

Abstract
Acute Kidney Injury (AKI) is a common clinical syndrome that involves renal tubular epithelial cell death, which leads to acute decline in renal function. Improper tubular regeneration following AKI often leads to chronic kidney disease (CKD). We discuss the role of a serine/threonine protein kinase called glycogen synthase kinase −3 (GSK3) in renal tubular injury and renal fibrosis. We also highlight the importance of GSK3 as a potential drug target in AKI patients and molecular mechanisms promoting tissue regeneration.
Keywords: GSK3, ischemia/reperfusion, AKI, TDZD-8, apoptosis, renal fibrosis, regeneration
Background:
AKI affects over 13 million people and causes 1.7 million deaths every year worldwide. Longterm consequences of AKI include prolonged renal dysfunction, cardiovascular effects, progression to CKD and death [1]. AKI is commonly caused by nephrotoxic agents, infection, drug toxicity or ischemia-reperfusion (I/R), and is often seen in hospitalized patients. AKI involves apoptotic or necrotic cell death of renal tubular epithelial cells, especially of the proximal tubules. Although the injury to the nephron is often reversible, approximately 15% of AKI patients can advance to CKD within 24 months. AKI to CKD progression is mainly attributed to faulty tubular repair and the development of renal fibrosis after AKI. Hence it is important to develop therapies to ensure proper repair of tubules and minimize the chances of AKI progressing to CKD. This review examines the progress made in understanding the role of a serine-threonine protein kinase called GSK3 in renal tubular injury, repair and renal fibrosis after AKI.
GSK3 regulates apoptotic cell death of renal tubular epithelial cells in AKI.
Mammalian GSK3 consists of α and β isoforms which share 97% sequence homology in their kinase domain and were first cloned by James R Woodgett. Although initially discovered as a key kinase in the insulin signaling pathway, a flood of studies in the 1990s and 2000s revealed that GSK3 acts as a gatekeeper of many cellular signaling pathways including Wnt, mTOR, sonic hedgehog and notch signaling. GSK3 is inhibited by phosphorylation of Serine-21 and Serine-9 respectively on GSK3α and GSK3β isoforms or by their cytoplasmic sequestration. Many of the above-mentioned pathways when active, lead to inhibition of GSK3, and consequently GSK3 is active when these signaling pathways are inactive, as in fully differentiated cells. GSK3 thus plays an important role during embryonic development and during injury/ repair processes in adult tissues [2].
A pro-apoptotic role of GSK3β in kidney cells was first shown by our study in which overexpression of a constitutively active GSK3β induced apoptosis, while GSK3 inhibition using small molecule inhibitors SB216763 and SB415286, or LiCl reduced apoptosis following hypertonic stress [3]. The first studies examining the role of GSK3 in AKI [4] came from the hypothesis that GSK-3 inhibition could have anti-inflammatory effects. This hypothesis was based on prior findings that global GSK3β gene deleted mice died post-birth and showed a similar phenotype to mice with suppressed NF-κB activation, due to p65 or IκB kinase 2 gene deletion [2]. Pre-treatment with GSK3 inhibitor protected rodents from lipopolysaccharide-induced endotoxemic renal failure, and specifically proximal tubular injury [4, 5]. Following this, multiple in vivo studies showed that GSK3 plays an important role in apoptosis in AKI models including I/R [6, 7] or nephrotoxicity induced by diclofenac, cisplatin, mercuric chloride, gentamicin or paraquat [8–11]. Isoform non-specific pharmacologic inhibitors of GSK3 such as LiCl, TDZD-8, SB216763, SB415286 or BIO reduced apoptosis of renal tubular epithelial cells in the above-mentioned studies. SB216763 acts as an ATP competitive inhibitor, whereas TDZD is a non-ATP competitive inhibitor of GSK3. TDZD has been hypothesized to interact with cysteine 199, an important amino acid residue located in the active site of GSK3. GSK3 is inhibited by BIO which interacts with its ATP binding pocket, and by LiCl which competes with Mg2+ [12, 13].
The main mechanism by which GSK3 promotes apoptosis is by the regulation of mitochondrial permeability transition. Plotnikov et al. observed swollen and fragmented renal proximal tubular mitochondria with reduced membrane potential following renal I/R in rats, which was partially restored by LiCl treatment [14]. Rogung Gong’s group demonstrated the presence of GSK3β in mitochondria of cultured cells. They also showed that elevated reactive oxygen species (ROS) in AKI increased GSK3β activity and GSK3 mediated phosphorylation of cyclophilin F and voltage-dependent anion channel in the mitochondria, leading to mitochondrial permeability transition and cell death. Consistently, GSK3 inhibition reduced mitochondrial permeability transition and apoptosis in in vitro and in vivo AKI models [8, 11, 7]. Wang et al. found that overexpression of constitutively active GSK3β led to activated Bax and cleaved caspase-3, and increased apoptosis after ATP depletion in vitro, while GSK3 inhibition by TDZD-8 reduced tubular cell apoptosis in rats following I/R [6]. The role of the GSK3β isoform in survival was further shown by our study employing a proximal tubule specific GSK3β gene deleted mouse, which showed reduced tubular injury and preserved renal function in a mercuric chloride induced AKI mouse model. Reduced renal tubular cell apoptosis in the GSK3β knockout mice was accompanied by reduced levels of activated Bax6A7 and caspase-3 [9]. In addition, multiple in vitro and in vivo studies not discussed here have observed that increased phosphor-GSK3β Ser9 levels correlate with reduced apoptosis and kidney damage in various AKI and CKD models. Thus, pre-treatment with GSK3 inhibitors could confer renal protection during AKI.
GSK3 regulates renal tubular repair and fibrosis after AKI:
Renal tubules are capable of regenerating after injury, mainly by proliferation of surviving tubular epithelial cells. However, repair is often incomplete, which along with persistent inflammation could lead to the development of fibrosis and CKD. Since pre-treatment with GSK3 inhibitors or GSK3β gene deletion prior to AKI could significantly reduce injury, studies examining the role of GSK3 in repair have used post-AKI GSK3 inhibitor treatment protocols. We found that treatment with TDZD-8 starting 2 days after mercuric chloride induced nephrotoxic AKI in mice showed significantly better restoration of renal structure and function compared to vehicle treated AKI group. TDZD-8 treatment post-AKI increased renal levels of pro-proliferative factors such as cyclin D1, c-myc and β-catenin (previously known GSK3β targets for phosphorylation and degradation) and significantly increased cell proliferation of tubular epithelial cells [9]. Increased tubular cell proliferation was also observed in GSK3β knockout mice subjected to similar nephrotoxic AKI [9]. Consistently, LiCl treatment following cisplatin or I/R-induced AKI was found to significantly improve kidney repair in mice [7]. In vitro studies in murine renal epithelial cell lines exposed to cisplatin showed that GSK3β interacts with cyclin D1, c-Myc, and hypoxia inducible factor-1α (HIF-1α), resulting in their phosphorylation, nuclear exit and subsequent proteasomal degradation [7]. In a recent study, we have found that GSK3β suppresses tubular cell proliferation after AKI by inhibiting FoxM1, a crucial regulator of cell-cycle progression (revised manuscript under review in FASEB journal).
Progressive renal fibrosis is a major problem associated with AKI and is closely linked to persistent inflammation. GSK3 is a known regulator of NF-kB activity [2]. In a model of severe acute pancreatitis- induced AKI, GSK3 inhibition reduced NF-kB activity and proinflammatory factors [15]. We demonstrated that inhibition of GSK3β using TDZD-8 starting two days after I/R induced AKI reduced macrophage infiltration, TNFα, IL6, IL1β, CCl2, CCL3 and ICAM1 levels [16]. TDZD-8 treatment also reduced renal fibrosis, myofibroblasts, extracellular matrix proteins such as like collagen-1 and fibronectin, mRNA levels of TGF-β1, PAI-1 and TGF-β signaling [16]. In vitro, GSK3 inhibitor treatment abolished TGF-β mediated fibroblast to myofibroblast differentiation [16]. Similarly, in a mouse model of chronic alcohol feeding followed by I/R induced AKI, GSK3 inhibitor pre-treatment before I/R reduced renal fibrosis, ECM, myofibroblasts and TGF-β signaling, and improved renal function [17]. These studies suggest that GSK3 could promote renal fibrosis after AKI by activating the TGF-β signaling.
In spite of the extensive knowledge that we have gained on the role of GSK3 in AKI, how GSK3 is regulated during renal injury, repair and fibrosis it is still unclear.
Conclusions and Future Directions:
GSK3β plays a critical role in AKI by promoting tubular epithelial cell apoptosis, inflammation and fibrosis, and suppressing repair (Figure-1). Pharmacological GSK3 inhibition or GSK3β gene deletion in animal models of AKI have proven to improve outcomes after various types of AKI. Since AKI is often detected after the injury has occurred, delayed pharmacologic intervention using GSK3 inhibitors after AKI could be a useful strategy to accelerate repair, prevent fibrosis and minimize the chance of AKI progressing to CKD.
Figure 1: Pathways regulated by GSK3β in AKI:
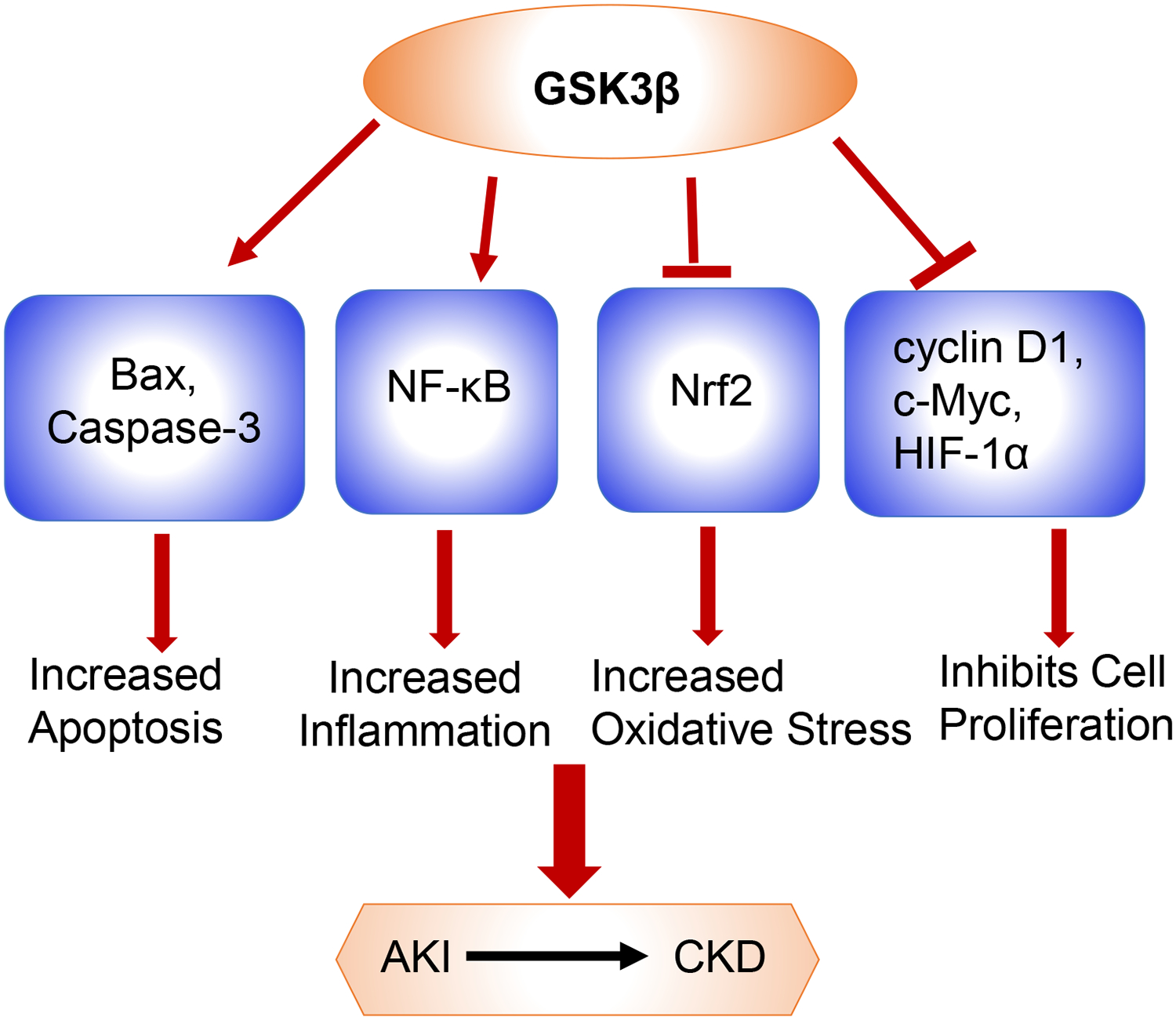
GSK3β induces mitochondrial injury and apoptosis by directly phosphorylating and activating pro-apoptotic proteins Bax and Caspase-3. GSK-3β also phosphorylates and activates the p65 subunit of NF-κB and enhances its transcriptional response leading to inflammation. Similarly, Nrf2, which plays an important role in antioxidant defense mechanism is suppressed by GSK3β. Nrf2 is phosphorylated by GSK3β, which facilitates its nuclear exclusion, resulting in persistent oxidative stress. GSK3β also regulates cell proliferation by phosphorylating cyclin D1, c-Myc, and HIF-1α resulting in their nuclear exit and subsequent proteasomal degradation. All these pathways slows down tubular recovery and aids in AKI to CKD transition.
Acknowledgement:
This work was supported by NIH R01-DK083525 to RR.
Footnotes
Disclosure: The authors have no conflicts of interest to declare.
References:
- 1.Agarwal A, Dong Z, Harris R, Murray P, Parikh SM, Rosner MH, et al. Cellular and Molecular Mechanisms of AKI. J Am Soc Nephrol. 2016. May;27(5):1288–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Patel P, Woodgett JR. Glycogen Synthase Kinase 3: A Kinase for All Pathways? Curr Top Dev Biol. 2017;123:277–302. [DOI] [PubMed] [Google Scholar]
- 3.Rao R, Hao CM, Breyer MD. Hypertonic stress activates glycogen synthase kinase 3betamediated apoptosis of renal medullary interstitial cells, suppressing an NFkappaB-driven cyclooxygenase-2-dependent survival pathway. J Biol Chem. 2004. February 6;279(6):3949–55. [DOI] [PubMed] [Google Scholar]
- 4.Dugo L, Collin M, Allen DA, Patel NS, Bauer I, Mervaala EM, et al. GSK-3beta inhibitors attenuate the organ injury/dysfunction caused by endotoxemia in the rat. Crit Care Med. 2005. September;33(9):1903–12. [DOI] [PubMed] [Google Scholar]
- 5.Wang Y, Huang WC, Wang CY, Tsai CC, Chen CL, Chang YT, et al. Inhibiting glycogen synthase kinase-3 reduces endotoxaemic acute renal failure by down-regulating inflammation and renal cell apoptosis. Br J Pharmacol. 2009. July;157(6):1004–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Z, Havasi A, Gall J, Bonegio R, Li Z, Mao H, et al. GSK3{beta} promotes apoptosis after renal ischemic injury. J Am Soc Nephrol. 2010. February;21(2):284–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bao H, Ge Y, Wang Z, Zhuang S, Dworkin L, Peng A, et al. Delayed administration of a single dose of lithium promotes recovery from AKI. J Am Soc Nephrol. 2014. March;25(3):488–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bao H, Ge Y, Zhuang S, Dworkin LD, Liu Z, Gong R. Inhibition of glycogen synthase kinase-3beta prevents NSAID-induced acute kidney injury. Kidney Int. 2012. April;81(7):662–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Howard C, Tao S, Yang HC, Fogo AB, Woodgett JR, Harris RC, et al. Specific deletion of glycogen synthase kinase-3beta in the renal proximal tubule protects against acute nephrotoxic injury in mice. Kidney Int. 2012. November;82(9):1000–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Plotnikov EY, Grebenchikov OA, Babenko VA, Pevzner IB, Zorova LD, Likhvantsev VV, et al. Nephroprotective effect of GSK-3beta inhibition by lithium ions and delta-opioid receptor agonist dalargin on gentamicin-induced nephrotoxicity. Toxicol Lett. 2013. July 18;220(3):303–8. [DOI] [PubMed] [Google Scholar]
- 11.Wang Z, Ge Y, Bao H, Dworkin L, Peng A, Gong R. Redox-sensitive glycogen synthase kinase 3beta-directed control of mitochondrial permeability transition: rheostatic regulation of acute kidney injury. Free Radic Biol Med. 2013. December;65:849–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martinez A, Alonso M, Castro A, Perez C, Moreno FJ. First non-ATP competitive glycogen synthase kinase 3 beta (GSK-3beta) inhibitors: thiadiazolidinones (TDZD) as potential drugs for the treatment of Alzheimer’s disease. J Med Chem. 2002. March 14;45(6):1292–9. [DOI] [PubMed] [Google Scholar]
- 13.Eldar-Finkelman H, Martinez A. GSK-3 Inhibitors: Preclinical and Clinical Focus on CNS. Front Mol Neurosci. 2011;4:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Plotnikov EY, Kazachenko AV, Vyssokikh MY, Vasileva AK, Tcvirkun DV, Isaev NK, et al. The role of mitochondria in oxidative and nitrosative stress during ischemia/reperfusion in the rat kidney. Kidney Int. 2007. December;72(12):1493–502. [DOI] [PubMed] [Google Scholar]
- 15.Zhao K, Chen C, Shi Q, Deng W, Zuo T, He X, et al. Inhibition of glycogen synthase kinase-3beta attenuates acute kidney injury in sodium taurocholateinduced severe acute pancreatitis in rats. Mol Med Rep. 2014. December;10(6):3185–92. [DOI] [PubMed] [Google Scholar]
- 16.Singh SP, Tao S, Fields TA, Webb S, Harris RC, Rao R. Glycogen synthase kinase-3 inhibition attenuates fibroblast activation and development of fibrosis following renal ischemia-reperfusion in mice. Dis Model Mech. 2015. August 1;8(8):931–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang L, Zhu Y, Wang L, Hou J, Gao Y, Shen L, et al. Effects of chronic alcohol exposure on ischemia-reperfusion-induced acute kidney injury in mice: the role of beta-arrestin 2 and glycogen synthase kinase 3. Exp Mol Med. 2017. June 23;49(6):e347. [DOI] [PMC free article] [PubMed] [Google Scholar]