

Abstract
Diffuse large B-cell lymphoma (DLBCL) is an aggressive B-cell non-Hodgkin lymphoma that affects patients of all ages with a wide range of clinical presentations. Although DLBCL is curable even in advanced stages, up to one-third of patients will not achieve cure with initial therapy. In the modern era of rituximab-based therapy as the first-line treatment, the prognoses of patients who require salvage therapy are poor and most will eventually succumb to their disease. Insight into the complex molecular circuitry of DLBCL reveals a diverse range of somatic mutations and aberrant intracellular signalling pathways that characterize distinct molecular subsets of the disease. The next major breakthrough in DLBCL therapy during this ‘molecular era’ of disease definition will be the identification of combinations of novel agents that target the oncogenic drivers of these subsets. Well-conducted clinical trials, with translational molecular investigations, will be essential to achieve the goal of precision medicine and expand the number of patients with DLBCL who achieve a cure.
Introduction
Diffuse large B-cell lymphoma (DLBCL) is the most common B-cell non-Hodgkin lymphoma (NHL) throughout the world, comprising 30–35% of all NHLs.1 DLBCL is biologically aggressive, but can be cured in >50% of cases, even in advanced stages.2 However, up to one-third of patients have refractory disease or relapse after treatment.3 The standard salvage treatment for patients with relapsed or refractory DLBCL that remains sensitive to chemotherapy is autologous stem cell transplantation (ASCT), but success rates are poor in the current era,4 highlighting the urgent need for novel therapeutic approaches for these patients.
DLBCL was first cured with combination chemotherapy in the 1970s with anthracycline-based regimens.5 Cyclophosphamide, doxorubicin, vincristine and prednisone (CHOP) chemotherapy has been the standard of care for patients with DLBCL since the landmark randomized phase III study of 899 patients with advanced intermediate-grade and high-grade lymphomas. In that study, equivalent response rates and survival were demonstrated, but with lower toxicity for CHOP compared with third-generation anthracycline-based regimens.6 The most recent breakthrough in the treatment of DLBCL has been the development of rituximab, a chimeric monoclonal antibody targeting the CD20 receptor, which has shown benefit for all DLBCL subgroups.7–11 The first study to show the benefit of rituximab was performed by Croupe d’Etude des Lymphomes de FAdulte (GELA), in which patients >60 years of age were randomly assigned to receive CHOP plus rituximab (R-CHOP) or CHOP alone.7 In this study, the complete remission rate (76% versus 63%, P = 0.005) and 2-year event-free survival (EFS) rate (57% versus 39%, P<0.001) were improved with R-CHOP, ultimately translating to an improvement of overall survival at 10 years (43.5% versus 27.6%, P = 0.005).9 R-CHOP also demonstrated an almost 20% improvement in the 6-year EFS rate compared with CHOP alone when administered to younger patients with DLBCL who had good prognostic features (74.3% versus 55.8%, P<0.0001).10 On the basis of these results, R-CHOP became the de facto standard of care in patients of all ages with DLBCL.
The development of other more-effective chemotherapy platforms has been limited. For example, randomized studies of ASCT in first remission12 and dose-dense strategies13 have failed to demonstrate improvement over R-CHOP. A randomized study from GELA in 380 patients between the ages of 18–59 years with newly diagnosed DLBCL and with low-risk features showed that rituximab, doxorubicin, cyclophosphamide, vindesine, bleomycin and prednisone (R-ACVBP) was superior to R-CHOP in terms of both 3-year EFS rates (81% versus 67%, P = 0.0035) and overall survival (92% versus 84%, P = 0.007).14 However, the haematological toxicity of R-ACVBP limits its use in older patients.14 The pharmaco-dynamically derived infusional regimen of dose-adjusted etoposide, doxorubicin, cyclophosphamide, vincristine, prednisone and rituximab (DA-EPOCH-R) has proven to be highly effective for certain molecular subtypes of DLBCL,15,16 and is currently being compared with R-CHOP in a randomized phase III study that has completed accrual.17 Overall, R-CHOP remains the most commonly used regimen for newly diagnosed DLBCL.
DLBCL has significant molecular heterogeneity within morphologically indistinguishable tumours and can be subdivided by gene-expression profiling (GEP) into three distinct molecular cell-of-origin subtypes: germinal centre B-cell (GCB), activated B-cell (ABC) and primary mediastinal B-cell lymphoma (PMBL)18–22 (Figure 1). We are currently in the molecular era of disease definition, and discovery of new signalling pathways—through GEP, transcriptome sequencing, RNA interference screens and DNA sequencing—has identified an array of new therapeutic targets in DLBCL (Table 1). Targeting specific oncogene addictions within the DLBCL subgroups offers a more-precise approach to therapy23 compared with the standard chemotherapy-based approaches. Analysis of DLBCL primary tumour samples using whole-genome and exome sequencing has revealed tremendous molecular complexity,24–27 indicating that the development of precision personalized medicine in DLBCL will not only require identification of mutations that drive tumorigenesis and disease progression, but will also involve characterization of cooperating mutations that confer drug resistance in patients. It is likely that the next major breakthrough in DLBCL therapy will spawn from a principled understanding of how to manage the subgroups at highest risk of initial treatment failure. In this Review, we describe the current molecular understanding of the DLBCL subtypes and discuss promising targeted approaches for each specific subtype.
Figure 1 |.
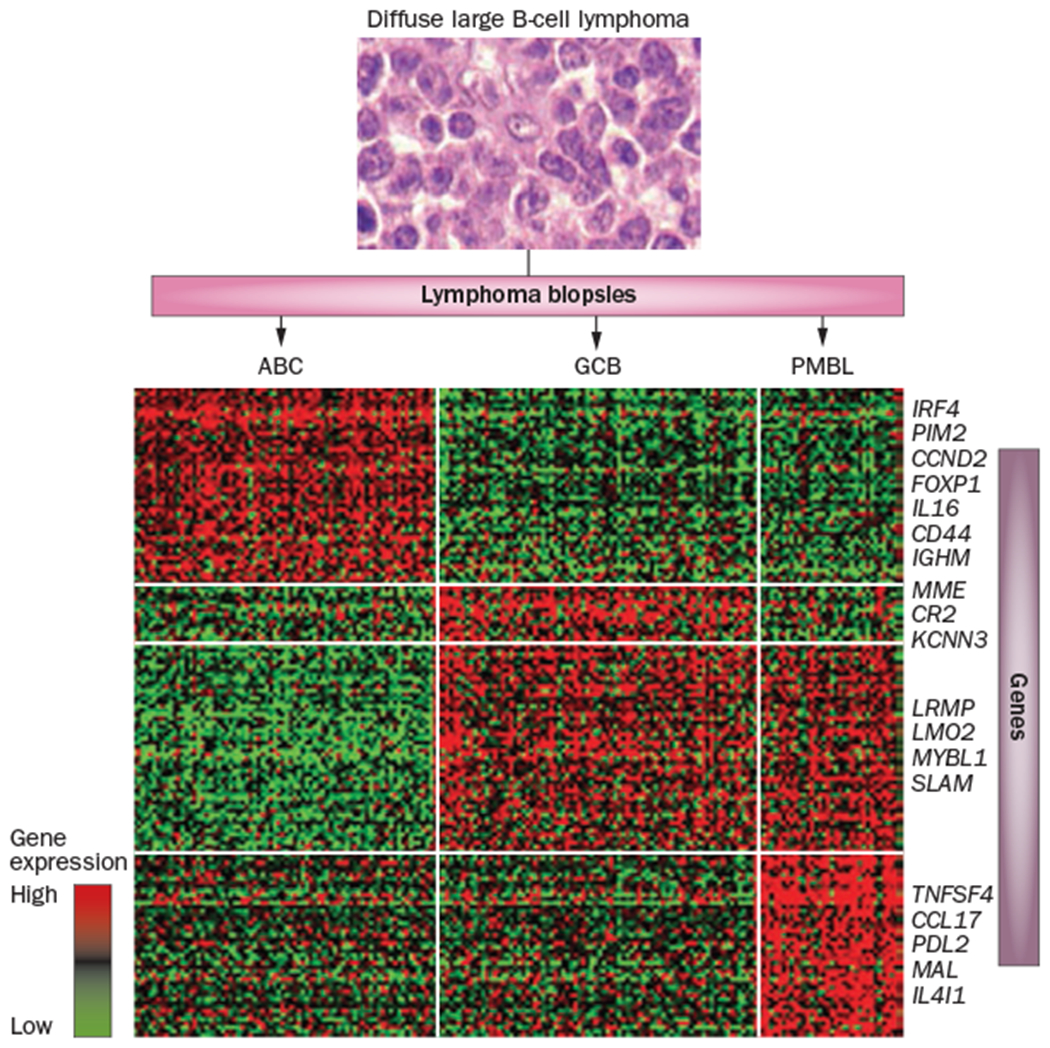
Gene-expression profiling subdivides morphologically indistinguishable DLBCL tumours into three distinct cell-of-origin subtypes.18 The genes overexpressed within these subtypes correspond to the putative developmental stage of the B cell from which the tumour originated. The GCB subtype is derived from a B cell that continues to undergo somatic hypermutation and demonstrates overexpression of genes associated with the germinal centre reaction, such as LRMP and LMO2.21 The ABC subtype is likely derived from a post-germinal centre B cell and is characterized by the overexpression of genes that regulate the plasmacytic differentiation programme, such as IRF4, PIM2 and FOXP1.21 The PMBL subtype is likely derived from the rare post-thymic B cell that has a unique gene-expression signature with more similarities with classic Hodgkin lymphoma than with other DLBCL subtypes.22,128 Abbreviations: ABC, activated B cell; DLBCL, diffuse large B-cell lymphoma; FOXP1, forkhead box protein P1; GCB, germinal centre B cell; IRF-4, interferon regulatory factor 4; LMO2, LIM domain only 2; LRMP, lymphoid-restricted membrane protein; PIM2, proviral integrations of moloney virus 2; PMBL, primary mediastinal B-cell lymphoma.
Table 1 |.
Oncogenic mechanisms and potential targets in DLBCL subtypes
| DLBCL subtype | Cell of origin | Oncogenic mechanisms | Potential targets |
|---|---|---|---|
| GCB | Germinal centre B-cell |
BCL2 translocation* EZH2 mutations‡ PTEN deletions§ Loss of PTEN expression |
BCL6 EZH2 PI3K/Akt |
| ABC | Post-germinal centre B-cell | NF-κB activation∥ CARD11 mutations MYD88 mutations CD79B mutations A20 deletions |
BCR CBM complex IRAK-4 JAK-STAT |
| PMBL | Post-thymic B-cell | NF-κB activation¶ 9p24 amplification¶ REL amplification JAK2 mutations CIITA translocations# |
JAK-STAT PD-1# |
GCB DLBCL frequently has BCL2 translocations63 and most result in activation of BCL-6, the master transcriptional regulator of the germinal centre.70
Mutations in EZH2 (21% of GCB DLBCL cases) are specific for this subtype.43
Loss of PTEN expression (55% of GCB DLBCL cases)53 results in activation of the PI3K/Akt pathway for which multiple inhibitors are currently in development.
ABC DLBCL is defined by constitutive NF-κB pathway activation75 and BCR signalling pathways are oncogenically activated in this subtype:81 mutations in MYD88, CARD11 and CD79B are found in ABC DLBCL along with deletions and mutations of TNFAIP3.80,82
PMBL is characterized by amplification of chromosome 9p24 and NF-κB pathway activation.136
PDL1 and PDL2 are overexpressed in PMBL22 making their receptor, PD-1, a potential target; recurrent CIITA translocations also result in PD-1 pathway activation in PMBL.141
Abbreviations: ABC, activated B-cell; BCR, B-cell receptor; CBM, CARD11-MALT1-BCL-10; DLBCL, diffuse large B-cell lymphoma; GCB, germinal centre B-cell; MALT1, mucosa-associated lymphoid tissue lymphoma translocation protein 1; PMBL, primary mediastinal B-cell lymphoma.
Differential outcome of DLBCL subtypes
DLBCL is a widely heterogeneous disease associated with a variety of clinical presentations and tremendous underlying molecular diversity. The International Prognostic Index is the most robust clinical prognostic tool used to predict the overall treatment outcome in patients with DLBCL,28 but does not capture the genetic and molecular heterogeneity of the disease. The three cell-of-origin DLBCL subtypes have distinct mechanisms of oncogenic activation and are associated with very different prognoses (Table 2). Most patients with PMBL can be cured with an effective chemotherapeutic regimen, such as DA-EPOCH-R and, in most cases, without mediastinal radiotherapy.16 Retrospective analysis by the Lymphoma/Leukemia Molecular Profiling Project (LLMPP) demonstrated that patients with GCB DLBCL have better prognoses than those with ABC DLBCL when treated with R-CHOP.29 Similar discordant results regarding survival in different DLBCL subtypes have been observed after treatment with DA-EPOCH-R. In a phase II study of 69 patients with previously untreated DLBCL, treatment with DA-EPOCH-R resulted in progression-free survival (PFS) after 5 years of 100% for patients with GCB DLBCL compared with 67% for patients with ABC DLBCL (P = 0.008).15 These observations suggest that most patients who fail to respond to, or relapse after, therapy likely have ABC DLBCL and identifying the driver mutations and aberrant signalling activity in this subset of patients is of the highest priority.
Table 2 |.
PFS and overall survival for each DIBCI molecular suhtvpe
| Molecular subtype | Regimen | 3-year PFS rate | 3-year overall survival rate | Reference |
|---|---|---|---|---|
| ABC DLBCL | R-CHOP | 40% | Approximately 45% | Lenz et al. (2008)29 |
| GCB DLBCL | R-CHOP | 74% | Approximately 80% | Lenz et al. (2008)29 |
| PMBL | DA-EPOCH-R | 100%* | 97%* | Dunleavy et al. (2013)16 |
At 5 years.
Abbreviations: ABC, activated B-cell; DA-EPOCH-R, dose-adjusted etoposide, doxorubicin and cyclophosphamide with vincristine, prednisone and rituximab; DLBCL, diffuse large B-cell lymphoma; GCB, germinal centre B-cell; PFS, progression-free survival; PMBL, primary mediastinal B-cell lymphoma; R-CHOP, rituximab, cylophosphamide, doxorubicin, vincristine and prednisone.
Approximately 10% of patients with newly diagnosed DLBCL harbour an underlying MYC rearrangement (MYC+) within a complex karyotype, and these patients are at high risk of treatment failure with R-CHOP.30 Although Burkitt lymphoma is also defined molecularly by the presence of a MYC translocation, MYC+ DLBCL tumours are pathogenetically distinct from this type of lymphoma.31 Whether MYC rearrangement is sufficient to confer the poor prognosis of MYC+ DLBCL, or if other genetic aberrations are also necessary, remains unclear. Retrospective analyses have shown that a subset of patients with DLBCL whose tumours have a MYC rearrangement also harbour additional oncogenic rearrangements involving BCL2, BCL6 or CCND1; these ‘double-hit’ lymphomas carry a dismal prognosis when treated with R-CHOP.32–34 Preliminary data suggest that DA-EPOCH-R might be an effective chemotherapy platform for the treatment of MYC+ DLBCL,35 and these findings are currently being prospectively evaluated in an ongoing multicentre phase II study of patients with newly diagnosed MYC+ DLBCL and Burkitt lymphoma.36 Importantly, the results from the CORAL study demonstrate that patients with MYC rearrangement at relapse do not benefit from ASCT.37 In this study, 396 patients with relapsed or refractory DLBCL were treated with dexamethasone, cytarabine, cisplatin and rituximab (R-DHAP) or rituximab, ifosfamide, carboplatin and etoposide (R-ICE) prior to undergoing ASCT.4 Of the 161 patients with tissue evaluable for MYC rearrangement prior to therapy, 28 (17%) were positive. Compared with patients without a detectable MYC rearrangement, patients with MYC+ DLBCL at relapse had a worse PFS (18% versus 42%, P = 0.0322) and overall survival (29% versus 62%, P = 0.0113) independent of the salvage regimen used. Multiple reports have demonstrated that poor prognosis with DLBCL is associated with the overexpression of Bcl-2 and Myc, which can be assessed using immunohistochemistry on formalin-fixed paraffin-embedded tissue.38–41 However, whether patients with concomitant Myc and Bcl-2 expression are a molecularly distinct group has not been demonstrated.
Targeted therapy in GCB DLBCL
EZH2 inhibition in GCB DLBCL
GCB DLBCL has a better prognosis than ABC DLBCL, but some patients will relapse after initial immunochemotherapy.42 Next-generation genomic techniques such as whole-exome sequencing,25 genome-wide copy number analysis20 and direct DNA and RNA sequencing24,26,27 have facilitated the identification of individual mutations specific to GCB DLBCL. Somatic heterozygous point mutations have been identified within exon 15 of the EZH2 gene, which encodes histone-lysine N-methyltransferase EZH2 (EZH2) in GCB DLBCL, resulting in the replacement of a single tyrosine (Tyr641) in the SET domain of the protein.43 Multiple samples of DLBCL and follicular lymphoma were analysed using Sanger sequencing to determine the prevalence of this point mutation. Of the 83 GCB DLBCL samples, 18 (22%) were found to harbour the EZH2 mutation whereas none of the 42 ABC DLBCL samples analysed had the mutation.43 The EZH2 protein is the catalytic component of the polycomb-repressive complex 2 (PRC2) and is responsible for adding methyl groups to Lys27 of histone H3 (H3K27).44 EZH2Y641 acts as a dominant gain-of-function mutation in DLBCL,45 and studies of B-cell lymphoma cell lines harbouring this mutation show an increase in trimethylation of H3K27.45,46 Through experiments in DLBCL cell lines, as well as in vivo mouse experiments, EZH2 has been shown to act as a master regulator of the GCB DLBCL phenotype and promotes lymphomagenesis through transcriptional silencing of key antiproliferative and tumour suppressor genes involved in cell cycle regulation, such as CDKN1A.47,48
DLBCL cell lines with EZH2 mutations seem to be dependent on the increased histone methyltransferase activity for proliferation49 and selective EZH2 inhibitors, such as GSK126 and El1, decrease proliferation and induce cell-cycle arrest and apoptosis in DLBCL cell lines and xenograft mouse models.50,51 An oral EZH2 histone methyltransferase inhibitor, E7438, is in early clinical testing for relapsed DLBCL of all subtypes.52
PI3K/Akt/mTOR inhibition in GCB DLBCL
The PI3K signalling pathway is involved in many cellular processes that are critical for cancer progression, including cell metabolism, growth, migration, survival and angiogenesis. This pathway has been shown to be activated by various mechanisms across B-cell malignancies, and somatic mutations of downstream effectors, such as deletions of tumour suppressor gene PTEN on chromosome 10 and amplification of the microRNA (miR)-17–92 (MIHG1 locus cluster on chromosome 13), likely promote cell proliferation in GCB DLBCL20 (Figure 2). The deletion of PTEN is found in 11% of GCB DLBCL cases, but is never observed in ABC DLBCL or PMBL.20,55 PTEN inactivation has been postulated to be linked with activation of the Akt pathway in DLBCL harbouring the t(14;18) translocation.20,54
Figure 2 |.
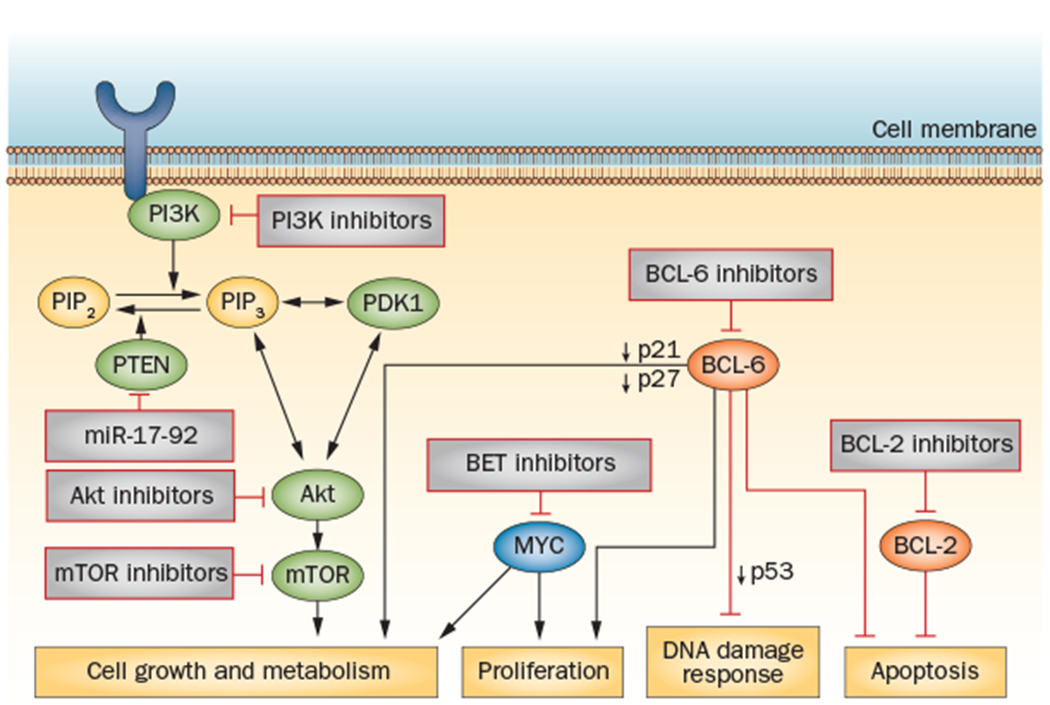
The key signalling pathways implicated in GCB DLBCL with targeted novel agents in clinical development. Loss of PTEN expression is found in 55% of cases of GCB DLBCL, resulting in activation of the PI3K/Akt/mTOR signalling pathway;53 small-molecule inhibitors of this pathway can be effective in cases of GCB DLBCL with decreased PTEN expression. BCL-6 is frequently activated in GCB DLBCL; BCL6 deregulation results in enhanced tumour proliferation via decreased expression of the cell-cycle checkpoint proteins p21 and p27, impaired DNA damage response through decreased p53 expression, impaired cellular metabolism and resistance to apoptosis.70 Inhibitors that target key co-repressor proteins of BCL-6 are in preclinical development.72 In normal B cells, BCL-6 suppresses transcription of the MYC oncogene; in 10–15% cases of DLBCL this mechanism is bypassed through MYC oncogene translocation, resulting in uncontrolled cellular metabolism and growth.31 BET bromodomain inhibitors are in early clinical development and represent a novel strategy of epigenetic regulation of MYC-driven tumours.143 BCL2 translocations are observed in up to 35% of GCB DLBCL cases, resulting in inhibition of apoptosis.63 Inhibitors of BCL-2, such as ABT-263, have demonstrated early clinical activity in B-cell malignancies.65 Abbreviations: BCL-2, apoptosis regulator Bcl-2; BCL-6, B-cell lymphoma 6; DLBCL, diffuse large B-cell lymphoma; GCB, germinal centre B-cell; mTOR, mammalian target of rapamycin; PI3K, phosphatidylinositol 3-kinase.
The functional consequence of PTEN inactivation was investigated in 248 samples from patients with primary DLBCL.53 The loss of PTEN expression, measured by immunohistochemical techniques, was assessed in 55% of GCB DLBCL cases, but in only 14% of non-GCB DLBCL cases. PTEN status was inversely correlated with activation of the PI3K/Akt pathway in GCB DLBCL cell lines, suggesting constitutive activation of the pathway and oncogenic addiction specific for this molecular subset of DLBCL.53 Inhibition of PI3K—using the pan-PI3K inhibitor LY294002—demonstrated selective toxicity in PTEN-deficient GCB DLBCL cell lines that was not observed in PTEN-positive GCB DLBCL cell lines.53 These results give a strong indication that PTEN-negative GCB DLBCL will respond to therapy that inhibits the PI3K/Akt pathway.
Three classes of PI3Ks have been described, with class IA being the most clearly implicated in cancer. Class IA PI3Ks are activated by growth factors through numerous receptor tyrosine kinases and the PI3K catalytic subunit isoforms are potentially amenable to inhibition by small molecules. Indeed, many inhibitors of the PI3K/Akt/mTOR pathway are in clinical development.55 Idelalisib (formerly CAL-101) is a potent small-molecule inhibitor of the PI3K isoform p110δ that blocks constitutive PI3K signalling in vitro.56 In a phase I study in nine patients with DLBCL, idelalisib was well-tolerated, but did not invoke clinical responses.57 Everolimus and temsirolimus are inhibitors of mammalian target of rapamycin (mTOR) that have demonstrated modest activity of 25–30% across a variety of aggressive lymphoma subtypes, such as DLBCL, grade 3 follicular lymphoma, mantle-cell lymphoma and transformed lymphomas,58,59 but no molecular evidence is available to identify which subset of patients benefit most from these inhibitors. Small-molecule inhibitors of Akt, such as MK-2206, have been explored in DLBCL cell lines as a method for overcoming mTOR inhibitor resistance60 and are undergoing early phase clinical testing as single agents in all subtypes of relapsed DLBCL.61,62
BCL-2 inhibition in GCB DLBCL
The apoptosis regulator Bcl-2 protein (BCL-2) might represent another therapeutic target in GCB DLBCL. Its gene, BCL2, is deregulated in DLBCL through multiple mechanisms that differ among the molecular subtypes. The BCL2 translocation t(14;18) is found in 34% of GCB DLBCL cases63 and next-generation sequencing studies have shown that BCL2 is the most mutated gene in GCB DLBCL.64
Navitoclax (ABT-263) is a BCL-2 inhibitor that has demonstrated significant clinical activity in chronic lymphocytic leukaemia, but produced dose-limiting thrombocytopenia because of co-inhibition of Bcl-xL (B-cell lymphoma-extra large).65 ABT-199 is a second-generation inhibitor of BCL-2 that lacks significant Bcl-xL binding and has demonstrated activity in early patient studies in a variety of B-cell lymphomas, including DLBCL,66,67 with less impact on platelets than navito-clax. ABT-199 is currently being tested in a phase II study that includes patients with relapsed B-cell lymphomas, including DLBCL of all molecular subtypes, in combination with bendamustine and rituximab.68
BCL-6 inhibition in GCB DLBCL
Treatment resistance in GCB DLBCL might also be due to the activation of the transcription factor B-cell lymphoma 6 protein (BCL-6), which is a master regulator of the germinal centre reaction and transcriptional repressor of many target genes involved in proliferation, survival, cell growth and metabolism (Figure 2).69,70 Targeting BCL-6 directly is difficult as it is a transcription factor, but the use of small-molecule inhibitors to target the protein-protein interactions required to mediate its effects might be a more promising approach. The small-molecule inhibitor 79-6 specifically disrupts the activity of BCL-6 by binding in the lateral groove of the BCL-6 BTB domain and selectively inhibiting the co-repressor proteins encoded by BCOR, NCOR1 and Ncor2, inducing apoptosis in DLBCL cell lines in vitro.71 Tumour shrinkage was observed in a DLBCL xenograft mouse model following treatment with 79-6, supporting the notion that BCL-6 inhibition is a rational therapeutic strategy in GCB DLBCL.72
NF-κB pathway activation in ABC DLBCL
A unique characteristic of B cells is the expression of both an antigen-specific B-cell receptor (BCR) and one or more germline-encoded receptors of the innate immune system, known as Toll-like receptors (TLR).73 This dual expression receptor pattern provides B cells the ability to display an integrated response to a variety of stimuli and engage downstream transcription factors, such as the NF-κB pathway. The NF-κB family of transcription factors control multiple cellular processes involved in tumour development and progression, including cytokine secretion, inflammation, cellular proliferation, angiogenesis, invasion and metastasis.74 The pathogenic hallmark of ABC DLBCL is the constitutive activation of the NF-κB pathway through a variety of mechanisms with resultant upregulation of the transcription factor interferon regulatory factor 4 (IRF-4).75,76 (Figure 3). IRF-4 expression propels B cells towards plasmacytic differentiation and the dependence of ABC DLBCL on IRF-4 is an example of non-oncogene addiction.77 IRF-4 is a direct target of NF-κB transcription factors and can be induced by both the BCR and TLR signalling pathways.78
Figure 3 |.
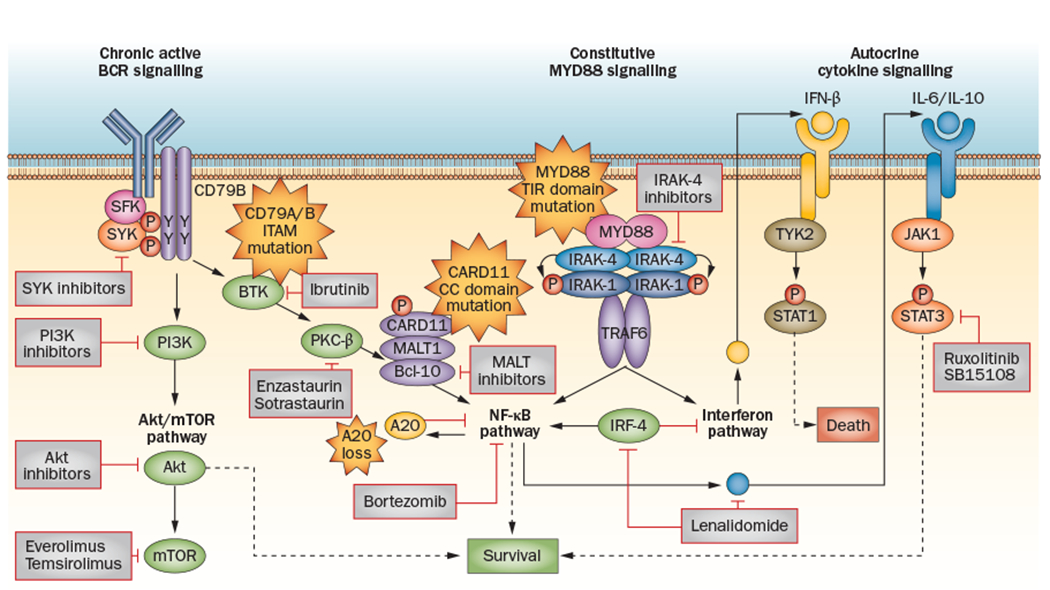
The key signalling pathways implicated in ABC DLBCL with targeted novel agents in clinical development. Upstream inhibitors of the NF-κB pathway—such as ibrutinib, enzastaurin, SYK inhibitors and PI3K inhibitors—target specific tyrosine kinases involved in BCR signalling.101 These inhibitors will likely be effective only in DLBCL subsets that rely on chronic active BCR signalling.81 Everolimus, temsirolimus and Akt inhibitors target the mTOR/Akt pathway, which is also involved in BCR signalling.58–60 Specific inhibitors of IRAK-4 and MALT1 have shown promising activity in vitro.118,119,145 Downstream inhibitors of the NF-κB pathway have shown activity in ABC DLBCL and include bortezomib86 (a proteasome inhibitor) and lenalidomide.150 Lenalidomide has multiple inhibitory mechanisms, including direct targeting of IRF-4 and augmentation of the interferon pathway.104 JAK–STAT signalling is also a promising target in ABC DLBCL and in PMBL; ruxolitinib and SB1518 target this pathway and are in early clinical development.125,126 Abbreviations: ABC, activated B cell; BCR, B-cell receptor; DLBCL, diffuse large B-cell lymphoma; IRAK-4, interleukin-1 receptor-associated kinase 4; IRF-4, interferon regulatory factor 4; JAK, Janus activating kinase; mTOR, mammalian target of rapamycin; PMBL, primary mediastinal B-cell lymphoma; SYK, spleen tyrosine kinase.
The most-effective method of targeting the NF-κB pathway is likely dependent on the presence of specific activating mutations in subsets of patients with ABC DLBCL (Figure 4). Caspase recruitment domain-containing protein 11 (CARD11), B-cell lymphoma/ leukaemia 10 (BCL-10) and mucosa-associated lymphoid tissue lymphoma translocation protein 1 (MALT1) form a signalling complex of adaptor proteins that leads to the BCR-dependent activation of NF-κB upon antigen stimulation.79 Mutations in CARD11 are found in up to 10% of cases of ABC DLBCL,80 and patients with ABC DLBCL harbouring such mutations are likely to require inhibition of downstream targets of NF-κB for effective treatment. Conversely, patients with wild-type CARD11 engage NF-κB through ‘chronic active’ BCR signalling and ABC DLBCL cell lines with wild-type CARD11 seem to be more sensitive to the inhibition of targets upstream of NF-κB, such as kinases involved in BCR signalling,81 than models with mutated CARD11. Point mutations in the tyrosine residues of the B-cell co-receptor CD79B result in chronically active BCR signalling in 21% of cases of ABC DLBCL and might also be associated with selective sensitivity to individual therapeutic agents.81
Figure 4 |.
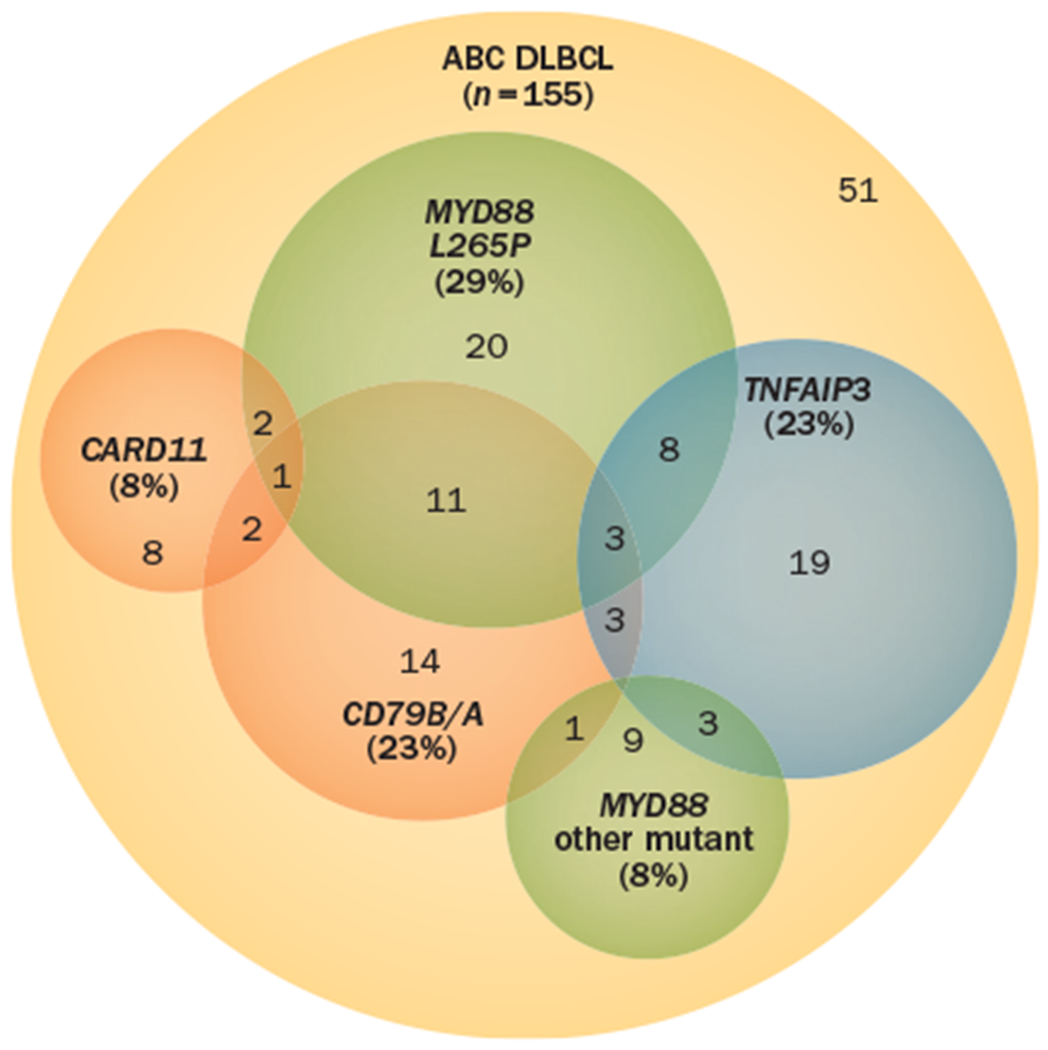
Constitutive activation of the NF-κB pathway is the hallmark of ABC DLBCL and occurs through a variety of mechanisms.82 Recurrent genetic alterations have been described, including mutations in MYD88, CARD11, CD79B/A and biallelic deletions of TNFAIP3. The overlap of known mutations in ABC DLBCL that result in constitutive activation of the NF-κB pathway are shown. The presence of individual mutations might inform treatment decisions in the future, but the absence of an individual mutation does not necessarily rule out a potential treatment response. Combinations of agents that target cooperating mutations or cooperating pathway activity might be required to target this pathway. Mechanistic understanding of these cooperating pathways is the focus of translational research in ABC DLBCL. Abbreviations: ABC, activated B cell; CARD11, caspase recruitment domain-containing protein 11; DLBCL, diffuse large B-cell lymphoma.
Highly recurrent oncogenic mutations in MYD88, which encodes an adaptor protein involved in TLR signalling, have been identified in ABC DLBCL, and a specific point mutation (the amino acid substitution L265P) has been observed in almost 30% of cases of ABC DLBCL, but is rarely observed in GCB DLBCL.82 MYD88 mutations activate a complex of IL-1-associated kinases (IRAK-1 and IRAK-4) that engage the NF-κB and MAP kinase pathways leading to the production of IL-6 and IL-10 and autocrine activation of the JAK pathway.82
Tumour necrosis factor α-induced protein 3 (TNFAIP3, also known as A20) is a negative regulator of the NF-κB pathway that prevents excessive stimulation in response to various external stimuli, such as tumour necrosis factor (TNF). TNFAIP3 was first implicated as a tumour suppressor involved in lymphomagenesis inactivated by somatic mutation in a variety of B-cell lymphomas.83 In ABC DLBCL, biallelic inactivation of TNFAIP3 (by mutations, deletions or both) occurs in 30% of cases and can coexist with mutations in both MYD88 and CD79B, suggesting that inactivation of TNFAIP3 can enhance both BCR and TLR signalling pathways.76
Inhibition of NF-κB pathway in ABC DLBCL
The genetic ablation of key NF-κB signalling molecules results in arrest of B-cell differentiation and prevents B-cell activation.74 The validation of the NF-κB pathway as a therapeutic target in ABC DLBCL was first demonstrated when small-molecule inhibitors of the IκB kinase (IKK) complex demonstrated selective inhibition of ABC DLBCL cell lines.84 Targeting the NF-κB pathway via proteasome inhibition has demonstrated selective efficacy in patients with ABC DLBCL. Bortezomib reversibly targets the chymotrypsin-like activity of the proteasome and blocks the degradation of the NF-κB inhibitory protein IκBα.85 A landmark study in 49 patients with relapsed or refractory DLBCL revealed that bortezomib sensitized patients with ABC DLBCL—but not patients with GCB DLBCL—to chemotherapy.86 Patients were treated with bortezomib alone or in combination with DA-EPOCH. Bortezomib plus DA-EPOCH resulted in higher response rates and survival in patients with ABC DLBCL compared with GCB DLBCL; bortezomib alone had no activity in any patients with DLBCL. The overall response rate (ORR) with bortezomib plus DA-EPOCH was 83% versus 13% (P <0.001) and overall survival was 10.8 months versus 3.5 months (P = 0.003) in patients with ABC DLBCL and GCB DLBCL, respectively. Bortezomib was subsequently investigated in combination with R-CHOP in a phase II study of 40 patients with previously untreated DLBCL.87 The 2-year PFS of patients with GCB DLBCL and non-GCB DLBCL (subtypes determined by immunohistochemistry) treated with bortezomib plus R-CHOP were similar, indicating that bortezomib helps to overcome the poorer prognoses of patients with ABC DLBCL compared with GCB DLBCL.29 Taken together, these findings suggest that ABC DLBCL is selectively sensitive to inhibition of the ubiquitin-proteasome system. This finding prompted a randomized phase II study of R-CHOP with or without bortezomib in patients with previously untreated non-GCB DLBCL.88
Carfilzomib is a second-generation proteasome inhibitor that is more selective and better tolerated than first-generation inhibitors. It demonstrated promising preclinical activity in DLBCL cell lines, both as a single agent and in combination with other agents. As a single agent, carfilzomib induced cell-cycle arrest in DLBCL cell lines, including those resistant to rituximab.89 The combination of carfilzomib with vorinostat, a nonselective histone deacetylease (HDAC) inhibitor, was tested in GCB and ABC DLBCL models and found to have synergistic activity in vitro as well as in vivo.90 Carfilzomib was also tested in combination with ACY-1215, an HDAC inhibitor with selectivity for HDAC6, and demonstrated synergistic killing in both GCB and ABC DLBCL cell lines.91 Finally, carfilzomib has shown promising in vitro and in vivo activity in combination with obatoclax, a small molecule that binds to BCL-2, Bcl-xL and induced myeloid leukaemia cell differentiation protein Mcl-1 (Mcl-1).92 Carfilzomib in combination with vorinostat is currently undergoing phase I testing in relapsed and refractory B-cell lymphomas, including DLBCL.93
Lenalidomide is an oral immunomodulatory agent that exerts anticancer effects through multiple mechanisms, postulated to include inhibition of angiogenesis, recruitment of natural killer cells, upregulation of CD80 and CD40, impairment of inflammatory cytokines and effects on the tumour microenvironment.94 Lenalidomide selectively kills ABC DLBCL cells by targeting IRF-4 directly leading to an increase in IFN-β production (Figure 3).95 In ABC DLBCL cell lines, lenalidomide seems to work via downregulation of IRF-4 and requires the expression of the E3 ubiquitin ligase complex co-receptor protein cereblon.96 In a study of patients with relapsed or refractory DLBCL (n = 40), single-agent lenalidomide demonstrated differential efficacy for non-GCB DLBCL, with an ORR of 52.9% in patients with non-GCB DLBCL compared with only 8.7% (P = 0.004) in patients with GCB DLBCL.97
NF-κB pathway regulation in ABC DLBCL
BCR signalling pathway
BCR signal transduction is a complex process involving multiple interconnected pathways of effector molecules responsible for signal initiation, propagation, integration and modulation that culminates in transcription factor activation and gene expression.98 BCR signalling thereby represents an important connection between intracellular signalling pathways and regulation of gene expression in the nucleus via NF-κB. Normal B cells require a continuous (tonic) survival signal through the BCR, independent of antigen binding and that is mediated by PI3K.99 By contrast, antigen-induced BCR signalling (‘chronic active’ signalling) initiates a signalling transduction cascade involving several tyrosine kinases and adapter molecules—including p72-Syk, p53Lyn, Bruton tyrosine kinase (BTK), PI3K, PKC-β and mTOR—to promote downstream survival pathways.98,100 ABC DLBCL exhibits oncogene addiction to mutations that engage the classical NF-κB pathway and are susceptible to inhibition of the BCR pathway.101
BTK inhibition in ABC DLBCL
Ibrutinib, formerly PCI-32765, is an oral small-molecule inhibitor that selectively and irreversibly inhibits BTK through covalent binding via cysteine-481.102 Ibrutinib induced objective clinical responses in dogs with spontaneous B-cell lymphoma, prompting its clinical development for the treatment of human B-cell malignancies.103 In a study of BCR signalling in DLBCL, knockdown of BTK using short hairpin RNAs (shRNAs) was found to be highly toxic to an ABC DLBCL cell line with wildtype CARD11, but not for one with mutant CARD11.81 Similar results were observed in survival assays: a BTK shRNA was toxic for ABC DLBCL lines with wildtype CARD11, but had no effect on cell lines with CARD11 mutations. Since ibrutinib has been shown to selectively kill DLBCL cells,104 these results suggest that its mechanism of action might be reliant on constitutive BCR signalling.
In a phase I study of 56 patients with relapsed or refractory B-cell malignancies, patients with DLBCL responded to treatment with ibrutinib at various dose levels,105 and preliminary results from a phase II study of ibrutinib in 70 patients with relapsed or refractory DLBCL of all molecular subtypes revealed an overall response rate of 40% in the first 25 patients with ABC DLBCL treated with ibrutinib at 560 mg daily.106 Only one of 20 patients with GCB DLBCL responded to ibrutinib, supporting its selectivity for ABC DLBCL. Mutational analysis of pretreatment and post-treatment biopsies showed that clinical responses were observed in patients with ABC DLBCL with wild-type CD79B or mutated CD79B, indicating that ibrutinib does not require a BCR mutation to be efficacious, which suggests that alternative mechanisms of BCR signalling exist. Patients with a MYD88 mutation, but without a CD79B, mutation did not respond (n = 4), whereas all patients who had mutations in both MYD88 and CD79B responded (n = 4), suggesting alternative activation mechanisms.
These results are highly suggestive that BTK is an important target in ABC DLBCL and that its inhibition might be dependent on the presence (or absence) of specific mutations. A multi-institutional phase IB trial testing the safety of ibrutinib in combination with R-CHOP for patients with untreated B-cell lymphomas— including DLBCL—has reported results from the first 17 patients, with encouraging results of no unanticipated toxicities observed.107 A large international randomized phase III study of R-CHOP with or without ibrutinib in patients with newly diagnosed non-GCB DLBCL has begun enrollment.108
PKC-β inhibition in DLBCL
PKC-β is a serine/threonine kinase that has an essential role in the propagation of BCR signalling and activation of the NF-κB pathway;109 in vitro evidence supports PKC-β as a rational therapeutic target in DLBCL.110 Enzastaurin is a potent oral inhibitor of PKC-β that has been studied in patients with DLBCL. In a phase II study, 55 patients with relapsed or refractory DLBCL were treated with oncedaily enzastaurin until disease progression or unacceptable toxicity.111 Overall, twelve of 55 patients (22%; 95% CI 13–46%) experienced freedom from progression (FFP) for two cycles, and four patients (7%; 95% CI 2–18%) experienced significant FFP of >20 months. Preliminary analysis of a randomized phase II study in 100 patients with high-risk or intermediate-risk DLBCL treated with R-CHOP with or without enzastaurin demonstrated a 1-year PFS of 71% (95% CI 0.58–0.84) in patients treated with the enzastaurin combination compared with 52% (95% CI 0.35–0.69) in those treated with R-CHOP alone.112 The molecular DLBCL subgroups that benefited most from enzastaurin were not reported in this study.
Sotrastaurin, another selective PKC-β inhibitor, has also been evaluated in ABC DLBCL cell lines and mouse xenograft models.113 A number of cell lines showed sensitivity to sotrastaurin and the growth inhibitory effects of PKC-β inhibition correlated with inhibition of the NF-κB pathway. In addition, the nature of the mutations in components of the NF-κB pathway predicted responsiveness to sotrastaurin. Mutations in CD79B correlated with sensitivity to sotrastaurin, whereas the presence of CARD11 mutations resulted in resistance to the inhibitor. Sotrastaurin is currently being tested in an international multi-institutional phase I study in patients with relapsed or refractory DLBCL that harbour either a CD79A or CD79B mutation.114
MALT1 inhibition in ABC DLBCL
MALT1 is the active signalling component essential for upstream activation of NF-κB following antigen stimulation of BCR. MALT1 has proteolytic activity that is required for the survival of ABC DLBCL cells115,116 and, therefore, represents a new therapeutic target.117 Irreversible inhibition of MALT1 with small-molecule inhibitors selectively suppresses the protease function of MALT1 in ABC DLBCL cell lines and xenograft models with very little toxicity.118 Phenothiazine derivatives have also demonstrated selective inhibition of the proteolytic activity of MALT1 in ABC DLBCL cell lines.119 These results suggest inhibitors of MALT1 are targeted agents with promising potential in patients with DLBCL that rely on MALT1 for BCR signalling.120
JAK-STAT inhibition in ABC DLBCL
JAKs are nonreceptor protein tyrosine kinases that act downstream of cytokine signals to activate the STAT proteins, a family of transcription factors that translocate to the nucleus when activated and regulate cellular events, such as proliferation and survival. In some cases, abnormal activation of JAK-STAT3 signalling is driven by activating mutations of MYD88 through an autocrine feed-forward loop involving IL-6, IL-10 and IFN-β (Figure 3).104,121 STAT3 expression and activation are significantly higher in ABC DLBCL cell lines and these cell lines demonstrate higher NF-κB activity than those with low STAT3 activity.121,122 Knockdown of STAT3 expression, using shRNAs, in mouse models suppresses the growth of ABC DLBCL tumours, validating STAT3 as a therapeutic target in this subtype of DLBCL.123
STAT3 lacks intrinsic enzymatic activity, which means that direct therapeutic targeting of the protein is challenging; however, small-molecule inhibitors of the JAK family are in clinical development. Ruxolitinib is an oral inhibitor of JAK-1 and JAK-2 that has been approved for use in primary myelofibrosis,124 and a multicentre phase II study of ruxolitinib in patients with relapsed or refractory DLBCL is ongoing.125 Pacritinib (formerly SB1518) is an oral small molecule that selectively and potently inhibits JAK-2, showing promising in vitro activity in JAK-2-dependent DLBCL cell lines independent of JAK-2 mutational status. In a phase I study in 34 patients with relapsed or refractory lymphomas, including DLBCL, pacritinib demonstrated a favourable toxicity profile.126
Primary mediastinal B-cell lymphoma
PMBL is a molecularly distinct subtype of DLBCL with clinical and biological features that overlap with nodular sclerosing Hodgkin lymphoma (NSHL).127–129 Frequently, PMBL and NSHL tumours contain thymic remnants, an observation that led to the hypothesis that these cancers are derived from a post-thymic B cell.130,131 PMBL and NSHL can be distinguished by gene-expression profiling: PMBL genes are expressed in mature B cells but not in NSHL.132 Both PMBL and NSHL rely on the NF-κB and JAK-STAT signalling pathways for tumour proliferation.133,134
A genetic hallmark of PMBL is amplification of a region on chromosome 9p24, observed in 70% of PMBL cases.135 The 9p24 amplicon contains several key targets, such as CD274 (encoding PD-L1), PDCD1LG2 (encoding PD-L2) and JAK2.136,137 PD-L1 and PD-L2 are ligands of the programmed cell death-1 (PD-1) receptor that normally delivers inhibitory signals to regulate the balance between T-cell activation and immune tolerance.138,139 PD-L1 is expressed on PMBL tumour cells, suggesting they harbour an ineffective T-cell immune response.139,140 JAK2 amplification on chromosome 9p24 also induces PD-1 ligands and is associated with sensitivity to JAK-2 inhibitors.136,137 Recurrent unbalanced rearrangements involving the MHC class II transactivator (CIITA), with multiple fusion partners, were reported in 38% of cases of PMBL and these CIITA gene fusions were found to result in overexpression of PD-1 ligands.141 These results further implicate the PD-1 pathway as a potential target in PMBL.
PMBL is generally curable if the most-effective immunochemotherapy regimen, DA-EPOCH-R, is used. In a prospective phase II study, 51 patients with PMBL were treated with six cycles of DA-EPOCH-R. At 5 years of follow-up, the overall survival rate was 97% (95% CI 81–99%) and the EFS rate was 93% (95% CI 81–98%), whereas only two of the 51 patients treated with DA-EPOCH-R required mediastinal radiation.16 Inhibitors of the JAK-STAT pathway or neutralizing antibodies of PD-1, such as pidilizumab,142 are rational therapeutic approaches for the treatment of PMBL.
DLBCL with MYC translocations
The Myc oncoproteins have generally been considered ‘undruggable’ because the protein structures are not amenable to inhibition by small molecules. Epigenetic manipulation of the BET bromodomain protein BRD4 by the compound JQ1 has demonstrated promise in inhibiting Myc in murine models of multiple myeloma.143 As bromodomain proteins serve as regulatory factors for Myc, targeting these proteins might provide an indirect method to alter MYC gene expression.
One mechanism by which Myc promotes lymphomagenesis is by suppressing the transcription of the tumour suppressor tristetraprolin (encoded by ZFP36).144 Tristetraprolin is an AU-binding protein (AUBP) and ZFP36 transcription is suppressed in cancers that express Myc; restoring tristetraprolin expression impairs Myc-induced lymphomagenesis and abolishes the malignant state.144 Targeting bromodomain proteins or reactivation of tristetraprolin expression both represent novel approaches for indirect targeting of Myc. These strategies could potentially be combined with chemotherapy to avoid the development of resistance to treatments.
Functional pathway interactions
Given the complex molecular heterogeneity observed across DLBCL tumour samples,24–27 it is probably an oversimplification to conceptualize singular driver mutations or activated pathways within given subsets of patients with DLBCL. A more likely scenario is that rational combination therapy will be required to effectively target the cooperating pathways with functional interactions that exist within tumours. Preclinical investigations of DLBCL cell lines and animal models support the notion that targeted combination therapy might overcome drug resistance mechanisms. In ABC DLBCL cell lines, cooperation between the BCR and TLR signalling pathways has been demonstrated. Simultaneous RNA silencing of both CD79A and MYD88 in ABC DLBCL cell lines is more toxic than knockdown of either one alone104 and additional inhibition of IKK, with either the selective inhibitor MLN120B or ibrutinib, results in undetectable IRF-4 expression.104 Inhibitors of IRAK-4 show strong synergy with ibrutinib in ABC DLBCL cell lines,145 and the combination of lenalidomide and ibrutinib is highly effective in arresting the growth of ABC DLBCL tumours in mouse models.95 Additional evidence for the cooperation between the BCR and TLR signalling pathways has been observed in a broad range of lymphoma cell lines in which PI3K inhibition is enhanced with concomitant knockdown of the PAK1 gene.146 In mouse models of DLBCL tumours with MYC rearrangements, co-treatment with inhibitors of >NF-κB, STAT3 and PI3K yielded an additive effect on the inhibition of proliferation,147 and BEZ235 (a pan-PI3K, dual-mTOR inhibitor) demonstrated preclinical activity in DLBCL cell lines with MYC translocations.148 Indeed, the rational combination of targeted agents that exhibit synthetic lethality149 offers a promising method to overcome drug resistance in patients with DLBCL. The current paradigmatic challenge is to define mechanism-based synergistic combinations with acceptable toxicities that can be translated to the clinic.
Conclusions
DLBCL is a molecularly heterogeneous disease with identifiable subsets that are at high risk for treatment failure with standard immunochemotherapy. DLBCL remains curable in advanced stages, but up to one-third of patients will ultimately fail to respond to initial therapy and the efficacy of salvage options are diminished. Indeed, anthracycline-based chemotherapy and rituximab have been historical breakthroughs in the management of DLBCL, resulting in a notable survival increase. However, we have now entered the molecular era of defining DLBCL, whereby the goal is to pinpoint driver mutations and pathway addictions within distinct molecular subsets of DLBCL and target them therapeutically. Numerous small molecules and pathway inhibitors are in various stages of investigation and demonstrate promise in that regard. It is highly probable that the next major breakthrough in DLBCL therapy will involve novel agents that simultaneously target both the driver mutations and the cooperating mutations that confer drug resistance within subsets of DLBCL. Clinical trials of novel therapies that do not take the molecular subtypes of DLBCL into account will not inform identification of the optimal therapy for this disease. To expeditiously achieve the goal of personalized precision therapy in patients with DLBCL, novel agents must be tested in patients with mutational profiles that predict poor response to standard treatment. Correlative investigations, such as pretreatment and post-treatment biopsies, are also essential to understand mechanisms of both sensitivity and resistance to therapy.
Key points.
Molecular analyses have led to the definition of diffuse large B-cell lymphoma (DLBCL) subtypes, with differential therapeutic responses, and identification of driver mutations that represent the ‘Achilles Heel' of these tumours
DLBCL can be classified into three different molecular cell-of-origin subtypes: germinal centre B-cell (GCB), activated B-cell (ABC) and primary mediastinal B-cell lymphoma (PMBL)
Patients with DLBCL at the highest risk for disease relapse after standard immunochemotherapy are patients with ABC DLBCL and those whose tumours harbour MYC translocations
Constitutive activation of the NF-κB pathway is the hallmark of ABC DLBCL; sensitivity to upstream versus downstream inhibition of NF-κB is likely determined by specific mutations found in ABC DLBCL
Downstream NF-κB pathway inhibitors include those targeting the ubiquitin-proteasome complex; upstream inhibitors include inhibitors of B-cell receptor signalling and inhibitors targeting other aberrant signalling pathways in ABC DLBCL
Overcoming drug resistance in DLBCL will ultimately require identification of cooperating mutations and rational combination therapies targeting the signalling pathways implicated in the pathogenesis of this disease
Acknowledgements
The authors would like to acknowledge support from the intramural research programme of the NIH.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Siegel R, Naishadham D & Jemal A Cancer statistics, 2013. CA Cancer J. Clin 63, 11–30 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Armitage JO My treatment approach to patients with diffuse large B-cell lymphoma. Mayo Clin. Proc 87, 161–171 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Friedberg JW Relapsed/refractory diffuse large B-cell lymphoma. Hematology Am. Soc. Hematol. Educ. Program 2011, 498–505 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Gisselbrecht C et al. Salvage regimens with autologous transplantation for relapsed large B-cell lymphoma in the rituximab era. J. Clin. Oncol 28, 4184–4190 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DeVita VT Jr et al. Advanced diffuse histiocytic lymphoma, a potentially curable disease. Lancet 1, 248–250 (1975). [DOI] [PubMed] [Google Scholar]
- 6.Fisher RI et al. Comparison of a standard regimen (CHOP) with three intensive chemotherapy regimens for advanced non-Hodgkin’s lymphoma. N. Engl. J. Med 328, 1002–1006 (1993). [DOI] [PubMed] [Google Scholar]
- 7.Coiffier B et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N. Engl. J. Med 346, 235–242 (2002). [DOI] [PubMed] [Google Scholar]
- 8.Pfreundschuh M et al. CHOP-like chemotherapy plus rituximab versus CHOP-like chemotherapy alone in young patients with good-prognosis diffuse large-B-cell lymphoma: a randomised controlled trial by the MabThera International Trial (MInT) Group. Lancet Oncol. 7, 379–391 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Coiffier B et al. Long-term outcome of patients in the LNH-98.5 trial, the first randomized study comparing rituximab-CHOP to standard CHOP chemotherapy in DLBCL patients: a study by the Groupe d’Etudes des Lymphomes de l’Adulte. Blood 116, 2040–2045 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pfreundschuh M et al. CHOP-like chemotherapy with or without rituximab in young patients with good-prognosis diffuse large-B-cell lymphoma: 6-year results of an open-label randomised study of the MabThera International Trial (MInT) Group. Lancet Oncol. 12, 1013–1022 (2011). [DOI] [PubMed] [Google Scholar]
- 11.Fu K et al. Addition of rituximab to standard chemotherapy improves the survival of both the germinal center B-cell-like and non-germinal center B-cell-like subtypes of diffuse large b-cell lymphoma. J. Clin. Oncol 26, 4587–4594 (2008). [DOI] [PubMed] [Google Scholar]
- 12.Schmitz N et al. Conventional chemotherapy (CHOEP-14) with rituximab or high-dose chemotherapy (MegaCHOEP) with rituximab for young, high-risk patients with aggressive B-cell lymphoma: an open-label, randomised, phase 3 trial (DSHNHL 2002–1). Lancet Oncol. 13, 1250–1259 (2012). [DOI] [PubMed] [Google Scholar]
- 13.Cunningham D et al. Rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisolone in patients with newly diagnosed diffuse large B-cell non-Hodgkin lymphoma: a phase 3 comparison of dose intensification with 14-day versus 21-day cycles. Lancet 381, 1817–1826 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Recher C et al. Intensified chemotherapy with ACVBP plus rituximab versus standard CHOP plus rituximab for the treatment of diffuse large B-cell lymphoma (LNH03–2B): an open-label randomised phase 3 trial. Lancet 378, 1858–1867 (2011). [DOI] [PubMed] [Google Scholar]
- 15.Wilson WH et al. A Cancer and Leukemia Group B multi-center study of DA-EPOCH-rituximab in untreated diffuse large B-cell lymphoma with analysis of outcome by molecular subtype. Haematologica 97, 758–765 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dunleavy K et al. Dose-adjusted EPOCH-rituximab therapy in primary mediastinal B-cell lymphoma. N. Engl. J. Med 368, 1408–1416 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.US National Library of Medicine. ClinicalTrials.gov [online], http://www.clinicaltrials.gov/ct2/show/NCT00118209 (2013). [DOI] [PubMed] [Google Scholar]
- 18.Alizadeh AA et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 403, 503–511 (2000). [DOI] [PubMed] [Google Scholar]
- 19.Rosenwald A et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N. Engl. J. Med 346, 1937–1947 (2002). [DOI] [PubMed] [Google Scholar]
- 20.Lenz G et al. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc. Natl Acad. Sci. USA 105, 13520–13525 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wright G et al. A gene expression-based method to diagnose clinically distinct subgroups of diffuse large B cell lymphoma. Proc. Natl Acad. Sci. USA 100, 9991–9996 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rosenwald A et al. Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J. Exp. Med 198, 851–862 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Steinhardt JJ & Gartenhaus RB Promising personalized therapeutic options for diffuse large B-cell lymphoma subtypes with oncogene addictions. Clin. Cancer Res 18, 4538–4548 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pasqualucci L et al. Analysis of the coding genome of diffuse large B-cell lymphoma. Nat. Genet 43, 830–837 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lohr JG et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc. Natl Acad. Sci. USA 109, 3879–3884 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang J et al. Genetic heterogeneity of diffuse large B-cell lymphoma. Proc. Natl Acad. Sci. USA 110, 1398–1403 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morin RD et al. Frequent mutation of histonemodifying genes in non-Hodgkin lymphoma. Nature 476, 298–303 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.[No authors listed] A predictive model for aggressive non-Hodgkin’s lymphoma. The International Non-Hodgkin’s Lymphoma Prognostic Factors Project. N. Engl. J. Med 329, 987–994 (1993). [DOI] [PubMed] [Google Scholar]
- 29.Lenz G et al. Stromal gene signatures in large-B-cell lymphomas. N. Engl. J. Med 359, 2313–2323 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Savage KJ et al. MYC gene rearrangements are associated with a poor prognosis in diffuse large B-cell lymphoma patients treated with R-CHOP chemotherapy. Blood 114, 3533–3537 (2009). [DOI] [PubMed] [Google Scholar]
- 31.Jaffe ES & Pittaluga S Aggressive B-cell lymphomas: a review of new and old entities in the WHO classification. Hematology Am. Soc. Hematol. Educ. Program 2011, 506–514 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnson NA et al. Lymphomas with concurrent BCL2 and MYC translocations: the critical factors associated with survival. Blood 114, 2273–2279 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aukema SM et al. Double-hit B-cell lymphomas. Blood 117, 2319–2331 (2011). [DOI] [PubMed] [Google Scholar]
- 34.Snuderl M et al. B-cell lymphomas with concurrent IGH-BCL2 and MYC rearrangements are aggressive neoplasms with clinical and pathologic features distinct from Burkitt lymphoma and diffuse large B-cell lymphoma. Am. J. Surg. Pathol 34, 327–340 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dunleavy K et al. MYC+ aggressive B-cell lymphomas: novel therapy of untreated Burkitt lymphoma (BL) and MYC+ diffuse large B-cell lymphoma (DLBCL) with DA-EPOCH-R [abstract]. Ann. Oncol 22 (Suppl. 4), a071 (2011). [Google Scholar]
- 36.US National Library of Medicine. ClinicalTrials.gov [online], http://www.clinicaltrials.gov/ct2/show/NCT01092182 (2013). [DOI] [PubMed] [Google Scholar]
- 37.Cuccuini W et al. MYC+ diffuse large B-cell lymphoma is not salvaged by classical R-ICE or R-DHAP followed by followed by BEAM plus autologous stem cell transplantation. Blood 119, 4619–4624 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Green TM et al. Immunohistochemical double-hit score is a strong predictor of outcome in patients with diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J. Clin. Oncol 30, 3460–3467 (2012). [DOI] [PubMed] [Google Scholar]
- 39.Johnson NA et al. Concurrent expression of MYC and BCL2 in diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J. Clin. Oncol 30, 3452–3459 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hu S et al. MYC/BCL2 protein coexpression contributes to the inferior survival of activated B-cell subtype of diffuse large B-cell lymphoma and demonstrates high-risk gene expression signatures: a report from The International DLBCL Rituximab-CHOP Consortium Program. Blood 121, 4021–4031 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Horn H et al. MYC status in concert with BCL2 and BCL6 expression predicts outcome in diffuse large B-cell lymphoma. Blood 121, 2253–2263 (2013). [DOI] [PubMed] [Google Scholar]
- 42.Thieblemont C et al. The germinal center/ activated B-cell subclassification has a prognostic impact for response to salvage therapy in relapsed/refractory diffuse large B-cell lymphoma: a bio-CORAL study. J. Clin. Oncol 29, 4079–4087 (2011). [DOI] [PubMed] [Google Scholar]
- 43.Morin RD et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat. Genet 42, 181–185 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kirmizis A et al. Silencing of human polycomb target genes is associated with methylation of histone H3 Lys 27. Genes Dev. 18, 1592–1605 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yap DB et al. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood 117, 2451–2459 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sneeringer CJ et al. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc. Natl Acad. Sci. USA 107, 20980–20985 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Béguelin W et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell 23, 677–692 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Velichutina I et al. EZH2-mediated epigenetic silencing in germinal center B cells contributes to proliferation and lymphomagenesis. Blood 116, 5247–5255 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Knutson SK et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat. Chem. Biol 8, 890–896 (2012). [DOI] [PubMed] [Google Scholar]
- 50.McCabe MT et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 492, 108–112 (2012). [DOI] [PubMed] [Google Scholar]
- 51.Qi W et al. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc. Natl Acad. Sci. USA 109, 21360–21365 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.US National Library of Medicine. ClinicalTrials.gov [online], http://clinicaltrials.gov/ct2/show/NCT01897571 (2013). [DOI] [PubMed] [Google Scholar]
- 53.Pfeifer M et al. PTEN loss defines a PI3K/AKT pathway-dependent germinal center subtype of diffuse large B-cell lymphoma. Proc. Natl Acad. Sci. USA 110, 12420–12425 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Siebert R et al. Deletions in the long arm of chromosome 10 in lymphomas with t(14;18): a pathogenetic role of the tumor supressor genes PTEN/MMAC1 and MXI1? Blood 92, 4487–4489 (1998). [PubMed] [Google Scholar]
- 55.Rodon J, Dienstmann R, Serra V & Tabernero J Development of PI3K inhibitors: lessons learned from early clinical trials. Nat. Rev. Clin. Oncol 10, 143–153 (2013). [DOI] [PubMed] [Google Scholar]
- 56.Lannutti BJ et al. CAL-101, a p110delta selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood 117, 591–594 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Furman RR et al. CAL-101, an isoform-selective inhibitor of phosphatidylinositol 3-kinase P110{delta}, demonstrates clinical activity and pharmacodynamic effects in patients with relapsed or refractory chronic lymphocytic leukemia [abstract]. Blood 116, a55 (2010). [Google Scholar]
- 58.Smith SM et al. Temsirolimus has activity in non-mantle cell non-Hodgkin’s lymphoma subtypes: the University of Chicago phase II consortium. J. Clin. Oncol 28, 4740–4746 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Witzig TE et al. A phase II trial of the oral mTOR inhibitor everolimus in relapsed aggressive lymphoma. Leukemia 25, 341–347 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Petrich AM et al. Akt inhibitors MK-2206 and nelfinavir overcome mTOR inhibitor resistance in diffuse large B-cell lymphoma. Clin. Cancer Res 18, 2534–2544 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.US National Library of Medicine. ClinicalTrials.gov [online], http://www.clinicaltrials.gov/ct2/show/NCT01466868 (2013). [DOI] [PubMed] [Google Scholar]
- 62.US National Library of Medicine. ClinicalTrials.gov [online], http://www.clinicaltrials.gov/ct2/show/NCT01481129 (2013). [DOI] [PubMed] [Google Scholar]
- 63.Iqbal J et al. BCL2 translocation defines a unique tumor subset within the germinal center B-cell-like diffuse large B-cell lymphoma. Am. J. Pathol 165, 159–166 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schuetz JM et al. BCL2 mutations in diffuse large B-cell lymphoma. Leukemia 26, 1383–1390 (2012). [DOI] [PubMed] [Google Scholar]
- 65.Wilson WH et al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: a phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. 11, 1149–1159 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Souers AJ et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med 19, 202–208 (2013). [DOI] [PubMed] [Google Scholar]
- 67.Davids MS et al. The BCL-2-specific BH3-mimetic ABT-199 (GDC-0199) is active and well-tolerated in patients with relapsed non-Hodgkin lymphoma: interim results of a phase I study [abstract]. Blood 120, a304 (2012). [Google Scholar]
- 68.US National Library of Medicine. ClinicalTrials.gov [online], http://www.clinicaltrials.gov/ct2/show/NCT01594229 (2013). [DOI] [PubMed] [Google Scholar]
- 69.Phan RT & Dalla-Favera R The BCL6 proto-oncogene suppresses p53 expression in germinal-centre B cells. Nature 432, 635–639 (2004). [DOI] [PubMed] [Google Scholar]
- 70.Basso K & Dalla-Favera R BCL6: master regulator of the germinal center reaction and key oncogene in B cell lymphomagenesis. Adv. Immunol 105, 193–210 (2010). [DOI] [PubMed] [Google Scholar]
- 71.Cerchietti LC et al. A peptomimetic inhibitor of BCL6 with potent antilymphoma effects in vitro and in vivo. Blood 113, 3397–3405 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cerchietti LC et al. A small-molecule inhibitor of BCL6 kills DLBCL cells in vitro and in vivo. Cancer Cell 17, 400–411 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rawlings DJ, Schwartz MA, Jackson SW & Meyer-Bahlburg A Integration of B cell responses through Toll-like receptors and antigen receptors. Nat. Rev. Immunol 12, 282–294 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lim KH, Yang Y & Staudt LM Pathogenetic importance and therapeutic implications of NF-κB in lymphoid malignancies. Immunol. Rev 246, 359–378 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Davis RE, Brown KD, Siebenlist U & Staudt LM Constitutive nuclear factor κB activity is required for survival of activated B cell-like diffuse large B cell lymphoma cells. J. Exp. Med 194, 1861–1874 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Compagno M et al. Mutations of multiple genes cause deregulation of NF-κB in diffuse large B-cell lymphoma. Nature 459, 717–721 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Luo J, Solimini NL & Elledge SJ Principles of cancer therapy: oncogene and non-oncogene addiction. Cell 136, 823–837 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rui L, Schmitz R, Ceribelli M & Staudt LM Malignant pirates of the immune system. Nat. Immunol 12, 933–940 (2011). [DOI] [PubMed] [Google Scholar]
- 79.Rawlings DJ, Sommer K & Moreno-García ME The CARMA1 signalosome links the signalling machinery of adaptive and innate immunity in lymphocytes. Nat. Rev. Immunol 6, 799–812 (2006). [DOI] [PubMed] [Google Scholar]
- 80.Lenz G et al. Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science 319, 1676–1679 (2008). [DOI] [PubMed] [Google Scholar]
- 81.Davis RE et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature 463, 88–92 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ngo VN et al. Oncogenically active MYD88 mutations in human lymphoma. Nature 470, 115–119 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kato M et al. Frequent inactivation of A20 in B-cell lymphomas. Nature 459, 712–716 (2009). [DOI] [PubMed] [Google Scholar]
- 84.Lam LT et al. Small molecule inhibitors of IκB kinase are selectively toxic for subgroups of diffuse large B-cell lymphoma defined by gene expression profiling. Clin. Cancer Res 11, 28–40 (2005). [PubMed] [Google Scholar]
- 85.Strauss SJ et al. The proteasome inhibitor bortezomib acts independently of p53 and induces cell death via apoptosis and mitotic catastrophe in B-cell lymphoma cell lines. Cancer Res. 67, 2783–2790 (2007). [DOI] [PubMed] [Google Scholar]
- 86.Dunleavy K et al. Differential efficacy of bortezomib plus chemotherapy within molecular subtypes of diffuse large B-cell lymphoma. Blood 113, 6069–6076 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ruan J et al. Bortezomib plus CHOP-rituximab for previously untreated diffuse large B-cell lymphoma and mantle cell lymphoma. J. Clin. Oncol 29, 690–697 (2011). [DOI] [PubMed] [Google Scholar]
- 88.US National Library of Medicine. ClinicalTrials.gov [online], http://www.clinicaltrials.gov/ct2/show/NCT00931918 (2013). [DOI] [PubMed] [Google Scholar]
- 89.Gu JJ et al. The novel proteasome inhibitor carfilzomib induces cell cycle arrest, apoptosis and potentiates the anti-tumour activity of chemotherapy in rituximab-resistant lymphoma. Br. J. Haematol 162, 657–669 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dasmahapatra G et al. The pan-HDAC inhibitor vorinostat potentiates the activity of the proteasome inhibitor carfilzomib in human DLBCL cells in vitro and in vivo. Blood 115, 4478–4487 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 91.Dasmahapatra G et al. The irreversible proteasome inhibitor carfilzomib interacts synergistically with the selective HDAC6 inhibitor ACY1215 in ABC- and GC-DLBCL and mantle cell lymphoma sensitive or resistant to bortezomib [abstract]. Blood 120, a2765 (2012). [Google Scholar]
- 92.Dasmahapatra G et al. Obatoclax interacts synergistically with the irreversible proteasome inhibitor carfilzomib in GC- and ABC-DLBCL cells in vitro and in vivo. Mol. Cancer Ther 11, 1122–1132 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 93.US National Library of Medicine. ClinicalTrials.gov [online], http://www.clinicaltrials.gov/ct2/show/NCT01276717 (2013). [DOI] [PubMed] [Google Scholar]
- 94.Tageja N Lenalidomide - current understanding of mechanistic properties. Anticancer Agents Med. Chem 11, 315–326 (2011). [DOI] [PubMed] [Google Scholar]
- 95.Yang Y et al. Exploiting synthetic lethality for the therapy of ABC diffuse large B cell lymphoma. Cancer Cell 21, 723–737 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhang LH et al. Lenalidomide efficacy in activated B-cell-like subtype diffuse large B-cell lymphoma is dependent upon IRF4 and cereblon expression. Br. J. Haematol 160, 487–502 (2012). [DOI] [PubMed] [Google Scholar]
- 97.Hernandez-Ilizaliturri FJ et al. Higher response to lenalidomide in relapsed/refractory diffuse large B-cell lymphoma in nongerminal center B-cell-like than in germinal center B-cell-like phenotype. Cancer 117, 5058–5066 (2011). [DOI] [PubMed] [Google Scholar]
- 98.Dal Porto JM et al. B cell antigen receptor signaling 101. Mol. Immunol 41, 599–613 (2004). [DOI] [PubMed] [Google Scholar]
- 99.Srinivasan L et al. PI3 kinase signals BCR-dependent mature B cell survival. Cell 139, 573–586 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gauld SB, Dal Porto JM & Cambier JC B cell antigen receptor signaling: roles in cell development and disease. Science 296, 1641–1642 (2002). [DOI] [PubMed] [Google Scholar]
- 101.Young RM & Staudt LM Targeting pathological B cell receptor signalling in lymphoid malignancies. Nat. Rev. Drug Discov 12, 229–243 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Winer ES, Ingham RR & Castillo JJ PCI-32765: a novel Bruton’s tyrosine kinase inhibitor for the treatment of lymphoid malignancies. Expert Opin. Investig. Drugs 21, 355–361 (2012). [DOI] [PubMed] [Google Scholar]
- 103.Honigberg LA et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc. Natl Acad. Sci. USA 107, 13075–13080 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yang Y et al. Exploiting synthetic lethality for the therapy of ABC diffuse large B cell lymphoma. Cancer Cell 21, 723–737 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Advani RH et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J. Clin. Oncol 31, 88–94 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wilson WH et al. The Bruton’s tyrosine kinase (BTK) inhibitor, ibrutinib (PCI-32765), has preferential activity in the ABC subtype of relapsed/refractory de novo diffuse large B-cell lymphoma (DLBCL): interim results of a multicenter, open-label, phase 2 study [abstract]. Blood 120, a686 (2012). [Google Scholar]
- 107.Younes A et al. Phase Ib study combining ibrutinib with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) in patients with CD20-positive B-cell non-Hodgkin lymphoma (NHL) [abstract]. J. Clin. Oncol 31, a8502 (2013). [Google Scholar]
- 108.US National Library of Medicine. ClinicalTrials.gov [online], http://www.clinicaltrials.gov/ct2/show/NCT01855750 (2013). [DOI] [PubMed] [Google Scholar]
- 109.Saijo K et al. Protein kinase C β controls nuclear factor κB activation in B cells through selective regulation of the IkB kinase α. J. Exp. Med 195, 1647–1652 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kim SW et al. Protein kinase C-associated kinase is required for NF-κB signaling and survival in diffuse large B-cell lymphoma cells. Blood 111, 1644–1653 (2008). [DOI] [PubMed] [Google Scholar]
- 111.Robertson MJ et al. Phase II study of enzastaurin, a protein kinase C β inhibitor, in patients with relapsed or refractory diffuse large B-cell lymphoma. J. Clin. Oncol 25, 1741–1746 (2007). [DOI] [PubMed] [Google Scholar]
- 112.Hainsworth JD et al. Randomized phase II study of R-CHOP plus enzastaurin versus R-CHOP in the first line treatment of patients with intermediate and high-risk diffuse large B-cell lymphoma (DLBCL)—preliminary analysis [abstract]. Ann. Oncol 22 (Suppl. 4), a074 (2011). [Google Scholar]
- 113.Naylor TL et al. Protein kinase C inhibitor sotrastaurin selectively inhibits the growth of CD79 mutant diffuse large B-cell lymphomas. Cancer Res. 71, 2643–2653 (2011). [DOI] [PubMed] [Google Scholar]
- 114.US National Library of Medicine. ClinicalTrials.gov [online], http://www.clinicaltrials.gov/ct2/show/NCT01402440 (2013). [DOI] [PubMed] [Google Scholar]
- 115.Ferch U et al. Inhibition of MALT1 protease activity is selectively toxic for activated B cell-like diffuse large B cell lymphoma cells. J. Exp. Med 206, 2313–2320 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hailfinger S et al. Essential role of MALT1 protease activity in activated B cell-like diffuse large B-cell lymphoma. Proc. Natl Acad. Sci. USA 106, 19946–19951 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.McAllister-Lucas LM, Baens M & Lucas PC MALT1 protease: a new therapeutic target in B lymphoma and beyond? Clin. Cancer Res 17, 6623–6631 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Fontan L et al. MALT1 small molecule inhibitors specifically suppress ABC-DLBCL in vitro and in vivo. Cancer Cell 22, 812–824 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Nagel D et al. Pharmacologic inhibition of MALT1 protease by phenothiazines as a therapeutic approach for the treatment of aggressive ABC-DLBCL. Cancer Cell 22, 825–837 (2012). [DOI] [PubMed] [Google Scholar]
- 120.Young RM & Staudt LM A new “brew” of MALT1 inhibitors. Cancer Cell 22, 706–707 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lam LT et al. Cooperative signaling through the signal transducer and activator of transcription 3 and nuclear factor-κB pathways in subtypes of diffuse large B-cell lymphoma. Blood 111, 3701–3713 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ding BB et al. Constitutively activated STAT3 promotes cell proliferation and survival in the activated B-cell subtype of diffuse large B-cell lymphomas. Blood 111, 1515–1523 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Scuto A et al. STAT3 inhibition is a therapeutic strategy for ABC-like diffuse large B-cell lymphoma. Cancer Res. 71, 3182–3188 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Mascarenhas J & Hoffman R Ruxolitinib: the first FDA approved therapy for the treatment of myelofibrosis. Clin. Cancer Res 18, 3008–3014 (2012). [DOI] [PubMed] [Google Scholar]
- 125.US National Library of Medicine. ClinicalTrials.gov [online], http://www.clinicaltrials.gov/ct2/show/NCT01431209 (2013). [DOI] [PubMed] [Google Scholar]
- 126.Younes A et al. Phase I study of a novel oral Janus kinase 2 inhibitor, SB1518, in patients with relapsed lymphoma: evidence of clinical and biologic activity in multiple lymphoma subtypes. J. Clin. Oncol 30, 4161–4167 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Grant C et al. Primary mediastinal large B-cell lymphoma, classic Hodgkin lymphoma presenting in the mediastinum, and mediastinal gray zone lymphoma: what is the oncologist to do? Curr. Hematol. Malig. Rep 6, 157–163 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Savage KJ et al. The molecular signature of mediastinal large B-cell lymphoma differs from that of other diffuse large B-cell lymphomas and shares features with classical Hodgkin lymphoma. Blood 102, 3871–3879 (2003). [DOI] [PubMed] [Google Scholar]
- 129.Eberle FC et al. Methylation profiling of mediastinal gray zone lymphoma reveals a distinctive signature with elements shared by classical Hodgkin’s lymphoma and primary mediastinal large B-cell lymphoma. Haematologica 96, 558–566 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Isaacson PG, Norton AJ & Addis BJ The human thymus contains a novel population of B lymphocytes. Lancet 2, 1488–1491 (1987). [DOI] [PubMed] [Google Scholar]
- 131.Steidl C & Gascoyne RD The molecular pathogenesis of primary mediastinal large B-cell lymphoma. Blood 118, 2659–2669 (2011). [DOI] [PubMed] [Google Scholar]
- 132.Lenz G & Staudt LM Aggressive lymphomas. N. Engl. J. Med 362, 1417–1429 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Feuerhake F et al. NFκB activity, function, and target-gene signatures in primary mediastinal large B-cell lymphoma and diffuse large B-cell lymphoma subtypes. Blood 106, 1392–1399 (2005). [DOI] [PubMed] [Google Scholar]
- 134.Guiter C et al. Constitutive STAT6 activation in primary mediastinal large B-cell lymphoma. Blood 104, 543–549 (2004). [DOI] [PubMed] [Google Scholar]
- 135.Joos S et al. Primary mediastinal (thymic) B-cell lymphoma is characterized by gains of chromosomal material including 9p and amplification of the REL gene. Blood 87, 1571–1578 (1996). [PubMed] [Google Scholar]
- 136.Green MR et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood 116, 3268–3277 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Rui L et al. Cooperative epigenetic modulation by cancer amplicon genes. Cancer Cell 18, 590–605 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Keir ME, Butte MJ, Freeman GJ & Sharpe AH PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol 26, 677–704 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Iwai Y et al. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl Acad. Sci. USA 99, 12293–12297 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Chen BJ et al. PD-L1 expression is characteristic of a subset of aggressive B-cell lymphomas and virus-associated malignancies. Clin. Cancer Res 19, 3462–3473 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Steidl C et al. MHC class II transactivator CIITA is a recurrent gene fusion partner in lymphoid cancers. Nature 471, 377–381 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Berger R et al. Phase I safety and pharmacokinetic study of CT-011, a humanized antibody interacting with PD-1, in patients with advanced hematologic malignancies. Clin. Cancer Res 14, 3044–3051 (2008). [DOI] [PubMed] [Google Scholar]
- 143.Delmore JE et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 146, 904–917 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Rounbehler RJ et al. Tristetraprolin impairs myc-induced lymphoma and abolishes the malignant state. Cell 150, 563–574 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lim K-H, Romero DL, Chaudhary D, Robinson SD & Staudt LM IRAK4 kinase as a novel therapeutic target in the ABC subtype of diffuse large B cell lymphoma [abstract]. Blood 120, a62 (2012). [Google Scholar]
- 146.Walsh K et al. PAK1 mediates resistance to PI3K inhibition in lymphomas. Clin. Cancer Res 19, 1106–1115 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Han SS et al. NF-κB/STAT3/PI3K signaling crosstalk in iMyc E mu B lymphoma. Mol. Cancer 9, 97 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Shortt J et al. Combined inhibition of PI3K-related DNA-damage response kinases and mTORC1 induces apoptosis in MYC-driven B-cell lymphomas. Blood 121, 2964–2974 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kaelin WG Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer 5, 689–698 (2005). [DOI] [PubMed] [Google Scholar]
- 150.Hernandez-Ilizaliturri FJ et al. Higher response to lenalidomide in relapsed/refractory diffuse large B-cell lymphoma in nongerminal center B-cell-like than in germinal center B-cell-like phenotype. Cancer 117, 5058–5066 (2011). [DOI] [PubMed] [Google Scholar]