

Abstract
Cellular antioxidants protect against hyperoxic lung injury. The role of the glutathione (GSH) system in lung development and bronchopulmonary dysplasia (BPD) pathogenesis has not been systematically investigated. The current study utilized GSH reductase-deficient (Gsr-KO) neonatal mice to test the hypothesis that early disruption of the GSH system negatively impacts lung development and hyperoxic responses. Lungs from wild-type (Gsr-WT) and Gsr-KO mice were analyzed for histopathology, developmental markers, redox indices, and transcriptome profiling at different developmental stages following exposure to room air or hyperoxia (85% O2) for up to 14 d. Lungs from Gsr-KO mice exhibited alveolar epithelial dysplasia in the embryonic and neonatal periods with relatively normal lung architecture in adulthood. GSH and its oxidized form (GSSG) were 50–70% lower at E19-PND14 in Gsr-KO lungs than in age-matched Gsr-WT. Differential gene expression between Gsr-WT and Gsr-KO lungs was analyzed at discrete developmental stages. Gsr-KO lungs exhibited downregulated cell cycle and DNA damage checkpoint genes at E19, as well as lung lipid metabolism and surfactant genes at PND5. In addition to abnormal baseline lung morphometry, Gsr-KO mice displayed a blunted response to hyperoxia. Hyperoxia caused a more robust upregulation of the lung thioredoxin system in Gsr-KO compared to Gsr-WT. Gsr-dependent, hyperoxia-responsive genes were highly associated with abnormal cytoskeleton, skeletal-muscular function, and tissue morphology at PND5. Overall, our data in Gsr-KO mice implicate the GSH system as a key regulator of lung development, cellular differentiation, and hyperoxic responses in neonatal mice.
Keywords: Glutathione reductase, Mice, Hyperoxia, Bronchopulmonary dysplasia, Thioredoxin, Neonate, Embryo, Microarray
Abbreviations: AT1, alveolar type 1 epithelial cell; AT2, alveolar type 2 epithelial cell; BPD, bronchopulmonary dysplasia; GSH, glutathione; GSR, glutathione reductase; (Gsr-KO, glutathione reductase-deficient; Gsr-WT, wild-type mice; GSSG, glutathione disulfide; PND, postnatal day; SP-C, surfactant protein C; T1a, T1-alpha; TXN, thioredoxin; TXNRD, thioredoxin reductase
Graphical abstract
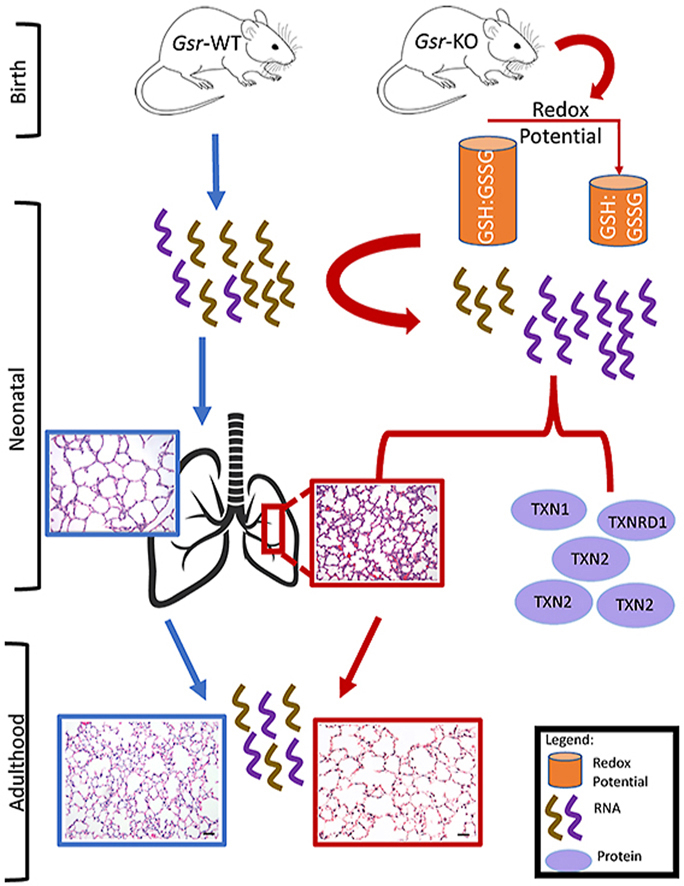
Highlights
-
•
Glutathione reductase deficiency alters lung development in the neonatal period.
-
•
Thioredoxin system compensates for glutathione reductase deficiency.
-
•
Significant transcriptomic differences occur from glutathione reductase deficiency.
-
•
Lung structure and transcriptome normalizes by adulthood.
-
•
More genes differentially expressed from glutathione reductase deficiency than oxygen.
1. Introduction
In preterm infants, oxygen toxicity is a frequent consequence of prolonged supplemental oxygen exposure that contributes to the development of bronchopulmonary dysplasia (BPD) [1,2]. The mechanisms that mediate oxygen-induced lung injury are primarily mediated through the actions of reactive oxygen intermediates including superoxide (O2•−), hydrogen peroxide (H2O2), peroxynitrite (ONOO−), and hydroxyl radical (HO•) [3]. Glutathione (GSH) peroxidases (GPx) catalyze the GSH-dependent reduction of H2O2 and other oxidants resulting in the oxidation of GSH to glutathione disulfide (GSSG). Glutathione reductase (GSR) catalyzes the reduction of GSSG to GSH using NADPH as an electron donor (NADPH + H+ + GSSG → NADP+ + 2 GSH). We and others have shown that Gsr deficiency appears to be well tolerated in adult mice [[4], [5], [6], [7], [8], [9]].
Thioredoxin (TXN) is a dithiol/disulfide oxidoreductase that is catalytically maintained in a reduced state by the enzyme thioredoxin reductase (TXNRD). A series of preclinical studies by our group has established pharmacologic TXNRD inhibition as a promising therapeutic strategy to prevent neonatal and adult lung injury [[10], [11], [12], [13], [14], [15]]. Nuclear factor (erythroid-derived 2)-like 2 (NRF2)-mediated upregulation of GSH-dependent antioxidant defenses are necessary for the efficacy of TXNRD inhibitors to prevent lung injury [8,10]. Compensatory upregulation of the TXN system contributes to the resistance of adult Gsr-deficient (Gsr-KO) mice to hyperoxic lung injury [8].
The role of GSR on lung development and hyperoxic responses in neonatal mice has not been formally investigated. The current study utilized neonatal Gsr-KO mice to test the hypothesis that disruption of the GSH system negatively impacts lung development and alters hyperoxia-induced lung injury in neonates.
2. Methods
2.1. Animals, timed mating, and exposure
Animal studies were performed at The Abigail Wexner Research Institute at Nationwide Children's Hospital using protocols approved by the Institutional Animal Care and Use Committee. Wild-type (Gsr-WT, C3H/HeN) mice were purchased from Harlan (Indianapolis, IN). Gsr-KO mice were generated as described previously [16,17]. Mice were bred and dated as to time of pregnancy. Time-pregnant dams were euthanized at E19 by cervical dislocation and the embryos were removed. For exposure studies, newborn mice were exposed to room air (FiO2 0.21) or hyperoxia (FiO2 0.85) beginning within 12 h of life on postnatal day 0.5 (PND 0.5) for up to 14 d. Dams were rotated every 24 h to prevent oxygen toxicity. Pups were weighed daily. Subsets of hyperoxia-exposed mice were returned to room air for up to PND56. In both prenatal and perinatal studies, lungs were harvested at designated times and either inflation fixed with 10% formalin or were snap frozen in liquid nitrogen and stored at −80 °C for molecular analyses.
2.2. Lung glutathione measurements
The concentrations of reduced (GSH) and disulfide (GSSG) forms of glutathione were measured by HPLC and were determined using simultaneously run standard curves [18,19]. Redox potentials of GSSG/GSH [Eh(GSSG/GSH)] were calculated according to the Nernst equation [18].
2.3. Pulmonary function testing
Pulmonary function analyses were performed in adult mice using Flexivent system (Scireq, Montreal, QC, Canada) [20].
2.4. Western blotting
Lungs were homogenized in radioimmunoprecipitation assay (RIPA) buffer. The following antibodies were used: mouse canonical alveolar type 1 cell (AT1) marker T1-alpha (T1α, hamster monoclonal, clone 8.1.1, University of Iowa Hybridoma Bank), alveolar type 2 cell (AT2) cell marker surfactant protein C (SP–C, goat polyclonal, Santa Cruz Biotechnologies, Dallas, TX), and β-actin (goat polyclonal, Santa Cruz Biotechnologies). The membranes were developed with ECL + reagent (GE Healthcare, Buckinghamshire, UK) and read with a phosphoimager (GE Healthcare, Buckinghamshire, UK) and band images (n=3/group) were quantitated using ImageQuant software (Molecular Dynamics, Sunnyvale, CA).
2.5. Genome-wide transcriptomic analyses
Lungs were collected from Gsr-WT and Gsr-KO embryos at E19, or from animals exposed to room air (FiO2=0.21) or hyperoxia (FiO2=0.85) from birth to PND5. Lungs were collected from a separate set of animals exposed to room air or hyperoxia from PND0-14 then raised in room air through PND56. RNA was isolated from lung homogenates of each mouse (n = 4/group for embryos, n = 3/group for others) using the RNeasy Mini-Plus kit (Qiagen, Valencia, CA). Aliquots of total lung RNA (200 ng) were applied to mouse 430 2.0 array (Affymetrix, Inc., Santa Clara, CA) in the NIEHS Microarray Core Facility as described previously [21]. Using GeneSpring GX software (Agilent Technologies, Inc., Santa Clara, CA), independent experiments were created using raw data of E19 (Gsr-WT, Gsr-KO), PND5 (Gsr-WT/0.21, Gsr-WT/0.85, Gsr-KO/0.21, Gsr-KO/0.85), and PND56 (Gsr-WT/0.21, Gsr-WT/0.85, Gsr-KO/0.21, Gsr-KO/0.85). Array raw data were filtered by lower expression percentile (at least 1 sample had values within 20% cut-off rage) and the expression levels were normalized to mean value of experimental control (i.e., Gsr-WT in E19, Gsr-WT/0.21 in PND5, Gsr-WT/0.21 in PND56) for each gene by quantile algorithm. Moderated t-test (E19/Gsr-WT vs E19/Gsr-WT, PND5- Gsr-WT/0.21 vs PND5-Gsr-WT/0.21, PND56-Gsr-WT/0.21 vs PND56- Gsr-WT/0.21, PND5-Gsr-KO/0.21 vs PND5- Gsr-KO/0.85) or 2-way ANOVA followed by Student-Newman-Keuls comparison (interaction between genotype and exposure in Gsr-WT or in Gsr-KO at PND5 and PND56) identified statistically significant genes (p value cut-off at < 0.01). Ingenuity Pathway Analysis (IPA, Qiagen Inc., Valencia, CA) predicted underlying molecular mechanisms including upstream modulators and downstream effectors as well as molecular interactions. Microarray data are deposited in Gene Expression Omnibus (GEO, accession number GSE116485).
2.6. Statistical analysis
Non-microarray data were analyzed by unpaired t-test or by ANOVA followed by post hoc testing using GraphPad Prism Version 8 (San Diego, CA). Significance was accepted at p < 0.05. Microarray data analysis is described above.
3. Results
3.1. Lung glutathione contents in Gsr-KO mice
Lung GSH and GSSG contents were significantly lower in Gsr-KO lungs when compared to age-matched Gsr-WT controls at PND0, PND1, PND3, PND7 and PND14 (Fig. 1A and B). In embryonic lungs, GSSG but not GSH contents were lower in Gsr-KO vs Gsr-WT controls (Fig. 1B). Effects of Gsr deletion on lung GSH:GSSG ratios were inconsistent (Fig. 1C). Redox potentials (Eh) were calculated using the Nernst equation in order to express the redox potential of the GSH/GSSG thiol/disulfide couple [22]. GSSG/GSH potential was significantly lower (4–13%) in Gsr-KO compared to age matched Gsr-WT at all embryonic and neonatal timepoints (Fig. 1D).
Fig. 1.

Lung glutathione contents in embryo and newborn wild-type (Gsr-WT) and Gsr-deficient (Gsr-KO) mice. (A) Independent effects of genotype and day of life as well as interaction on oxidized (GSH) and (B) reduced disulfide (GSSG) glutathione contents were detected in lung homogenates from newborn Gsr-WT and Gsr-KO mice. (C) GSH/GSSG ratios were subsequently calculated. (D) Calculated GSH redox potential demonstrated independent effects of genotype and day of life as well as interaction between the two. Data are expressed as mean +SEM (*p < 0.05 vs Gsr-WT, n=3–6) and were analyzed by 2-way ANOVA with Tukey's multiple comparison test.
3.2. Role of GSR in developmental mouse lung
Body weights were not significantly different between Gsr-WT and Gsr-KO mice between PND0-PND70 (Supplemental Fig. 1). Canalicular stage lungs from Gsr-KO mice at E19 display septal thickening and epithelial dysplasia when compared to Gsr-WT mice (Fig. 2A). Persistent septal thickening and epithelial cell dysplasia were also evident at PND3 in Gsr-KO mice (Fig. 2B). There was little histologic difference between mature Gsr-KO and Gsr-WT lungs at PND56 (Fig. 2C). Pulmonary function analyses revealed baseline differences in adult function between genotypes. Gsr-KO mice had decreased pulmonary resistance and total lung capacity compared to Gsr-WT mice (Fig. 2D, G), but no difference in airway resistance or compliance (Fig. 2E and F). In addition, Gsr-KO exhibited significantly enhanced airway responsiveness to methacholine starting at the 5 mg/ml dosage when compared to Gsr-WT (Fig. 2H).
Fig. 2.

Lung morphology of wild-type (Gsr-WT) and glutathione reductase-deficient (Gsr-KO) mouse lungs from canalicular to matured stages. H&E-stained lungs from Gsr-KO mice (200x) display evidence of septal thickening and alveolar epithelial dysplasia compared to Gsr-WT at embryonic day E19 (A) and persistent septal thickening alveolar epithelial cell dysplasia at PND3 (B) which has essentially normalized by PND56 (C). Bars = 50 μm. Pulmonary function assessment in PND56 mice for pulmonary resistance (D), compliance (E), total lung capacity (F) and airway resistance (G) were assessed. Pulmonary resistance and total lung capacity were significantly decreased in Gsr-KO mice compared to Gsr-WT. (H) Methacholine responses reveal enhanced airway responsiveness in Gsr-KO compared to Gsr-WT at PND 56. Data are expressed as mean +SEM (*p < 0.05 vs Gsr-WT; n=14–18).
3.3. Lung T1 alpha (T1α) and surfactant protein C (SP–C) expression
To further explore the alveolar cell dysplasia found in Gsr-KO mice, western blotting was performed on whole lung homogenates using antibodies for canonical alveolar type 1 cells (AT1; T1α) and alveolar type 2 cells (AT2; SP-C). Lung T1α protein levels increased as a function of age between E19 and PND70 in Gsr-WT mice (Fig. 3A). In contrast to findings of increased T1α expression at PND70 in Gsr-WT mice, T1α expression in Gsr-KO mice did not increase above the levels observed at PND28. There were no consistent differences in SP-C expression in either Gsr-WT or Gsr-KO mice (Fig. 3B).
Fig. 3.

Western Blot analyses for T1-alpha (T1α) and surfactant protein C (SP–C) in glutathione reductase-deficient (Gsr-KO) and -sufficient (Gsr-WT) mouse lungs. (A) Independent effects of genotype and day of life on lung T1α expression and an interaction between day of life and genotype were detected. (B) No effects or interactions were detected. Data are expressed as mean +SEM (n=3) and were assessed by two-way ANOVA followed by Newman-Keuls post hoc. P < 0.05 *vs E19-Gsr-WT, #vs PND14 Gsr-WT, &vs PND28-Gsr-WT, ∧vs PND70-Gsr-WT, %vs E19-Gsr-KO, @vs PND14-Gsr-KO.
3.4. Role of Gsr in developmental lung transcriptomics
Overall, pulmonary transcriptome variation between Gsr-WT and Gsr-KO mice was more predominant in developing lungs (E19 and PND5) than in mature lungs at PND56 (Fig. 4A). Many of the genes known to have Gsr-dependent regulation were highly suppressed (up to 40 fold) in Gsr-KO lungs versus Gsr-WT lungs at E19 and PND5 [e.g., cytochrome P450, family 2 (e.g., Cyp2d26), alpha-2-macroglobulin (A2m), apolipoprotein family (e.g., Apoc2), serine (or cysteine) peptidase inhibitor, clade A family (e.g., Serpina1a), fibrinogen alpha chain (Fga), albumin (Alb), alpha fetoprotein (Afp)] (Tables 1, S1, S2). GSR may play roles in unique pathways at different lung developmental stages, as only a small number of genes were commonly modulated by Gsr (black block in Fig. 4A) between canalicular (E19), saccular-alveolar (PND5), and mature lungs (PND56).
Fig. 4.

Effect of glutathione reductase (Gsr) deficiency on embryonic and postnatal lung transcriptomics. (A) Venn Diagram analysis determined Gsr-dependently expressed genes at embryonic day E19 and postnatal days 5 (PND5) and PND56. Common Gsr-dependent genes at different developmental ages are shown as same color/pattern blocks in the bar graphs. (B) At E19 (canalicular stage), lungs in Gsr-deficient (Gsr-KO) embryo displayed significantly lowered expression of genes for cell cycle control of chromosomal replication and DNA damage checkpoint regulation and heightened expression of inflammatory and cell death response genes. (C) At PND5 (entering alveolar stage), Gsr-KO lungs showed significantly suppressed transcriptome for lipid metabolism and transport and coagulation system. (D) In mature lung (PND56), Gsr deficiency modulated genes to promote abnormal cardiovascular system function and connective tissue disorders. Analyses were done using GeneSpring (moderated t-test followed by Benjamini-Hochberg multiple testing correction, p < 0.01) and Ingenuity Pathway Analysis software. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
In the canalicular lung (E19), Gsr-dependent lung transcriptome variation was greatest (Fig. 4A). Among the significantly varied genes (n=908, p < 0.01; Tables 1 and S1), a cluster of genes involved in cell cycle control of chromosomal replication and DNA damage checkpoint regulation were suppressed in Gsr-KO lungs compared to Gsr-WT lungs (Fig. 4B, Table 1). Cell division cycle (e.g., Cdc6) and cyclin and related (e.g., Ccnb1, Cdk1) genes were highly represented (Tables 1 and S1). These data suggest that embryonic Gsr deficiency may interrupt cell cycle in the developing lung and may contribute to the histopathologic septal thickening and alveolar epithelial dysplasia observed in Gsr-KO lungs at E19 (Fig. 2A). In contrast, Gsr-KO embryonic lung had markedly heightened expression of inflammatory and cell death response genes versus Gsr-WT (Fig. 4B, Tables 1 and S1). These genes include interferon-induced protein family (Ifit1), toll like receptor signal transducers (Tlr4, Cd14), and complement (C6). Several cytochrome P450 (Cyp2d26), GSH homeostasis, and redox balance (Ggt1, Gstt1, Ugt2b5) genes were also relatively suppressed in Gsr-KO embryonic lungs (Tables 1 and S1). Gsr deficiency may thus predispose the animals to altered pulmonary immune and inflammatory responses.
Table 1.
Representative glutathione reductase (Gsr)-dependent genes in embryo to adult mouse lungs.
| Category (Age) | RefSeq ID | †FD | Gene Symbol | Gene Title | Functions |
|---|---|---|---|---|---|
| Cell cycle, DNA damage checkpoint regulation (E19) | NM_001025779 | −3.14 | Cdc6 | cell division cycle 6 | DNA replication initiation |
| NM_007659 | −2.26 | Cdk1 | cyclin-dependent kinase 1 | protein complex assembly | |
| NM_001302540 | −2.66 | Mcm5 | minichromosome maintenance deficient 5 | DNA replication initiation | |
| NM_001291185 | −2.41 | Aurka | aurora kinase A | meiotic spindle organization | |
| NM_172301 | −2.36 | Ccnb1 | cyclin B1 | mitotic cell cycle | |
| NM_001195298 | −2.24 | Kifc1 | kinesin family member C1 | mitotic sister chromatid segregation | |
| Redox and metabolism (E19) | NM_029562 | −32.87 | Cyp2d26 | cytochrome P450, family 2, subfamily d, polypeptide 26 | xenobiotic metabolic process |
| NM_001039555 | −25.90 | Cyp2c68 | cytochrome P450, family 2, subfamily c, polypeptide 68 | xenobiotic metabolic process | |
| NM_019775 | −19.54 | Cpb2 | carboxypeptidase B2 (plasma) | fibrinolysis | |
| NM_001304800 | −18.34 | Hsd3b1 | hydroxy-delta-5-steroid dehydrogenase, 3 beta- and steroid delta-isomerase 1 | lipid metabolism | |
| NM_001277944 | −15.45 | Apoc2 | apolipoprotein C-II | lipid metabolism | |
| NM_009467 | −12.76 | Ugt2b5 | UDP glucuronosyltransferase 2 family, polypeptide B5 | phase 2 conjugation | |
| NM_001305992 | −3.25 | Ggt1 | gamma-glutamyltransferase 1 | glutamate metabolic process | |
| NM_008185 | −2.88 | Gstt1 | glutathione S-transferase, theta 1 | glutathione metabolic process | |
| Inflammatory and immunological response (E19) | NM_009892 | 3.14 | Chil3 | chitinase-like 3 | polysaccharide catabolic process |
| NM_001164724 | 2.43 | Il33 | interleukin 33 | leukocyte migration | |
| NM_133871 | 3.66 | Ifi44 | interferon-induced protein 44 | immune response | |
| NM_008331 | 6.73 | Ifit1 | interferon-induced protein with tetratricopeptide repeats 1 | immune system process, response to virus | |
| NM_001099217 | 4.35 | Ly6c1 | lymphocyte antigen 6 complex, locus C1 | Activation of lymphocytes | |
| NM_022983 | 5.03 | Lpar3 | lysophosphatidic acid receptor 3 | activation of MAPK activity | |
| NM_144559 | 2.51 | Fcgr4 | Fc receptor, IgG, low affinity IV | NK T cell proliferation | |
| NM_021297 | 2.42 | Tlr4 | toll-like receptor 4 | innate immune response | |
| NM_016704 | 3.37 | C6 | complement component 6 | in utero embryonic development | |
| Coagulation system/Acute phase response signaling (PND5) | NM_175628 | −8.24 | A2m | alpha-2-macroglobulin | negative regulation of complement activation |
| NM_009244 | −21.46 | Serpina1b | serine (or cysteine) preptidase inhibitor, clade A, member 1 B | protein N-linked glycosylation | |
| NM_001111048 | −39.04 | Fga | fibrinogen alpha chain | adaptive immune response | |
| NM_008407 | −9.09 | Itih3 | inter-alpha trypsin inhibitor, heavy chain 3 | peptidase inhibitor activity | |
| NM_017371 | −4.13 | Hpx | hemopexin | positive regulation of immunoglobulin production | |
| NM_031164 | −6.00 | F13b | coagulation factor XIII, beta subunit | blood coagulation pathway | |
| NM_013697 | −23.5 | Ttr | transthyretin | retinol metabolic process, thyroid hormone transport | |
| Lipid metabolism (PND5) | NM_001277944 | −4.54 | Apoc2 | apolipoprotein C-II | lipid transport |
| NM_009692 | −16.65 | Apoa1 | apolipoprotein A-I | lipid transporter activity | |
| NM_009654 | −27.57 | Alb | albumin | fatty acid binding, transport | |
| NM_007443 | −8.44 | Ambp | alpha 1 microglobulin | protein-chromophore linkage | |
| Angiogenesis, connective tissue disorder (PND56) | NM_172399 | −2.54 | Ndnf | neuron-derived neurotrophic factor | angiogenesis |
| NM_054077 | −2.25 | Prelp | proline arginine-rich end leucine-rich repeat | axonogenesis | |
| NM_007734 | −1.87 | Col4a3 | collagen, type IV, alpha 3 | negative regulation of angiogenesis | |
| NM_009371 | −1.86 | Tgfbr2 | transforming growth factor, beta receptor II | patterning of blood vessels |
†Fold difference of baseline lung gene expression between Gsr-WT and Gsr-KO mice at the designated age (negative values indicate lowered expression in Gsr-KO than in Gsr-WT, positive values indicate heightened expression in Gsr-KO than in Gsr-WT). Full lists of the significantly varied genes between two genotypes determined by moderated t-test (p < 0.01) are in Supplementary Tables S1–S3. E=embryonic day. PND=postnatal day.
At the saccular to alveolar transition (PND5), Gsr deficiency significantly affected the expression of 448 transcripts (p < 0.01). Most predominantly, the transcripts involved in lipid metabolism and process (e.g., apolipoprotein family, albumin), coagulation system (e.g., fibrinogens, serine (or cysteine) peptidase inhibitor family), and acute phase responses (alpha-2-macroglobulin) were suppressed in Gsr-KO lungs versus Gsr-WT lungs (Fig. 4C, Tables 1 and S2). These data suggest that GSR is likely essential in surfactant production and other lipid-related lung developmental processes as well as blood coagulation during the saccular-to-alveolar transition stage. This may explain the histologic interruption of alveolar budding beginning at this stage in Gsr-KO lungs (Fig. 2B).
In the mature (PND56) lung, the effect of Gsr deficiency on the transcriptome was relatively marginal (n=318, p < 0.01). Genes differentially expressed between two genotypes (Tables 1 and S3) were mainly involved in connective tissue development, angiogenesis, and cardiovascular function (Fig. 4D). Transcripts constitutively lower in Gsr-KO than in Gsr-WT at PND56 included collagens (e.g., collagen type IV), transforming growth factor, beta receptor II (Tgfbr2), neuron-derived neurotrophic factor (Ndnf), and vinculin (Vcl) (Tables 1 and S3).
3.5. Differential effects of neonatal hyperoxic exposure on Gsr-WT and Gsr-KO mice
Characteristic alveolar simplification was present in Gsr-WT/0.85 mice at PND14 (Fig. 5B) as evidenced by decreased alveolar number (Fig. 5E) and increased alveolar perimeter (Fig. 5F). In Gsr-KO/0.21 mice at PND14, evidence of persistent alveolar epithelial dysplasia remained (Fig. 5C) and was quantitatively reflected by decreased alveolar number (Fig. 5E). Morphometric analyses indicated independent effects of and an interaction between hyperoxia and genotype on alveolar number and perimeter (Fig. 5E and F). The effects of hyperoxia on lung architecture in Gsr-WT mice were comparatively blunted in Gsr-KO mice (Fig. 5E and F).
Fig. 5.

Effects of neonatal hyperoxic exposure on glutathione reductase-sufficient (Gsr-WT) and -deficient (Gsr-KO) mice. (A) H&E-stained lung sections (200x) from PND14 Gsr-WT and Gsr-KO neonates exposed to room air (FiO2 0.21) or hyperoxia (FiO2 0.85) for 14 d (PND0-14). Characteristic alveolar simplification is present in hyperoxia-exposed Gsr-WT mice while evidence of persistent alveolar epithelial dysplasia remains in Gsr-KO mice after hyperoxia exposure. (B) Independent effects of and an interaction between hyperoxia and genotype on alveolar number and perimeter. Data are expressed as mean +SEM (n=3–5). *p < 0.05 vs Gsr-WT/Room Air. **p < 0.05 vs Gsr-KO/Room Air. Bars = 50 μm.
3.6. Compensatory effects within the TXN system in Gsr-KO mice with and without oxygen exposure
By PND7 in Gsr-WT animals, the effects of hyperoxia on the pulmonary TXN system are variable with no change in TXN1, but increase in TXN2 expression, and a trend toward an increase in TXNRD1 (Fig. 6). In contrast, Gsr deficiency altered the baseline expression of the TXN system with increased TXN1 and TXN2 expression. Exposure to hyperoxia potentiated this effect relative to Gsr-KO/0.21, as well as both Gsr-WT/0.21 and Gsr-WT/0.85 mice (Fig. 6A, C). Similarly, TXNRD1 expression was significantly upregulated in Gsr-KO/0.85 mice when compared to all other groups (Gsr-KO/0.21, Gsr-WT/0.21 and Gsr-WT/0.85), although not significantly increased at baseline in Gsr-KO/0.21 compared to Gsr-WT/0.21 mice (Fig. 6B). Western blot analysis indicated an effect of genotype and exposure on the expression of TXN1, TXN2 and TXNRD1, as well as an interaction for TXNRD1 and TXN2 expression based on genotype and exposure.
Fig. 6.

Effects of glutathione reductase (Gsr) deficiency and hyperoxia on the pulmonary thioredoxin (TXN) system at PND7. Protein levels of TXN1, TXN reductase 1 (Txnrd1), and TXN2 normalized by actin levels in wild-type (Gsr-WT) and Gsr-deficient (Gsr-KO) neonatal mice. 2-Way ANOVA followed by Tukey's post hoc revealed an effect of genotype and exposure on the expression of all three proteins, as well as an interaction on TXNRD1 and TXN2 expression. Data expressed as mean +SEM (n=3), p < 0.05 *vs Gsr-WT/0.21, #vs Gsr-KO/0.21, &vs Gsr-WT/0.85.
3.7. Effect of neonatal hyperoxic exposure on lung transcriptomics
At the saccular to alveolar transition (PND5), hyperoxic exposure alone caused 1129 genes to be differentially expressed in Gsr-WT mice, whereas Gsr deficiency modulated these changes to only 663 genes (Fig. 7A). A total of 303 transcripts were significantly (p < 0.01) modulated by hyperoxia in a Gsr-dependent manner (Table 2 and S4). Interestingly, many of these genes were basally suppressed in Gsr-KO neonates relative to Gsr-WT neonates while they were upregulated by hyperoxia only in Gsr-KO mice as depicted in the heat map from hierarchical clustering analysis (Fig. 7A). Venn diagram analysis determined that only 96 common genes were modulated by hyperoxia in both genotypes (Fig. 7A, Tables S5 and S6). The Gsr-dependent, hyperoxia-responsive genes included multiple keratins, myosins, actins, and calcium channels (Fig. 7B, Tables 2 and S4), which were highly associated with abnormal cytoskeleton and skeletal-muscular function and tissue morphology (Fig. 7C). On the other hand, these genes were also predicted to inhibit respiratory failure and neonatal death through activation of upstream myogenic differentiation 1 (MYOD1) and β-catenin (CTNNB1) in hyperoxia-exposed Gsr-KO neonates (Fig. 7D). There was marked upregulation of genes involved in neuronal nitric oxide synthase signaling (e.g., Cacng1, Nos1, Ryr1), calcium signaling (Acta1, Caso1, Myh, Tnnc2), and glycolysis/ketolysis (e.g., Fbp2, Eno3, Pfkm, Bdh1, Acat1) as well as keratins and myosins (Table S6), in the neonate hyperoxia-exposed Gsr-KO lungs. These transcriptome changes suggeste that compromise of the morphology and function in connective tissue and muscle may explain the morphologic features of Gsr-KO neonates after hyperoxia exposure (Fig. 5).
Fig. 7.

Effects of glutathione reductase (Gsr) deficiency and hyperoxia (85% O2) on lung transcriptomics. (A) Heat map from hierarchical clustering analysis depicts lung expression profiles of Gsr-dependently hyperoxia (0.85 FiO2)-responded genes at postnatal day 5 (PND5) after hyperoxia exposure (PND0-PND5, n = 303, 2-way ANOVA with p < 0.01). Color bar indicates average expression intensity (n = 3/group) normalized to wild-type (Gsr-WT)-Air (0.21 FiO2) group. Venn Diagram analysis depicted number of genotype-specific hyperoxia responsive genes determined by moderated t-test in Gsr-WT and Gsr-deficient (Gsr-KO) neonates. (B) Hyperoxia altered lung genes involved predominantly in cytoskeleton and skeletal/muscular development and tissue and cell morphology in Gsr-KO neonates. (C) The key molecular network of the Gsr-dependently altered neonatal lung genes by hyperoxia was cytoskeleton and skeletal-muscular function and tissue morphology and development. (D) Hyperoxia-altered genes in Gsr-KO neonates may inhibit respiratory failure and neonatal death through activation of upstream molecules such as myogenic differentiation 1 (Myod1) and β-catennin (CTNNB1). (E) Venn Diagram analysis depicted number of genotype-specific hyperoxia responsive genes following recovery from neonatal hyperoxia recovery within Gsr-WT and Gsr-KO adult mice. (F) Neonatally exposed hyperoxia was predicted to inhibit genes involved in lipid metabolism (e.g., bile acid, cholesterol) and enhance genes involved in transcription of adulthood lungs in Gsr-KO mice. Analysis was done by GeneSpring and Ingenuity Pathway Analysis software. Molecules colored by expression levels of Gsr-KO/0.85 FiO2 at PND5 (vs Gsr-WT/0.85 FiO2). (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Table 2.
Representative glutathione reductase (Gsr)-dependent neonate lung genes regulated by hyperoxia exposed during postnatal days (PND0-PND5).
| RefSeq ID | p | aFC WT | aFC KO | ¶FD | Gene Symbol | Gene Title |
|---|---|---|---|---|---|---|
| NM_001313949 | 0.001502453 | −5.40 | 34.61 | 36.03 | Krt13 | keratin 13 |
| NM_008475 | 7.66E-04 | −4.69 | 20.55 | 21.29 | Krt4 | keratin 4 |
| NM_177369 | 0.0081382 | −1.69 | 48.45 | 19.19 | Myh8 | myosin, heavy polypeptide 8, skeletal muscle, perinatal |
| NM_027416 | 0.001268343 | −2.70 | 7.90 | 9.26 | Calml3 | calmodulin-like 3 |
| NM_001272041 | 0.005731548 | −1.50 | 21.62 | 7.56 | Acta1 | actin, alpha 1, skeletal muscle |
| NM_001081123 | 3.51E-04 | −1.34 | 8.05 | 5.92 | Arhgap36 | Rho GTPase activating protein 36 |
| NM_028798 | 0.001839206 | −2.15 | 4.21 | 5.20 | Crct1 | cysteine-rich C-terminal 1 |
| NM_028216 | 0.008994052 | −2.29 | 4.52 | 4.92 | Psca | prostate stem cell antigen |
| NM_001101605 | 0.0049872 | −1.91 | 4.34 | 4.90 | Ifit1bl1 | interferon induced protein with tetratricpeptide repeats 1 B like 1 |
| NM_013456 | 0.006323175 | −1.12 | 6.94 | 4.67 | Actn3 | actinin alpha 3 |
| NM_001164787 | 0.007458202 | −2.65 | 4.38 | 4.64 | Sprr2a1, etc. | small proline-rich protein 2A1, etc. |
| NM_001033239 | 0.002655688 | −1.90 | 5.58 | 4.51 | Csta1 | cystatin A1 |
| NM_173385 | 0.0084611 | −1.54 | 3.61 | 4.50 | Cilp | cartilage intermediate layer protein, nucleotide pyrophosphohydrolase |
| NM_007582 | 0.001477798 | −1.80 | 3.55 | 4.22 | Dsc2 | desmocollin 2 |
| NM_013505 | 0.001027153 | −1.15 | 4.35 | 4.05 | Cacng1 | calcium channel, voltage-dependent, gamma subunit 1 |
| NM_008657 | 0.008563787 | −1.37 | 3.87 | 3.54 | Myf6 | myogenic factor 6 |
| NM_001081375 | 0.003077287 | −1.92 | 3.02 | 3.29 | Cnfn | cornifelin |
| NM_001081157 | 0.004109589 | −1.44 | 2.83 | 3.27 | Lmod3 | leiomodin 3 (fetal) |
| NM_001033131 | 0.0016160 | −1.41 | 2.92 | 3.17 | Krtdap | keratinocyte differentiation associated protein |
| NM_009109 | 0.004861059 | −1.15 | 3.82 | 3.12 | Ryr1 | ryanodine receptor 1, skeletal muscle |
| NM_007812 | 0.0023872 | −1.31 | 3.12 | 2.90 | Cyp2a4 | cytochrome P450, family 2, subfamily a, polypeptide 4 |
| NM_007376 | 0.004647014 | −9.79 | −1.18 | −1.59 | Pzp | pregnancy zone protein |
| NM_008725 | 0.008516206 | 1.42 | −19.17 | −1.52 | Nppa | natriuretic peptide type A |
| NM_001012766 | 0.0036717 | 1.20 | −1.30 | −1.36 | Ear1 | eosinophil-associated, ribonuclease A family, member 1 |
| NM_007707 | 0.006182605 | 1.41 | −1.01 | −1.32 | Socs3 | suppressor of cytokine signaling 3 |
| NM_001150749 | 0.007889654 | −5.05 | 1.04 | −1.32 | Rdh7 | retinol dehydrogenase 7 |
| NM_030110 | 0.0063489 | 1.27 | −1.01 | −1.27 | Micu3 | mitochondrial calcium uptake family, member 3 |
¶Fold difference of gene expression between Gsr-WT and Gsr-KO mice at the end of 5-day hyperoxia exposure at PND5 (negative values indicate lower expression in Gsr-KO than in Gsr-WT, positive values indicate higher expression in Gsr-KO than in Gsr-WT). Full lists of the Gsr-dependently varied neonatal lung genes by hyperoxia (n=303, 2-way ANOVA p < 0.01) are in Supplementary Table S4.
Fold change after 5-day hyperoxia exposure in Gsr-WT or in Gsr-KO over genotype-matched air-exposed controls (negative values indicate decreased expression by hyperoxia, positive values indicate increased expression by hyperoxia).
At 8 weeks of age, 6 weeks after termination of hyperoxic exposure, 403 genes (p < 0.01) were significantly different between neonatal hyperoxia-exposed and neonatal air exposed Gsr-WT mice (Table S7). Chemokine and other inflammatory genes (e.g., Cxcl5, Cxcl3, Ccl17, Mmp12) were a distinguished cluster of genes that were more abundantly expressed in neonatal hyperoxia-exposed lungs than in air-exposed lungs with prediction of activated upstream regulators including tumor necrosis factor and nuclear factor-kappa B (Supplementary Fig. S2A). Dysregulation of tissue and vessel development and increased mortality and lymphocytic tumorigenesis were also identified by pathway analysis (Supplementary Figs. S2–B). These data indicate that ongoing adverse pulmonary molecular events into adulthood may adversely influence the persistent BPD-like phenotype. Between Gsr-WT and Gsr-KO mice, 491 lung genes were significantly varied (p < 0.01) at PND56 and after neonatal exposure although expression difference of these genes were marginal (<2-folds, Table 3 and S8). Of the 491 genes differentially expressed between the genotypes at PND56 following oxygen exposure, 56 genes were similarly altered by oxygen exposure in the Gsr-WT animals leaving 435 genes that were uniquely altered by Gsr deficiency (Fig. 7E). Interestingly, up or downregulation trends of many genes were opposite in Gsr-WT and Gsr-KO mice at this time point (Tables 3 and S8). Pathway analysis demonstrated that lack of Gsr may drive transcriptome changes in nerve and tissue development and morphology, cell-to-cell signaling, RNA transcription, and lipid metabolism (e.g., bile acid, cholesterol) in adulthood lungs exposed to neonatal hyperoxia (Fig. 7F).
Table 3.
Representative glutathione reductase (Gsr)-dependent lung genes at postnatal day 56 (PND56) after neonatal hyperoxia exposure.
| RefSeq ID | p | aFC WT | aFC KO | ¶FD | Gene Symbol | Gene Title |
|---|---|---|---|---|---|---|
| NM_001177713 | 0.00457 | −1.73 | 2.26 | 4.13 | Cyp26b1 | cytochrome P450, family 26, subfamily b, polypeptide 1 |
| NM_001166627 | 0.001644 | −1.30 | 1.03 | 1.49 | Dynlt1-ps1 | dynein light chain Tctex-type 1, pseuodogene 1///dynein light chain Tctex-type 1A |
| NM_010731 | 4.38E-04 | −1.91 | 1.12 | 1.49 | Zbtb7a | zinc finger and BTB domain containing 7a |
| NM_144879 | 5.83E-04 | −1.21 | 1.30 | 1.42 | Vash2 | vasohibin 2 |
| NM_146244 | 4.96E-05 | −1.15 | 1.32 | 1.36 | Rps6kl1 | ribosomal protein S6 kinase-like 1 |
| NM_022033 | 0.004434 | −1.30 | 1.22 | 1.35 | Oxct2a | 3-oxoacid CoA transferase 2A |
| NM_203320 | 0.005966 | 5.21 | 1.32 | −1.81 | Cxcl3 | chemokine (C-X-C motif) ligand 3 |
| NM_010861 | 0.005792 | 1.09 | −3.33 | −1.76 | Myl2 | myosin, light polypeptide 2, regulatory, cardiac, slow |
| NM_010780 | 6.16E-04 | 1.36 | −1.15 | −1.47 | Cma1 | chymase 1, mast cell |
| XM_006504243 | 0.00933 | 1.42 | −1.26 | −1.47 | Tmem156 | transmembrane protein 156 |
| NM_019514 | 6.26E-04 | 1.35 | −1.16 | −1.43 | Astn2 | astrotactin 2 |
| NM_201638 | 0.002129 | 1.18 | −1.41 | −1.40 | Mettl14 | methyltransferase like 14 |
| NM_009140 | 0.003614 | 2.65 | 1.24 | −1.27 | Cxcl2 | chemokine (C-X-C motif) ligand 2 |
| NM_019810 | 0.006028 | 1.88 | 1.23 | −1.36 | Slc5a1 | solute carrier family 5 (sodium/glucose cotransporter), member 1 |
| NM_009100 | 0.008386 | 1.59 | −1.09 | −1.35 | Rptn | repetin |
| NM_001099774 | 0.008755 | 1.08 | −1.63 | −1.13 | Krtap17-1 | keratin associated protein 17-1 |
| NM_009162 | 3.89E-06 | 1.18 | −1.48 | −1.12 | Scg5 | secretogranin V |
¶Fold difference of lung gene expression between Gsr-WT and Gsr-KO mice at PND56 after neonatal hyperoxia exposure (negative values indicate lower expression in hyperoxia-PND56 Gsr-KO than in hyperoxia-PND56 Gsr-WT, positive values indicate higher expression in hyperoxia-PND56 Gsr-KO than in hyperoxia-PND56 Gsr-WT). Full lists of the Gsr-dependently varied lung genes during recovery after neonate hyperoxia exposure (n=491, 2-way ANOVA p < 0.01) are in Supplementary Table S8.
Fold change in PND56 lung after neonate-hyperoxia exposure compared to neonate-air exposure in each genotype (negative values indicate decreased expression in neonate hyperoxia-exposed lung than in neonate air-exposed lung, positive values indicate increased expression in neonate hyperoxia-exposed lung than in neonate air-exposed lung).
4. Discussion
The effect of Gsr deficiency in the neonatal period has not been previously described. We quantified (compared) lung GSH, GSSG and GSH/GSSG levels in 6-week old adult Gsr-WT and Gsr-KO mice and found an increase in GSSG levels and a dramatic decrease in the GSH/GSSG ratio in Gsr-KO mice [8]. In contrast, our current neonatal studies show a significant decrease in all components in the glutathione redox cycle suggesting that the increased GSSG levels detected in adult animals build up over time as the animal is unable to reduce GSSG to GSH in order to recycle reducing equivalents. Neonatal mammals are known to have immature redox systems which place them at risk for oxidative stress and damage, especially when born prematurely or exposed to postnatal stressors [[23], [24], [25], [26], [27]].
The GSH and GSSG deficits within our Gsr-KO mice reveal an inability to stimulate this intracellular antioxidant system in the neonatal period as was observed in the Gsr-WT mice. Our data also suggest a progressive decline in redox potential in Gsr-KO mice as evidenced by the decreasing GSH:GSSG Eh during the neonatal period. GSH + GSSG levels were lower in Gsr-KO mice though the mechanism for this finding is not entirely clear. A review of transcriptomic data revealed no strain-dependent differences in the expression of genes required for de novo GSH synthesis, including glutamate-cysteine ligase, glutamate-cysteine ligase catalytic subunit, and glutathione synthetase. It is possible that the cysteine pool is lower in Gsr-KO mice when compared to wild-type mice, though these investigations are beyond the scope of the present study.
Our previous experience with Gsr-KO mice indicated that no lung phenotype was present in adulthood [6,8]. We first investigated the maturation of the lung during the late canalicular and late saccular phases of murine lung development. These stages of lung development are prior to alveolarization, when standard, objective morphometric quantification is valid [28]; Gross examination of canalicular phase lungs at E19 revealed the presence of septal thickening and epithelial dysplasia. Similar epithelial abnormalities persisted into the saccular phase at PND3 with thickened septal walls and disorganization of the developing saccules. In the absence of formal morphometric analyses at the canalicular and saccular phase of lung development, lung samples were assessed by a murine pathologist who found the Gsr-KO lungs to be significantly different than WT controls with the presence of epithelial cell dysplasia and decreased transdifferentiation of AT2 to AT1 cells. With the onset of the alveolar lung development, there is the exponential expansion of the alveoli due to transdifferentiation of AT2 to AT1 cells which is represented in the doubling of the lung weight and alveoli numbers between PND5 and 7 [[29], [30], [31], [32]]. By examining AT2 and AT1 cell markers via Western blot at this time point, our data supported deficits in the ability of AT2 cells to transdifferentiate into AT1 cells as described by the pathologist. By PND14, there were still abnormalities of the alveolar architecture when quantified by formal morphometric analyses. Gsr-KO mice exhibited a more homogeneous lung phenotype as there were less complete alveoli per high power field and those that could be quantified had a smaller perimeter. Usually, these two morphology parameters track together with decreased numbers correlating with significantly enlarged airway perimeters. We speculate that the Gsr-KO animals have many enlarged air-sacs that could not be counted due to the field of view of the microscope.
In accordance with our previous experience, the lung architecture was relatively normal by adulthood in Gsr-KO mice. Looks can be deceiving, however, as functional assessments identified pulmonary function deficits and airway hypersensitivity that had not previously been appreciated. Physiologically, airway resistance comes from tethering of the lung parenchyma by alveolar segments. Thus, decreased resistance in for Gsr-KO mice might reflect subtle alveolar abnormalities not appreciated under gross structural examination [33] or decreases in matrix deposition that affect lung structure itself. Similarly, increased sensitivity to methacholine indicates an airway hyperactivity to stimuli similar to that seen in childhood for patients with a history of BPD and in other models with altered reactive oxygen species [[34], [35], [36], [37]].
To determine potential mechanisms driving structural abnormalities in Gsr-KO lungs, we next investigated the effects of Gsr-deficiency on gene expression and cell signaling pathways. Whole lung transcriptome analysis identified differentially expressed genes over time due to aging between the genotypes. Consistent with the pronounced histopathologic differences between Gsr-WT and Gsr-KO at earlier timepoints, the most dramatic transcriptomic changes were present in the embryonic tissues during the canalicular phase of lung development. The differences decreased by almost half with the transition to the late saccular stage at PND5. Similar to our histopathologic findings, the transcriptomic phenotype was less distinct between the genotypes in adulthood.
Genes with differential expression in Gsr-deficiency were not static throughout lung development and our data revealed unique sets of genes and differential expression at embryonic and neonatal timepoints. Decreased expression of genes related to cell cycle pathways and cell division at E19 are plausible explanations for the histopathologically dysplastic epithelium present at these early timepoints. At PND5, the Gsr-KO mouse lung transcriptome was reflective of impaired lipid metabolism and surfactant production preceding the beginning of alveolarization. Lipid metabolism, including specifically surfactant production, is a highly oxidative process that occurs within the endoplasmic reticulum [38,39]. The glutathione redox potential within the endoplasmic reticulum depends in GSH import [40]. Given our finding of decreased intracellular GSH levels in the Gsr-KO animals, we speculate that there is decreased reducing equivalents to buffer these processes within the ER. Surfactant production is mediated by AT2 cells. In the rodent, surfactant production begins in the canalicular phase and increases through the saccular alveolar stage [32,38]. Though our data suggests that the total quantity of AT2 cells is not affected by Gsr deficiency, the AT2 cell function may be compromised. Poor Gsr-KO AT2 function is supported by the finding of decreased transdifferentiation into AT1 cells and the transcriptomic changes in surfactant production. The compensatory increase of TXN to maintain intracellular redox balance may not be effective within the endoplasmic reticulum or even be transported into the endoplasmic reticulum, and therefore, unable to compensate for oxidative stress during lipid transport. By adulthood, the pathways that remained active in the Gsr-KO animals were less connected to the pulmonary epithelium and more related to angiogenesis and cardiovascular function. Interestingly, fibrotic genes, such as collagens and Tgfbr2 traditionally associated with hyperoxic lung injury models and BPD were significantly decreased in the Gsr-KO adult mice at baseline; changes in expression of these genes might contribute to the resistance towards hyperoxic lung injury previously reported in this genotype as adults [8,20,41,42].
As previously seen in our adult Gsr-KO studies, neonatal Gsr-KO mice had a blunted injury response to hyperoxia. Although the Gsr-KO mice had an abnormal baseline lung structure on morphometric analysis (as discussed above), the relative change between unexposed and oxygen exposed groups was comparatively less than in Gsr-WT animals. Following oxygen exposure, our data indicate an upregulation of the TXN system in Gsr-KO mice. It is likely that this compensatory response contributes to relative hyperoxic insensitivity to lung injury; however, such studies are beyond the scope of the present manuscript.
Similar to previous reports [21,[43], [44], [45]], we found a high number of genes to be differentially expressed following oxygen exposure in wild type animals. Conversely, this response was blunted in Gsr-KO mice and the directionality of the change in gene expression was inconsistent. Activation of pathways like myogenic differentiation and β-catenin in the Gsr-KO animals may contribute the blunted response to hyperoxia. Alterations in neuronal nitric oxide synthase signaling, glycolysis/ketolysis and keratins are predicted to coalesce into pathways that compromise morphology and structure of connective tissue which may explain the functional abnormalities that were present in the lungs.
Other groups have examined differential gene expression following recovery from neonatal hyperoxia exposure and found a similar decline in the number of differentially expressed genes [46]. As expected, in Gsr-WT animals we found persistent signals consistent with BPD-like phenotypes, such as dysregulation of tissue and vessel development and inflammation. Activation of tumor necrosis factor and nuclear factor-kappa B have been extensively described in murine models for BPD [47,48]. Interestingly, there was not a significant overlap of the differentially expressed genes between the genotypes after recovery from hyperoxia exposure. Although Gsr deficiency may disrupt cell-to-cell signaling or lipid metabolism in adult lungs of mice exposed to hyperoxia during the neonatal period, there does not appear to be a persistent inflammatory or vascular phenotype.
The current studies support our initial hypothesis that GSR expression is necessary for normal perinatal lung maturation and development. Differences in lung redox status in Gsr-KO mice are evident shortly after birth and likely contribute to altered lung development, airway reactivity, and differential gene expression profiles. Furthermore, Gsr-deficient mice are less sensitive to the effects of hyperoxia, likely via compensatory upregulation of TXN-dependent cytoprotective responses as was similarly observed in adult mice. Our data reveal that alterations in GSH system functionality may drive epithelial dysregulation and altered lung development in the neonatal lung. Strategies that enhance GSH-dependent redox responses, such as TXN system modulation, may attenuate oxidative injury in the neonatal lung and reduce the incidence of BPD.
Author contributions
TET designed and supervised the study. MER, JMM, JRL, MLL, and MV conducted experiments. HYC and SRK performed microarray and bioinformatic analysis. TET, MER, LKR, and HYC wrote the manuscript.
Conflicts of interest
The authors declare that they have no conflict of interest.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The authors wish to thank Cynthia Hill and Stephanie Wall for excellent technical assistance. Funding for this work was provided by National Institue of Health/National Heart Lung Blood Institute (NIH/NHLBI )(K08HL093365 and R01HL119280 to T.E.T.) and NIH/National Institue of Environmental Health Sciences (NIEHS) (ZIAES100513 to H.C. and S.R.K.), United States. Additional funding was provided by the Abigail Wexner Research Institute at Nationwide Children's Hospital, United States (M.E.R., L.K.R. and T.E.T.) and The University of Alabama at Birmingham, United States.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2020.101797.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Crapo J.D. Morphologic changes in pulmonary oxygen toxicity. Annu. Rev. Physiol. 1986;48:721–731. doi: 10.1146/annurev.ph.48.030186.003445. PubMed PMID: 3518622. [DOI] [PubMed] [Google Scholar]
- 2.Barrios R., Shi Z.Z., Kala S.V., Wiseman A.L., Welty S.E., Kala G., Bahler A.A., Ou C.N., Lieberman M.W. Oxygen-induced pulmonary injury in gamma-glutamyl transpeptidase-deficient mice. Lung. 2001;179(5):319–330. doi: 10.1007/s004080000071. PubMed PMID: 11976899. [DOI] [PubMed] [Google Scholar]
- 3.Nordberg J., Arner E.S. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radic. Biol. Med. 2001;31(11):1287–1312. doi: 10.1016/s0891-5849(01)00724-9. PubMed PMID: 11728801. [DOI] [PubMed] [Google Scholar]
- 4.Eriksson S., Prigge J.R., Talago E.A., Arner E.S., Schmidt E.E. Dietary methionine can sustain cytosolic redox homeostasis in the mouse liver. Nat. Commun. 2015;6:6479. doi: 10.1038/ncomms7479. Epub 2015/03/21. PubMed PMID: 25790857; PMCID: PMC4369796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iverson S.V., Eriksson S., Xu J., Prigge J.R., Talago E.A., Meade T.A., Meade E.S., Capecchi M.R., Arner E.S., Schmidt E.E. A Txnrd1-dependent metabolic switch alters hepatic lipogenesis, glycogen storage, and detoxification. Free Radic. Biol. Med. 2013 doi: 10.1016/j.freeradbiomed.2013.05.028. Epub 2013/06/08. PubMed PMID: 23743293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rogers L.K., Tamura T., Rogers B.J., Welty S.E., Hansen T.N., Smith C.V. Analyses of glutathione reductase hypomorphic mice indicate a genetic knockout. Toxicol. Sci. 2004;82(2):367–373. doi: 10.1093/toxsci/kfh268. PubMed PMID: 15342956. [DOI] [PubMed] [Google Scholar]
- 7.Schirmer R.H., Krauth-Siegel R.L., Schulz G.E. Coenzymes and cofactors. In: Dolphin D., Poulson R., Avramovic O., editors. Coenzymes and Cofactors. John Wiley & Sons, Inc; New York: 1989. pp. 553–596. [Google Scholar]
- 8.Tipple T.E., Welty S.E., Rogers L.K., Hansen T.N., Choi Y.E., Kehrer J.P., Smith C.V. Thioredoxin-related mechanisms in hyperoxic lung injury in mice. Am. J. Respir. Cell Mol. Biol. 2007;37(4):405–413. doi: 10.1165/rcmb.2006-0376OC. Epub 2007/06/19. doi: 2006-0376OC [pii] 10.1165/rcmb.2006-0376OC. PubMed PMID: 17575077; PMCID: 2176120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Williams C.H. In: Chemistry and Biochemistry of Flavoenzymes. Muller F., editor. CRC Press, Inc; Boca Raton, FL: 1992. pp. 121–211. [Google Scholar]
- 10.Britt R.D., Jr., Velten M., Locy M.L., Rogers L.K., Tipple T.E. The thioredoxin reductase-1 inhibitor aurothioglucose attenuates lung injury and improves survival in a murine model of acute respiratory distress syndrome. Antioxidants Redox Signal. 2014;20(17):2681–2691. doi: 10.1089/ars.2013.5332. PubMed PMID: 24295151; PMCID: 4026403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dunigan K., Li Q., Li R., Locy M.L., Wall S.B., Tipple T.E. The thioredoxin reductase inhibitor auranofin induces heme oxygenase-1 in lung epithelial cells via nrf2-dependent mechanisms. Am. J. Physiol. Lung Cell Mol. Physiol. 2018 doi: 10.1152/ajplung.00214.2018. Epub 2018/07/20. PubMed PMID: 30024305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li Q., Li R., Wall S.B., Dunigan K., Ren C., Jilling T., Rogers L.K., Tipple T.E. Aurothioglucose does not improve alveolarization or elicit sustained Nrf2 activation in C57BL/6 models of bronchopulmonary dysplasia. Am. J. Physiol. Lung Cell Mol. Physiol. 2018;314(5):L736–L742. doi: 10.1152/ajplung.00539.2017. Epub 2018/01/26. PubMed PMID: 29368550; PMCID: PMC6008122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li Q., Wall S.B., Ren C., Velten M., Hill C.L., Locy M.L., Rogers L.K., Tipple T.E. Thioredoxin reductase inhibition attenuates neonatal hyperoxic lung injury and enhances Nrf2 activation. Am. J. Respir. Cell Mol. Biol. 2016 doi: 10.1165/rcmb.2015-0228OC. PubMed PMID: 27089175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Locy M.L., Rogers L.K., Prigge J.R., Schmidt E.E., Arner E.S., Tipple T.E. Thioredoxin reductase inhibition elicits nrf2-mediated responses in Clara cells: implications for oxidant-induced lung injury. Antioxidants Redox Signal. 2012 doi: 10.1089/ars.2011.4377. Epub 2012/05/23. PubMed PMID: 22607006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tindell R., Wall S.B., Li Q., Li R., Dunigan K., Wood R., Tipple T.E. Selenium supplementation of lung epithelial cells enhances nuclear factor E2-related factor 2 (Nrf2) activation following thioredoxin reductase inhibition. Redox Biol. 2018;19:331–338. doi: 10.1016/j.redox.2018.07.020. Epub 2018/09/14. PubMed PMID: 30212802; PMCID: PMC6134185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pretsch W. Glutathione reductase activity deficiency in homozygous Gr1a1Neu mice does not cause haemolytic anaemia. Genet. Res. 1999;73(1):1–5. doi: 10.1017/s0016672398003590. PubMed PMID: 10218442. [DOI] [PubMed] [Google Scholar]
- 17.Yan J., Meng X., Wancket L.M., Lintner K., Nelin L.D., Chen B., Francis K.P., Smith C.V., Rogers L.K., Liu Y. Glutathione reductase facilitates host defense by sustaining phagocytic oxidative burst and promoting the development of neutrophil extracellular traps. J. Immunol. 2012;188(5):2316–2327. doi: 10.4049/jimmunol.1102683. PubMed PMID: 22279102; PMCID: PMC3480216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Halvey P.J., Watson W.H., Hansen J.M., Go Y.M., Samali A., Jones D.P. Compartmental oxidation of thiol-disulphide redox couples during epidermal growth factor signalling. Biochem. J. 2005;386(Pt 2):215–219. doi: 10.1042/BJ20041829. Epub 2005/01/14. PubMed PMID: 15647005; PMCID: PMC1134784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jones D.P. Redox potential of GSH/GSSG couple: assay and biological significance. Methods Enzymol. 2002;348:93–112. doi: 10.1016/s0076-6879(02)48630-2. Epub 2002/03/12. PubMed PMID: 11885298. [DOI] [PubMed] [Google Scholar]
- 20.Velten M., Heyob K.M., Rogers L.K., Welty S.E. Deficits in lung alveolarization and function after systemic maternal inflammation and neonatal hyperoxia exposure. J. Appl. Physiol. 2010;108(5):1347–1356. doi: 10.1152/japplphysiol.01392.2009. Epub 2010/03/13. doi: 01392.2009 [pii] 10.1152/japplphysiol.01392.2009. PubMed PMID: 20223995; PMCID: 2867533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cho H.Y., van Houten B., Wang X., Miller-Degraff L., Fostel J., Gladwell W., Perrow L., Panduri V., Kobzik L., Yamamoto M., Bell D.A., Kleeberger S.R. Targeted deletion of Nrf2 impairs lung development and oxidant injury in neonatal mice. Antioxidants Redox Signal. 2012 doi: 10.1089/ars.2011.4288. Epub 2012/03/10. PubMed PMID: 22400915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Flohe L. The fairytale of the GSSG/GSH redox potential. Biochim. Biophys. Acta. 2013;1830(5):3139–3142. doi: 10.1016/j.bbagen.2012.10.020. Epub 2012/11/07. PubMed PMID: 23127894. [DOI] [PubMed] [Google Scholar]
- 23.Berkelhamer S.K., Kim G.A., Radder J.E., Wedgwood S., Czech L., Steinhorn R.H., Schumacker P.T. Developmental differences in hyperoxia-induced oxidative stress and cellular responses in the murine lung. Free Radic. Biol. Med. 2013;61:51–60. doi: 10.1016/j.freeradbiomed.2013.03.003. PubMed PMID: 23499839; PMCID: PMC3723750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lavoie J.C., Chessex P. Development of glutathione synthesis and gamma-glutamyltranspeptidase activities in tissues from newborn infants. Free Radic. Biol. Med. 1998;24(6):994–1001. doi: 10.1016/s0891-5849(97)00384-5. PubMed PMID: 9607610. [DOI] [PubMed] [Google Scholar]
- 25.Berkelhamer S.K., Farrow K.N. Developmental regulation of antioxidant enzymes and their impact on neonatal lung disease. Antioxidants Redox Signal. 2014;21(13):1837–1848. doi: 10.1089/ars.2013.5515. PubMed PMID: 24295375; PMCID: PMC4203145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lindeman J.H., Lentjes E.G., Berger H.M. Diminished protection against copper-induced lipid peroxidation by cord blood plasma of preterm and term infants. JPEN - J. Parenter. Enter. Nutr. 1995;19(5):373–375. doi: 10.1177/0148607195019005373. PubMed PMID: 8577014. [DOI] [PubMed] [Google Scholar]
- 27.Lindeman J.H., Lentjes E.G., van Zoeren-Grobben D., Berger H.M. Postnatal changes in plasma ceruloplasmin and transferrin antioxidant activities in preterm babies. Biol. Neonate. 2000;78(2):73–76. doi: 10.1159/000014252. PubMed PMID: 10970997. [DOI] [PubMed] [Google Scholar]
- 28.Hsia C.C., Hyde D.M., Ochs M., Weibel E.R., Structure AEJTFoQAoL An official research policy statement of the American Thoracic Society/European Respiratory Society: standards for quantitative assessment of lung structure. Am. J. Respir. Crit. Care Med. 2010;181(4):394–418. doi: 10.1164/rccm.200809-1522ST. Epub 2010/02/05. PubMed PMID: 20130146; PMCID: PMC5455840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Amy R.W., Bowes D., Burri P.H., Haines J., Thurlbeck W.M. Postnatal growth of the mouse lung. J. Anat. 1977;124(Pt 1):131–151. Epub 1977/09/01. PubMed PMID: 914698; PMCID: PMC1235518. [PMC free article] [PubMed] [Google Scholar]
- 30.Mund S.I., Stampanoni M., Schittny J.C. Developmental alveolarization of the mouse lung. Dev. Dynam. 2008;237(8):2108–2116. doi: 10.1002/dvdy.21633. Epub 2008/07/25. PubMed PMID: 18651668. [DOI] [PubMed] [Google Scholar]
- 31.Herriges M., Morrisey E.E. Lung development: orchestrating the generation and regeneration of a complex organ. Development. 2014;141(3):502–513. doi: 10.1242/dev.098186. Epub 2014/01/23. PubMed PMID: 24449833; PMCID: PMC3899811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schittny J.C. Development of the lung. Cell Tissue Res. 2017;367(3):427–444. doi: 10.1007/s00441-016-2545-0. Epub 2017/02/02. PubMed PMID: 28144783; PMCID: PMC5320013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bates J.H.T. Systems physiology of the airways in health and obstructive pulmonary disease. Wiley Interdisciplinary Reviews: Systems Biology and Medicine. 2016;8(5):423–437. doi: 10.1002/wsbm.1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cheong J.L.Y., Doyle L.W. An update on pulmonary and neurodevelopmental outcomes of bronchopulmonary dysplasia. Semin. Perinatol. 2018;42(7):478–484. doi: 10.1053/j.semperi.2018.09.013. Epub 2018/11/08. PubMed PMID: 30401478. [DOI] [PubMed] [Google Scholar]
- 35.Narang I., Baraldi E., Silverman M., Bush A. Airway function measurements and the long-term follow-up of survivors of preterm birth with and without chronic lung disease. Pediatr. Pulmonol. 2006;41(6):497–508. doi: 10.1002/ppul.20385. Epub 2006/04/18. PubMed PMID: 16617446. [DOI] [PubMed] [Google Scholar]
- 36.Shepherd E.G., Clouse B.J., Hasenstab K.A., Sitaram S., Malleske D.T., Nelin L.D., Jadcherla S.R. Infant pulmonary function testing and phenotypes in severe bronchopulmonary dysplasia. Pediatrics. 2018;141(5) doi: 10.1542/peds.2017-3350. Epub 2018/04/07. PubMed PMID: 29622720. [DOI] [PubMed] [Google Scholar]
- 37.Barnes P.J. Reactive oxygen species and airway inflammation. Free Radic. Biol. Med. 1990;9(3):235–243. doi: 10.1016/0891-5849(90)90034-g. Epub 1990/01/01. PubMed PMID: 2272532. [DOI] [PubMed] [Google Scholar]
- 38.Orgeig S.M., Janna, Sullivan Lucy, Daniels Christopher. In: Chapter 9- the Development of the Pulmonary Surfactant System. Harding R., Pinkerton K., editors. Academic Press; 2014. [Google Scholar]
- 39.Batenburg J.J. Surfactant phospholipids: synthesis and storage. Am. J. Physiol. 1992;262(4 Pt 1):L367–L385. doi: 10.1152/ajplung.1992.262.4.L367. Epub 1992/04/01. PubMed PMID: 1566854. [DOI] [PubMed] [Google Scholar]
- 40.Ponsero A.J., Igbaria A., Darch M.A., Miled S., Outten C.E., Winther J.R., Palais G., D'Autreaux B., Delaunay-Moisan A., Toledano M.B. Endoplasmic reticulum transport of glutathione by Sec61 is regulated by Ero1 and bip. Mol Cell. 2017;67(6):962–973. doi: 10.1016/j.molcel.2017.08.012. e5. Epub 2017/09/19. PubMed PMID: 28918898; PMCID: PMC5772773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kotecha S., Wangoo A., Silverman M., Shaw R.J. Increase in the concentration of transforming growth factor beta-1 in bronchoalveolar lavage fluid before development of chronic lung disease of prematurity. J. Pediatr. 1996;128(4):464–469. doi: 10.1016/s0022-3476(96)70355-4. Epub 1996/04/01. PubMed PMID: 8618178. [DOI] [PubMed] [Google Scholar]
- 42.Mizikova I., Morty R.E. The extracellular matrix in bronchopulmonary dysplasia: target and source. Front. Med. 2015;2:91. doi: 10.3389/fmed.2015.00091. Epub 2016/01/19. PubMed PMID: 26779482; PMCID: PMC4688343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McGrath-Morrow S.A., Lauer T., Collaco J.M., Lopez A., Malhotra D., Alekseyev Y.O., Neptune E., Wise R., Biswal S. Transcriptional responses of neonatal mouse lung to hyperoxia by Nrf2 status. Cytokine. 2014;65(1):4–9. doi: 10.1016/j.cyto.2013.09.021. Epub 2013/10/22. PubMed PMID: 24139870; PMCID: PMC3875154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wagenaar G.T., ter Horst S.A., van Gastelen M.A., Leijser L.M., Mauad T., van der Velden P.A., de Heer E., Hiemstra P.S., Poorthuis B.J., Walther F.J. Gene expression profile and histopathology of experimental bronchopulmonary dysplasia induced by prolonged oxidative stress. Free Radic. Biol. Med. 2004;36(6):782–801. doi: 10.1016/j.freeradbiomed.2003.12.007. Epub 2004/03/03. PubMed PMID: 14990357. [DOI] [PubMed] [Google Scholar]
- 45.Lingappan K., Jiang W., Wang L., Moorthy B. Sex-specific differences in neonatal hyperoxic lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 2016;311(2):L481–L493. doi: 10.1152/ajplung.00047.2016. Epub 2016/06/28. PubMed PMID: 27343189; PMCID: PMC5504425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Coarfa C., Zhang Y., Maity S., Perera D.N., Jiang W., Wang L., Couroucli X., Moorthy B., Lingappan K. Sexual dimorphism of the pulmonary transcriptome in neonatal hyperoxic lung injury: identification of angiogenesis as a key pathway. Am. J. Physiol. Lung Cell Mol. Physiol. 2017;313(6):L991–L1005. doi: 10.1152/ajplung.00230.2017. Epub 2017/08/19. PubMed PMID: 28818871; PMCID: PMC5814706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alvira C.M. Nuclear factor-kappa-B signaling in lung development and disease: one pathway, numerous functions. Birth Defects Res A Clin Mol Teratol. 2014;100(3):202–216. doi: 10.1002/bdra.23233. Epub 2014/03/19. PubMed PMID: 24639404; PMCID: PMC4158903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kazzi S.N., Kim U.O., Quasney M.W., Buhimschi I. Polymorphism of tumor necrosis factor-alpha and risk and severity of bronchopulmonary dysplasia among very low birth weight infants. Pediatrics. 2004;114(2):e243–e248. doi: 10.1542/peds.114.2.e243. Epub 2004/08/03. PubMed PMID: 15286263. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.