase C delta (PKCdelta): Implications for neurodegeneration in Parkinson’s disease. Mol Brain 1, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson O, Berg C, Brittebo EB, and Lindquist NG (2009). Retention of the cyanobacterial neurotoxin beta-N-methylamino-l-alanine in melanin and neuromelanin-containing cells - a possible link between Parkinson-dementia complex and pigmentary retinopathy. Pigment cell & melanoma research 22(1), 120–130. [DOI] [PubMed] [Google Scholar]
- Keeney PM, Dunham LD, Quigley CK, Morton SL, Bergquist KE, and Bennett JP Jr. (2009). Cybrid models of Parkinson’s disease show variable mitochondrial biogenesis and genotype-respiration relationships. Experimental neurology 220(2), 374–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima T, Naoi M, Wakabayashi K, Sugimura T, and Nagatsu T (1990). 3-Amino-1-Methyl-5h-Pyrido[4,3-B]Indole (Trp-P-2) and Other Heterocyclic Amines as Inhibitors of Mitochondrial Monoamine Oxidases Separated from Human Brain Synaptosomes. Neurochem Int 16(1), 51–57. [DOI] [PubMed] [Google Scholar]
- Kowal SL, Dall TM, Chakrabarti R, Storm MV, and Jain A (2013). The current and projected economic burden of Parkinson’s disease in the United States. Movement disorders : official journal of the Movement Disorder Society 28(3), 311–8. [DOI] [PubMed] [Google Scholar]
- Kuhn W, Muller T, Grosse H, Dierks T, and Rommelspacher H (1995a). Plasma levels of the beta-carbolines harman and norharman in Parkinson’s disease. Acta Neurol Scand 92(6), 451–4. [DOI] [PubMed] [Google Scholar]
- Kuhn W, Muller T, Grosse H, and Rommelspacher H (1996). Elevated levels of harman and norharman in cerebrospinal fluid of parkinsonian patients. J Neural Transm (Vienna) 103(12), 1435–40. [DOI] [PubMed] [Google Scholar]
- Kuhn W, Muller T, Grosse H, and Rommelspacher H (1995b). Plasma harman and norharman in Parkinson’s disease. Journal of neural transmission. Supplementum 46, 291–5. [PubMed] [Google Scholar]
- Langston JW, Forno LS, Tetrud J, Reeves AG, Kaplan JA, and Karluk D (1999). Evidence of active nerve cell degeneration in the substantia nigra of humans years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine exposure. Annals of Neurology 46(4), 598–605. [DOI] [PubMed] [Google Scholar]
- Lawana V, Singh N, Sarkar S, Charli A, Jin H, Anantharam V, Kanthasamy AG, and Kanthasamy A (2017). Involvement of c-Abl Kinase in Microglial Activation of NLRP3 Inflammasome and Impairment in Autolysosomal System. J Neuroimmune Pharmacol 12(4), 624–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawana V, Um SY, Rochet JC, Turesky RJ, Shannahan JH, and Cannon JR (2020). Neuromelanin Modulates Heterocyclic Aromatic Amine-Induced Dopaminergic Neurotoxicity. Toxicological sciences : an official journal of the Society of Toxicology 173(1), 171–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee VMY, and Trojanowski JQ (2006). Mechanisms of Parkinson’s disease linked to pathological alpha-synuclein: New targets for drug discovery. Neuron 52(1), 33–38. [DOI] [PubMed] [Google Scholar]
- Lindquist NG, Larsson BS, and Lydensokolowski A (1988). Autoradiography of [C-14] Paraquat or [C-14] Diquat in Frogs and Mice - Accumulation in Neuromelanin. Neurosci Lett 93(1), 1–6. [DOI] [PubMed] [Google Scholar]
- Logroscino G, Marder K, Cote L, Tang MX, Shea S, and Mayeux R (1996). Dietary lipids and antioxidants in Parkinson’s disease: a population-based, case-control study. Ann. Neurol 39(1), 89–94. [DOI] [PubMed] [Google Scholar]
- Louis ED, Michalec M, Jiang W, Factor-Litvak P, and Zheng W (2014). Elevated blood harmane (1-methyl-9H-pyrido[3,4-b]indole) concentrations in Parkinson’s disease. Neurotoxicology 40, 52–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann DMA, and Yates PO (1983). Possible Role of Neuromelanin in the Pathogenesis of Parkinsons-Disease. Mech Ageing Dev 21(2), 193–203. [DOI] [PubMed] [Google Scholar]
- Matsunaga J, Riley PA, Solano F, and Hearing VJ (2002). Biosynthesis of neuromelanin and melanin: The potential involvement of macrophage inhibitory factor and dopachrome tautomerase as rescue enzymes. Adv Behav Biol 53, 273–276. [Google Scholar]
- Miranda M, Botti D, Bonfigli A, Ventura T, and Arcadi A (1984). Tyrosinase-Like Activity in Normal Human Substantia Nigra. Gen Pharmacol 15(6), 541–544. [DOI] [PubMed] [Google Scholar]
- Moore DJ, West AB, Dawson VL, and Dawson TM (2005). Molecular pathophysiology of Parkinson’s disease. Annual review of neuroscience 28, 57–87. [DOI] [PubMed] [Google Scholar]
- Moriyama Y, Takano T, and Ohkuma S (1982). Acridine orange as a fluorescent probe for lysosomal proton pump. J Biochem 92(4), 1333–6. [DOI] [PubMed] [Google Scholar]
- Neafsey EJ, Albores R, Gearhart D, Kindel G, Raikoff K, Tamayo F, and Collins MA (1995). Methyl-beta-carbolinium analogs of MPP+ cause nigrostriatal toxicity after substantia nigra injections in rats. Brain research 675(1–2), 279–88. [DOI] [PubMed] [Google Scholar]
- Ostergren A, Lindquist NG, and Brittebo EB (2007). Differential effects of dopamine melanin on norharman-induced toxicity in PC12 cells. J Neural Transm 114(7), 909–918. [DOI] [PubMed] [Google Scholar]
- Panicker N, Saminathan H, Jin H, Neal M, Harischandra DS, Gordon R, Kanthasamy K, Lawana V, Sarkar S, Luo J, et al. (2015). Fyn Kinase Regulates Microglial Neuroinflammatory Responses in Cell Culture and Animal Models of Parkinson’s Disease. J Neurosci 35(27), 10058–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder RL, Double KL, and Gerber JP (2015). Using Sepia melanin as a PD model to describe the binding characteristics of neuromelanin - A critical review. J Chem Neuroanat 64–65, 20–32. [DOI] [PubMed] [Google Scholar]
- Shamoto-Nagai M, Maruyama W, Akao Y, Osawa T, Tribl F, Gerlach M, Zucca FA, Zecca L, Riederer P, and Naoi M (2004). Neuromelanin inhibits enzymatic activity of 26S proteasome in human dopaminergic SH-SY5Y cells. J Neural Transm 111(10–11), 1253–1265. [DOI] [PubMed] [Google Scholar]
- Shamoto-Nagai M, Maruyama W, Yi H, Akao Y, Tribl F, Gerlach M, Osawa T, Riederer P, and Naoi M (2006). Neuromelanin induces oxidative stress in mitochondria through release of iron: mechanism behind the inhibition of 26S proteasome. J Neural Transm 113(5), 633–644. [DOI] [PubMed] [Google Scholar]
- Singh N, Lawana V, Luo J, Phong P, Abdalla A, Palanisamy B, Rokad D, Sarkar S, Jin H, Anantharam V, et al. (2018). Organophosphate pesticide chlorpyrifos impairs STAT1 signaling to induce dopaminergic neurotoxicity: Implications for mitochondria mediated oxidative stress signaling events. Neurobiol Dis 117, 82–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skog KI, Johansson MAE, and Jagerstad MI (1998). Carcinogenic heterocyclic amines in model systems and cooked foods: A review on formation, occurrence and intake. Food Chem Toxicol 36(9–10), 879–896. [DOI] [PubMed] [Google Scholar]
- Turesky RJ (2007). Formation and biochemistry of carcinogenic heterocyclic aromatic amines in cooked meats. Toxicology letters 168(3), 219–227. [DOI] [PubMed] [Google Scholar]
- Zecca L, Fariello R, Riederer P, Sulzer D, Gatti A, and Tampellini D (2002). The absolute concentration of nigral neuromelanin, assayed by a new sensitive method, increases throughout the life and is dramatically decreased in Parkinson’s disease. Febs Lett 510(3), 216–220. [DOI] [PubMed] [Google Scholar]
- Zecca L, Pietra R, Goj C, Mecacci C, Radice D, and Sabbioni E (1994). Iron and Other Metals in Neuromelanin, Substantia-Nigra, and Putamen of Human Brain. Journal of Neurochemistry 62(3), 1097–1101. [DOI] [PubMed] [Google Scholar]
- Zecca L, Tampellini D, Rizzio E, Giaveri G, and Gallorini M (2001). The determination of iron and other metals by INAA in Cortex, Cerebellum and Putamen of human brain and in their neuromelanins. J Radioanal Nucl Ch 248(1), 129–131. [Google Scholar]
- Zhang W, Phillips K, Wielgus AR, Liu J, Albertini A, Zucca FA, Faust R, Qian SY, Miller DS, Chignell CF, et al. (2011). Neuromelanin Activates Microglia and Induces Degeneration of Dopaminergic Neurons: Implications for Progression of Parkinson’s Disease. Neurotox Res 19(1), 63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucca FA, Segura-Aguilar J, Ferrari E, Munoz P, Paris I, Sulzer D, Sarna T, Casella L, and Zecca L (2017). Interactions of iron, dopamine and neuromelanin pathways in brain aging and Parkinson’s disease. Progress in neurobiology 155, 96–119. [DOI] [PMC free article] [PubMed] [Google Scholar]