

Abstract
Background
Crenolanib (crenolanib besylate, 4-piperidinamine, 1-[2-[5-[(3-methyl-3-oxetanyl)methoxy]-1H-benzimidazol-1-yl]-8-quinolinyl]-, monobenzenesulfonate) is a potent and specific type I inhibitor of fms-like tyrosine kinase 3 (FLT3) that targets the active kinase conformation and is effective against FLT3 with internal tandem duplication (ITD) with point mutations induced by, and conferring resistance to, type II FLT3 inhibitors in acute myeloid leukemia (AML) cells. Crenolanib is also an inhibitor of platelet-derived growth factor receptor alpha and beta and is in clinical trials in both gastrointestinal stromal tumors and gliomas.
Methods
We tested crenolanib interactions with the multidrug resistance-associated ATP-binding cassette proteins ABCB1 (P-glycoprotein), ABCG2 (breast cancer resistance protein) and ABCC1 (multidrug resistance-associated protein 1), which are expressed on AML cells and other cancer cells and are important components of the blood-brain barrier.
Results
We found that crenolanib is a substrate of ABCB1, as evidenced by approximate five-fold resistance of ABCB1-overexpressing cells to crenolanib, reversal of this resistance by the ABCB1-specific inhibitor PSC-833 and stimulation of ABCB1 ATPase activity by crenolanib. In contrast, crenolanib was not a substrate of ABCG2 or ABCC1. Additionally, it did not inhibit substrate transport by ABCB1, ABCG2 or ABCC1, at pharmacologically relevant concentrations. Finally, incubation of the FLT3-ITD AML cell lines MV4-11 and MOLM-14 with crenolanib at a pharmacologically relevant concentration of 500 nM did not induce upregulation of ABCB1 cell surface expression.
Conclusions
Thus ABCB1 expression confers resistance to crenolanib and likely limits crenolanib penetration of the central nervous system, but crenolanib at therapeutic concentrations should not alter cellular exposure to ABC protein substrate chemotherapy drugs.
Keywords: Crenolanib, ABCB1, FLT3, Platelet-derived growth factor receptor, Acute myeloid leukemia, Glioma
Introduction
Diverse kinase inhibitors are being tested alone and in combination with chemotherapy drugs with the goal of improving treatment outcomes in a variety of malignancies. Crenolanib (crenolanib besylate, 4-piperidinamine, 1-[2-[5-[(3-methyl-3-oxetanyl)methoxy]-1H-benzimidazol-1-yl]-8-quinolinyl]-,monobenzenesulfonate) (Fig. 1) is undergoing clinical testing in acute myeloid leukemia (AML) as a potent and specific inhibitor of the class III receptor tyrosine kinase (RTK) fms-like tyrosine kinase 3 (FLT3) [1–4] and in gastrointestinal stromal tumors (GIST) [5] and gliomas [6–8] as an inhibitor of class III RTK platelet-derived growth factor receptor (PDGFR)alpha. It also has promising activity in melanoma [9].
Fig. 1.
Chemical structure of crenolanib
The type 3 tyrosine kinase fms-like tyrosine kinase 3 (FLT3) is expressed on AML cells in most AML patients [10], and is mutated, most commonly by internal tandem duplication (ITD), in up to a third [11–15]. FLT3-ITD mutations result in constitutive and aberrant FLT3 signaling, promoting survival and proliferation of AML cells with these mutations [16]. FLT3-ITD is associated with a high relapse rate and short-disease free survival following both chemotherapy and allogeneic hematopoietic stem cell transplantation [11–15]. Given the high frequency of FLT3-ITD mutations in AML and the poor treatment outcomes of patients whose AML cells harbor these mutations, much work has focused on identifying and developing inhibitors of FLT3 signaling and testing them both as single agents and in conjunction with AML chemotherapy regimens.
Diverse FLT3 inhibitors have been tested preclinically and in clinical trials. First-generation FLT3 inhibitors, including midostaurin (PKC412) [17] and lestaurtinib (CEP-701) [18], did not have optimal potency, specificity or pharmacokinetic properties. Sorafenib, a multikinase inhibitor approved for treatment of renal, hepatic and thyroid carcinomas, is an effective FLT3 inhibitor and is used off-label in AML with FLT3 mutations, but its activity is frequently lost over time, sometimes due to development of resistance-inducing point mutations in FLT3-ITD [19]. Quizartinib (AC220) is the most potent and specific FLT3 inhibitor identified to date [20], but its clinical activity is also not durable, and, like sorafenib, it induces mutations in FLT3-ITD that disrupt binding of the drug to the target and have been demonstrated to be a mechanism of acquired resistance [21, 22].
The importance of identifying new compounds that inhibit FLT3-ITD with acquired point mutations associated with resistance to current FLT3 inhibitors has become apparent. Quizartinib and sorafenib are type II FLT3 inhibitors that bind to the inactive conformation of the kinase and prevent its activation. In contrast, type I inhibitors target the active conformation of the kinase and may be effective against FLT3-ITD with point mutations conferring resistance to type II FLT3 inhibitors. Ponatinib, approved as a BCR-ABL inhibitor, is also a potent type I FLT3 inhibitor with activity against FLT3 with induced point mutations [23, 24], but it is not being developed as a FLT3 inhibitor due to increased risk of vascular adverse events. Crenolanib is a type I FLT3 inhibitor that is active at nanomolar concentrations against FLT3-ITD, FLT3 D835 mutations, which are less common, wild-type FLT3 and FLT3-ITD with resistance-conferring point mutations induced by quizartinib or sorafenib [1–4].
In addition to inhibiting FLT3, crenolanib also inhibits platelet-derived growth factor receptor alpha and beta (PDGFRA, PDGFRB), and is in clinical trials in gastrointestinal stromal cell tumors (GIST) with PDGFRA mutations associated with imatinib resistance [5] and in gliomas [6], which are characterized by PDGFRA amplification and mutations in both children and adults [7, 8]. Moreover, it may also have efficacy in other solid tumors, such as melanoma [9].
In a phase I clinical trial, the recommended dose of crenolanib was 100 mg administered orally twice daily with food, yielding a mean maximum plasma concentration of 225 ng/ml, or approximately 0.5 μM (molecular weight 443.54), on Day 1, with mean maximum plasma concentration increasing to approximately 1 μM on Day 15 [25]. These concentrations greatly exceed the predicted concentration for target inhibition. Crenolanib is being administered orally at 100 mg three times daily in a phase II clinical trial in AML due to its half-life of 8 to 9 h, and has been well tolerated and shown promising clinical activity [26]. Crenolanib exhibits little plasma protein binding [4].
Little is known yet about mechanisms of resistance to crenolanib [4]. Of note, many other kinase inhibitors in current use or in clinical trials in cancer therapy are substrates and/or inhibitors [27–29] of the ATP-binding cassette (ABC) proteins ABCB1 [P-glycoprotein (Pgp); MDR1], ABCG2 [breast cancer resistance protein (BCRP)] and ABCC1 [multidrug resistance-associated protein 1 (MRP1)], drug efflux proteins that are frequently expressed on AML cells and other cancer cells [30]. Notably, the first-generation FLT3 inhibitors midostaurin [31], lestaurtinib [32], tandutinib [33, 34], sorafenib [35–37] and sunitinib [37, 38] are all substrates and/or inhibitors of ABCB1 and ABCG2, and our group previously demonstrated that both quizartinib [39] and ponatinib [40] are ABCG2 inhibitors.
ABCB1, ABCG2 and ABCC1 are also important components of the blood-brain barrier [41]. As such, drugs that are substrates of these ABC proteins have limited penetration of the central nervous system (CNS), and drugs that inhibit the transport function of these proteins have the potential to increase CNS penetration of their substrate drugs.
We sought to characterize interactions of crenolanib with the ABC proteins ABCB1, ABCG2 and ABCC1.
Materials and methods
Cell lines
Vincristine-selected HL60/VCR cells [42], overexpressing ABCB1, were obtained from Dr. Ahmad R. Safa, Indiana University, Indianapolis, IN, doxorubicin-selected HL60/ADR cells, overexpressing ABCC1 [43], from Dr. Kapil Bhalla, University of Kansas Cancer Center, Kansas City, KS, and mitoxantrone-selected 8226/MR20 myeloma cells [44], overexpressing wild-type ABCG2 [45], from Dr. William Dalton, Moffitt Cancer Center, Tampa, FL. HL60/VCR cells were maintained in drug-free RPMI 1640 medium with 10 % fetal bovine serum (FBS) and 8226/MR20 in RPMI 1640 medium with 10 % FBS and 20 nM mitoxantrone. Transfected K562 cells stably overexpressing ABCB1 [46] or wild-type ABCG2 [47] were gifts from Dr. Michael Gottesman, National Cancer Institute, Bethesda, MD and Dr. Yoshikazu Sugimoto, Kyoritsu University of Pharmacy, Tokyo, Japan, respectively. They were cultured in RPMI 1640, pH 7.4, supplemented with 10 % FBS at 37 °C in a humidified atmosphere containing 5 % CO2. Parental HL60 and K562 cells and MV4-11 and MOLM-14 cells, with FLT3-ITD [48], were obtained from the American Type Culture Collection (ATCC, Manassas, VA).
Materials
Crenolanib was purchased from Selleck Chemicals, Houston, TX, and was stored at −80 °C as a 100 mM stock solution in dimethyl sulfoxide. Cell Proliferation Reagent WST-1 was purchased from Roche Diagnostics (Indianapolis, IN). The fluorescent ABCB1 substrate 3,3′-diethyloxacarbocyanine iodide [DiOC2(3)] was purchased from Sigma-Aldrich (St Louis, MO), the fluorescent ABCG2 substrate pheophorbide A (PhA) from Frontier Scientific (Logan, VT) [40], and the ABCC1 protein substrate rhodamine 123 (RH 123) from Sigma-Aldrich [40]. The ABCB1 inhibitor PSC-833 was obtained from Novartis Pharmaceutical Corporation (East Hanover, NJ), the ABCG2 inhibitor fumitremorgin C (FTC) was purchased from Sigma-Aldrich and the ABCC1 inhibitor p-[dipropylsulfamoyl] benzoic acid (probenecid) from Sigma-Aldrich [40]. [125I]-Iodoarylazidoprazosin ([125I]-IAAP) (2200 Ci/mmol) was purchased from PerkinElmer Life and Analytical Sciences (Waltham, MA).
Cell viability assay
Viable cell numbers following drug treatment were measured using the WST-1 assay [40]. Briefly, 1×103 cells were seeded in 100 μL complete medium per well in 96-well tissue culture plates and incubated with crenolanib (0–10 μM) at 37°C in 5% CO2 for 96 h. 10 μL WST-1 reagent was then added to each well, incubation was continued for two additional hours and the color developed was quantified according to the manufacturer’s instructions. Each experiment was performed in triplicate. IC50 concentrations were calculated by the least square fit of dose-response inhibition in a non-linear regression model using GraphPad Prism V software (GraphPad Software, Inc., La Jolla, CA).
ABCB1 ATPase assay
High-Five insect cells were infected with recombinant baculovirus encoding ABCB1-His6. Membranes were isolated using hypotonic lysis and differential centrifugation, and the ATPase activity of vanadate (Vi)-sensitive ABCB1 expressed in membrane vesicles was measured in the presence of crenolanib at serial concentrations, as previously described [49]. Briefly, crude membrane protein (100 μg protein/ml) from ABCB1-expressing High-Five insect cells was incubated at 37 °C with crenolanib in increasing concentrations with and without 0.3 mM sodium orthovanadate, and the amount of inorganic phosphate released and the Vi-sensitive ATPase activity were measured.
ABCB1 cell surface expression
To detect ABCB1 cell surface expression, cells were incubated with MRK16 antibody for 1 h, washed twice with PBS, and then incubated with phycoerythrin (PE)-conjugated anti-human antibody for 30 min. Cells were acquired on a FACSCanto II (BD Biosciences, San Jose, CA) and analyzed with FlowJo software (Tree Star, Inc., Ashland, OR), as previously described [40].
Uptake of fluorescent ABC protein substrates
To measure the effect of crenolanib on uptake of fluorescent ABC protein substrates, HL60/VCR and K562/ABCB1 cells (1× 106), expressing ABCB1, were incubated for 30 min at 37 °C with DiOC2(3) (0.6 ng/ml) and crenolanib (0–10 μM) or PSC-883 (2.5 μM) as a positive control, 8226/MR20 and K562/ABCG2 cells, expressing ABCG2, with PhA (1 μM) and crenolanib (0–10 μM) or FTC (10 μM) as a positive control, and HL60/ADR cells, expressing ABCC1, with RH 123 (0.5 μg/mL) and crenolanib (0–10 μM) or probenecid (1 mmol/L) as a positive control, as previously described [40]. Cells were then washed twice, resuspended in phosphate-buffered saline (PBS) and kept on ice until analysis. They were then acquired on a FACSCanto II flow cytometer and analyzed using FlowJo software. Substrate content after uptake with and without modulator was compared using the Kolmogorov-Smirnov statistic, expressed as a D-value ranging from 0 (no difference) to 1 (no overlap) [50], with D-values ≥0.2 indicating significant modulation, based on previous work [51].
Photoaffinity labeling of ABCB1 and ABCG2 with [125I]-Iodoarylazidoprazosin (IAAP)
High-Five insect cell membrane vesicles expressing ABCB1 (50–70 μg protein) were incubated with 0–20 μM crenolanib for 5 min at 21–23 °C in 50 mM Tris-HCl, pH 7.5. [125I]-IAAP (2200 Ci/mmole), 3–6 nM, was added and incubation was continued for 5 additional minutes under subdued light. ABCB1 crosslinked with [125I]-IAAP was separated on 7 % SDS-PAGE gels and the amount of [125I]-IAAP incorporated into ABCB1 was quantified as previously described [52].
Results
Crenolanib resistance is conferred by ABCB1, but not ABCG2 or ABCC1
To determine whether crenolanib is a substrate for the multidrug resistance (MDR)-associated ABC proteins ABCB1, ABCC1 and ABCG2, we studied cytotoxicity of crenolanib in HL60/VCR and HL60/ADR cells, expressing ABCB1 and ABCC1, respectively, with parental HL60 as a control, as well as transfected K562/ABCB1 and K562/ABCG2 cells, with parental K562 cells as a control. HL60/VCR and K562/ABCB1 cells, overexpressing ABCB1, were 6.9- and 3.6-fold resistant to crenolanib, respectively, in relation to parental HL60 and K562 cells (Fig. 2a and b and Table 1). In contrast, K562/ABCG2 and HL60/ADR cells, overexpressing ABCG2 and ABCC1, respectively, were not resistant to crenolanib, in relation to parental K562 and HL60 cells (Fig. 2b and c and Table 1). These data are consistent with crenolanib being a substrate of ABCB1, but not ABCC1 or ABCG2. To confirm that ABCB1 mediates crenolanib resistance, we studied crenolanib cytotoxicity in HL60/VCR and K562/ABCB1 cells in the presence and absence of 2.5 μM PSC-833, a specific inhibitor of ABCB1-mediated substrate transport. PSC-833 fully reversed resistance to crenolanib in both HL60/VCR and K562/ABCB1 cells (Fig. 2d and e and Table 1).
Fig. 2.
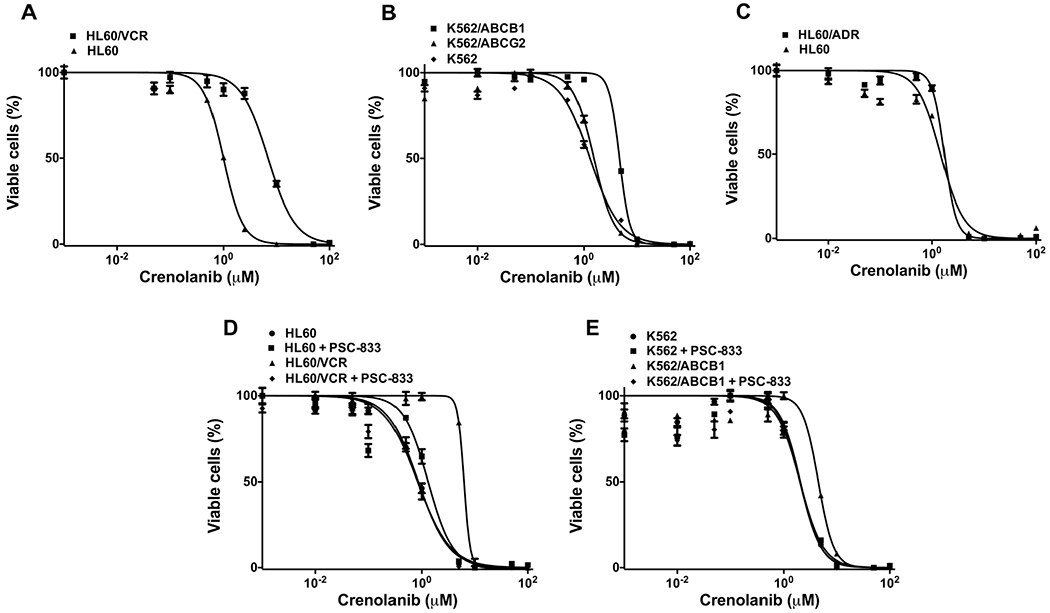
Cells overexpressing ABCB1 are approximately 5-fold resistant to crenolanib, while ABCC1 and ABCG2 do not confer crenolanib resistance. a, b, c. Crenolanib cytotoxicity in cells overexpressing ABCB1 (HL60/VCR, K562/ABCB1), ABCG2 (K562/ABCG2) and ABCC1 (HL60/ADR) and parental cells (HL60, K562) is shown. Multidrug resistant cells overexpressing ABCB1, ABCC1 or ABCG2 and parental cells were cultured in 96-well tissue culture plates and incubated with crenolanib (0–10 μM) at 37 °C in 5 % CO2 for 96 h. Viability of crenolanib-treated cells was evaluated using the WST-1 assay, and IC50s were calculated as described in (Materials and Methods). d, e. Crenolanib resistance of ABCB1-overexpressing cells is reversed by the ABCB1-specific inhibitor PSC-833. Viability of crenolanib-treated HL60/VCR and K562/ABCB1 cells, as well as HL60 and K562 parental cells, was measured in the presence and absence of the ABCB1-specific inhibitor PSC-833 at 2.5 μM. Mean and standard error values from three independent experiments are shown
Table 1.
Crenolanib IC50 values of ABC protein-overexpressing and parental cell lines
| Cell line | IC50 (μM) | Standard error |
|---|---|---|
| HL60 | 1.00 | 0.03 |
| HL60/VCR | 6.93 | 0.03 |
| K562 | 1.30 | 0.03 |
| K562/ABCB1 | 4.67 | 0.01 |
| K562/ABCG2 | 1.54 | 0.03 |
| HL60 | 1.46 | 0.04 |
| HL60/ADR | 1.72 | 0.06 |
| HL60 | 0.86 | 0.02 |
| HL60+PSC-833 | 1.32 | 0.06 |
| HL60/VCR | 6.27 | 0.02 |
| HL60/VCR+PSC-833 | 0.84 | 0.04 |
| K562 | 2.02 | 0.05 |
| K562+PSC-833 | 2.02 | 0.08 |
| K562/ABCB1 | 4.49 | 0.04 |
| K562/ABCB1+PSC-833 | 2.06 | 0.08 |
Crenolanib stimulates ABCB1 ATPase activity in a concentration-dependent manner
To further study the interaction of crenolanib with ABCB1, we studied its effect on ATP hydrolysis mediated by ABCB1, as described in (Materials and Methods). Crenolanib was found to stimulate ABCB1 ATPase activity in a concentration-dependent manner (Fig. 3), suggesting that it interacts as a typical substrate of ABCB1 at the drug-binding pocket of the transporter.
Fig. 3.
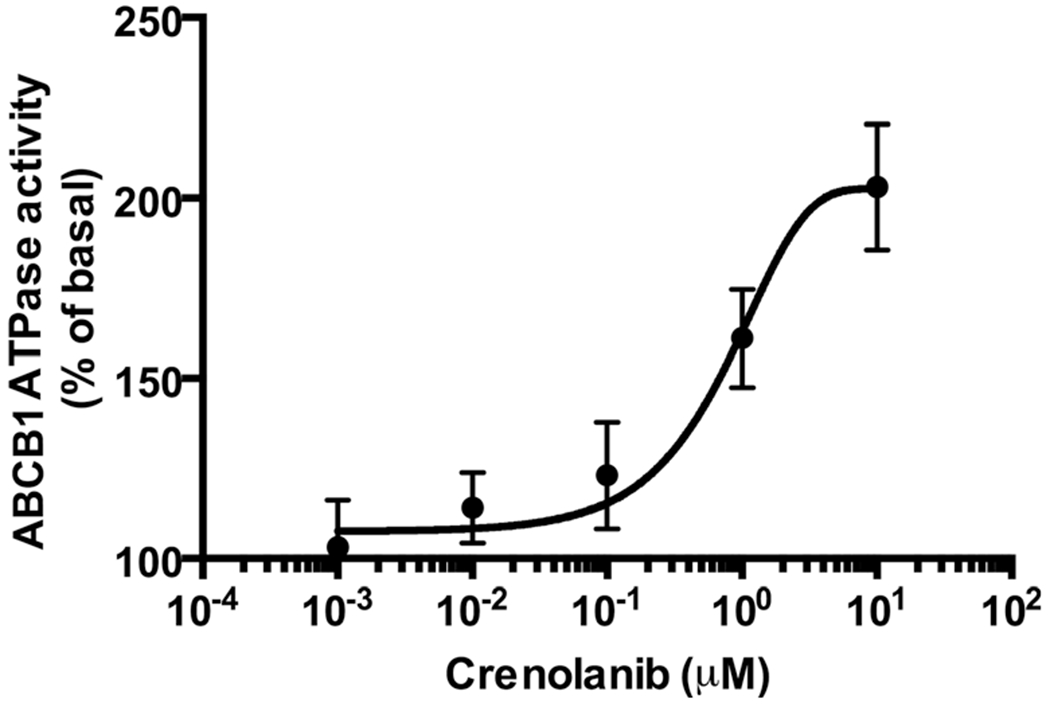
Crenolanib stimulates ABCB1 ATPase activity, consistent with transport by ABCB1. Crude membrane preparations from ABCB1-expressing High-Five insect cells were incubated with crenolanib in increasing concentrations with and without 0.3 mM sodium orthovanadate. The amount of inorganic phosphate released and the vanadate-sensitive ATPase activity were measured. The basal ATPase activity was considered as 100 % and the stimulation of ATP hydrolysis by varying concentrations of crenolanib (calculated as % increase of basal activity) (Y-axis) was plotted as a function of crenolanib concentrations (X-axis). The average from independent duplicate experiments is shown, with error bars representing standard error
Crenolanib treatment of FLT3-ITD cells does not upregulate ABCB1 cell surface expression
We then sought to determine whether treatment of cells with crenolanib induced expression of ABCB1. To this end, we cultured MV4-11 and MOLM-14 cells, both human cell lines with FLT3-ITD, with crenolanib at the pharmacologically relevant concentration of 500 nM [25]. Cells were counted at 48 and 96 h and tested for viability and for surface expression of ABCB1, measured by flow cytometry. Cell concentrations after 96-hour culture with 500 nM crenolanib and DMSO control were 0.73×105/ml and 6.75×105/ml, respectively. No induction of ABCB1 was seen on cells treated with crenolanib, in relation to DMSO control. It appeared that cell surface expression actually decreased on both MV4-11 (Fig. 4a) and MOLM-14 (data not shown) cells. This occurred in conjunction with a decrease in both forward and side scatter (Fig. 4b) and morphologic changes consistent with monocytic maturation in surviving crenolanib-treated cells (Fig. 4c).
Fig. 4.
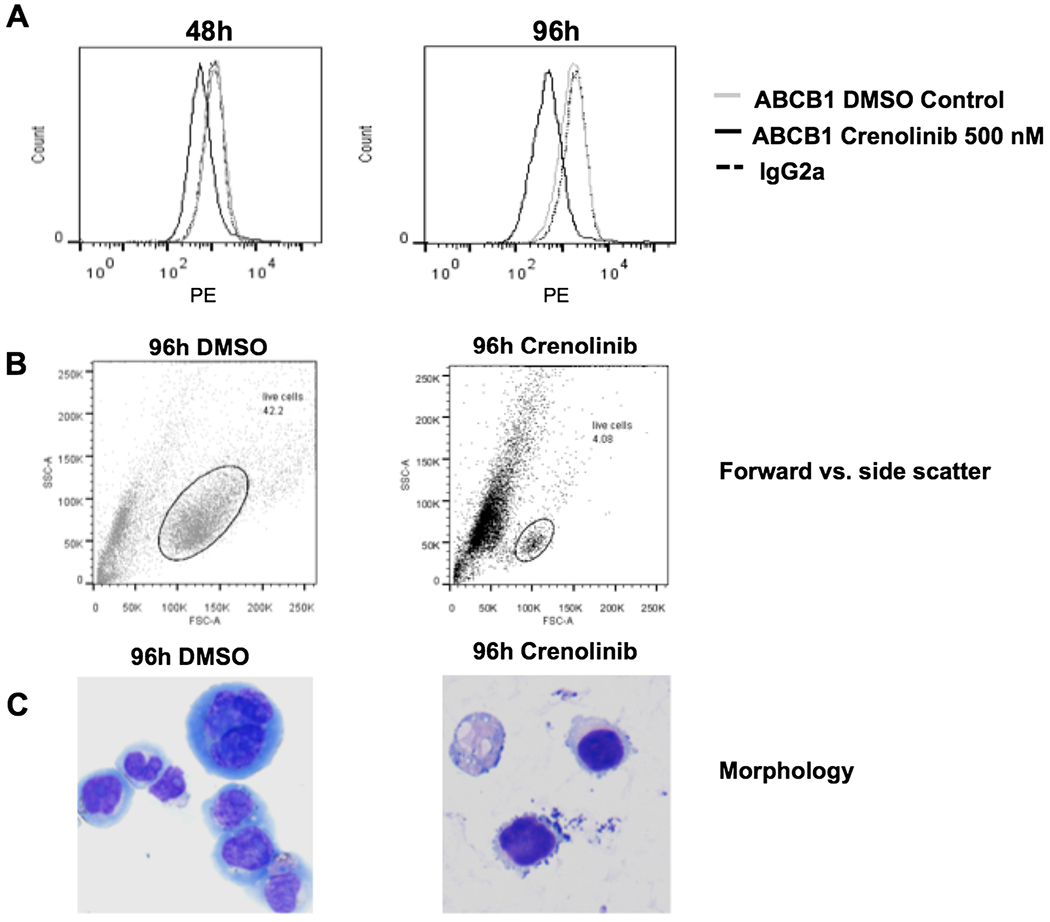
Crenolanib treatment does not increase the cell surface expression of ABCB1. MV4-11, human myeloid leukemia cells expressing FLT3-ITD, were treated with 500 nM crenolanib or DMSO control and cell surface expression of ABCB1 was measured by flow cytometry at 48 and 96 h as described in (Materials and Methods). No increase in ABCB1 cell surface expression was seen in crenolanib-treated cells, and, of note, a progressive decrease in ABCB1 cell surface expression was actually seen, in association with flow cytometric and morphologic changes indicative of cellular maturation. a. Decreased ABCB1 cell surface expression on MV4-11 cells treated for 48 and 96 h with crenolanib, in relation to DMSO control. b. Decreased forward and side scatter of MV4-11 cells treated for 96 h with crenolanib, in relation to DMSO control. c. Cellular maturation of MV4-11 cells treated for 96 hwith crenolanib, in relation to DMSO control. Cytospin preparations (Wright-Giemsa stain x 400) of MV4-11 cells treated with DMSO control for 96 h (left) shows intermediate to large cells with a monocytoid appearance, including some multinucleate cells, with ‘blastic’ fine chromatin and prominent nucleoli. In contrast the same cells after incubation with 500 nM crenolanib for 96 h (right) are smaller and have dense chromatin, and their morphology more closely resembles that of mature monocytes
Crenolanib does not alter substrate transport by ABCB1, ABCG2 or ABCC1 at pharmacologically relevant concentration
We next sought to determine whether crenolanib alters substrate transport by ABCB1, ABCG2 or ABCC1. To this end, we measured uptake of fluorescent substrates of these transport proteins in the presence of crenolanib at concentrations of 0 to 10 μM. Cell lines studied included HL60/VCR and K562/ABCB1, overexpressing ABCB1, K562/ABCG2 and 8226/MR20 cells, overexpressing ABCG2, and HL60/ADR, overexpressing ABCC1. Significant effect on ABCB1, ABCC1 or ABCG2 substrate transport was not seen with crenolanib at concentrations up to 1 μM, but progressive effect was seen at 5 and 10 μM (Fig. 5), consistent with crenolanib inhibiting the activity of ABCB1 only at high concentrations.
Fig. 5.
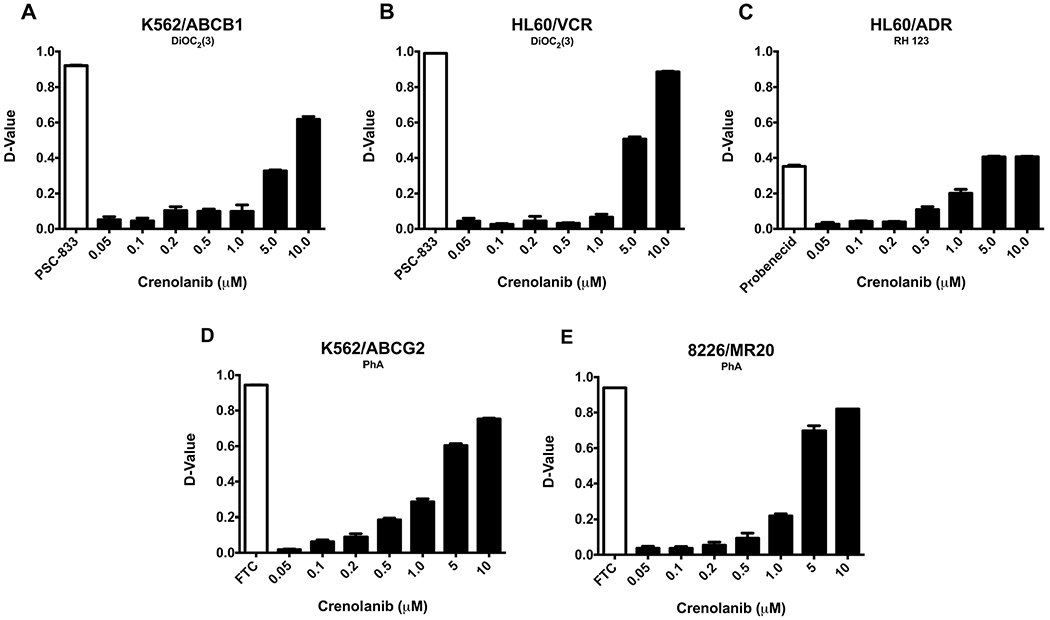
Crenolanib does not alter substrate transport by ABCB1, ABCC1 or ABCG2 at pharmacologically relevant concentrations. Uptake of the fluorescent substrates of ABCB1, ABCC1 and ABCG2 3,3′-diethyloxacarbocyanine iodide [DiOC2(3)], rhodamine 123 (RH 123) and pheophorbide A (PhA), respectively, was measured in the presence of crenolanib at concentrations of 0–10 μM, as described in (Materials and Methods). Crenolanib at concentrations up to 1 μM did not affect ABCB1, ABCC1 or ABCG2 substrate transport, but a concentration-dependent decrease in transport was seen at 5 and 10 μM. Mean and standard error values from three independent experiments are shown
Crenolanib inhibits [125I]-IAAP photocrosslinking of ABCB1 only at high concentrations
We further examined the effect of crenolanib on substrate binding of ABCB1 by studying its effect on the photocrosslinking of ABCB1 with a photoactivatable substrate analog, [125I]-IAAP. Crenolanib inhibited [125I]-IAAP photocrosslinking of ABCB1 at high concentrations, with 50 % inhibition at 10 μM, but had little effect at lower concentrations, below 1 μM, that are pharmacologically relevant in patients (Fig. 6).
Fig. 6.
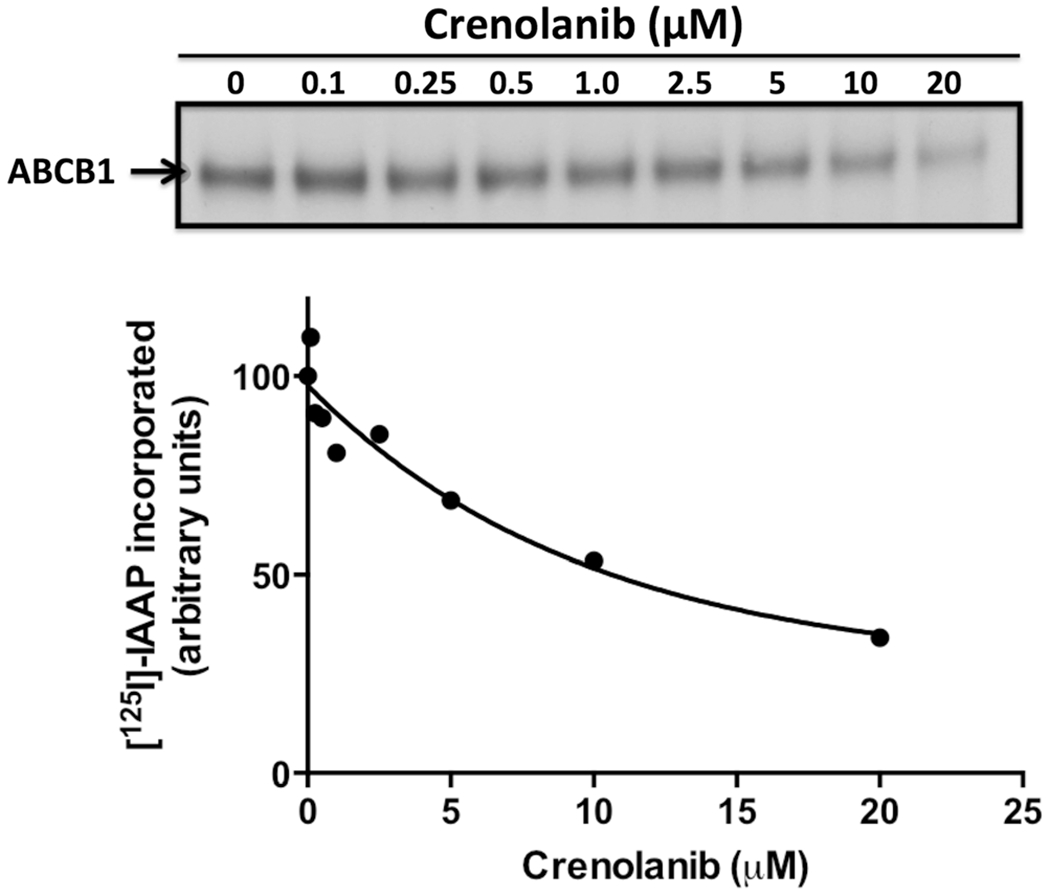
Crenolanib does not alter[125I]-IAAP photocrosslinking of ABCB1 at pharmacologically relevant concentrations. The effect of crenolanib (0–20 μM) on photocrosslinking of ABCB1 with [125I]-IAAP was studied as described in (Materials and Methods). The upper panel shows a representative autoradiogram. The average values from two independent experiments are shown in the graph. The amount of [125I]-IAAP incorporated into ABCB1 in the absence of crenolanib was considered as 100 % and the percent decrease in [125I]-IAAP incorporation (Y-axis) at the indicated concentrations of crenolanib was plotted as a function of crenolanib concentration used (X-axis)
Discussion
Crenolanib is a type I FLT3 inhibitor with activity at nanomolar concentrations against FLT3-ITD, FLT3 with D835 mutation, wild-type FLT3, and FLT3-ITD with resistance-conferring point mutations induced by quizartinib or sorafenib; it is being tested in clinical trials in patients with AML with FLT3 mutations [1–4]. It is also an inhibitor of PDGFRA, as well as PDGFRB, and is being tested in clinical trials in patients with GIST [5] and gliomas [6–8]. Its efficacy in gliomas is also dependent upon its ability to cross the blood-brain barrier.
We studied interactions of crenolanib with the ABC proteins ABCB1, ABCG2 and ABCC1, which are expressed on AML cells and other cancer cells and are associated with multidrug resistance, and are also important components of the blood–brain barrier. We found that cells expressing ABCB1 were resistant to crenolanib, while cells expressing ABCG2 or ABCC1 were equally sensitive to crenolanib as parental cells not expressing these transporters. Additionally, while crenolanib had no effect on ABCB1, ABCG2 or ABCC1 substrate transport at the less than 1 μM concentrations expected in patients, concentration-dependent inhibition of substrate transport by crenolanib at higher concentrations was observed in cells expressing these transporters.
Crenolanib transport by ABCB1 was evidenced by higher IC50 concentrations in cells overexpressing ABCB1, in relation to parental cells, reversal of crenolanib resistance in ABCB1-expressing cells by the ABCB1-specific transport inhibitor PSC-833, and crenolanib stimulation of ABCB1 ATPase activity. Crenolanib also inhibits ABCB1 substrate transport activity, but only at concentrations in the range of 5 μM and above, which are higher than pharmacologically relevant concentrations in patients. Of note, crenolanib was previously identified as an ABCB1 inhibitor in a high-throughput screen, but concentration-dependence and relationship to pharmacologically relevant concentrations were not addressed [53]. Ambudkar et al. defined three classes of ABCB1 inhibitors based on their effects on ABCB1 ATPase activity [54]. Class I agents stimulate ATPase activity at low concentrations but inhibit it at high concentrations, while Class II compounds stimulate ATPase activity in a concentration-dependent manner without any inhibition, and Class III compounds inhibit ATPase activity. We found crenolanib to be a Class II agent.
The clinical importance of ABCB1-mediated efflux as a mechanism of resistance to crenolanib in AML with FLT3-ITD and in GIST and gliomas remains to be determined. ABCB1 is frequently overexpressed on AML cells and association of its overexpression with inferior treatment outcomes is well established [55]. Of note, however, in one report, presence of FLT3-ITD and ABCB1 overexpression were found to be adverse prognostic factors that were usually mutually exclusive in untreated AML [56]. In contrast, it is not known whether ABCB1 is upregulated in relapsed or refractory FLT3-ITD AML. ABCB1 is not infrequently overexpressed in GIST [57, 58], while data on ABCB1 expression in gliomas are inconsistent [59, 60].
The fact that crenolanib appears to be a transport substrate of ABCB1 raises concern not only about crenolanib resistance in cancer cells overexpressing ABCB1, but also induction of ABCB1 in the setting of crenolanib treatment, as ABCB1 has been shown to be induced by substrate transport [61]. Induction of ABCB1 by FLT3 inhibitors has not been studied previously. We tested for induction of ABCB1 cell surface expression in two human AML cell lines with FLT3-ITD, MV4-11 and MOLM-14, treated with crenolanib at a pharmacologically relevant concentration, and found no induction of ABCB1 cell surface expression. Of note, crenolanib actually induced a decrease in ABCB1 cell surface expression on MV4-11 and MOLM-14 cells. This was associated with, and likely caused by, cellular maturation analogous to the myeloid differentiation of AML cells described in patients treated with the FLT3 inhibitor quizartinib [62]. Patient samples were not studied here, and cellular maturation of patient cells has not been studied with crenolanib, but similar findings may be expected.
In addition to ABCB1 expression on gliomas, its expression on the intact blood-brain barrier could limit access of crenolanib to gliomas. However, in a preclinical study evaluating crenolanib penetration in a spontaneous glioblastoma murine model, crenolanib showed increased penetration in tumor-bearing brain as compared to normal brain, indicating a disrupted blood-brain barrier in tumor-bearing mice [63]. Relevance to human central nervous system tumors is unknown. A clinical trial of crenolanib in gliomas is ongoing, and results are awaited.
Since crenolanib is an ABCB1 substrate, there is a potential for competitive inhibition of transport of co-administered ABCB1 substrate medications. However we demonstrated that crenolanib does not inhibit the transport function of this multidrug transporter at the plasma concentrations that are achieved in patients, and it is therefore unlikely to alter transport of co-administered chemotherapy drugs in cancer cells. In contrast, higher concentrations are expected to be achieved in the gastrointestinal tract, and could alter absorption of orally co-administered ABC drug transporter substrate chemotherapy drugs and other medications, as we previously showed for quizartinib [64].
Acknowledgments
This work was funded by a Leukemia and Lymphoma Society Translational Research Award (M.R. Baer), University of Maryland, Baltimore UMMG Cancer Research Grant #CH 649 CRF, State of Maryland Department of Health and Mental Hygiene (DHMH) under the Cigarette Restitution Fund Program (M.R. Baer), and NCI Cancer Center Support Grant P30 CA134274 (UMGCC). Drs. S. Shukla and S.V. Ambudkar were supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Footnotes
Conflict of interest The authors declare that they have no conflict of interest.
Contributor Information
Trevor J. Mathias, University of Maryland Greenebaum Cancer Center, 22 South Greene Street, Baltimore, MD 21201, USA
Karthika Natarajan, University of Maryland Greenebaum Cancer Center, 22 South Greene Street, Baltimore, MD 21201, USA; Department of Medicine, University of Maryland School of Medicine, Baltimore, MD, USA.
Suneet Shukla, Laboratory of Cell Biology, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD, USA.
Kshama A. Doshi, University of Maryland Greenebaum Cancer Center, 22 South Greene Street, Baltimore, MD 21201, USA
Zeba N. Singh, Department of Pathology, University of Maryland School of Medicine, Baltimore, MD, USA
Suresh V. Ambudkar, Laboratory of Cell Biology, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD, USA
Maria R. Baer, University of Maryland Greenebaum Cancer Center, 22 South Greene Street, Baltimore, MD 21201, USA; Department of Medicine, University of Maryland School of Medicine, Baltimore, MD, USA
References
- 1.Zimmerman EI, Turner DC, Buaboonnam J,Hu S, Orwick S, Roberts MS, Janke LJ, Ramachandran A, Stewart CF, Inaba H, Baker SD (2013) Crenolanib is active against models of drug-resistant FLT3-ITD-positive acute myeloid leukemia. Blood 122:3607–3615. doi: 10.1182/blood-2013-07-513044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Galanis A, Ma H, Rajkhowa T, Ramachandran A, Small D, Cortes J, Levis M(2014) Crenolanib is a potent inhibitor of FLT3 with activity against resistance-conferring point mutants. Blood 123:94–100. doi: 10.1182/blood-2013-10-529313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang W, Gao C, Konopleva M, Chen Y, Jacamo RO, Borthakur G, Cortes JE, Ravandi F, Ramachandran A, Andreeff M(2014) Reversal of acquired drug resistance in FLT3-mutated acute myeloid leukemia cells via distinct drug combination strategies. Clin Cancer Res 20: 2363–2374. doi: 10.1158/1078-0432.CCR-13-2052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith CC, Lasater EA, Lin KC, Wang Q, McCreery MQ, Stewart WK, Damon LE, Perl AE, Jeschke GR, Sugita M, Carroll M, Kogan SC, Kuriyan J, Shah NP (2014) Crenolanib is a selective type I pan-FLT3 inhibitor. Proc Natl Acad Sci U S A 111:5319–5324. doi: 10.1073/pnas.1320661111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heinrich MC, Griffith D, McKinley A, Patterson J, Presnell A, Ramachandran A, Debiec-Rychter M (2012) Crenolanib inhibits the drug-resistant PDGFRA D842V mutation associated with imatinib-resistant gastrointestinal stromal tumors. Clin Cancer Res 18:4375–4384. doi: 10.1158/1078-0432.CCR-12-0625 [DOI] [PubMed] [Google Scholar]
- 6.Wetmore C, Broniscer A, Turner D, Wright KD, Pai-Panandiker A, Kun LE, Ramachandran A, Onar-Thomas A, Huang J, Gajjar AJ, Baker S, Stewart CF (2014) First-in-pediatrics phase I study of crenolanib besylate (CP-868,596-26) administered during and after radiation therapy (RT) in newly diagnosed diffuse intrinsic pontine glioma (DIPG) and recurrent high-grade glioma (HGG). J Clin Oncol 32:5s (suppl; abstr 10064) [Google Scholar]
- 7.Ozawa T, Brennan CW, Wang L, Squatrito M, Sasayama T, Nakada M, Huse JT, Pedraza A, Utsuki S,Yasui Y, Tandon A, Fomchenko EI, Oka H, Levine RL, Fujii K, Ladanyi M, Holland EC (2010) PDGFRA gene rearrangements are frequent genetic events in PDGFRA-amplified glioblastomas. Genes Dev 24:2205–2218. doi: 10.1101/gad.1972310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paugh BS, Zhu X, Qu C, Endersby R, Diaz AK, Zhang J, Bax DA, Carvalho D, Reis RM, Onar-Thomas A, Broniscer A, Wetmore C, Zhang J, Jones C, Ellison DW, Baker SJ (2013) Novel oncogenic PDGFRA mutations in pediatric high-grade gliomas. Cancer Res 73: 6219–6229. doi: 10.1158/0008-5472.CAN-13-1491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rebecca VW, Wood E, Fedorenko IV, Paraiso KH, Haarberg HE, Chen Y, Xiang Y, Sarnaik A, Gibney GT, Sondak VK, Koomen JM, Smalley KS (2014) Evaluating melanoma drug response and therapeutic escape with quantitative proteomics. Mol Cell Proteomics 13:1844–1854. doi: 10.1074/mcp.M113.037424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rosnet O, Bühring HJ, Marchetto S, Rappold I, Lavagna C, Sainty D, Arnoulet C, Chabannon C, Kanz L, Hannum C, Birnbaum D (1996) Human FLT3/FLK2 receptor tyrosine kinase is expressed at the surface of normal and malignant hematopoietic cells. Leukemia 10:238–248 [PubMed] [Google Scholar]
- 11.Kottaridis PD, Gale RE, Frew ME, Harrison G, Langabeer SE, Belton AA, Walker H, Wheatley K, Bowen DT, Burnett AK, Goldstone AH, Linch DC (2001) The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients from the United Kingdom Medical Research Council AML 10 and 12 trials. Blood 98:1752–1759 [DOI] [PubMed] [Google Scholar]
- 12.Fröhling S, Schlenk RF, Breitruck J, Benner A, Kreitmeier S, Tobis K, Döhner H, Döhner K (2002) Prognostic significance of activating FLT3mutations in younger adults (16 to 60 years)with acutemyeloid leukemia and normal cytogenetics: a study of the AML Study Group Ulm. Blood 100:4372–4380 [DOI] [PubMed] [Google Scholar]
- 13.Schnittger S, Schoch C, Dugas M, Kern W, Staib P, Wuchter C, Löffler H, Sauerland CM, Serve H, Büchner T, Haferlach T, Hiddemann W (2002) Analysis of FLT3 length mutations in 1003 patients with acute myeloid leukemia: correlation to cytogenetics, FAB subtype, and prognosis in the AMLCG study and usefulness as a marker for the detection of minimal residual disease. Blood 100: 59–66 [DOI] [PubMed] [Google Scholar]
- 14.Schlenk RF, Döhner K, Krauter J, Fröhling S, Corbacioglu A, Bullinger L, Habdank M, Späth D, Morgan M, Benner A, Schlegelberger B, Heil G, Ganser A, Döhner H (2008) Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med 358:1909–1918. doi: 10.1056/NEJMoa074306 [DOI] [PubMed] [Google Scholar]
- 15.Patel JP, Gönen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, Van Vlierberghe P, Dolgalev I, Thomas S, Aminova O, Huberman K, Cheng J, Viale A, Socci ND, Heguy A, Cherry A, Vance G, Higgins RR, Ketterling RP, Gallagher RE, Litzow M, van den Brink MR, Lazarus HM, Rowe JM, Luger S, Ferrando A, Paietta E, Tallman MS, Melnick A, Abdel-Wahab O, Levine RL (2012) Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med 366:1079–1089. doi: 10.1056/NEJMoa1112304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hayakawa F, Towatari M, Kiyoi H, Tanimoto M, Kitamura T, Saito H, Naoe T (2000) Tandem-duplicated Flt3 constitutively activates STAT5 and MAP kinase and introduces autonomous cell growth in IL-3-dependent cell lines. Oncogene 19:624–631 [DOI] [PubMed] [Google Scholar]
- 17.Fischer T, Stone RM, Deangelo DJ, Galinsky I, Estey E, Lanza C, Fox E, Ehninger G, Feldman EJ, Schiller GJ,Klimek VM, Nimer SD, Gilliland DG, Dutreix C, Huntsman-Labed A, Virkus J, Giles FJ (2010) Phase IIB trial of oral midostaurin (PKC412), the FMS-like tyrosine kinase 3 receptor (FLT3) and multi-targeted kinase inhibitor, in patients with acute myeloid leukemia and high-risk myelodysplastic syndrome with either wild-type or mutated FLT3. J Clin Oncol 28:4339–4345. doi: 10.1200/JCO.2010.28.9678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Levis M, Ravandi F, Wang ES, Baer MR, Perl A, Coutre S, Erba H, Stuart RK, Baccarani M, Cripe LD, Tallman MS, Meloni G, Godley LA, Langston AA, Amadori S, Lewis ID, Nagler A, Stone R, Yee K, Advani A,Douer D,Wiktor-Jedrzejczak W, Juliusson G, Litzow MR, Petersdorf S, Sanz M, Kantarjian HM, Sato T, Tremmel L, Bensen-Kennedy DM, Small D, Smith BD (2011) Results from a randomized trial of salvage chemotherapy followed by lestaurtinib for patients with FLT3 mutant AML in first relapse. Blood 117:3294–3301. doi: 10.1182/blood-2010-08-301796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Man CH, Fung TK, Ho C, Han HH, Chow HC, Ma AC, Choi WW, Lok S, Cheung AM, Eaves C, Kwong YL, Leung AY (2012) Sorafenib treatment of FLT3-ITD+ acute myeloid leukemia: favorable initial outcome and mechanisms of subsequent non-responsiveness associated with a D835 mutation. Blood 119:5133–5143. doi: 10.1182/blood-2011-06-363960 [DOI] [PubMed] [Google Scholar]
- 20.Zarrinkar PP, Gunawardane RN, Cramer MD, Gardner MF, Brigham D, Belli B, Karaman MW, Pratz KW, Pallares G, Chao Q, Sprankle KG, Patel HK, Levis M, Armstrong RC, James J, Bhagwat SS (2009) AC220 is a uniquely potent and selective inhibitor of FLT3 for the treatment of acute myeloid leukemia (AML). Blood 114:2984–2992. doi: 10.1182/blood-2009-05-222034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith CC,Wang Q, Chin CS, Salerno S, Damon LE, Levis MJ, Perl AE, Travers KJ,Wang S,Hunt JP, Zarrinkar PP, Schadt EE,Kasarskis A, Kuriyan J, Shah NP (2012) Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature 485:260–263. doi: 10.1038/nature11016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pauwels D, Sweron B, Cools J (2012) The N676D and G697R mutations in the kinase domain of FLT3 confer resistance to the inhibitor AC220. Haematologica 97:1773–1774. doi: 10.3324/haematol.2012.069781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zirm E, Spies-Weisshart B, Heidel F, Schnetzke U, Böhmer FD, Hochhaus A, Fischer T, Scholl S (2012) Ponatinib may overcome resistance of FLT3-ITD harbouring additional point mutations, notably the previously refractory F691I mutation. Br J Haematol 157: 483–492. doi: 10.1111/j.1365-2141.2012.09085.x [DOI] [PubMed] [Google Scholar]
- 24.Smith CC, Lasater EA, Zhu X, Lin KC, Stewart WK, Damon LE, Salerno S, Shah NP (2013) Activity of ponatinib against clinically-relevant AC220-resistant kinase domain mutants of FLT3-ITD. Blood 121:3165–3171. doi: 10.1182/blood-2012-07-442871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lewis NL, Lewis LD, Eder JP, Reddy NJ, Guo F, Pierce KJ, Olszanski AJ, Cohen RB (2009) Phase I study of the safety, tolerability, and pharmacokinetics of oral CP-868,596, a highly specific platelet-derived growth factor receptor tyrosine kinase inhibitor in patients with advanced cancers. J Clin Oncol 27:5262–5269. doi: 10.1200/JCO.2009.21.8487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Collins R, Kantarjian HM, Levis MJ, Perl AE, Ramachandran A, Ravandi F, Ku N, Cortes JE (2014) Clinical activity of crenolanib in patients with D835 mutant FLT3-positive relapsed/refractory acute myeloid leukemia (AML). J Clin Oncol 32:5s (suppl; abstr 7027) [Google Scholar]
- 27.Wang XK, Fu LW (2010) Interaction of tyrosine kinase inhibitors with the MDR- related ABC transporter proteins. Curr Drug Metab 11:618–628 [DOI] [PubMed] [Google Scholar]
- 28.Brózik A, Hegedüs C, Erdei Z, Hegedus T, Özvegy-Laczka C, Szakács G, Sarkadi B (2011) Tyrosine kinase inhibitors as modulators of ATP binding cassette multidrug transporters: substrates, chemosensitizers or inducers of acquired multidrug resistance? Expert Opin Drug Metab Toxicol 7:623–642. doi: 10.1517/17425255.2011.562892 [DOI] [PubMed] [Google Scholar]
- 29.Shukla S, Chen ZS, Ambudkar SV (2012) Tyrosine kinase inhibitors as modulators of ABC transporter-mediated drug resistance. Drug Resist Updat 15:70–80. doi: 10.1016/j.drup.2012.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Szakács G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM (2006) Targeting multidrug resistance in cancer. Nat Rev Drug Discov 5:219–234 [DOI] [PubMed] [Google Scholar]
- 31.Hunter HM, Pallis M, Seedhouse CH, Grundy M, Gray C, Russell NH (2004) The expression of P-glycoprotein in AML cells with FLT3 internal tandem duplications is associated with reduced apoptosis in response to FLT3 inhibitors. Br J Haematol 127:26–33 [DOI] [PubMed] [Google Scholar]
- 32.Robey RW, Shukla S, Steadman K, Obrzut T, Finley EM, Ambudkar SV, Bates SE (2007) Inhibition of ABCG2-mediated transport by protein kinase inhibitors with a bisindolylmaleimide or indolocarbazole structure. Mol Cancer Ther 6:1877–1885 [DOI] [PubMed] [Google Scholar]
- 33.Yang JJ, Milton MN, Yu S, Liao M, Liu N,Wu JT, Gan L, Balani SK, Lee FW, Prakash S, Xia CQ (2010) P-glycoprotein and breast cancer resistance protein affect disposition of tandutinib, a tyrosine kinase inhibitor. Drug Metab Lett 4:201–212 [DOI] [PubMed] [Google Scholar]
- 34.Zhao XQ, Dai CL, Ohnuma S, Liang YJ, Deng W, Chen JJ, Zeng MS, Ambudkar SV, Chen ZS, Fu LW (2013) Tandutinib (MLN518/CT53518) targeted to stem-like cells by inhibiting the function of ATP-binding cassette subfamily G member 2. Eur J Pharm Sci 49: 441–450. doi: 10.1016/j.ejps.2013.04.015 [DOI] [PubMed] [Google Scholar]
- 35.Lagas JS, van Waterschoot RA, Sparidans RW,Wagenaar E, Beijnen JH, Schinkel AH (2010) Breast cancer resistance protein and P-glycoprotein limit sorafenib brain accumulation. Mol Cancer Ther 9:319–326. doi: 10.1158/1535-7163.MCT-09-0663 [DOI] [PubMed] [Google Scholar]
- 36.Agarwal S, Sane R, Ohlfest JR, Elmquist WF (2011) The role of the breast cancer resistance protein (ABCG2) in the distribution of sorafenib to the brain. J Pharmacol Exp Ther 336:223–233. doi: 10.1124/jpet.110.175034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hu S, Chen Z, Franke R, Orwick S, Zhao M, Rudek MA, Sparreboom A, Baker SD (2009) Interaction of the multikinase inhibitors sorafenib and sunitinib with solute carriers and ATP-binding cassette transporters. Clin Cancer Res 15:6062–6069. doi: 10.1158/1078-0432.CCR-09-0048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shukla S, Robey RW, Bates SE, Ambudkar SV (2009) Sunitinib (Sutent, SU11248), a small-molecule receptor tyrosine kinase inhibitor, blocks function of the ATP-binding cassette (ABC) transporters P-glycoprotein (ABCB1) and ABCG2. Drug Metab Dispos 37:359–365. doi: 10.1124/dmd.108.024612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bhullar J, Natarajan K, Shukla S, Mathias TJ, Sadowska M, Ambudkar SV, Baer MR (2013) The FLT3 inhibitor quizartinib inhibits ABCG2 at pharmacologically relevant concentrations, with implications for both chemosensitization and adverse drug interactions. PLoS One 8:e71266. doi: 10.1371/journal.pone.0071266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sen R, Natarajan K, Bhullar J, Shukla S, Fang HB, Cai L, Chen ZS, Ambudkar SV, Baer MR (2012) The novel BCR-ABL and FLT3 inhibitor ponatinib is a potent inhibitor of the MDR-associated ATP-binding cassette transporter ABCG2. Mol Cancer Ther 11: 2033–2044. doi: 10.1158/1535-7163.MCT-12-0302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shen S, Zhang W (2010) ABC transporters and drug efflux at the blood–brain barrier. Rev Neurosci 21:29–53 [DOI] [PubMed] [Google Scholar]
- 42.Ogretmen B, Safa AR (2000) Identification and characterization of the MDR1 promoter-enhancing factor 1 (MEF1) in the multidrug resistant HL60/VCR human acute myeloid leukemia cell line. Biochemistry 39:194–204 [DOI] [PubMed] [Google Scholar]
- 43.Marsh W, Sicheri D, Center MS (1986) Isolation and characterization of adriamycin-resistant HL-60 cells which are not defective in the initial intracellular accumulation of drug. Cancer Res 46:4053–4057 [PubMed] [Google Scholar]
- 44.Hazlehurst LA, Foley NE, Gleason-Guzman MC, Hacker MP, Cress AE, Greenberger LW, De Jong MC, Dalton WS (1999) Multiple mechanisms confer drug resistance to mitoxantrone in the human 8226 myeloma cell line. Cancer Res 59:1021–1028 [PubMed] [Google Scholar]
- 45.Suvannasankha A, Minderman H, O’Loughlin KL, Nakanishi T, Greco WR, Ross DD, Baer MR (2004) Breast cancer resistance protein (BCRP/MXR/ABCG2) in acute myeloid leukemia: discordance between expression and function. Leukemia 18:1252–1257 [DOI] [PubMed] [Google Scholar]
- 46.Hafkemeyer P, Licht T, Pastan I, Gottesman MM (2000) Chemoprotection of hematopoietic cells by a mutant P-glycoprotein resistant to a potent chemosensitizer of multidrug-resistant cancers. Hum Gene Ther 11:555–565 [DOI] [PubMed] [Google Scholar]
- 47.Yanase K, Tsukahara S, Asada S, Ishikawa E, Imai Y, Sugimoto Y (2004) Gefitinib reverses breast cancer resistance protein-mediated drug resistance. Mol Cancer Ther 3:1119–1125 [PubMed] [Google Scholar]
- 48.Quentmeier H, Reinhardt J, Zaborski M, Drexler HG (2003) FLT3 mutations in acute myeloid leukemia cell lines. Leukemia 17:120–124 [DOI] [PubMed] [Google Scholar]
- 49.Ambudkar SV (1998) Drug-stimulatable ATPase activity in crude membranes of humanMDR1-transfected mammalian cells. Methods Enzymol 292:504–514 [DOI] [PubMed] [Google Scholar]
- 50.Young IT (1977) Proof without prejudice: use of the Kolmogorov-Smirnov test for the analysis of histograms from flow systems and other sources. J Histochem Cytochem 25:935–941 [DOI] [PubMed] [Google Scholar]
- 51.Minderman H, Suvannasankha A, O’Loughlin KL, Scheffer GL, Scheper RJ, Robey RW, Baer MR (2002) Flow cytometric analysis of breast cancer resistance protein expression and function. Cytometry 48:59–65 [DOI] [PubMed] [Google Scholar]
- 52.Sauna ZE, Ambudkar SV (2000) Evidence for a requirement for ATP hydrolysis at two distinct steps during a single turnover of the catalytic cycle of human P-glycoprotein. Proc Natl Acad Sci U S A 97: 2515–2520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ansbro MR, Shukla S, Ambudkar SV, Yuspa SH, Li L (2013) Screening compounds with a novel high-throughput ABCB1-mediated efflux assay identifies drugs with known therapeutic targets at risk for multidrug resistance interference. PLoS One 8:e60334. doi: 10.1371/journal.pone.0060334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ambudkar SV, Dey S, Hrycyna CA, Ramachandra M, Pastan I, Gottesman MM (1999) Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu Rev Pharmacol Toxicol 39:361–398 [DOI] [PubMed] [Google Scholar]
- 55.Shaffer BC, Gillet JP, Patel C, Baer MR, Bates SE, Gottesman MM (2012) Drug resistance: still a daunting challenge to the successful treatment of AML. Drug Resist Updat 15:62–69. doi: 10.1016/j.drup.2012.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marzac C, Teyssandier I, Calendini O, Perrot JY, Faussat AM, Tang R, Casadevall N, Marie JP, Legrand O (2006) Flt3 internal tandem duplication and P-glycoprotein functionality in 171 patients with acute myeloid leukemia. Clin Cancer Res 12:7018–7024 [DOI] [PubMed] [Google Scholar]
- 57.Plaat BE, Hollema H, Molenaar WM, Torn Broers GH, Pijpe J, Mastik MF, Hoekstra HJ, van den Berg E, Scheper RJ, van der Graaf WT (2000) Soft tissue leiomyosarcomas and malignant gastrointestinal stromal tumors: differences in clinical outcome and expression of multidrug resistance proteins. J Clin Oncol 18:3211–3220 [DOI] [PubMed] [Google Scholar]
- 58.Théou N, Gil S, Devocelle A, Julié C, Lavergne-Slove A, Beauchet A, Callard P, Farinotti R, Le Cesne A, Lemoine A, Faivre-Bonhomme L, Emile JF (2005) Multidrug resistance proteins in gastrointestinal stromal tumors: site-dependent expression and initial response to imatinib. Clin Cancer Res 11:7593–7598 [DOI] [PubMed] [Google Scholar]
- 59.von Bossanyi P, Diete S, Dietzmann K,Warich-Kirches M, Kirches E (1997) Immunohistochemical expression of P-glycoprotein and glutathione S-transferases in cerebral gliomas and response to chemotherapy. Acta Neuropathol (Berlin) 94:605–611 [DOI] [PubMed] [Google Scholar]
- 60.Fruehauf JP, Brem H, Brem S, Sloan A, Barger G, Huang W, Parker R (2006) In vitro drug response and molecular markers associated with drug resistance inmalignant gliomas. Clin Cancer Res 12:4523–4532 [DOI] [PubMed] [Google Scholar]
- 61.Hu XF, Slater A, Wall DM, Parkin JD, Kantharidis P, Zalcberg JR (1996) Cyclosporin A and PSC 833 prevent up-regulation of MDR1 expression by anthracyclines in a human multidrug-resistant cell line. Clin Cancer Res 2:713–720 [PubMed] [Google Scholar]
- 62.Sexauer A, Perl A, Yang X, Borowitz M, Gocke C, Rajkhowa T, Thiede C, Frattini M, Nybakken GE, Pratz K, Karp J, Smith BD, Levis M (2012) Terminal myeloid differentiation in vivo is induced by FLT3 inhibition in FLT3/ITD AML. Blood 120:4205–4214. doi: 10.1182/blood-2012-01-402545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Elmeliegy MA, Bai F, Juel S, Throm S, Ramachandran A, Stewart CF (2011) Microdialysis for evaluation of crenolanib penetration in spontaneous glioblastoma murine model using a sensitive liquid chromatography mass spectrometry (LC-MS/MS) method. Proc Am Assoc Cancer Res abstract 5474 [Google Scholar]
- 64.Tachibana T, Kato M, Takano J, Sugiyama Y (2010) Predicting drugdruginteractions involving the inhibition of intestinal CYP3A4 and P-glycoprotein. Curr Drug Metab 11:762–777 [DOI] [PubMed] [Google Scholar]