

Summary
STAT3 is a transcription factor involved in several cellular activities including inflammation, proliferation, and survival, but it also plays a non-transcriptional role in modulating mitochondrial metabolism. Given its diverse functions in human cancers, it is an emerging therapeutic target. Here we show that OPB-51602, a small molecule inhibitor of STAT3, is highly toxic in a STAT3-dependent manner. Specifically, drug toxicity depends on mitochondrial STAT3 as tumor cells expressing only a mitochondrially restricted form of STAT3 are sensitive to the compound, whereas STAT3-null cells are protected. OPB-51602 inhibited complex I activity and led to increased ROS production, which in turn induced mitophagy, actin rearrangements, and cell death. Cells undergoing reduced oxidative phosphorylation or expressing NDI1 NADH dehydrogenase from Saccharomyces cerevisiae, which bypasses mammalian complex I, were resistant to OPB-51602 toxicity. These results show that targeting mitochondrial STAT3 function causes synthetic lethality through complex I inhibition that could be exploited for cancer chemotherapy.
Subject Areas: Cell Biology, Cancer
Graphical Abstract
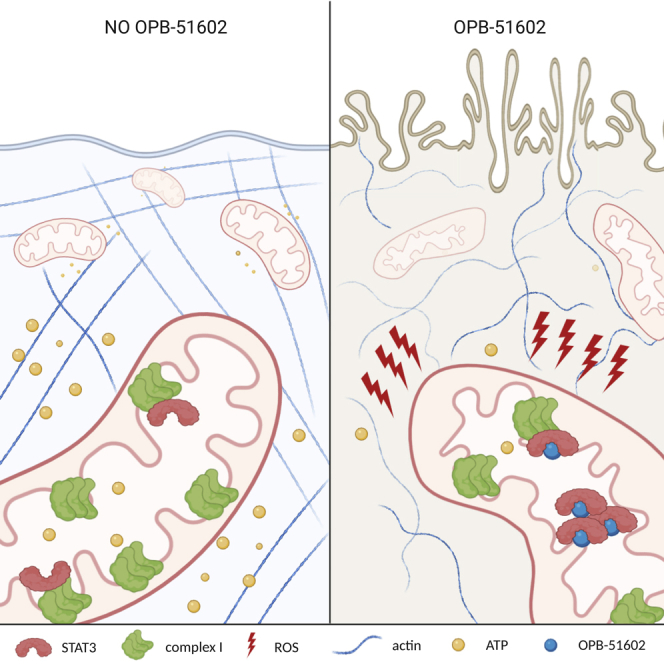
Highlights
-
•
OPB-51602 is cytotoxic to human tumor cell lines in a STAT3-dependent manner
-
•
Cytotoxicity depends on ROS induction and leads to mitophagy and actin remodeling
-
•
OPB-51602 affects oxidative phosphorylation by inhibiting complex I via STAT3
-
•
Expression of a STAT3-independent form of complex I is cytoprotective
Cell Biology; Cancer
Introduction
Signal transducer and activator of transcription (STAT) proteins are latent cytoplasmic transcription factors that can be activated by receptor-mediated tyrosine phosphorylation to regulate cell growth, survival, and differentiation (Levy and Darnell, 2002). There are seven STAT family members, STAT1, 2, 3, 4, 5a, 5b, and 6, and they all share extensive structural homology consisting of six basic motifs. The N-terminal domain (NTD) and the coiled-coil domain (CCD) are required for receptor recruitment, nuclear translocation, tetramerization, and protein-protein interactions, whereas the DNA-binding domain (DBD) is needed to recognize specific elements in the promoters of target genes (Vinkemeier et al., 1996; Vogt et al., 2011; Zhang and Darnell, 2001). The Src homology 2 (SH2) domain, the most conserved one, is involved in receptor recruitment and in the formation of dimers via interaction with specific phosphotyrosine residues in the C-terminal transactivation domain (CTD) of another STAT monomer (Becker et al., 1998; Chakraborty et al., 1999; Hemmann et al., 1996). STAT3 is a member of this family best known for transducing extracellular signals to promote cell proliferation, survival, angiogenesis and immuno-evasion (Horvath, 2000; Yu et al., 2009). Usually catalyzed by members of the Janus kinase (JAK) family, phosphorylation at Tyr705 stabilizes STAT3 dimerization via its interaction with the SH2 domain of another STAT3 monomer, leading to nuclear accumulation and transcription of specific target genes (Levy and Darnell, 2002).
Numerous studies have documented roles for activated STAT3 in human cancer, both intrinsically and in the tumor microenvironment. Accumulating evidence also indicates that STAT3 mediates metabolic processes in tumor cells, some of which require its accumulation in mitochondria in a pTyr705-independent manner (Avalle and Poli, 2018; Yu et al., 2014). In particular, mitochondrially localized STAT3 (mito-STAT3) has been shown to play a key role in the survival of RAS-transformed cancers, contributing to aerobic glycolysis and oxidative phosphorylation (OXPHOS) (Avalle and Poli, 2018; Demaria et al., 2010; Garama et al., 2015; Gough et al., 2009, Gough et al., 2012, 2014; Szczepanek et al., 2012a; Wegrzyn et al., 2009; Zhang et al., 2013). Owing to its pivotal roles in oncogenic processes, STAT3 is an attractive cancer therapeutic target (Yu et al., 2009; Yue and Turkson, 2009). Numerous direct small-molecule STAT3 inhibitors (STAT3i) have been developed, some of which have shown encouraging activity in preclinical models and have progressed to ongoing clinical trials (Gelain et al., 2019; Yoo et al., 2019). Much of the attention has been focused on the inhibition of nuclear and transcriptional functions of STAT3. However, blocking pTyr705 would not reverse the metabolic consequences mediated by mito-STAT3. The emerging understanding of mitochondria as central organelles in metabolic adaptation, drug resistance, and stemness in human cancers makes this issue timely and relevant (Lee et al., 2019).
In this study, we investigated how a novel small-molecule STAT3i, OPB-51602 (also referred to as OPB), currently in clinical trials for human cancer, interferes with STAT3 metabolic functions in cancer cells (Genini et al., 2017; Hirpara et al., 2019; Ogura et al., 2015; Wong et al., 2015). Our data show that OPB-51602 is toxic for human non-small cell lung carcinoma (NSCLC) and triple negative breast cancer (TNBC) cells in a STAT3-dependent manner, despite the ability of these cells to survive in the absence of STAT3. Indeed, OPB-51602 appears to induce a synthetic lethality dependent on its ability to impair OXPHOS through a STAT3-dependent inhibition of respiratory complex I, leading to cell death.
Results
Depletion of STAT3 Reduces OPB-51602 Toxicity, which Is Restored by Mitochondrial STAT3
To extend previous data that showed that OPB-51602 represses the proliferation of numerous human cancer cell lines (Genini et al., 2017), we assessed its effect on four human NSCLC cell lines, H522, H2228, H23, and A549, and two TNBC cell lines, MDA-MB-231 and MDA-MB-468. Similar to data obtained from other cell types, OPB-51602 was cytotoxic at nanomolar concentrations, with an IC50 between 0.5 and 2.8 nM (Figure 1A). Cytotoxicity by OPB-51602 was observed as decreased metabolic activity by measuring reductive capacity using the CCK8 assay, decreased cell adhesion by using crystal violet staining, and decreased cell viability by using trypan blue exclusion (Figure S1A). Notably, although there were some quantitative differences among the three methods, overall results were comparable. Previous work showed that OPB-51602 directly binds to the STAT3 SH2 domain, inducing the formation of STAT3-containing protein aggregates that eventually lead to functional depletion of STAT3 from cells (Genini et al., 2017). To directly assess the role of STAT3 in toxicity, we tested the hypothesis that STAT3 depletion from cells prior to treatment would lead to decreased sensitivity to OPB-51602 toxicity, compared with their wild-type (WT) counterparts. To this end, we targeted STAT3 expression by RNA interference (Figure 1C) and observed that STAT3-depleted cells were protected from drug-induced cytotoxicity, although to varying degrees depending on the cell line (Figure 1B). To exclude the possibility that residual adverse effects of OPB-51602 could have been dependent on the remaining levels of STAT3, we produced STAT3 knockout A549, MDA-MB-231, and MDA-MB-468 cells by using the CRISPR-Cas9 system (Figures 1E and 1I). We tested OPB-51602 toxicity in the resulting stable STAT3 KO cell pool (KOp) in comparison with control WT cells (Figures 1D and 1H) and observed that STAT3 KO cells were highly resistant but still retained some residual sensitivity to the compound. We next derived multiple single clones from STAT3 KO pool (e.g., Clone 1; Figure 1G), which were also extremely resistant even to high concentrations of the inhibitor (Figure 1F). These data suggest that STAT3 is the primary mediator of OPB-51602 toxicity but that there is also residual STAT3-independent toxicity. We also measured the IC50 across multiple tumorigenic cell lines (Figure S1E) and several individual A549 clones selected for drug resistance (Figure S1C). Interestingly, drug sensitivity did not correlate with basal expression levels of STAT3 (Figures S1B, S1D, S1F and S1G). Although STAT3 is a cytokine-activated transcription factor, it also mediates functions in mitochondria in the absence of cytokine stimulation (Avalle and Poli, 2018). To test that mitochondrial STAT3 was responsible for OPB-51602 toxicity, we complemented STAT3-KO cells, both pool and individual clones, with either full-length STAT3 (WT) or a mitochondrially restricted form (MTS) (Figures 1K, M, and S1H) and measured drug sensitivity. Re-expression of STAT3 restored sensitivity to the drug to levels, which was comparable with that of WT cells (Figures 1J and L). Notably, mitochondrially restricted STAT3 that accumulates exclusively in mitochondria and is unable to mediate nuclear function was as effective as the wild-type protein in terms of restoring sensitivity to OPB-51602 toxicity. Since mitochondrial STAT3 has been documented in non-transformed cells and to mediate a variety of functions (Szczepanek et al., 2012b; Mantel et al., 2012), we evaluated the effect of OPB-51602 also on normal lung and mammary epithelial cell lines. HBEC3-KT and MCF 10A treated with OPB-51602 were less sensitive compared with A549 and MDA-MB-231, respectively (Figure 1N), in spite of expressing STAT3 protein to similar levels. Lastly, owing to the homology between STAT1 and STAT3, particularly within their SH2 domains, we tested whether absence of STAT1 would also protect cells from OPB-51602 toxicity. However, we observed no protection by deleting STAT1, confirming that STAT3 is the major target (Figure 1O and 1P).
Figure 1.

Depletion of STAT3 Reduces OPB-51602 Toxicity, which Is Restored by Mitochondrial STAT3
(A) Cell viability of human NSCLC and TNBC cell lines incubated with OPB-51602 for 16 h and analyzed with CCK8.
(B) Cell viability of NSCLC cell lines, STAT3 WT versus KD, incubated with OPB-51602 for 16 h and analyzed with CCK8.
(C) Knockdown of STAT3 in NSCLC analyzed by WB. Cell viability of A549 STAT3 WT versus KO pool (D) or Clone 1 (F) and of MDA-MB-231 and MDA-MB-468 STAT3 WT versus KO pool (H) incubated with OPB-51602 for 16 h analyzed with CCK8. Knockout of STAT3 in A549 (E: pool and (G) Clone 1) and MDA-MB-231 and MDA-MB-468 cells (I) analyzed by WB. Cell viability of A549 STAT3 KO pool (J) and Clone 1 (L) reconstituted with STAT3-WT and STAT3-MTS incubated with OPB-51602 for 16 h analyzed with CCK8. Re-expression of STAT3 in A549 KO pool (K) and Clone 1 (M) analyzed by WB.
(N) Cell viability in HBEC3-KT and MCF 10A compared with A549 and MDA-MB-231 analyzed with crystal violet.
(O) Cell viability in HT1080 STAT1 WT and KO analyzed with CCK8. In (A), (B), (D), (F), (H), (J), (L), (N), and (O) data are represented as mean ± SEM; n ≥ 3. Unpaired two-tailed Student's t test; ∗p < 0.05, ∗∗p < 0.002, ∗∗∗∗p < 0.0001. See also Figure S1. Molecular weight is expressed in kDa.
OPB-51602 Induces ROS Production Resulting in Mitophagy and Cell Death in a STAT3-Dependent Manner
Since MTS-STAT3 is unable to mediate transcriptional responses (Gough et al., 2009) but mediates OPB-51602 toxicity, we were prompted to investigate direct mitochondrial actions of the compound. ROS production, a measure of damaged mitochondria (Fan et al., 2019), was significantly increased following 2-h incubation with the drug (Figure 2A). Interestingly, ROS production was not stimulated by OPB-51602 treatment in the absence of STAT3 (Figure 2A) and it was partially quenchable by a pre-treatment with the cell-permeable form of glutathione (GSH-EE), an effective anti-oxidant (Figure 2B). Notably, the addition of the antioxidant was sufficient to greatly increase WST-8 dye reduction after OPB-51602 treatment (Figure 2C), confirming that ROS production induced by the compound contributes to cell death. The cytotoxic effect of OPB-51602 on STAT3-expressing cells is time dependent (Figure 2D), and dead cells are visible as early as 4 h after treatment. Moreover, consistent with a reduced cell toxicity (Figures 1D, 1F and 1H), cell death greatly diminished (3-fold less) in drug-treated A549 STAT3 KO cells, confirming that the presence of STAT3 is important to mediate OPB-51602 effects (Figure 2E). Compound-treated cells became Annexin V positive and permeable to PI, consistent with apoptosis/necrosis. Notably, OPB-51602 treatment did not affect cell cycle parameters (Figure S2A). As damaged mitochondria are physiologically removed by mitophagy (Pickles et al., 2018), we next assessed the effects of OPB-51602 on this process. We hypothesized that, if OPB-51602 functioned as a STAT3-dependent mitochondrial toxin, drug treatment would increase mitophagy. We measured mitophagy in living cells using the fluorescent mt-mKeima system (Lazarou et al., 2015). As shown in Figure 2F, treatment with OPB-51602 induced mitophagy to the same extent as fully decoupled mitochondria following FCCP treatment, a known mitophagy inducer (Berezhnov et al., 2016). However, pre-treatment with chloroquine, an autophagy inhibitor, did not alter the effects of OPB-51602 (Figure S2B), suggesting that mitophagy is probably a cellular response to drug-induced mitochondria damage rather than an attempt to prevent cell death. We also observed that prolonged exposure to the inhibitor induced an actin rearrangement resulting in the formation of actin protrusions resembling tunneling nanotubes (Rustom et al., 2004) (Figure 2G and S2C). These actin formations were reduced in STAT3 KO cells treated with the compound, suggesting that disruption of the actin cytoskeleton could be involved in the initiation of OPB-51602 STAT3-dependent toxicity (Figure 2H and S2C). These actin filaments did not appear to be signs of loss of cell adhesion or retraction fibers, as they were packed with f-actin (Figure S2D). Interference reflection microscopy (IRM) showed these protrusions only loosely pressed against the attachment substrate and the cells, which acquired a balled-up appearance, displayed long actin-filled structures resembling extended filopods that were not attachment points to the substrate. OPB-51602 also reduced cell migration in A549 STAT3 WT (Figure 2I) and, to a lesser extent, in STAT3 KO (Figure S2E), confirming previous reports of a role for STAT3 in the migratory capacity of cells (Verma et al., 2009; Akira, 2000). Lastly, we previously showed that OPB-51602 treatment leads to the formation of STAT3 aggregates, which also included other proteins such as autophagy-related protein p62/Sqstm1 (Moscat et al., 2016) and HDAC6 (Genini et al., 2017). We observed aggregate formation also in NSCLC cells treated with OPB-51602, as demonstrated by depletion of STAT3 and p62 from soluble extracts and their translocation to an insoluble fraction (Figure 2J). However, OPB-51602 treatment of STAT3 KO cells also induced the translocation of p62 to an insoluble fraction (Figure 2J, right lanes). This result suggests that aggregate formation, at least as defined by insolubilization of p62, represents a STAT3-independent action of OPB-51602. Moreover, aggregate formation per se may not be related to, or at least not sufficient for, drug-induced toxicity, since it occurred in STAT3 KO cells, which were protected from toxicity. Lastly, we assessed whether OPB-51602 treatment affected mitochondrial morphology. We examined the mitochondrial network by confocal microscopy using A549 cells expressing a mitochondrially restricted red fluorescent protein, but no significant differences were observed between WT and KO cells (Figure S2F).
Figure 2.

OPB-51602 Induces ROS Production Resulting in Mitophagy and Cell Death in a STAT3-Dependent Manner
(A) Cellular ROS measured with DCFDA in A549 WT and KO (pool and Clone 1) treated with 50 nM OPB-51602 for 2 h (n = 6).
(B) Mitochondrial ROS measured with MitoSOX after OPB-51602 (50 nM) treatment with or without 2 h pre-treatment of GSH-EE (1 mM) (n = 11).
(C and D) (C) Cell viability of A549 incubated with OPB-51602 10 nM alone or in combination with 1 mM GSH-EE for 16 h and analyzed with crystal violet (n = 15). Cell death measured by Annexin V-PI staining in A549 treated with OPB-51602 at 50 nM for different time points (D, n = 7) and in A549 STAT3 WT and KO (pool and Clone 1) treated with 50 nM OPB-51602 for 16 h (E, n = 5).
(F) Increase in mitophagy measured in A549 treated with 5 nM OPB-51602 and 50 μM FCCP for 16 h (n = 13).
(G) Actin rearrangement in A549, MDA-MB-231, and MDA-MB-468 STAT3 WT cells treated with OPB-51602 at 10 nM for 16h.
(H) Actin rearrangement in A549 WT and KO pool treated with OPB-51602 at 10 nM for 16 h.
(I) Cell migration of A549 treated with 10 nM OPB-51602 (n = 11).
(J) Proteotoxic aggregates in A549 WT and KO pool treated with OPB-51602 at 5 and 50 nM for 16 h. The graph shows quantification of p62 levels relative to the control in the soluble fraction (n = 4).
In (A)–(F) and (I) and (J) data are represented as mean ± SEM. Unpaired two-tailed Student's t test; ∗p < 0.05, ∗∗p < 0.001, ∗∗∗p < 0.0005, ∗∗∗∗p < 0.0001. In (G) and (H) scale bar is 10 μm. See also Figure S2. Molecular weight is expressed in kDa.
OPB-51602 Toxicity Depends on Mitochondrial Respiration
We previously showed that OPB-51602 interferes with mitochondrial functions, resulting in reduced basal respiration, ATP production, and mitochondrial membrane potential (Genini et al., 2017). We measured cellular respiration parameters through Seahorse experiments in A549 (Figure 3A) as well as MDA-MB-231 and MDA-MB-468 (Figure S3A), which confirmed abrogation of respiration by OPB-51602. Therefore, we examined the requirement for respiration in OPB-51602-induced toxicity by impairing OXPHOS efficiency by chemically inhibiting different components of the mitochondrial electron transport chain. In brief, we pre-treated A549 cells with either rotenone, metformin (complex I inhibitors), or antimycin A (complex III inhibitor) and challenged them with OPB-51602. As shown in Figure 3B, all three mitochondrial inhibitors effectively reduced cell staining with crystal violet, even at higher concentrations of OPB-51602. Next, we diminished cellular respiration by growing cells under reduced oxygen tension (3% O2) and compared them with cells grown under normoxic conditions (20% O2). Notably, cells exposed to mild hypoxia were fully protected from OPB-51602 toxicity (Figure 3C) and showed only a slight impairment of cell proliferation (Figure S3B). To further examine the hypothesis that mitochondrial respiration is fundamental to OPB-51602 action, we produced A549 Rho0 cells by depleting functional mitochondria through inhibition of mitochondrial DNA replication (Figure S3C). We found that A549 Rho0 cells were fully protected from OPB-51602 toxicity (Figure 3D). Notably, since we grew Rho0 cells in the presence of uridine and pyruvate to meet their energy and biosynthetic demands (Kukat et al., 2008), we tested whether these added nutrients were involved in OPB-51602 resistance but found no effect on WT cells (Figure S3D). Therefore, it is likely that the absence of functional mitochondria and thus the absence of active OXPHOS renders Rho0 cells resistant to the drug.
Figure 3.

OPB-51602 Toxicity Depends on Mitochondrial Respiration
(A) Oxygen Consumption Rate (OCR) in A549 cells treated with 50 nM OPB-51602 for 2 h (n = 12).
(B) Cell viability of A549 cells grown in the presence or absence of 1 nM rotenone, 100 nM antimycin A, 2 mM metformin and incubated with OPB-51602 for 16 h, analyzed with crystal violet.
(C) Cell viability of A549 cells grown in high (20%) and low (3%) O2 and treated with OPB-51602 for 16 h, analyzed with CCK8.
(D) Cell viability of A549 parental and A549 Rho0 cells incubated with OPB-51602 for 16 h, analyzed with CCK8. In (A)–(D) data are represented as mean ± SEM; n ≥ 3. Unpaired two-tailed Student's t test; ∗p < 0.05, ∗∗p < 0.001, ∗∗∗p < 0.0005, ∗∗∗∗p < 0.0001. See also Figure S3.
OPB-51602 Targets Complex I in a STAT3-Dependent Manner
Induction of ROS in response to OPB-51602 treatment and the protection conferred by the complex I inhibitors rotenone and metformin prompted us to directly examine the role of complex I, a major generator of mitochondrial ROS (Kussmaul and Hirst, 2006) and the rate-limiting step in respiration (Kerscher et al., 2008). As shown in Figure 4A, we observed a dramatic loss of complex I activity after 4 h of OPB-51602 treatment in all STAT3-expressing cell lines. This reduction in activity was STAT3 dependent, since it was attenuated in STAT3-KO cells at 10 nM OPB-51602 (Figure 4B). We also tested complex II activity after OPB-51602 treatment, but no significant effects were observed, suggesting that the compound specifically targets complex I (Figure S4A). Mammalian complex I is a multi-subunit NADH:ubiquinone oxidoreductase that simultaneously pumps protons across the inner mitochondrial membrane and maintains NADH/NAD+ homeostasis (Ohnishi et al., 2018). In contrast, the yeast type II enzyme NDI1 is a single component NADH dehydrogenase that is capable of complementing mammalian cellular defects caused by complex I deficiencies (Seo et al., 1998). We expressed yeast NDI1 in both A549 STAT3 WT and KO cells and measured OPB-51602 toxicity. NDI1-expressing clones were insensitive to OPB-51602 (Figure 4C), and even the partial sensitivity of STAT3-KO cells was eliminated. Distinct from the mammalian complex I, NDI1 is insensitive to inhibition by rotenone (Martinez-Reyes et al., 2016) so we directly assessed NADH dehydrogenase activity in the presence and absence of OPB-51602. The compound failed to inhibit the exogenous dehydrogenase activity in NDI1-expressing cells (Figure 4D), suggesting that this enzyme is also resistant to OPB-51602. To confirm that OPB-51602 toxicity is a consequence of an uncontrolled ROS production due to complex I inhibition, we measured OPB-induced ROS generation in A549 cells after a short pre-treatment with rotenone. As shown in Figure 4E, the compound was not able to induce ROS if complex I was already inhibited by rotenone treatment. Moreover, stimulation of NDI1-expressing cells with OPB-51602 did not translate in ROS production (Figure 4F) since, as demonstrated before (Figure 4D), NDI1 is insensitive to OPB-51602 activity. Since we had established that actin rearrangement and induction of STAT3-containing aggregates were phenotypes associated with OPB-51602 treatment, we tested their formation in NDI1-expressing cells. No proteotoxic aggregates or actin filament rearrangements were observed in NDI1-expressing cells after OPB-51602 treatment (Figures 4G and 4H), suggesting that these responses are related to inhibition of complex I activity. Interestingly, however, complex I inhibition by rotenone treatment of wild-type A549 cells did not induce actin rearrangements (Figure 4I) or proteotoxic aggregates (Figure 4J) and was not cytotoxic under the conditions employed, suggesting that OPB-51602 toxicity is more complex than simply inhibiting mitochondrial respiration. Lastly, we analyzed STAT3 mitochondrial localization after OPB-51602 treatment in A549 cells and found that mitochondrial STAT3 also translocates to the insoluble fraction (Figure 4K). However, no subunits of complex I were found in these aggregates suggesting that inhibition of complex I activity after OPB-51602 treatment is not simply due to the sequestration of complex I components. These data suggest that, although OPB-51602 is a potent complex I inhibitor, its cytotoxic effects are multifactorial and rely on the inhibition of additional mitochondrial STAT3 functions to achieve cellular toxicity.
Figure 4.

OPB-51602 Targets Complex I in a STAT3-Dependent Manner
(A) Activity of Complex I in A549, MDA-MB-231, and MDA-MB-468 STAT3 WT cells treated with OPB-51602 at 50 nM for 4 h (n = 4).
(B) Activity of Complex I in A549 STAT3 WT and KO pool cells treated with 5, 10, and 50 nM OPB-51602 for 4 h (n ≥ 5).
(C) Cell viability of A549 STAT3 WT or KO pool cells overexpressing NDI1 and incubated with OPB-51602 for 16 h, analyzed with CCK8.
(D) Activity of Complex I in A549 cells overexpressing NDI1 and treated with 5 and 50 nM OPB-51602 for 4 h (n = 5).
(E) Mitochondrial ROS measured with MitoSOX in A549 cells treated with 50 nM OPB-51602 for 2 h, 50 nM rotenone for 2 h, or both (n = 8).
(F) Mitochondrial ROS measured with MitoSOX in NDI1-expressing cells treated with 50 nM OPB-51602 for 2 h (n = 9).
(G) Actin rearrangement in A549 STAT3 WT and NDI1-overexpressing cells treated with 10 nM OPB-51602 for 16 h.
(H) Proteotoxic aggregates in A549 STAT3 WT and NDI1-overexpressing cells treated with 5 and 50 nM OPB-51602 for 16 h.
(I) Actin rearrangement in A549 cells treated with 10 nM OPB-51602 or 50 nM rotenone for 16 h
(J) Proteotoxic aggregates in A549 cells treated with 10 nM OPB-51602 and 50 nM rotenone for 16 h.
(K) Subcellular localization of STAT3 in A549 cells treated with 10 nM OPB-51602 for 16 h.
In (A)–(F) data are represented as mean ± SEM; n ≥ 3. Unpaired two-tailed Student's t test; ∗p < 0.05, ∗∗p < 0.001, ∗∗∗p < 0.0005, ∗∗∗∗p < 0.0001. In (G) and (I) scale bar is 10 μm. See also Figure S4. Molecular weight is expressed in kDa.
Discussion
Here we report that the STAT3i OPB-51602 disrupts cellular respiration in cancer cells in a STAT3-dependent manner by decreasing the enzymatic activity of complex I, resulting in cell toxicity. Loss of STAT3 largely protected cells from OPB-51602 treatment, and toxicity could be restored to STAT3-null cells through re-expression of a mitochondrially restricted, transcriptionally inert form of STAT3. Moreover, high respiratory flux appeared to be necessary for cytotoxicity, since mild hypoxia, inhibition of respiration, or depletion of functional mitochondria eliminated drug effects. Finally, we found that ectopic expression of a yeast NADH dehydrogenase, which does not depend on STAT3, was capable of bypassing the need for endogenous complex I and conferred complete protection against OPB-51602 toxicity. Of note, all the effects of OPB-51602 treatment occurred in the low nanomolar range, consistent with its high affinity for STAT3 (Genini et al., 2017).
However, the exact mechanisms of STAT3 inhibition and/or cellular toxicity have remained unclear. Short-term effects of OPB-51602 treatment include inhibition of both tyrosine and serine phosphorylation, whereas one of the long-term consequences is the complete depletion of STAT3 protein through recruitment into aggregates (Genini et al., 2017). Inhibition of respiration also occurs very rapidly (4 h), whereas cell death requires a more extended treatment time (16 h). Interestingly, although OPB-51602 toxicity was dependent on STAT3 (Figure 1), this effect was not directly or solely due to inhibition of STAT3 function or depletion of STAT3 protein, since STAT3 KO cells do not show impaired viability in the absence of OPB-51602. Rather, it is likely that an acquired attribute of STAT3 or the drug is involved in drug action, either due to an alteration of the protein or possibly to a STAT3:OPB-51602 protein-drug complex, which could acquire a neomorphic function not possessed by either molecule alone, or through the production of a drug metabolite dependent on the activity of complex 1. For instance, induction of STAT3-containing protein aggregates might be proteotoxic and this hypothesis is supported by the observation that depletion of STAT3 largely protected from OPB-51602-induced toxicity. However, at least p62-containing aggregates still formed in STAT3-null cells, in the absence of toxicity, which would suggest a requirement for STAT3 in the aggregates to be proteotoxic. It is possible that induction of STAT1-containing aggregates in STAT3 KO cells (Figure 2G) was responsible for residual toxicity in these cells. Notably, all STAT family members share high structural similarity, particularly in the SH2 domain, which contains the binding site for OPB-51602 and similar molecules (Brambilla et al., 2015; Genini et al., 2017). Moreover, the idea that a STAT3 inhibitor might bind to STAT1 has been explored and led to the development of isoform-selective STAT inhibitors (Lai et al., 2015; Szelag et al., 2015).
As mentioned before, these insoluble aggregates contain additional proteins, such as p62, a well-characterized player in autophagy, particularly in macroautophagy or aggrephagy (Sun et al., 2020), which could be related to the induction of mitophagy observed in OPB-51602-treated cells. Moreover, studies conducted in Drosophila associated the macroautophagy process with cytoplasmic remodeling, including induction of the formation of actin filaments involved in hemocytes spreading (Kadandale et al., 2010). In our study, cell death was preceded by significant rearrangements of the actin cytoskeleton (Figure 2F). It is likely that the same metabolic processes that resulted in cellular toxicity by OPB-51602 in a STAT3- and respiration-dependent manner also caused the cytoskeletal rearrangements. This phenotype could be mechanistically related to a process that has been observed in macrophages, where an active actin cytoskeleton remodeling process is required for the cells to survey the surrounding microenvironment in response to bacterial signals, such as lipopolysaccharide, and depends on metabolic alterations (Venter et al., 2014). Although the specifics of the underlying molecular mechanisms of actin rearrangements observed after treatment of cancer cells with OPB-51602 are likely distinct from macrophage responses, a similar involvement of metabolic changes, due to alterations of mitochondrial functions, remains an attractive assumption for both processes.
An alternative hypothesis for the mechanism of drug toxicity is that a neomorphic STAT3:OPB-51602 complex is the active cytotoxic agent, given that STAT3 inhibition by OPB-51602 produces a distinct response compared with STAT3 loss. Such a STAT3:drug complex likely initiates apoptosis prior to protein aggregate formation. In support of this notion, we observed significant cell death induced after only 4 h of incubation with the compound (Figure 2B), whereas STAT3 relocalization into p62-containing aggregates was not observed until 16 h. Given the rapid decrease of complex I activity after OPB-51602 treatment, one explanation could be that STAT3 facilitates targeting of the drug to complex I, since the presence of STAT3 in mitochondria has been shown to affect complex I activity (Wegrzyn et al., 2009) and STAT3 may directly associate with complex I components (Tammineni et al., 2013). Again, however, simple inhibition of complex I is insufficient to explain OPB-51602 toxicity, since other complex I inhibitors (e.g., rotenone) are not cytotoxic to the same extent as OPB-51602, nor do they induce actin filament formation.
A third possibility is that OPB-51602 becomes metabolized to a toxic compound, presumably in a STAT3-dependent and complex I-dependent manner, through a process that results in complex I inhibition. This possibility might explain why reduced respiration protects against OPB-51602 toxicity. If true, it would appear that an intrinsic activity of complex I is involved in OPB-51602-mediated cell death, rather than simply high enzymatic activity, since complementing cells with the OPB-51602-resistent NDI1 dehydrogenase did not enhance toxicity but rather fully protected them against all drug-induced phenotypes (Figure 4). Further experiments are needed to distinguish among these various hypotheses.
In summary, we have shown that OPB-51602, a STAT3-interacting small molecule, impairs cancer cell viability in a STAT3-dependent manner, but not simply through inhibition of canonical STAT3 functions. Instead, cell death depends on active respiration, correlates with profound inhibition of respiratory complex I, and can be prevented through the action of complex I enzymatic activity that is not dependent on STAT3. Although it remains unclear why cancer cells are selectively sensitive to OPB-51602-induced toxicity relative to non-transformed cells, these results identify a unique cancer cell vulnerability that might be exploited therapeutically, particularly for cancers that are highly dependent on oxidative phosphorylation for growth. Moreover, OPB-51602 could be used as a new tool to study STAT3-dependent complex I functions and to expand our knowledge of non-transcriptional roles of mitochondrial STAT3.
Limitation of the Study
This study characterized the mechanism of action of a cytotoxic direct STAT3 inhibitor. All the experiments have been done entirely in human cancer cell lines grown in culture, which may not reflect exactly what happens in tumors. Moreover, actin rearrangement has been described as a consequence of the compound treatment, but further investigation is required to address the underlying mechanism.
Resources Availability
Lead Contact
Further information and requests should be directed to the Lead Contact, Dr. David E. Levy (David.levy@med.nyu.edu).
Materials Availability
Any unique reagents are available from the Lead Contact on request.
Data and Code Availability
This study did not generate/analyze datasets/code.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank Drs. Navdeep Chandel (Northwestern University), Seth Parker, Alec Kimmelman, Joseph Puccini, Dafna Bar-Sagi, Richard Possemato, Agnel Sfeir, Ian Ahearn, Antonio Marzio, Isabelle Marié, and Mark Philips (NYU School of Medicine) for gifts of valuable reagents, advice, and helpful discussions. We thank Dusan Kostic, Matthew Harlin, and Agnes Elekes for support and helpful discussions. We also thank the cell cytometry core and the microscopy core of the NYU Division of Applied Research Technologies for technical support. Graphical abstract created with BioRender.com. This work was supported in part by Otsuka Pharmaceutical Development & Commercialization, Inc., which owns intellectual property interests in OPB-51602, and by the NIH (R01 AI28900).
Author Contributions
Conceptualization, L.B. and D.E.L.; Methodology, L.B., T.L., M.C., and D.E.L.; Investigation, L.B., T.L., and M.C.; Writing L.B. and D.E.L.; Funding Acquisition, D.E.L.; Supervision, D.E.L.
Declaration of Interests
The authors declare no competing interests.
Published: December 18, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101822.
Supplemental Information
References
- Akira S. Roles of STAT3 defined by tissue-specific gene targeting. Oncogene. 2000;19:2607–2611. doi: 10.1038/sj.onc.1203478. [DOI] [PubMed] [Google Scholar]
- Avalle L., Poli V. Nucleus, mitochondrion, or reticulum? STAT3 a La Carte. Int. J. Mol. Sci. 2018;19:2820. doi: 10.3390/ijms19092820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker S., Groner B., Müller C.W. Three-dimensional structure of the Stat3b homodimer bound to DNA. Nature. 1998;394:145–151. doi: 10.1038/28101. [DOI] [PubMed] [Google Scholar]
- Berezhnov A.V., Soutar M.P., Fedotova E.I., Frolova M.S., Plun-Favreau H., Zinchenko V.P., Abramov A.Y. Intracellular pH modulates autophagy and mitophagy. J. Biol. Chem. 2016;291:8701–8708. doi: 10.1074/jbc.M115.691774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambilla L., Genini D., Laurini E., Merulla J., Perez L., Fermeglia M., Carbone G.M., Pricl S., Catapano C.V. Hitting the right spot: mechanism of action of OPB-31121, a novel and potent inhibitor of the signal transducer and activator of transcription 3 (STAT3) Mol. Oncol. 2015;9:1194–1206. doi: 10.1016/j.molonc.2015.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty A., Dyer K.F., Cascio M., Mietzner T.A., Tweardy D.J. Identification of a novel Stat3 recruitment and activation motif within the granulocyte colony-stimulating factor receptor. Blood. 1999;93:15–24. [PubMed] [Google Scholar]
- Demaria M., Giorgi C., Lebiedzinska M., Esposito G., D'Angeli L., Bartoli A., Gough D.J., Turkson J., Levy D.E., Watson C.J. A STAT3-mediated metabolic switch is involved in tumour transformation and STAT3 addiction. Aging. 2010;2:823–842. doi: 10.18632/aging.100232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan P., Xie X.H., Chen C.H., Peng X., Zhang P., Yang C., Wang Y.T. Molecular regulation mechanisms and interactions between reactive oxygen species and mitophagy. DNA Cell Biol. 2019;38:10–22. doi: 10.1089/dna.2018.4348. [DOI] [PubMed] [Google Scholar]
- Garama D.J., Harris T.J., White C.L., Rosello F.J., Abdul-Hay M., Gough D.J., Levy D.E. A synthetic lethal interaction between glutathione synthesis and mitochondrial reactive oxygen species provides a tumor specific vulnerability dependent on STAT3. Mol. Cell Biol. 2015;35:3646–3656. doi: 10.1128/MCB.00541-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelain A., Mori M., Meneghetti F., Villa S. Signal transducer and activator of transcription protein 3 (STAT3): an update on its direct inhibitors as promising anticancer agents. Curr. Med. Chem. 2019;26:5165–5206. doi: 10.2174/0929867325666180719122729. [DOI] [PubMed] [Google Scholar]
- Genini D., Brambilla L., Laurini E., Merulla J., Civenni G., Pandit S., D'Antuono R., Perez L., Levy D.E., Pricl S. Mitochondrial dysfunction induced by a SH2 domain-targeting STAT3 inhibitor leads to metabolic synthetic lethality in cancer cells. Proc. Natl. Acad. Sci. U S A. 2017;114:E4924–E4933. doi: 10.1073/pnas.1615730114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gough D.J., Corlett A., Schlessinger K., Wegrzyn J., Larner A.C., Levy D.E. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science. 2009;324:1713–1716. doi: 10.1126/science.1171721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gough D.J., Marie I.J., Lobry C., Aifantis I., Levy D.E. STAT3 supports experimental K-RasG12D-induced murine myeloproliferative neoplasms dependent on serine phosphorylation. Blood. 2014;124:2252–2261. doi: 10.1182/blood-2013-02-484196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gough D.J., Sehgal P.B., Levy D.E. Nongenomic functions of STAT3. In: Decker T., Müller M., editors. Jak-Stat Signaling: From Basics to Disease. Springer-Verlag Gmbh; Wien: 2012. pp. 91–98. [Google Scholar]
- Hemmann U., Gerhartz C., Heesel B., Sasse J., Kurapkat G., Grotzinger J., Wollmer A., Zhong Z., Darnell J.E., Graeve L. Differential activation of acute phase response factor/Stat3 and Stat1 via the cytoplasmic domain of the interleukin 6 signal transducer gp130. II. Src homology SH2 domains define the specificity of stat factor activation. J. Biol. Chem. 1996;271:12999–13007. doi: 10.1074/jbc.271.22.12999. [DOI] [PubMed] [Google Scholar]
- Hirpara J., Eu J.Q., Tan J.K.M., Wong A.L., Clement M.V., Kong L.R., Ohi N., Tsunoda T., Qu J., Goh B.C., Pervaiz S. Metabolic reprogramming of oncogene-addicted cancer cells to OXPHOS as a mechanism of drug resistance. Redox Biol. 2019;25:101076. doi: 10.1016/j.redox.2018.101076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath C.M. STAT proteins and transcriptional responses to extracellular signals. Trends Biochem. Sci. 2000;25:496–502. doi: 10.1016/s0968-0004(00)01624-8. [DOI] [PubMed] [Google Scholar]
- Kadandale P., Stender J.D., Glass C.K., Kiger A.A. Conserved role for autophagy in Rho1-mediated cortical remodeling and blood cell recruitment. Proc. Natl. Acad. Sci. U S A. 2010;107:10502–10507. doi: 10.1073/pnas.0914168107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerscher S., Drose S., Zickermann V., Brandt U. The three families of respiratory NADH dehydrogenases. Results Probl. Cell Differ. 2008;45:185–222. doi: 10.1007/400_2007_028. [DOI] [PubMed] [Google Scholar]
- Kukat A., Kukat C., Brocher J., Schafer I., Krohne G., Trounce I.A., Villani G., Seibel P. Generation of rho0 cells utilizing a mitochondrially targeted restriction endonuclease and comparative analyses. Nucleic Acids Res. 2008;36:e44. doi: 10.1093/nar/gkn124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kussmaul L., Hirst J. The mechanism of superoxide production by NADH:ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proc. Natl. Acad. Sci. U S A. 2006;103:7607–7612. doi: 10.1073/pnas.0510977103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai P.S., Rosa D.A., Magdy ALI A., Gomez-Biagi R.F., Ball D.P., Shouksmith A.E., Gunning P.T. A STAT inhibitor patent review: progress since 2011. Expert Opin. Ther. Pat. 2015;25:1397–1421. doi: 10.1517/13543776.2015.1086749. [DOI] [PubMed] [Google Scholar]
- Lazarou M., Sliter D.A., Kane L.A., Sarraf S.A., Wang C., Burman J.L., Sideris D.P., Fogel A.I., Youle R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524:309–314. doi: 10.1038/nature14893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M., Hirpara J.L., Eu J.Q., Sethi G., Wang L., Goh B.C., Wong A.L. Targeting STAT3 and oxidative phosphorylation in oncogene-addicted tumors. Redox Biol. 2019;25:101073. doi: 10.1016/j.redox.2018.101073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy D.E., Darnell J.E., Jr. STATs: transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 2002;3:651–662. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- Mantel C., Messina-Graham S., Moh A., Cooper S., Hangoc G., Fu X.Y., Broxmeyer H.E. Mouse hematopoietic cell-targeted STAT3 deletion: stem/progenitor cell defects, mitochondrial dysfunction, ROS overproduction, and a rapid aging-like phenotype. Blood. 2012;120:2589–2599. doi: 10.1182/blood-2012-01-404004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Reyes I., Diebold L.P., Kong H., Schieber M., Huang H., Hensley C.T., Mehta M.M., Wang T., Santos J.H., Woychik R. TCA cycle and mitochondrial membrane potential are necessary for diverse biological functions. Mol. Cell. 2016;61:199–209. doi: 10.1016/j.molcel.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscat J., Karin M., Diaz-Meco M.T. p62 in cancer: signaling adaptor beyond autophagy. Cell. 2016;167:606–609. doi: 10.1016/j.cell.2016.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura M., Uchida T., Terui Y., Hayakawa F., Kobayashi Y., Taniwaki M., Takamatsu Y., Naoe T., Tobinai K., Munakata W. Phase I study of OPB-51602, an oral inhibitor of signal transducer and activator of transcription 3, in patients with relapsed/refractory hematological malignancies. Cancer Sci. 2015;106:896–901. doi: 10.1111/cas.12683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnishi T., Ohnishi S.T., Salerno J.C. Five decades of research on mitochondrial NADH-quinone oxidoreductase (complex I) Biol. Chem. 2018;399:1249–1264. doi: 10.1515/hsz-2018-0164. [DOI] [PubMed] [Google Scholar]
- Pickles S., Vigie P., Youle R.J. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr. Biol. 2018;28:R170–R185. doi: 10.1016/j.cub.2018.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rustom A., Saffrich R., Markovic I., Walther P., Gerdes H.H. Nanotubular highways for intercellular organelle transport. Science. 2004;303:1007–1010. doi: 10.1126/science.1093133. [DOI] [PubMed] [Google Scholar]
- Seo B.B., Kitajima-Ihara T., Chan E.K., Scheffler I.E., Matsuno-Yagi A., Yagi T. Molecular remedy of complex I defects: rotenone-insensitive internal NADH-quinone oxidoreductase of Saccharomyces cerevisiae mitochondria restores the NADH oxidase activity of complex I-deficient mammalian cells. Proc. Natl. Acad. Sci. U S A. 1998;95:9167–9171. doi: 10.1073/pnas.95.16.9167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun D., Wu R., Li P., Yu L. Phase separation in regulation of aggrephagy. J. Mol. Biol. 2020;432:160–169. doi: 10.1016/j.jmb.2019.06.026. [DOI] [PubMed] [Google Scholar]
- Szczepanek K., Chen Q., Larner A.C., Lesnefsky E.J. Cytoprotection by the modulation of mitochondrial electron transport chain: the emerging role of mitochondrial STAT3. Mitochondrion. 2012;12:180–189. doi: 10.1016/j.mito.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczepanek K., Lesnefsky E.J., Larner A.C. Multi-tasking: nuclear transcription factors with novel roles in the mitochondria. Trends Cell Biol. 2012;22:429–437. doi: 10.1016/j.tcb.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szelag M., Czerwoniec A., Wesoly J., Bluyssen H.A. Identification of STAT1 and STAT3 specific inhibitors using comparative virtual screening and docking validation. PLoS One. 2015;10:e0116688. doi: 10.1371/journal.pone.0116688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tammineni P., Anugula C., Mohammed F., Anjaneyulu M., Larner A.C., Sepuri N.B. The import of the transcription factor STAT3 into mitochondria depends on GRIM-19, a component of the electron transport chain. J. Biol. Chem. 2013;288:4723–4732. doi: 10.1074/jbc.M112.378984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venter G., Oerlemans F.T., Wijers M., Willemse M., Fransen J.A., Wieringa B. Glucose controls morphodynamics of LPS-stimulated macrophages. PLoS One. 2014;9:e96786. doi: 10.1371/journal.pone.0096786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma N.K., Dourlat J., Davies A.M., Long A., Liu W.Q., Garbay C., Kelleher D., Volkov Y. STAT3-stathmin interactions control microtubule dynamics in migrating T-cells. J. Biol. Chem. 2009;284:12349–12362. doi: 10.1074/jbc.M807761200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinkemeier U., Cohen S.L., Moarefi I., Chait B.T., Kuriyan J., Darnell J.E. DNA binding of in vitro activated Stat1 alpha, Stat1 beta and truncated Stat1: interaction between NH2-terminal domains stabilizes binding of two dimers to tandem DNA sites. EMBO J. 1996;15:5616–5626. [PMC free article] [PubMed] [Google Scholar]
- Vogt M., Domoszlai T., Kleshchanok D., Lehmann S., Schmitt A., Poli V., Richtering W., Muller-Newen G. The role of the N-terminal domain in dimerization and nucleocytoplasmic shuttling of latent STAT3. J. Cell Sci. 2011;124:900–909. doi: 10.1242/jcs.072520. [DOI] [PubMed] [Google Scholar]
- Wegrzyn J., Potla R., Chwae Y.J., Sepuri N.B., Zhang Q., Koeck T., Derecka M., Szczepanek K., Szelag M., Gornicka A. Function of mitochondrial Stat3 in cellular respiration. Science. 2009;323:793–797. doi: 10.1126/science.1164551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong A.L., Soo R.A., Tan D.S., Lee S.C., Lim J.S., Marban P.C., Kong L.R., Lee Y.J., Wang L.Z., Thuya W.L. Phase I and biomarker study of OPB-51602, a novel signal transducer and activator of transcription (STAT) 3 inhibitor, in patients with refractory solid malignancies. Ann. Oncol. 2015;26:998–1005. doi: 10.1093/annonc/mdv026. [DOI] [PubMed] [Google Scholar]
- Yoo C., Kang J., Lim H.Y., Kim J.H., Lee M.A., Lee K.H., Kim T.Y., Ryoo B.Y. Phase I dose-finding study of OPB-111077, a novel STAT3 inhibitor, in patients with advanced hepatocellular carcinoma. Cancer Res. Treat. 2019;51:510–518. doi: 10.4143/crt.2018.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H., Lee H., Herrmann A., Buettner R., Jove R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat. Rev. Cancer. 2014;14:736–746. doi: 10.1038/nrc3818. [DOI] [PubMed] [Google Scholar]
- Yu H., Pardoll D., Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat. Rev. Cancer. 2009;9:798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue P., Turkson J. Targeting STAT3 in cancer: how successful are we? Expert Opin. Investig. Drugs. 2009;18:45–56. doi: 10.1517/13543780802565791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q., Raje V., Yakovlev V.A., Yacoub A., Szczepanek K., Meier J., Derecka M., Chen Q., Hu Y., Sisler J. Mitochondrial localized Stat3 promotes breast cancer growth via phosphorylation of serine 727. J. Biol. Chem. 2013;288:31280–31288. doi: 10.1074/jbc.M113.505057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Darnell J.E., Jr. Functional importance of Stat3 tetramerization in activation of the alpha 2-macroglobulin gene. J. Biol. Chem. 2001;276:33576–33581. doi: 10.1074/jbc.M104978200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate/analyze datasets/code.