

Abstract
Inherited genetic variations may alter drug sensitivity in patients with acute lymphoblastic leukemia (ALL), predisposing to adverse treatment side effects. In this review, we discuss evidence from children and young adults with ALL to review the available pharmacogenomic data with an emphasis on clinically actionable and emerging discoveries, for example genetic variants in TPMT and NUDT15 that alter 6-mercaptopurine dosing. We also highlight the need for ongoing pharmacogenomic research to validate the significance of recent findings. Further research in young adults, as well as with novel therapeutics, are needed to provide optimal therapy in future trials.
Keywords: Acute lymphoblastic leukemia, pharmacogenomics, precision medicine
Introduction
Improvements in outcomes for acute lymphoblastic leukemia (ALL) has been driven in part by the ability to better personalize therapy to reduce toxicity and improve efficacy. Key aspects of this personalization include risk adapted therapy based on both leukemia genetics, disease response, as well as adapting chemotherapeutic regimens to attempt to minimize toxicity to patients. Chemotherapy can be preemptively modified using pharmacogenomics on the basis of germline genetic variations that influence the responsiveness and toxicity of chemotherapy.(1) Because pharmacogenomics involves the interaction between chemotherapy and inherited genetic variation, it is necessary to understand both the patient-specific genetics influencing treatment response as well as the chemotherapeutic regimen being utilized.(2) In this review, we will highlight data demonstrating genetic variations which may affect the efficacy and toxicity of current treatment regimens for young adults with ALL. While we will highlight recent findings in adult pharmacogenomic studies, we will also note relevant data from pediatric studies using similar chemotherapeutic regimens. Increasing similarity between adult and pediatric trials makes these comparisons meaningful given overlapping chemotherapeutic agents.(3–5) We will divide our review according to the implicated chemotherapy agent and focus on the five drug groups with the most extensive data: 6-mercaptopurine, methotrexate, asparaginase, vincristine, and glucocorticoids.
6-Mercaptopurine
6-Mercaptopurine (6-MP) is a mainstay of maintenance therapy for patients with ALL. It is taken orally daily with doses adjusted based on myelosuppression, the primary toxicity of 6-MP. Mercaptopurine exerts its antileukemic effects through conversion to thioguanine nucleotides (TGNs) which are ultimately incorporated into DNA (DNA-TG).(6) There, they result in DNA damage and ultimately apoptosis (Figure 1). In addition to its use in ALL, mercaptopurine is used for patients with inflammatory bowel disease, as is its prodrug azathioprine. A related compound, 6-thioguanine, is also utilized in ALL, typically during intensification phases. While the primary toxicity of both mercaptopurine and thioguanine is myelosuppression, thioguanine is also associated with a risk of venoocclusive disease/ sinusoidal obstructive syndrome.(7) In contrast, medication induced transaminitis is more common with mercaptopurine and is reversible without long-term hepatic injury, likely involving methyl metabolites of thiopurines. Mercaptopurine is the best characterized pharmacokinetic drug-gene pairing in ALL. Inter-individual differences in tolerance to mercaptopurine resulted in typical dose ranges between 10 mg/m2 3 days a week to more than 75 mg/m2 daily, with most patients tolerating 50–75mg/m2 daily.(8) The first genetic variant identified which was associated with decreased tolerance to mercaptopurine was thiopurine methyltransferase (TPMT),(9) a gene whose activity is regulated primarily by coding variants within the gene itself.(10) This association has been identified across diseases treated with this drug, including inflammatory bowel disease as well as ALL.(11) TPMT methylates mercaptopurine and its intermediate metabolites; this results in lower thioguanine nucleotide levels.(12) While thioguanine nucleotides have been most directly associated with cellular cytotoxicity, methylated mercaptopurines have been associated with transaminitis during mercaptopurine therapy.(13) Measurement of red blood cell TGN levels is now used as a surrogate for these levels in blasts, as RBC and blast TPMT activity are highly correlated and red blood cell TGN levels are highly correlated with hematological toxicity.(9, 14, 15) While no clinical threshold has been establish for either efficacy or toxicity in patients with ALL, evaluation of red blood cell thioguanine and methyl mercaptopurine nucleotide levels have been used to monitor adherence to therapy in patients receiving 6-MP.(16–18)
Figure 1:
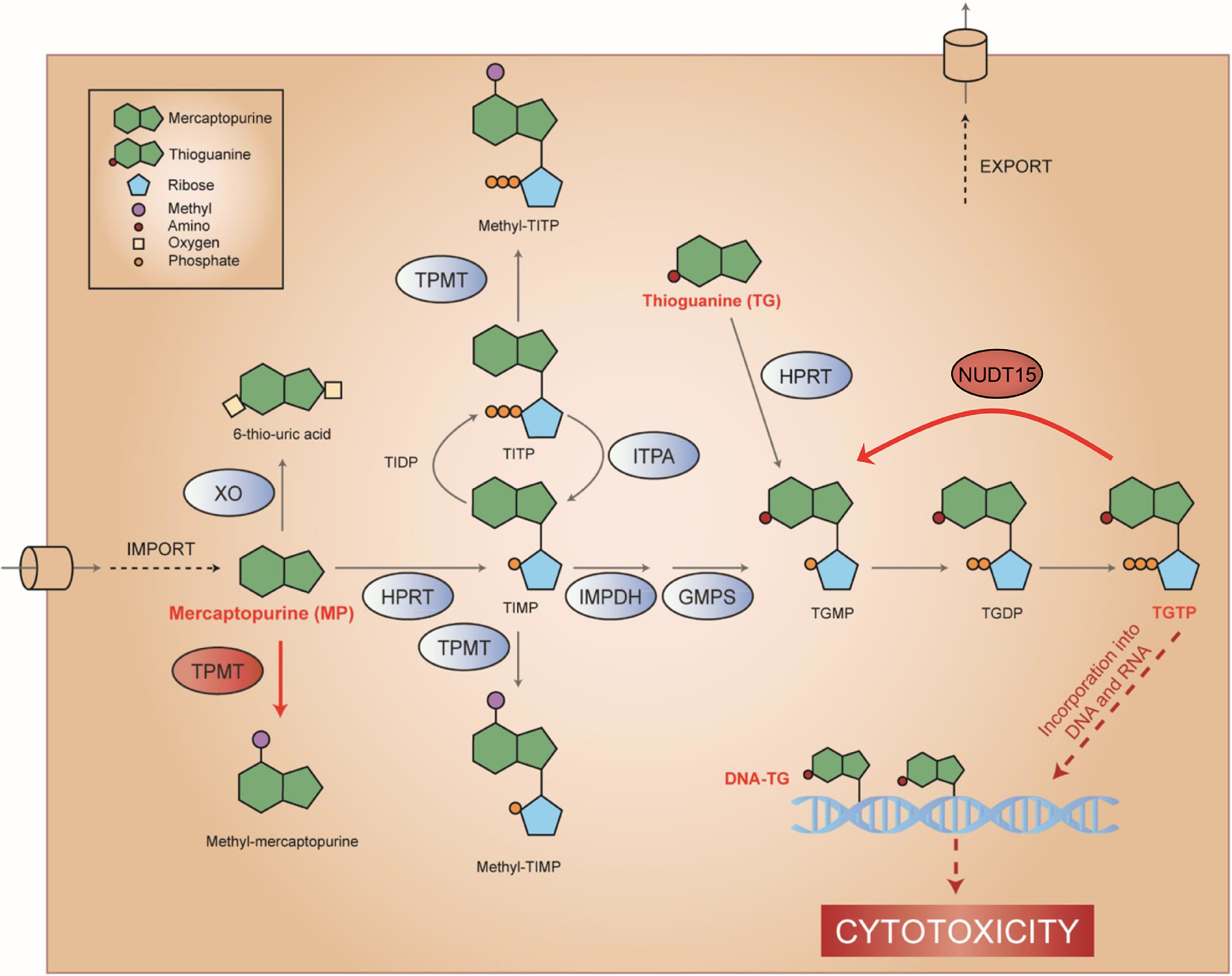
TPMT and NUDT15 inactivate thiopurines
As prodrugs, thiopurines (mercaptopurine [6MP] and thioguanine [TG]) are enzymatically metabolized to TGTP that is incorporated into DNA resulting in DNA damage and cytotoxicity. TPMT reduces MP cytotoxicity by converting it to inactive methyl-MP, whereas NUDT15 dephosphorylates TGTP and converts it to inactive TGMP.
TPMT and NUDT15 are highlighted in red ovals, while MP/TG and their active metabolites (TGTP and DNA.TG) are shown in red text.
Variants in TPMT are present in approximately 4.5% of patients with European ancestry, 7.7% of patients with African Americans, ~2% of Asians, and 5% of Hispanic patients.(19–21) The most common variant allele in European, Hispanic, and Asian patients is the 3A (C>T at rs1800460 and T>C at rs1142345), while the 3C allele (T>C at rs1142345) is the most common variant identified in African Americans (Table 1).(21) Patients with one known inactivating variant in TPMT (i.e. *3A, *3C, *2) are considered intermediate metabolizers, while patients with two inactivating alleles are considered poor metabolizers. All other patients are considered normal metabolizers. Preemptive identification of the genetic variants in children with ALL resulted in reduced periods of myelosuppression as well as a reduction in the risk of secondary acute myeloid leukemia.(9, 22, 23) Limited adult data also suggests that patients who experience severe hematological toxicity while receiving mercaptopurine frequently carries variations in TPMT.(24)
Table 1:
Frequently encountered low-function alleles in TPMT and NUDT15.
Data for the frequency of NUDT15 variant alleles in African Americans is unavailable at this time. Adapted from current Clinical Pharmacogenetics Implementation Consortium guidelines.(21)
| Gene/Haplotype | rsID | Allele Frequency | |||
|---|---|---|---|---|---|
| European | African American | Hispanic | East Asian | ||
| TPMT | |||||
| *3A | rs1800460, rs1142345 |
3.4% | 0.8% | 4.2% | <0.1% |
| *3C | rs1142345 | 0.5% | 2.4% | 0.6% | 1.6% |
| *2 | rs267607275 | 0.2% | 0.5% | 0.4% | <0.1% |
| Other non-reference | Various | 0.6% | 3.9% | 0.6% | 0.4% |
| NUDT15 | |||||
| *2 | rs869320766, rs116855232 |
<0.1% | 3.7% | 3.5% | |
| *3 | rs116855232 | 0.2% | 0.8% | 6.1% | |
| *9 | rs746071566 | <0.1% | <0.1% | <0.1% | |
The recent discovery of the association between NUDT15 variants and mercaptopurine intolerance has expanded the understanding of causal variants driving thiopurine induced myelosuppression. Historical demographic data has suggested that patients of Asian ancestry are more sensitive to 6-MP than patients of European ancestry, with mean tolerated dose approximately 65% of what is observed in other populations.(25) Genome-wide association studies (GWAS) identified variants in NUDT15 significantly associated with this dose intolerance.(25) These variants are present across genetic ancestries but are relatively less common in patients of European ancestry (0.7%) compared to those of Hispanic or Asian ancestry, in which they are observed in 4.5–6.4% and 10–12.1% of patients, respectively (Table 1).(21, 25) Similar to patients with only inactivating TPMT variations, patients with one loss-of-function NUDT15 variation (i.e. *2, *3, *9) are considered intermediate metabolizers and tolerate 6-mercaptopurine after a 20–30% dose reduction (i.e. 50–60mg/m2/day as a starting dose).(21, 25) While rare, NUDT15 variants in European adults are also associated with myelosuppression.(24) Again similar to TPMT variant patients, those with 2 inactivating mutations are considered poor metabolizers and tolerate less than 10% of the protocol specified dose (i.e. 10mg/m2/dose 3–7 days/ week; Figure 2).
Figure 2:
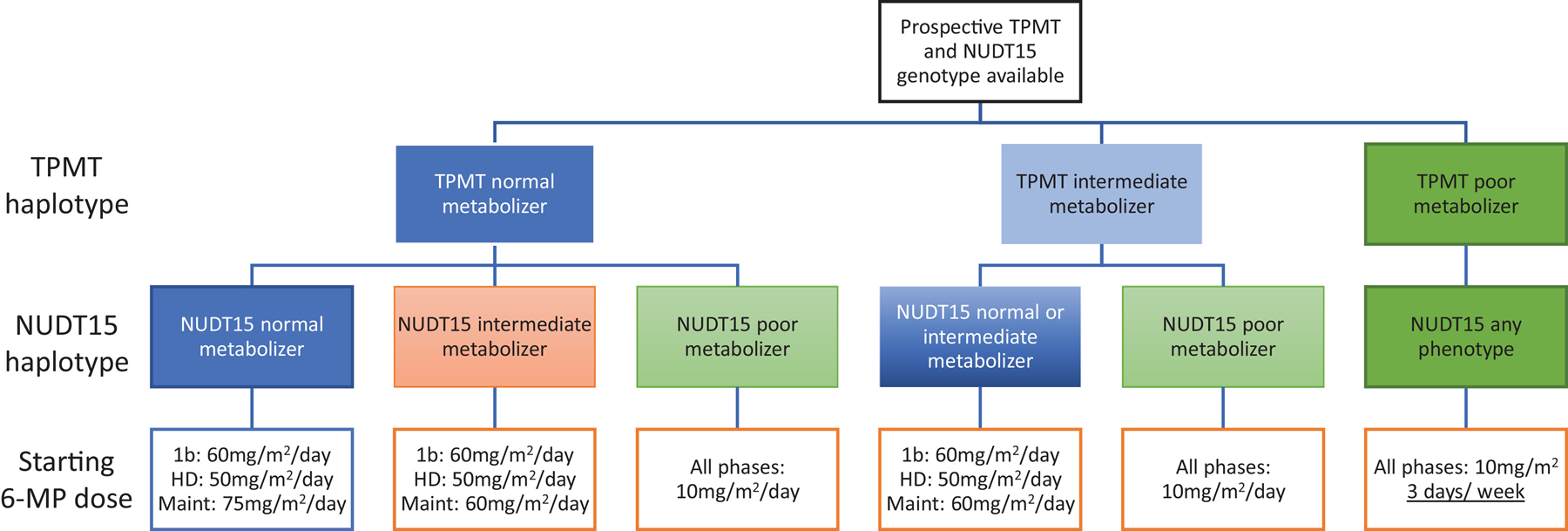
Genotype guided dose of 6-MP
Starting 6-MP doses in Total 17 (NCTgg03117751) vary by phase of therapy. Patients with one known inactivating variant in TPMT (i.e. *3A, *3C, *2) or NUDT15 (i.e. *2, *3, *9) are considered intermediate metabolizers for the respective enzyme, while patients with two inactivating alleles are considered poor metabolizers. All other patients are considered normal metabolizers. Phases of therapy include late induction (1b), consolidation with high-dose methotrexate (HD), and continuation (Maint). All doses are given daily with the exception of patients with TPMT poor metabolizer genotypes who receive 6-MP only 3 of 7 days each week.
NUDT15 acts by dephosphorylating thioguanine nucleotides prior to their incorporation into DNA.(26) Once thioguanine nucleotides are incorporated into DNA, they result in DNA damage and ultimately apoptosis. Damaging variants in NUDT15 result in increased incorporation of thioguanine nucleotides into DNA and thus increased sensitivity to the drug in the form of increased myelosuppression.
Because germline genetic variation in TPMT and NUDT15 are “inherited” by a patients’ ALL blasts, dose adjustments to reduce myelotoxicity can be made while maintaining therapeutic efficacy.(27, 28) Data from clinical trials in which prospective dose decreases in 6-mercaptopurine were made in patients with TPMT variation indicate similar therapeutic efficacy in patients with TPMT variant and wild-type alleles.(29) Preclinical data demonstrate the efficacy of these NUDT15-informed decreases on murine leukemias.(30, 31) Within the context of TPMT, host metabolism effects drug exposure such that even when wild-type TPMT leukemias are present in TPMT deficient hosts, therapeutic efficacy is maintained with dose decreased mercaptopurine.(27)
In summary, clinical and preclinical data strongly support prospective dose adjustments of 6-mercaptopurine in the context of TPMT or NUDT15 genetic variations. These recommendations are highlighted in a recently updated guideline from the Clinical Pharmacogenetics Implementation Consortium (CPIC).(21)
Methotrexate
Methotrexate exerts antileukemic efficacy through alterations in the folate metabolism pathway and subsequent alterations in purine generation and DNA synthesis. It is critical to antileukemic therapy and is used in intrathecal injections to control or prevent CNS leukemia, during interim maintenance periods either in the form of high-dose methotrexate (with leucovorin rescue) or in an escalating IV fashion when combined with asparaginase, the so-called Capizzi regiment. Finally, along with mercaptopurine, it is taken either orally or intravenously weekly as part of maintenance therapy.
The most commonly observed side effects of methotrexate include myelosuppression, mucositis, and hepatic toxicity.(4) In addition, acute renal insufficiency may also be observed, particularly after high-dose administration.(32) More rarely, an acute stroke-like syndrome is observed which is typically transient and resolves within 7 to 10 days.(33) Chronically, leukoencephalopathy and neurological impairment can be observed both during therapy and into survivorship.(34, 35) There have been no genetic variants which have been integrated into preemptive pharmacogenomics testing prior to methotrexate administration. Both GWAS and candidate gene approaches have been used to attempt to identify genetic variants which are associated with methotrexate sensitivity or toxicity. Variants in SLCO1B1 were first identified as being associated with methotrexate clearance and gastrointestinal toxicity in three St. Jude treatment cohorts.(36) Variants associated with slower methotrexate clearance were also associated with an increased risk of gastrointestinal toxicity/ mucositis in treatment regimens that did not adjust doses to target a predetermined drug exposure level. In contrast, this association with toxicity was not observed in regimens that specified the desired exposure (rather than a prespecified dose). This suggests that altering drug dosing to account for alterations in clearance mediated by SLCO1B1 variation can ameliorate toxicity. Although genetic variants in SLCO1B1 accounted for greater variation in clearance than sex, ancestry, or age, these variants explained only 11% of the variation in clearance,. By comparison, dosing regimen accounted for 17% of the variability in drug clearance.
The association between these variants alleles and methotrexate clearance was validated in an additional study of 1,279 patients with ALL. (37) In addition to the lead SLCO1B1 single nucleotide polymorphism (SNP), 2 additional SNPs in this gene were associated with methotrexate clearance after adjusting for the lead SNP. Within this cohort, the SNPs accounted for approximately 2% of the variation in methotrexate clearance in contrast to a 38% difference in clearance associated with a 4-hour versus 24-hour infusion. Notably, the variability associated with genetics was still greater than other treatment factors such as the presence of a delayed intensification phase, age, or sex.
Although the findings of these studies have not led to preemptive pharmacogenetics dose adjustments, they remain significant because they have highlighted the importance of the SLCO1B1 transporter in the clearance of methotrexate. This highlights potential drug-drug interactions that are particularly important around high-dose methotrexate infusions. Notably, proton pump inhibitors, aminopenicillins, and statins may all act as inhibitors of SLCO1B1 and therefore result in increased methotrexate exposure and associated toxicities.(38)
In summary, interpatient and intrapatient variability in dosing regimen result in greater differences in toxicity and clearance than genetic variants. Thus, careful monitoring of patients receiving high-dose methotrexate is needed to ensure optimal therapeutic efficacy and minimize toxicity.
Asparaginase
Asparaginase deaminates the amino acids asparagine and, to a lesser extent, glutamine to form aspartic acid and glutamic acid, respectively. This depletes serum asparagine. While most cells can generate asparagine using asparagine synthetase, ALL blasts are unable to do so because they frequently lack asparagine synthetase expression. The use of asparaginase in pediatric ALL regimens is a key difference when compared to conventional adult ALL regimens. The effectiveness of asparaginase in pediatric ALL regimens has been well documented,(39–41) and improvements in outcomes for adults with ALL receiving pediatric regimens have been attributed to the addition of asparaginase to these therapies.(3)
Common toxicities experienced by patients receiving asparaginase include allergic reaction, thrombosis, hepatic injury, and pancreatitis. Allergic reactions can be either clinically apparent or associated with a subclinical immune response that results in rapid asparaginase clearance (“silent inactivation”) and therefore inadequate asparagine depletion.
Asparaginase allergy occurs in between 5 and 15% of patients receiving multiple doses of pegylated asparaginase during ALL therapy.(42–44) Anti-asparaginase antibodies are highly correlated with the development of clinical allergy, with almost all patients who experience a clinical allergy having anti-asparaginase antibodies.(42) Silent inactivation can also occur in the presence of antibodies and is associated with an inferior therapeutic response.(41, 45) Candidate gene studies evaluating major histone compatibility regions have identified the human leukocyte antigen (HLA)DRB1*07:01 haplotype as being associated with the development of asparaginase allergy.(46, 47) Patients with this haplotype had a 60% increased risk of developing clinical hypersensitivity and a 2.9 fold increased risk of developing anti-asparaginase antibodies.(47) This association was maintained in both patients that received native asparaginase (which was used historically but is no longer commercially available in the United States) as well as pegylated asparaginase which is currently used in frontline trials around the world. The mechanism of this association appears to be increased binding due to alterations in the amino acid sequence in the binding pocket of the HLA-DR. While less frequently seen in the population studied, associations were also observed between HLA-DRB1*04:05 and *04:08 haplotypes. Evaluations in a European cohort confirmed associations between HLA loci and the development of asparaginase allergy.(48) In that cohort, HLA–DQA1 was associated with an increased risk of asparaginase allergy, although individual patient haplotype’s were not assigned. This study also associated a variation in CNOT3, a gene associated with major histocompatibility class II expression, with the development of asparaginase allergy. In a multiethnic cohort of patients treated in the United States, variants in NFATC2 were associated with an increased development of asparaginase allergy.(46) Interestingly, this gene is located on chromosome 21 and is paradoxically inhibited in Down’s syndrome due to overexpression of competing repressive genes. This analysis also demonstrated that patients with Down’s syndrome have a lower incidence of asparaginase allergy than patients without constitutional trisomy 21.
Asparaginase hepatotoxicity appears to be more common in adults than in children receiving similar ALL therapy.(3, 4, 49) Attempts to identify genetic associations with asparaginase induced hepatotoxicity have included evaluations of both adult and pediatric cohorts. In one adult cohort, the 55 patients with the rs4880 CC genotype in SOD2 had a 2.5-fold increase in their risk of grade 3/4 ALT/AST/bilirubin increase compared to the 135 patients with either a CT or TT genotype.(50) Separately, GWAS in pediatric patients identified a variant in PNPLA3 as being associated with the development of hepatotoxicity during ALL induction.(51) The identified variant (rs738409) has also been associated with the development of fatty liver disease in adult populations. Although not clinically actionable, these data, combined with data demonstrating fat accumulation as a mechanism of asparaginase induced hepatic injury,(52, 53) suggests potential pathways underlying therapy toxicity which may be amenable to intervention. Extensive work has been undertaken to identify genetic risk factors for the development of asparaginase associated pancreatitis. Identified variants linked to increased risk of pancreatitis have varied across cohorts. A candidate gene evaluation of French-Canadian children treated on Dana-Farber protocols identified the *1 (double-repeat) allele of asparagine synthetase as being a risk factor for pancreatitis development (HR 8.6, 95%CI 2–37.3).(54) A study of a multiethnic cohort from the Children’s Oncology group and St. Jude identified Native American ancestry as being associated with pancreatitis development, particularly in patients carrying rare nonsense variants in CPA2 (HR 587, 95%CI 67–5166).(55) Evaluation of pancreatitis in a Nordic cohort identified rs281366 near ULK2 as associated with pancreatitis (HR 6.7, 95%CI 3.2–14).(56) Finally, a combined analysis of 10 therapy groups evaluated pancreatitis in a case/control cohort and identified an association with pancreatitis in an expression-quantitative trait locus for trypsinogen (rs13228878 and rs10273639) which was replicated in a Children’s Oncology Group cohort.(57) Despite these exciting findings, the inability to replicate these genetic risks consistently across populations suggests further study is needed before altering therapy prospectively due to their presence.
Although thrombosis is a commonly observed toxicity in children and young adults treated with asparaginase, studies have not identified consistent genetic risk factors. Notably, “classic” thrombophilia variants (e.g. factor V Leiden, prothrombin G20210A, homozygosity for MTHFR variation) have not been consistently shown to be associated with an increased risk of thrombosis.(58–61) Thus, clinical factors, rather than the presence of prothrombotic variation, appear to be the most consistent drivers of thrombosis risk during ALL therapy.
Vincristine
The vinca alkaloid vincristine blocks microtubule polymerization and is used in multiple phases of ALL therapy, frequently in combination with glucocorticoids. The most frequently encountered toxicity is peripheral motor and/ or sensory neuropathy. This appears to be dose related, with patients receiving higher single and cumulative doses experiencing more neuropathy.(62) Neuropathy also appears to be more common in older vs. younger children, although no differences were observed between adults and children treated with identical therapy.(5, 62)
Numerous evaluations have been undertaken to identify predisposing genetic factors to vincristine induced neuropathy (VIN). Because vincristine is metabolized by cytochrome P450 3A4 (CYP3A4) and CYP3A5, the literature is rife with reports of increased VIN when used with CYP3A4/5 inhibitors, particularly azole antifungal. Evaluations of CYP3A5 genotype and expression have suggested that polymorphisms which drive higher CYP3A5 expression may be associated with lower rates of VIN,(63, 64) although that finding has not been replicated in all treatment groups.(65)
More recently, GWAS identified an association between a variant (TT at rs924607) in the centrosomal protein 72 (CEP72) promoter and the development of VIN in children with ALL.(66) This variant was associated with decreased CEP72 expression, increased sensitivity of nerve cells to disruption by vincristine, and increased in vitro sensitivity of lymphoblasts to vincristine mediated cytotoxicity. The variant allelic frequency is 8% in African Ancestry, 40% in Europeans, and ~30–40% in Asian and Hispanic populations. This differential frequency may partly underly lower incidence of VIN in African American populations. Notably, the association is weakened with higher cumulative doses of vincristine, suggesting that the variant lowers the threshold for development of VIN but that increased dose intensity may abrogate the significance of the variant allele. The association between this variant and VIN was also replicated in an adult cohort of ALL patients, in which 75% of patients with the TT genotypes developed VIN compared to 44% of those with either CC or CT genotypes.(67) Prospective genotyping and dose reduction for patients with CEP72 TT alleles is currently being testing in the St. Jude Total 17 study (NCT03117751).
Glucocorticoids
The glucocorticoids prednisone and dexamethasone are backbone components of all therapies for ALL. Common adverse effects from steroids include hyperglycemia, hyperlipidemia, hypertension, osteopenia, and osteonecrosis.(59) Although these symptoms are typically transient, patients with hyperglycemia are at increased risk of developing type II diabetes as survivors.(68) Leukemia survivors with osteonecrosis also suffer diminished quality of life.(69, 70)
GWAS have attempted to identify risk variants for osteonecrosis. An analysis of St. Jude Total XV patients identified variants near ACP as being associated with the development of osteonecrosis.(71) This variant was also associated with higher lipid levels, a feature associated with an increased risk of osteonecrosis in a separate cohort.(72) Analysis in a pediatric Québécois and Dana-Farber cohort identified an association between BCL2L11 variants and osteonecrosis development.(73)
Work from St. Jude and the Children’s Oncology group in contemporary trials identified associations between variants in or near glutamate receptors and increased osteonecrosis risk.(74, 75) Genes linked to these variants were also associated with an increased risk of arterial embolism and thrombosis in a large adult cohort. These data suggest that glucocorticoid induced osteonecrosis is driven by vascular factors, a theory supported by both clinical and preclinical data.(76–78) Unfortunately, the variants identified in these studies await validation in external cohorts, with a recent analysis only confirming the association between ACP and osteonecrosis in a Dana-Farber/ French-Canadian cohort.(79) Based on the available data, it appears that further replication is needed before preemptively intervening to address them.
Conclusions
Pharmacogenomics offers the promise of a future where chemotherapy for ALL is tailored to maximize anti-leukemic efficacy while minimizing therapy toxicity. While the current list of pharmacogenes which are clinically actionable are limited (TPMT and NUDT15), work is ongoing to assess the effectiveness of interventions on other variants (e.g. CEP72 for vincristine). Moreover, findings from pharmacogenomic studies have elucidated additional mechanisms of disease toxicity which are potentially amenable to targeted interventions. A summary of these are found in Table 2. While many of the associations identified in pediatric studies have been replicated in adults, further study in this population is needed to understand gene/environment/age interactions in therapy response and toxicity. Such studies will be needed to unlock the future promised by pharmacogenomics.
Table 2:
Most notable current pharmacogenomic interactions
| Drug | Most notable pharmacogenomic interactions | Clinical implications; actionability |
|---|---|---|
| 6-mercaptopurine | TPMT, NUDT15 | Increased myelosuppression; preemptive dose decrease indicated |
| Methotrexate | SLCO1B1 | Decreased clearance; avoidance of inhibitors during high-dose therapy |
| Asparaginase | HLA DRB1*07:01, NFATC2 | Increased risk of allergy; no intervention yet available |
| Vincristine | CEP72 | Increased neuropathy; prospective trial of preemptive dose decrease ongoing |
Acknowledgements
The authors thank Cyrine Haidar, PharmD, for assistance with the development of Figure 2. This work was supported by the American Lebanese Syrian Associated Charities (ALSAC), R01GM118578 (Yang), P50GM115279 (Yang), and K08CA250418 (Karol). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
All authors: Declarations of interest: none.
References
- 1.Relling MV, Evans WE. Pharmacogenomics in the clinic. Nature. 2015;526(7573):343–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Evans WE, Relling MV. Moving towards individualized medicine with pharmacogenomics. Nature. 2004;429(6990):464–8. [DOI] [PubMed] [Google Scholar]
- 3.Stock W, Luger SM, Advani AS, Yin J, Harvey RC, Mullighan CG, et al. A pediatric regimen for older adolescents and young adults with acute lymphoblastic leukemia: results of CALGB 10403. Blood. 2019;133(14):1548–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Larsen EC, Devidas M, Chen S, Salzer WL, Raetz EA, Loh ML, et al. Dexamethasone and High-Dose Methotrexate Improve Outcome for Children and Young Adults With High-Risk B-Acute Lymphoblastic Leukemia: A Report From Children’s Oncology Group Study AALL0232. J Clin Oncol. 2016;34(20):2380–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Toft N, Birgens H, Abrahamsson J, Griskevicius L, Hallbook H, Heyman M, et al. Results of NOPHO ALL2008 treatment for patients aged 1–45 years with acute lymphoblastic leukemia. Leukemia. 2018;32(3):606–15. [DOI] [PubMed] [Google Scholar]
- 6.Schmiegelow K, Nielsen SN, Frandsen TL, Nersting J. Mercaptopurine/Methotrexate maintenance therapy of childhood acute lymphoblastic leukemia: clinical facts and fiction. Journal of pediatric hematology/oncology. 2014;36(7):503–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stork LC, Matloub Y, Broxson E, La M, Yanofsky R, Sather H, et al. Oral 6-mercaptopurine versus oral 6-thioguanine and veno-occlusive disease in children with standard-risk acute lymphoblastic leukemia: report of the Children’s Oncology Group CCG-1952 clinical trial. Blood. 2010;115(14):2740–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Evans WE, Horner M, Chu YQ, Kalwinsky D, Roberts WM. Altered mercaptopurine metabolism, toxic effects, and dosage requirement in a thiopurine methyltransferase-deficient child with acute lymphocytic leukemia. J Pediatr. 1991;119(6):985–9. [DOI] [PubMed] [Google Scholar]
- 9.Relling MV, Hancock ML, Rivera GK, Sandlund JT, Ribeiro RC, Krynetski EY, et al. Mercaptopurine therapy intolerance and heterozygosity at the thiopurine S-methyltransferase gene locus. J Natl Cancer Inst. 1999;91(23):2001–8. [DOI] [PubMed] [Google Scholar]
- 10.Liu C, Yang W, Pei D, Cheng C, Smith C, Landier W, et al. Genomewide Approach Validates Thiopurine Methyltransferase Activity Is a Monogenic Pharmacogenomic Trait. Clin Pharmacol Ther. 2017;101(3):373–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dubinsky MC, Lamothe S, Yang HY, Targan SR, Sinnett D, Theoret Y, et al. Pharmacogenomics and metabolite measurement for 6-mercaptopurine therapy in inflammatory bowel disease. Gastroenterology. 2000;118(4):705–13. [DOI] [PubMed] [Google Scholar]
- 12.Krynetski EY, Krynetskaia NF, Yanishevski Y, Evans WE. Methylation of Mercaptopurine, Thioguanine, and Their Nucleotide Metabolites by Heterologously Expressed Human Thiopurine S-Methyltransferase. Mol Pharmacol. 1995;47(6):1141–7. [PubMed] [Google Scholar]
- 13.Gardiner SJ, Gearry RB, Burt MJ, Ding SL, Barclay ML. Severe hepatotoxicity with high 6-methylmercaptopurine nucleotide concentrations after thiopurine dose escalation due to low 6-thioguanine nucleotides. Eur J Gastroen Hepat. 2008;20(12):1238–42. [DOI] [PubMed] [Google Scholar]
- 14.Lennard L, Rees CA, Lilleyman JS, Maddocks JL. Childhood Leukemia - a Relationship between Intracellular 6-Mercaptopurine Metabolites and Neutropenia. Brit J Clin Pharmaco. 1983;16(4):359–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McLeod HL, Relling MV, Liu Q, Pui CH, Evans WE. Polymorphic thiopurine methyltransferase in erythrocytes is indicative of activity in leukemic blasts from children with acute lymphoblastic leukemia. Blood. 1995;85(7):1897–902. [PubMed] [Google Scholar]
- 16.Bhatia S, Landier W, Hageman L, Chen Y, Kim H, Sun CL, et al. Systemic Exposure to Thiopurines and Risk of Relapse in Children With Acute Lymphoblastic Leukemia: A Children’s Oncology Group Study. JAMA oncology. 2015;1(3):287–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Landier W, Hageman L, Chen YJ, Kornegay N, Evans WE, Bostrom BC, et al. Mercaptopurine Ingestion Habits, Red Cell Thioguanine Nucleotide Levels, and Relapse Risk in Children With Acute Lymphoblastic Leukemia: A Report From the Children’s Oncology Group Study AALL03N1. J Clin Oncol. 2017;35(15):1730–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nielsen SN, Grell K, Nersting J, Abrahamsson J, Lund B, Kanerva J, et al. DNA-thioguanine nucleotide concentration and relapse-free survival during maintenance therapy of childhood acute lymphoblastic leukaemia (NOPHO ALL2008): a prospective substudy of a phase 3 trial. Lancet Oncol. 2017;18(4):515–24. [DOI] [PubMed] [Google Scholar]
- 19.Sudmant PH, Rausch T, Gardner EJ, Handsaker RE, Abyzov A, Huddleston J, et al. An integrated map of structural variation in 2,504 human genomes. Nature. 2015;526(7571):75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Genomes Project C, Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491(7422):56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Relling MV, Schwab M, Whirl-Carrillo M, Suarez-Kurtz G, Pui CH, Stein CM, et al. Clinical Pharmacogenetics Implementation Consortium Guideline for Thiopurine Dosing Based on TPMT and NUDT15 Genotypes: 2018 Update. Clin Pharmacol Ther. 2019;105(5):1095–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Relling MV, Yanishevski Y, Nemec J, Evans WE, Boyett JM, Behm FG, et al. Etoposide and antimetabolite pharmacology in patients who develop secondary acute myeloid leukemia. Leukemia. 1998;12(3):346–52. [DOI] [PubMed] [Google Scholar]
- 23.Schmiegelow K, Bjork O, Glomstein A, Gustafsson G, Keiding N, Kristinsson J, et al. Intensification of mercaptopurine/methotrexate maintenance chemotherapy may increase the risk of relapse for some children with acute lymphoblastic leukemia. J Clin Oncol. 2003;21(7):1332–9. [DOI] [PubMed] [Google Scholar]
- 24.Schaeffeler E, Jaeger SU, Klumpp V, Yang JJ, Igel S, Hinze L, et al. Impact of NUDT15 genetics on severe thiopurine-related hematotoxicity in patients with European ancestry. Genet Med. 2019;21(9):2145–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang JJ, Landier W, Yang W, Liu C, Hageman L, Cheng C, et al. Inherited NUDT15 variant is a genetic determinant of mercaptopurine intolerance in children with acute lymphoblastic leukemia. J Clin Oncol. 2015;33(11):1235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moriyama T, Nishii R, Perez-Andreu V, Yang W, Klussmann FA, Zhao X, et al. NUDT15 polymorphisms alter thiopurine metabolism and hematopoietic toxicity. Nat Genet. 2016;48(4):367–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ramsey LB, Janke LJ, Edick MJ, Cheng C, Williams RT, Sherr CJ, et al. Host thiopurine methyltransferase status affects mercaptopurine antileukemic effectiveness in a murine model. Pharmacogenetics and genomics. 2014;24(5):263–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stanulla M, Schaeffeler E, Flohr T, Cario G, Schrauder A, Zimmermann M, et al. Thiopurine methyltransferase (TPMT) genotype and early treatment response to mercaptopurine in childhood acute lymphoblastic leukemia. JAMA. 2005;293(12):1485–9. [DOI] [PubMed] [Google Scholar]
- 29.Stocco G, Cheok MH, Crews KR, Dervieux T, French D, Pei D, et al. Genetic polymorphism of inosine triphosphate pyrophosphatase is a determinant of mercaptopurine metabolism and toxicity during treatment for acute lymphoblastic leukemia. Clin Pharmacol Ther. 2009;85(2):164–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nishii R, Moriyama T, Janke LJ, Yang W, Suiter CC, Lin TN, et al. Preclinical evaluation of NUDT15-guided thiopurine therapy and its effects on toxicity and antileukemic efficacy. Blood. 2018;131(22):2466–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moriyama T, Yang YL, Nishii R, Ariffin H, Liu C, Lin TN, et al. Novel variants in NUDT15 and thiopurine intolerance in children with acute lymphoblastic leukemia from diverse ancestry. Blood. 2017;130(10):1209–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Methotrexate. LiverTox: Clinical and Research Information on Drug-Induced Liver Injury. Bethesda (MD)2012. [PubMed] [Google Scholar]
- 33.Taylor OA, Brown AL, Brackett J, Dreyer ZE, Moore IK, Mitby P, et al. Disparities in Neurotoxicity Risk and Outcomes among Pediatric Acute Lymphoblastic Leukemia Patients. Clin Cancer Res. 2018;24(20):5012–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bhojwani D, Sabin ND, Pei D, Yang JJ, Khan RB, Panetta JC, et al. Methotrexate-induced neurotoxicity and leukoencephalopathy in childhood acute lymphoblastic leukemia. J Clin Oncol. 2014;32(9):949–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu W, Cheung YT, Conklin HM, Jacola LM, Srivastava D, Nolan VG, et al. Evolution of neurocognitive function in long-term survivors of childhood acute lymphoblastic leukemia treated with chemotherapy only. J Cancer Surviv. 2018;12(3):398–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Trevino LR, Shimasaki N, Yang W, Panetta JC, Cheng C, Pei D, et al. Germline genetic variation in an organic anion transporter polypeptide associated with methotrexate pharmacokinetics and clinical effects. J Clin Oncol. 2009;27(35):5972–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ramsey LB, Panetta JC, Smith C, Yang W, Fan Y, Winick NJ, et al. Genome-wide study of methotrexate clearance replicates SLCO1B1. Blood. 2013;121(6):898–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bezabeh S, Mackey AC, Kluetz P, Jappar D, Korvick J. Accumulating Evidence for a Drug-Drug Interaction Between Methotrexate and Proton Pump Inhibitors. Oncologist. 2012;17(4):550–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jeha S, Pei D, Choi J, Cheng C, Sandlund JT, Coustan-Smith E, et al. Improved CNS Control of Childhood Acute Lymphoblastic Leukemia Without Cranial Irradiation: St Jude Total Therapy Study 16. J Clin Oncol. 2019;37(35):3377–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vrooman LM, Stevenson KE, Supko JG, O’Brien J, Dahlberg SE, Asselin BL, et al. Postinduction dexamethasone and individualized dosing of Escherichia Coli L-asparaginase each improve outcome of children and adolescents with newly diagnosed acute lymphoblastic leukemia: results from a randomized study--Dana-Farber Cancer Institute ALL Consortium Protocol 00–01. J Clin Oncol. 2013;31(9):1202–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Silverman LB, Gelber RD, Dalton VK, Asselin BL, Barr RD, Clavell LA, et al. Improved outcome for children with acute lymphoblastic leukemia: results of Dana-Farber Consortium Protocol 91–01. Blood. 2001;97(5):1211–8. [DOI] [PubMed] [Google Scholar]
- 42.Liu Y, Smith CA, Panetta JC, Yang W, Thompson LE, Counts JP, et al. Antibodies Predict Pegaspargase Allergic Reactions and Failure of Rechallenge. J Clin Oncol. 2019;37(23):2051–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mondelaers V, Suciu S, De Moerloose B, Ferster A, Mazingue F, Plat G, et al. Prolonged versus standard native E. coli asparaginase therapy in childhood acute lymphoblastic leukemia and non-Hodgkin lymphoma: final results of the EORTC-CLG randomized phase III trial 58951. Haematologica. 2017;102(10):1727–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Henriksen LT, Harila-Saari A, Ruud E, Abrahamsson J, Pruunsild K, Vaitkeviciene G, et al. PEG-Asparaginase Allergy in Children With Acute Lymphoblastic Leukemia in the NOPHO ALL2008 Protocol. Pediatric blood & cancer. 2015;62(3):427–33. [DOI] [PubMed] [Google Scholar]
- 45.Kawedia JD, Liu C, Pei D, Cheng C, Fernandez CA, Howard SC, et al. Dexamethasone exposure and asparaginase antibodies affect relapse risk in acute lymphoblastic leukemia. Blood. 2012;119(7):1658–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fernandez CA, Smith C, Yang W, Mullighan CG, Qu C, Larsen E, et al. Genome-wide analysis links NFATC2 with asparaginase hypersensitivity. Blood. 2015;126(1):69–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fernandez CA, Smith C, Yang W, Date M, Bashford D, Larsen E, et al. HLA-DRB1*07:01 is associated with a higher risk of asparaginase allergies. Blood. 2014;124(8):1266–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hojfeldt SG, Wolthers BO, Tulstrup M, Abrahamsson J, Gupta R, Harila-Saari A, et al. Genetic predisposition to PEG-asparaginase hypersensitivity in children treated according to NOPHO ALL2008. Br J Haematol. 2019;184(3):405–17. [DOI] [PubMed] [Google Scholar]
- 49.Patel B, Kirkwood AA, Dey A, Marks DI, McMillan AK, Menne TF, et al. Pegylated-asparaginase during induction therapy for adult acute lymphoblastic leukaemia: toxicity data from the UKALL14 trial. Leukemia. 2017;31(1):58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alachkar H, Fulton N, Sanford B, Malnassy G, Mutonga M, Larson RA, et al. Expression and polymorphism (rs4880) of mitochondrial superoxide dismutase (SOD2) and asparaginase induced hepatotoxicity in adult patients with acute lymphoblastic leukemia. Pharmacogenomics J. 2017;17(3):274–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu Y, Fernandez CA, Smith C, Yang W, Cheng C, Panetta JC, et al. Genome-Wide Study Links PNPLA3 Variant With Elevated Hepatic Transaminase After Acute Lymphoblastic Leukemia Therapy. Clin Pharmacol Ther. 2017;102(1):131–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kamal N, Koh C, Samala N, Fontana RJ, Stolz A, Durazo F, et al. Asparaginase-induced hepatotoxicity: rapid development of cholestasis and hepatic steatosis. Hepatol Int. 2019;13(5):641–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu Y, Janke LJ, Li L, Relling MV. L-carnitine does not ameliorate asparaginase-associated hepatotoxicity in a C57BL6 mouse model. Leukemia & lymphoma. 2019;60(8):2088–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ben Tanfous M, Sharif-Askari B, Ceppi F, Laaribi H, Gagne V, Rousseau J, et al. Polymorphisms of asparaginase pathway and asparaginase-related complications in children with acute lymphoblastic leukemia. Clin Cancer Res. 2015;21(2):329–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu C, Yang W, Devidas M, Cheng C, Pei D, Smith C, et al. Clinical and Genetic Risk Factors for Acute Pancreatitis in Patients With Acute Lymphoblastic Leukemia. J Clin Oncol. 2016;34(18):2133–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wolthers BO, Frandsen TL, Abrahamsson J, Albertsen BK, Helt LR, Heyman M, et al. Asparaginase-associated pancreatitis: a study on phenotype and genotype in the NOPHO ALL2008 protocol. Leukemia. 2017;31(2):325–32. [DOI] [PubMed] [Google Scholar]
- 57.Wolthers BO, Frandsen TL, Patel CJ, Abaji R, Attarbaschi A, Barzilai S, et al. Trypsin-encoding PRSS1-PRSS2 variations influence the risk of asparaginase-associated pancreatitis in children with acute lymphoblastic leukemia: a Ponte di Legno toxicity working group report. Haematologica. 2019;104(3):556–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jarvis KB, LeBlanc M, Tulstrup M, Nielsen RL, Albertsen BK, Gupta R, et al. Candidate single nucleotide polymorphisms and thromboembolism in acute lymphoblastic leukemia - A NOPHO ALL2008 study. Thromb Res. 2019;184:92–8. [DOI] [PubMed] [Google Scholar]
- 59.Ramsey LB, Pounds S, Cheng C, Cao X, Yang W, Smith C, et al. Genetics of pleiotropic effects of dexamethasone. Pharmacogenetics and genomics. 2017;27(8):294–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mauz-Korholz C, Junker R, Gobel U, Nowak-Gottl U. Prothrombotic risk factors in children with acute lymphoblastic leukemia treated with delayed E. coli asparaginase (COALL-92 and 97 protocols). Thromb Haemost. 2000;83(6):840–3. [PubMed] [Google Scholar]
- 61.Wermes C, von Depka Prondzinski M, Lichtinghagen R, Barthels M, Welte K, Sykora KW. Clinical relevance of genetic risk factors for thrombosis in paediatric oncology patients with central venous catheters. Eur J Pediatr. 1999;158 Suppl 3:S143–6. [DOI] [PubMed] [Google Scholar]
- 62.Lew G, Chen Y, Lu X, Rheingold SR, Whitlock JA, Devidas M, et al. Outcomes after late bone marrow and very early central nervous system relapse of childhood B-Acute lymphoblastic leukemia: a report from the Children’s Oncology Group phase III study AALL0433. Haematologica. 2020. [DOI] [PMC free article] [PubMed]
- 63.Kocak U, Kayilioglu H, Karaer DK, Percin EP, Okur A, Karadeniz C. Relationship of Cyp3a5 Expression and Vincristine Neurotoxicity in Turkish Children with Malignancy. Pediatric blood & cancer. 2014;61:S349–S. [Google Scholar]
- 64.Egbelakin A, Ferguson MJ, MacGill EA, Lehmann AS, Topletz AR, Quinney SK, et al. Increased Risk of Vincristine Neurotoxicity Associated With Low CYP3A5 Expression Genotype in Children With Acute Lymphoblastic Leukemia. Pediatric blood & cancer. 2011;56(3):361–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ceppi F, Langlois-Pelletier C, Gagne V, Rousseau J, Ciolino C, De Lorenzo S, et al. Polymorphisms of the vincristine pathway and response to treatment in children with childhood acute lymphoblastic leukemia. Pharmacogenomics. 2014;15(8):1105–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Diouf B, Crews KR, Lew G, Pei D, Cheng C, Bao J, et al. Association of an inherited genetic variant with vincristine-related peripheral neuropathy in children with acute lymphoblastic leukemia. JAMA. 2015;313(8):815–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stock W, Diouf B, Crews KR, Pei D, Cheng C, Laumann K, et al. An Inherited Genetic Variant in CEP72 Promoter Predisposes to Vincristine-Induced Peripheral Neuropathy in Adults With Acute Lymphoblastic Leukemia. Clin Pharmacol Ther. 2017;101(3):391–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Williams HE, Howell CR, Chemaitilly W, Wilson CL, Karol SE, Nolan VG, et al. Diabetes mellitus among adult survivors of childhood acute lymphoblastic leukemia: A report from the St. Jude Lifetime Cohort Study. Cancer. 2020;126(4):870–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.DeFeo BM, Kaste SC, Li Z, Brinkman TM, Neel MD, Srivastava DK, et al. Long-Term Functional Outcomes Among Childhood Survivors of Cancer Who Have a History of Osteonecrosis. Phys Ther. 2020. [DOI] [PMC free article] [PubMed]
- 70.Girard P, Auquier P, Barlogis V, Contet A, Poiree M, Demeocq F, et al. Symptomatic osteonecrosis in childhood leukemia survivors: prevalence, risk factors and impact on quality of life in adulthood. Haematologica. 2013;98(7):1089–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kawedia JD, Kaste SC, Pei D, Panetta JC, Cai X, Cheng C, et al. Pharmacokinetic, pharmacodynamic, and pharmacogenetic determinants of osteonecrosis in children with acute lymphoblastic leukemia. Blood. 2011;117(8):2340–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Finch ER, Smith CA, Yang W, Liu Y, Kornegay NM, Panetta JC, et al. Asparaginase formulation impacts hypertriglyceridemia during therapy for acute lymphoblastic leukemia. Pediatric blood & cancer. 2020;67(1):e28040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Plesa M, Gagne V, Glisovic S, Younan M, Sharif-Askari B, Laverdiere C, et al. Influence of BCL2L11 polymorphism on osteonecrosis during treatment of childhood acute lymphoblastic leukemia. Pharmacogenomics J. 2019;19(1):33–41. [DOI] [PubMed] [Google Scholar]
- 74.Karol SE, Mattano LA Jr., Yang W, Maloney KW, Smith C, Liu C, et al. Genetic risk factors for the development of osteonecrosis in children under age 10 treated for acute lymphoblastic leukemia. Blood. 2016;127(5):558–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Karol SE, Yang W, Van Driest SL, Chang TY, Kaste S, Bowton E, et al. Genetics of glucocorticoid-associated osteonecrosis in children with acute lymphoblastic leukemia. Blood. 2015;126(15):1770–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Janke LJ, Van Driest SL, Portera MV, Atreya RV, Denny JC, Pei D, et al. Hypertension is a modifiable risk factor for osteonecrosis in acute lymphoblastic leukemia. Blood. 2019;134(12):983–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Janke LJ, Liu C, Vogel P, Kawedia J, Boyd KL, Funk AJ, et al. Primary epiphyseal arteriopathy in a mouse model of steroid-induced osteonecrosis. Am J Pathol. 2013;183(1):19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Saito S, Ohzono K, Ono K. Early arteriopathy and postulated pathogenesis of osteonecrosis of the femoral head. The intracapital arterioles. Clin Orthop Relat Res. 1992(277):98–110. [PubMed] [Google Scholar]
- 79.Gagne V, Aubry-Morin A, Plesa M, Abaji R, Petrykey K, St-Onge P, et al. Genes identified through genome-wide association studies of osteonecrosis in childhood acute lymphoblastic leukemia patients. Pharmacogenomics. 2019;20(17):1189–97. [DOI] [PMC free article] [PubMed] [Google Scholar]