

Abstract
Histone deacetylases (HDACs) are enzymes that remove acetyl groups from histones and have attracted attention as potential targets for cancer therapy. Several small molecule inhibitors have been developed to target HDACs; however, clinical trials of pan-HDAC inhibitors have found these types of inhibitors to be inefficient and to be relatively highly toxic. In the present study, the role of one HDAC isozyme, HDAC6, in urothelial cancer was investigated. Protein expression levels and subcellular localization of HDAC6 was identified in surgically resected bladder tumors using immunohistochemistry. The antitumor effects of 12 small molecule HDAC6 inhibitors were also examined in vitro using cultured urothelial cancer cells. The HDAC6 inhibitors decreased cell viability, with IC50 values in the low µM range, as low as 2.20 µM. HDACi D, E and F had the lowest IC50 values. HDAC6 has been previously reported to regulate programmed death-ligand 1 (PD-L1) and PD-L1 expression was found to be a predictor of decreased overall survival time. There was no association between the protein expression level of HDAC6 and PD-L1 in tumor tissues; however, HDAC6 inhibition by specific small molecule inhibitors resulted in decreased expression levels of membranous PD-L1 in cultured urothelial cancer cell lines. The results suggested that inhibition of HDAC6 could be a promising novel approach for the treatment of urothelial cancer.
Keywords: HDAC6, small molecule inhibitor, bladder cancer, apoptosis, cell cycle
Introduction
Bladder cancer is the second most common malignancy involving the urinary system (1) following prostate cancer. An estimated 549,393 new cases of bladder cancer were diagnosed in 2018 worldwide, making it the ninth most common type of cancer (2). Among the different types of bladder cancer, urothelial cancer (UC) is the predominant histological subtype in developed countries, accounting for 90% of all cases (3). Since the 1980s, methotrexate, vinblastine, doxorubicin, and cisplatin (M-VAC) have been used as standard chemotherapy treatments for metastatic urothelial cancer (4). Gemcitabine and cisplatin (GC) chemotherapy, which was introduced in the late 1990s (5), does not provide an overall survival advantage over M-VAC (6). These therapies are not curative and the median survival time of patients with metastatic disease was only ~13 months in the 1980s (4) and has not improved in the 1990s (7). Identification of new therapeutic targets is thus warranted for patients with metastatic UC. Histone deacetylase (HDAC) inhibition has been proposed as one alternative strategy for treatment of UC (8).
Histone acetylation and deacetylation are key processes of the epigenetic machinery that regulates gene expression. Increased levels of histone acetylation have been associated with increased transcriptional activity and decreased levels of acetylation have been associated with repression of gene expression (9). HDACs remove acetyl groups from lysine residues on histones, and 18 human HDACs, belonging to various classes of classic HDAC and SIR2 families, have been identified (10,11). HDACs are located in different subcellular compartments, such as the nucleus, cytoplasm, and mitochondria (12), are included in the transcription suppression complex, and recruitment of HDACs suppresses transcription of specific genes in cancer cells, such as p21, Bax and TGF-βRII (12). Thus, HDAC inhibition has been proposed as a strategy for the treatment of cancer (13). Several small molecule HDAC inhibitors have been developed and are generally classified as hydroxamates, benzamides, tetrapeptides/depsipeptides, or sirtuin inhibitors (14–16). Hematological malignancies, such as lymphoma, multiple myeloma, myelodysplastic syndrome and acute leukemia, have shown promising responses to HDAC inhibitors and 4 HDAC inhibitors (vorinostat, panobinostat, romidepsin and belinostat) have been approved by the United States Food and Drug Administration for the treatment of hematological malignancies (17,18). A high number of clinical trials, using structurally dissimilar pan-HDAC inhibitors, in solid tumors, as single agents or in combinations, are underway (13); however, these inhibitors have been ineffective. Furthermore, pan-HDAC inhibitors have been found to exhibit several significant dose-limiting toxicities (DLT) in clinical trials compared with that in other epigenetic agents, such as inhibitors of DNA methyltransferases (15,19). In particular, HDAC inhibitors have been associated with serious cardiotoxicities (19,20), as well as other common high-grade adverse events, such as hematological and gastrointestinal toxicities and fatigue/asthenia (15,19). Application of selective HDAC inhibitors might reduce the toxicity associated with broad inhibition of multiple HDAC family members and unwanted off-target effects of pan-HDAC inhibitors.
Class IIB HDACs, which include HDAC6 and HDAC10, are predominantly localized in the cytoplasm, controlling non-histone acetylation, but can also be found in the nucleus (15,21). HDAC6 is the largest of the HDACs and its structure differs from that of other members of the family, suggesting its early separation from other HDACs (21). It harbors dual deacetylase domains and a C-terminal ubiquitin-binding motif (21). HDAC6 has multiple nuclear and cytoplasmic targets and is involved in various physiological and disease processes, such as antigen presentation, cell migration and cell survival (22). HDAC6 also regulates the stability of various proteins, including α-tubulin, cortactin, HSP90, β-catenin and survivin, via the ubiquitin/proteasome system (23). It controls numerous important biological processes, including cell migration and regulation of the cytoskeleton through deacetylation of α-tubulin (24), cell-cell interactions and degradation of misfolded proteins (25). It has been reported that HDAC6 promotes the migration and invasion of bladder cancer cells by targeting the cytoskeletal protein, cortactin (26). In addition, HDAC6 supports progression through the cell cycle by interacting with Aurora kinases (27) and is involved in oncogenesis, by controlling endocytosis of oncogenic receptors, such as the epidermal growth factor receptor (28). It has also been reported that inhibition or depletion of HDAC6 suppresses PD-L1 expression via STAT3 (29). Thus, HDAC6 inhibition might sensitize tumor cells to the host immune response and several selective HDAC6 inhibitors have been synthesized (10).
In published expression array data (30), HDAC6 expression was either upregulated or unchanged in bladder cancer compared with that in benign controls. Furthermore, mRNA and protein HDAC6 expression has been confirmed in eight cultured UC cell lines using reverse transcription-quantitative PCR and western blot analysis (30). The expression of HDAC6 mRNA varies in different types of cancer tissues, and it remains unclear whether HDAC6 levels are elevated in UC and whether they are associated with clinicopathological characteristics and survival. In the present study, the protein expression level of HDAC6 in cultured UC cell lines was determined using western blot analysis and in bladder cancer specimens using immunohistochemistry (IHC). UC cells were treated with a panel of 12 small molecule HDAC6 inhibitors and a dose-dependent decrease in cancer cell proliferation was found using MTS assay with IC50 in the low µM range. Therefore, HDAC6 inhibition, using small molecule inhibitors could be a novel promising strategy for UC treatment.
Materials and methods
Patients and IHC
The present study was approved by the Ethical Committee of Niigata University (Institutional review board no. 2169) and consent to participate in the study was obtained using the opt-out method. A total of 97 UC surgical specimens and paired normal urothelium tissues (≥1 cm from the tumor) from 80 sequential patients (43 patients with solitary tumors and 37 patients with multiple tumors, from which a total of 54 tumors were used) who underwent transurethral resection of bladder tumor for BT at the Niigata University Hospital between January 2013 and March 2015 were included in the present study. A total of 59 patients had primary tumors and 38 patients had recurrent tumors. None of the patients had lymph node or distant metastasis (negative M status). The maximum follow-up period was 70 months (median, 46 months). Follow up data is missing for some patients who dropped out from follow-up and therefore the number of patients in the survival analysis is lower than that of total patients. Clinical characteristics are presented in Table I. The tumors were fixed in 10% buffered formalin and embedded in paraffin, and ID numbers were allocated to the samples for anonymity. Paraffin sections were routinely stained with hematoxylin and eosin, and pathological diagnoses of UC were made, according to the Union for International Cancer Control (UICC) TNM classification of malignant tumors (31), and pathological grades were assigned according to a system developed in 1997 by the Japanese Urological Association in 1997 on the basis of UICC criteria (32). The tissue microarrays, BL241a (BTs; 24 cores from 10 cases) and KD483 (48 cores from 48 cases; 13 cases were upper urinary tract UC) were purchased from US Biomax, Inc.
Table I.
Analysis of HDAC6 staining patterns with various clinicopathological characteristics and PD-L1 status.
| HDAC6 expression | |||||
|---|---|---|---|---|---|
| Clinicopathological characteristic | Total | Nuclear | Nuc+cyt | Cytoplasmic | Negative |
| Total (%) | 97 | 5 (5.2) | 36 (37.1) | 54 (55.7) | 2 (2.1) |
| T stage, n (%) | |||||
| pTa | 52 | 4 (7.7) | 21 (40.4) | 27 (51.9) | 0 (0.0) |
| pT1 | 25 | 1 (4.0) | 8 (32.0) | 14 (56) | 2 (8.0) |
| >pT2 | 14 | 0 (0.0) | 4 (28.6) | 10 (71.4) | 0 (0.0) |
| Tis | 6 | 0 (0.0) | 3 (50.0) | 3 (50.0) | 0 (0.0) |
| Grade, n (%) | |||||
| Low | 31 | 2 (6.4) | 13 (41.9) | 16 (51.6) | 0 (0.0) |
| High | 66 | 3 (4.6) | 23 (34.8) | 38 (57.6) | 2 (3.0) |
| PD-L1, n (%) | |||||
| Negative | 54 | 4 (7.4) | 18 (33.3) | 31 (57.4) | 1 (1.9) |
| Positive | 43 | 1 (2.3) | 18 (41.9) | 23 (53.5) | 1 (2.3) |
Nuc, nuclear; cyt, cytoplasmic; PD-L1, programmed death-ligand 1; HDAC, histone deacetylase.
The HDAC6 rabbit monoclonal antibody (D2E5; dilution 1:1,000) and the PD-L1 rabbit monoclonal antibody (E1L3N; dilution 1:200) (both Cell Signaling Technology, Inc.) were used for IHC analyses. IHC staining was performed as described previously (33). For each stain, two, 5-µm thick paraffin sections from different parts of each tumor, representative of the entire tumor, were mounted on silanized glass slides (Dako; Agilent Technologies, Inc.). After deparaffination and re-hydration, epitopes were reactivated by autoclaving sections in 10 mM citric buffer (pH 6.0) for 10 min. The slides were incubated with the primary antibodies for 1 h, at ambient temperature in a moist chamber. After washing with PBS, bound antibody was detected using the peroxidase method and the Histofine® simple stain MAX-PO (MULTI; Nichirei Corporation). The staining reaction was developed using 3,3′-diaminobenzidine in the presence of hydrogen peroxide for 10 min at room temperature. Nuclear counterstaining was performed using hematoxylin for 10 min at room temperature. Positive and negative controls were included in each staining series. Normal kidney and normal human placenta tissues served as positive controls for HDAC6 and PD-L1, respectively. The results were observed using an Olympus BX50 microscope equipped with an Olympus DP20 digital microscope camera. All slides were evaluated for immunostaining in a blinded manner. There were no inter- or intra-sample fluctuations in staining intensity. IHC evaluation was performed by two medical doctors trained in pathology blinded to the clinical data, with complete observer agreement. HDAC-6 immunoreactivity was scored according to the percentage of positive tumor cells (0, negative; 1, 0–4; 2, 5–24; 2, 25–49; 3, 50–100% of cells positive), and intensity (0, negative; 1, mild; 2, intermediate; 3, intense) (34). The HDAC IHC scores were calculated by multiplying intensity and percentage scores. The expression level of HDAC-6 was classified as low if the total score was <2 and high if the total score was ≥3. PD-L1 immunostaining was reported as positive if >1% of all tumor cells presented with any membrane PD-L1 staining (35). Cytoplasmic staining and inflammatory cell staining were not scored.
Cell culture and reagents
The established UC cell lines T24, HT1376, and RT4 were obtained from the American Type Culture Collection and were cultured as previously described (28). A panel of 12 HDAC6 inhibitors (Table II) was obtained from the Department of Medicinal Chemistry and Pharmacognosy, University of Illinois at Chicago. The cells were treated with the inhibitors at concentrations of 1, 5, 10 and 20 µM for 24, 48, 72 or 96 h at 37°C.
Table II.
Names, structure and IC50 values of the HDAC6 inhibitors in the 3 urothelial cancer cell lines.
| IC50 value | |||||
|---|---|---|---|---|---|
| Compound ID | Structure | T24 | HT1376 | RT4 | (Refs.) |
| Tubastatin A | 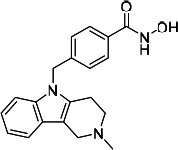 |
8.91 | 17.44 | 39.34 | commercial |
| Nexturastat | 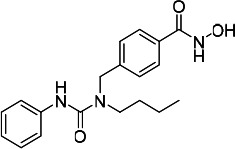 |
13.40 | 17.96 | 11.04 | commercial |
| HDACi A | 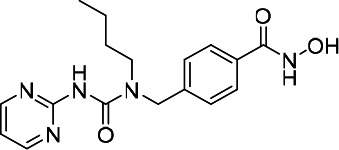 |
49.05 | Not convergeda | 109.10 | (46) |
| HDACi B | 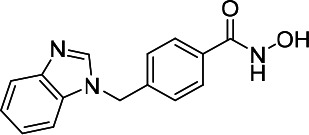 |
16.20 | 16.70 | 33.60 | (47) |
| HDACi C | 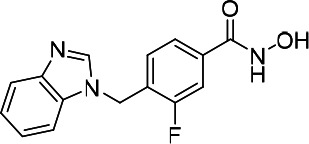 |
11.98 | 18.49 | 28.82 | (47) |
| HDACi D | 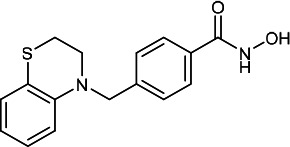 |
7.40 | 29.50 | 11.50 | (47,48) |
| HDACi E | 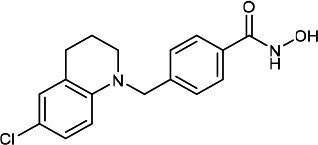 |
3.60 | 4.10 | 17.60 | (49,50) |
| HDACi F | 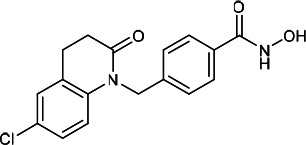 |
2.20 | 3.10 | 4.90 | (48) |
| HDACi G | 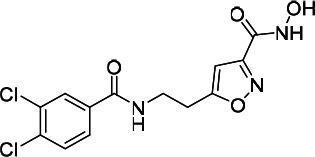 |
45.22 | 68.79 | 120.90 | (51) |
| HDACi H | 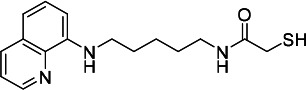 |
50.71 | 39.33 | 371.20 | (52) |
| HDACi I | 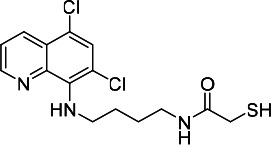 |
12.73 | 13.76 | 308.60 | (53) |
| HDACi J | 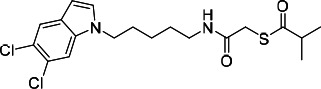 |
18.71 | 19.50 | 51.42 | (53) |
IC50 was not reached at the maximum tested dose. HDACi, histone deacetylase inhibitor.
Measurement of cell viability
Cell viability was measured with a colorimetric CellTiter 96® AQueous one solution cell proliferation assay (Promega Corporation) according to the manufacturer's instructions. Experiments were performed in three replicates using a flat-bottom 96-well plate (Corning, Inc.). Absorbance was measured at 490 nm. The graphs are presented as the mean ± SD. IC50, i.e. the drug concentration that inhibits the growth of cancer cells by 50%, was calculated using GraphPad Prism v7 (GraphPad Software Inc.).
Western blot analysis
Western blot analysis was performed as previously described (36). A HDAC6 (D2E5; cat. no. 7558; dilution 1:1,000) rabbit monoclonal antibody and a goat anti-rabbit HRP-labeled secondary antibody (cat. no. 7074; dilution 1:10,000) (both from Cell Signaling Technology, Inc.) were detected using a SuperSignal west pico substrate (Pierce; Thermo Fisher Scientific, Inc.) according to the manufacturer's instructions. A β-actin (13E5) rabbit monoclonal antibody (dilution 1:1,000; cat. no. 4970; Cell Signaling Technology, Inc.) was used as the loading control. Images were analyzed using AN UN-SCAN-IT gel automated digitizing system software (v5.1; Silk Scientific Inc.).
Flow cytometry
Cells were cultured in medium and then harvested by trypsinization. T24 cells were treated with HDACi F (5 and 10 µM) for 72 h. Protein expression of PD-L1 was detected using PD-L1 (E1L3N; cat. no. 14772) rabbit monoclonal antibody conjugated to Alexa Fluor 488 (Cell Signaling Technology, Inc.). The analysis was performed using a BD Accuri C6 flow cytometer system, equipped with a 488 nm argon laser light source and a 515 nm bandpass filter (FL1; Becton, Dickinson and Company) and analyzed using the C6 software (v1.0.264.21; Becton, Dickinson and Company).
Statistical analysis
The absolute and percentage values are presented as categorical variables. Fisher's exact test (including Fisher's exact test for larger tables) was used to determine the association between IHC staining and pathological or clinical characteristics, and between HDAC6 expression and PD-L1 staining. Survival curves were constructed using the Kaplan-Meier method and the differences between the curves were compared using the log-rank test. All tests were two-sided and P<0.05 were considered to indicate a statistically significant difference. Statistical analyses were performed using the Prism software package for Windows (GraphPad Software, Inc.).
Results
Expression and subcellular localization of HDAC6 in bladder cancer
In the tissue microarrays, HDAC6 was detected in the cytoplasm of high-grade bladder cancer, while low-grade bladder cancer and upper urinary tract UC did not stain positive for HDAC6 (data not shown). A subsequent IHC staining experiment, using a larger number of surgically resected UC tissues detected HDAC6 in the cytoplasm of normal urothelium (Fig. 1A) and in the nuclei and cytoplasm of UC (Fig. 1C and D). A total of 54 tumors had pure cytoplasmic staining without nuclear staining, 5 had only nuclear staining, and the remaining 36 had various combinations of nuclear and cytoplasmic staining (Fig. 1B-D and Table I). Nuclear staining was only observed in stages Ta and Ta; however, invasive tumors and Tis presented with either a combination of nuclear and cytoplasmic staining or cytoplasmic staining (Table I); however, this was not statistically significant. HDAC6 subcellular localization was not a predictor of overall or recurrence-free survival times in patients with UC (Fig. 2A and B). The expression of HDAC6 was classified as low or high based on the product of the intensity and percentage of positive tumor scores. Staining for HDAC6 was either low (Fig. 1B) or high (Fig. 1C and D). High HDAC6 expression was detected in 84 (86.6%) specimens (Fig. 1B and Table III) and was observed more frequently in multiple tumors compared with that in solitary ones (94.3 vs. 79.0%), as well as in all Tis samples; however, the difference was not statistically significant. Overall, HDAC6 expression was not associated with any pathological or clinical characteristics. There was no association between HDAC6 expression level and overall or recurrence-free survival (Fig. 2C and D).
Figure 1.

Immunohistochemical analysis of HDAC6 and PD-L1 in normal urothelium and UC. (A) HDAC 6 was detected in the cytoplasm of normal urothelium. Staining for HDAC6 was either (B) low or (C and D) high and was detected in the (C) cytoplasm or (D) nucleus and cytoplasm of UC. Membranous staining of PD-L1 was not detected in (E) normal urothelium; however, it was detected in (F) UC cells (indicated by the arrowheads). UC, urothelial cancer; PD-L1, programmed death-ligand 1; HDAC, histone deacetylase.
Figure 2.

Overall and recurrence-free survival times of patients categorized by (A and B) subcellular localization of HDAC6, (C and D) level of HDAC6 expression, and (E and F) positive and negative PD-L1 staining. Follow up data is missing for some patients who dropped out from follow up and therefore the number of patients in the survival analysis is lower than that of total patients. PD-L1, programmed death-ligand 1; HDAC, histone deacetylase.
Table III.
Immunohistochemical staining of HDAC6 and PD-L1, and the clinicopathological characteristics of the patients with urothelial cancer.
| HDAC6 | PD-L1 | ||||
|---|---|---|---|---|---|
| Clinicopathological characteristic | Value | Low | High | Negative | Positive |
| Total number | 97 tumors | 13 (13.4) | 84 (86.6) | 55 (56.7) | 42 (43.3) |
| Mean age ± SD, years | 75.48±8.79 | 75.46±8.79 | 75.49±8.84 | 76.65±8.75 | 75.26±8.94 |
| Sex, n (%) | |||||
| Male | 61 | 3 (15.8) | 54 (88.5) | 39 (64) | 22 (36.0) |
| Female | 19 | 7 (13.5) | 16 (84.2) | 10 (52.6) | 9 (47.4) |
| Number of tumors, n (%) | |||||
| Solitary | 43 | 9 (21.0) | 34 (79.0) | 24 (55.8) | 19 (44.1) |
| Multiple | 54 | 4 (7.5) | 50 (94.3) | 31 (58.4) | 23 (43.4) |
| Primary or recurrent, n (%) | |||||
| Primary | 59 | 7 (11.9) | 52 (88.1) | 34 (57.6) | 25 (42.4) |
| Recurrent | 38 | 6 (15.8) | 32 (84.2) | 18 (47.4) | 20 (52.6) |
| Tumor size, cm (%) | |||||
| <3 | 79 | 11 (14) | 68 (86) | 34 (43.0) | 45 (56.9) |
| >3 | 18 | 2 (11.2) | 16 (88.8) | 10 (55.6) | 8 (44.4) |
| Grade, n (%) | |||||
| Low | 31 | 4 (13) | 27 (87) | 18 (58.0) | 13 (41.9) |
| High | 66 | 9 (13.6) | 57 (86.4) | 37 (56.0) | 29 (43.9) |
| T stage, n (%) | |||||
| pTa | 52 | 7 (13.5) | 45 (86.5) | 32 (61.5) | 20 (38.5) |
| pT1 | 25 | 4 (19.3) | 21 (80.7) | 13 (53.8) | 12 (46.2) |
| >pT2 | 14 | 2 (14.3) | 12 (85.7) | 5 (35.7) | 9 (64.3) |
| Tis | 6 | 0 | 6 (100) | 5 (83.3) | 1 (16.7) |
PD-L1, programmed death-ligand 1; HDAC, histone deacetylase.
PD-L1 IHC staining and its prognostic significance
Normal urothelium was not stained by the anti-PD-L1 antibody (Fig. 1E). Membranous staining of PD-L1 was detected in 42 (43.3%) UC samples (Fig. 1F and Table III). The expression of PD-L1 was not associated with any pathological or clinical characteristics (Table III). There was also no association between HDAC6 expression and PD-L1 (Table I); however, PD-L1 positivity was a predictor of shorter overall survival times (Fig. 2E; P=0.039). In addition, patients with tumors positive for PD-L1 tended to have shorter recurrence-free survival times (Fig. 2F), but the difference was not statistically significant.
Expression of HDAC6 and PD-L1 in cultured cell lines and HDAC6 inhibition of PD-L1
Western blot analysis of HDAC6 protein expression was performed in the T24, HT1376, and RT4 bladder cancer cell lines (Fig. 3A). PD-L1 expression was also detected in the T24 cell line using flow cytometry (Fig. 3B). It has been reported that targeting HDAC6 downregulated the expression of PD-L1 via the STAT3 pathway in cell lines (29). Pharmacological inhibition of HDAC6 by HDACi F in T24 cells downregulated the expression of PD-L1 as detected by flow cytometry (Fig. 3B).
Figure 3.

(A) Protein expression level of HDAC6 was detected in three urothelial cancer cell lines using western blot analysis. A band at ~131 kDa corresponding to HDAC6 was detected. (B) Expression level of PD-L1 was detected using flow cytometry. T24 cells were treated with indicated concentrations of HDACi F, a specific HDAC6 small molecule inhibitor, and then stained with anti-PD-L1 antibody. PD-L1, programmed death-ligand 1; HDAC, histone deacetylase.
HDAC6 inhibitors induce growth inhibition of bladder cancer cell lines
A total of 12 selective HDAC6 inhibitors (Table II) were investigated in the T24, HT1376 and RT4 bladder cancer cell lines. Treatment with the inhibitors (HDACi B, D, E and F) decreased the viability (detected by MTS assay) of bladder cancer cells with the IC50 being as low as 2.20 µM (Figs. 4 and S1; Table II). The median IC50 (13.06) for T24, a high-grade invasive UC, was lower (range, 2.20–50.71) compared with that in HT1376, an intermediate grade cell line (median 17.96; range, 3.10–68.79) and in particular for RT4, a low-grade model cell line (median 36.47; range, 4.90–371.20) (Figs. 4 and S1; Table II).
Figure 4.

Cultured urothelial cancer cell lines were treated with DMSO or indicated doses of HDAC6 small molecule inhibitors for 24, 48, 72 and 96 h. Relative cell viability was measured using a MTS assay and results are presented as OD 490 nm. OD, optical density; HDAC, histone deacetylase.
Discussion
Histone acetylation and deacetylation are key processes of the epigenetic machinery regulating gene expression (9). Increased levels of histone acetylation have been associated with increased transcriptional activity, whereas decreased levels of acetylation have been associated with repression of gene expression (9). HDACs are included in the transcription suppression complex and recruitment of HDACs suppresses the transcription of specific genes in cancer cells. HDACs also regulate multiple nuclear and cytoplasmic proteins and, thus, play an essential role in protein degradation, cell survival and cell migration (21,37). A total of 18 members of the HDAC family have been discovered so far (11,16), among which HDAC6 belongs to the Class II HDACs, which can move in and out of the nucleus. HDAC6 is widely expressed in normal and tumor tissues, and is predominantly localized in the cytoplasm, where it controls non-histone acetylation (21,38) and regulates numerous important biological processes, including cell migration (16,30), while HDAC inhibitors are a new class of anti-cancer drugs, that can induce apoptosis and cell cycle arrest in cancer cells (16,39). The mechanism of HDAC inhibitors remains unknown; however, the effect could be either epigenetic or non-epigenetic (10).
HDACs are potential drug targets for chemotherapy; however, a previous study has shown only modest antineoplastic activity with pan-HDAC inhibitors in UC (30). The reported IC50 of vorinostat, a pan-HDAC inhibitor, in cultured UC cells is up to 6.0 µM, and sensitivity to vorinostat in UC is not associated with the expression levels of HDACs (30). Therefore, inhibition of specific HDAC isoenzymes might be more efficacious and tumor-specific. HDAC6 causes epigenetic alterations by deacetylation of histones but also controls cell metabolism by direct deacetylation of non-histone targets (10,22). Two bands of HDAC6 were detected in RT4 cells, which presumably corresponded to phosphorylated and dephosphorylated HDAC6 (40). To the best of our knowledge, the present study is the first report on the subcellular localization of HDAC6 in clinical UC samples, demonstrating that HDAC6 was predominantly a cytoplasmic protein, which is in agreement that it has non-histone targets (10,22). HDAC6 was previously found to be overexpressed in UC tissues, using reverse transcription-quantitative PCR (41); however, HDAC6 specific inhibitors were found to be active at high µM concentrations in UC cell lines (10–100 µM) (41). A different previous study concluded that targeting HDAC6 might not be effective in UC (8). In the present study, HDAC6 inhibitors reduced proliferation of cultured human UC cell lines at low µM concentrations, as low as 2.20 µM, and that high-grade invasive UC cells were more susceptible to pharmacological HDAC6 inhibition (Figs. 4 and S1; Table II). Thus, we hypothesized that HDAC6-specific inhibitors might be effective in bladder cancer; however, currently there are no biomarkers for HDAC inhibitors, and it has been reported that HDAC6 protein expression levels did not determine sensitivity to its inhibitors (41).
It has been reported that HDAC6 also induces upregulation of several tumor-associated antigens (gp100, MART1, TYRP1 and TYRP2) and major histocompatibility complex class I, suggesting a potential improvement in the immunogenicity of tumor cells (42). Immune checkpoint blockade with anti-CTLA-4, anti-PD-1 and anti-PD-L1 antibodies have changed the paradigm of cancer treatment, and it has been demonstrated that selective HDAC6 inhibition improves the antitumor activity of anti-PD-1 immune checkpoint blockade (42). PD-L1 is an important molecule expressed in cancer cells that activates the inhibitory PD-1 pathway in T-cells (43). HDAC6 is important for cytokine-mediated upregulation of PD-L1 protein expression, which is primarily mediated by the recruitment and activation of STAT3 (29). In the active state, HDAC6 forms a complex with STAT3 and pharmacological inhibition of HDAC6 results in disruption of this complex; this disruption leads to decreased STAT3 phosphorylation but no changes in STAT3 acetylation, as well as, presumably, diminished recruitment of STAT3 to the PD-L1 gene promoter region (29,44). In the present study, the pharmacological HDAC6 inhibition decreased PD-L1 protein expression in cultured T24 UC cells (Fig. 3B).
Experiments using HDAC6 knockout mouse embryonic fibroblasts (MEFs) have demonstrated that some effects of the HDAC6-specific inhibitor, tubacin were independent of HDAC6, whereas the effects of tubastatin A on MEF and in a cultured bladder cancer cell line were HDAC6-dependent (45). Therefore, further studies are required to clarify the effects of specific HDAC6 inhibitors in UC, which could be involved in the modulation of multiple intracellular pathways that are involved in apoptosis, tumor growth, antitumor immune responses and immune surveillance (10,12,16,22,44). The exact mechanism and factors influencing the modulation of PD-L1 by HDAC6 requires further investigation.
In conclusion, the present study showed cytoplasmic expression of HDAC6 in resected UC tumor specimens using IHC staining and investigated the effects of 12 selective HDAC6 small molecule inhibitors in vitro. It was found that pharmacological inhibition of HDAC6 reduced the proliferation of UC cells with some inhibitors (HDACi D, HDACi E and HDACi F) demonstrating low IC50 values. PD-L1 protein expression was also investigated using IHC staining of surgical specimens, and with flow cytometry and western blot analysis in cultured cell lines. The present study is a pilot study and demonstrated the efficacy of selective HDAC6 inhibitors in UC. Specific pharmacological HDAC6 inhibition could be a promising new strategy for the treatment of metastatic UC.
Supplementary Material
Acknowledgements
The authors would like to thank the Department of Medicinal Chemistry and Pharmacognosy (University of Illinois at Chicago), for providing the inhibitors. The authors would also like to thank Dr Irina Gaisina (Department of Medicinal Chemistry and Pharmacognosy, University of Illinois at Chicago) for the fruitful discussion and comments, which improved the manuscript.
Glossary
Abbreviations
- HDACs
histone deacetylases
- PD-L1
programmed death-ligand 1
- IHC
Immunohistochemistry
- UC
urothelial cancer
- BT
bladder tumor
- GC
gemcitabine and cisplatin
- M-VAC
methotrexate, vinblastine, doxorubicin, and cisplatin
Funding
The present study was supported by grants-in-aid from Niigata University (grant nos. CH28015, June 2018 and CH29018, December 2018).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
HK, TA, AK and YS acquired, analyzed and interpreted the patient data. HK drafted the manuscript and revised it critically for important intellectual content. TA, AK and YS participated in fruitful discussion on the manuscript. VB and YT made substantial contributions to conception and design. VB analyzed and interpreted the patient data, and drafted and revised the manuscript for important intellectual content. YT revised the manuscript for important intellectual content and gave final approval of the version to be published. All authors read and approved the final manuscript.
Ethics approval and consent to participate
This study was approved by the Human Research Ethics Committee of School of Medicine, Niigata University (Niigata, Japan). All the patients provided written informed consent according to the Declaration of Helsinki, and all experimental methods were conducted according to relevant guidelines.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Ebrahimi H, Amini E, Pishgar F, Moghaddam SS, Nabavizadeh B, Rostamabadi Y, Aminorroaya A, Fitzmaurice C, Farzadfar F, Nowroozi MR, et al. Global, regional and national burden of bladder cancer, 1990 to 2016: Results from the GBD Study 2016. J Urol. 2019;201:893–901. doi: 10.1097/JU.0000000000000025. [DOI] [PubMed] [Google Scholar]
- 2.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 3.Droller MJ. Bladder cancer: Current diagnosis and treatment (Current Clinical Urology) Humana. 2010 [Google Scholar]
- 4.Sternberg CN, Yagoda A, Scher HI, Watson RC, Herr HW, Morse MJ, Sogani PC, Vaughan ED, Jr, Bander N, Weiselberg LR, et al. M-Vac (methotrexate, vinblastine, doxorubicin and cisplatin) for advanced transitional cell carcinoma of the urothelium. J Urol. 1988;139:461–469. doi: 10.1016/S0022-5347(17)42494-3. [DOI] [PubMed] [Google Scholar]
- 5.Moore MJ, Winquist EW, Murray N, Tannock IF, Huan S, Bennett K, Walsh W, Seymour L. Gemcitabine plus cisplatin, an active regimen in advanced urothelial cancer: A phase II trial of the National cancer institute of Canada clinical trials group. J Clin Oncol. 1999;17:2876–2881. doi: 10.1200/JCO.1999.17.9.2876. [DOI] [PubMed] [Google Scholar]
- 6.Seront E, Machiels JP. Molecular biology and targeted therapies for urothelial carcinoma. Cancer Treat Rev. 2015;41:341–353. doi: 10.1016/j.ctrv.2015.03.004. [DOI] [PubMed] [Google Scholar]
- 7.Scher HI, Geller NL, Curley T, Tao Y. Effect of relative cumulative dose-intensity on survival of patients with urothelial cancer treated with M-VAC. J Clin Oncol. 1993;11:400–407. doi: 10.1200/JCO.1993.11.3.400. [DOI] [PubMed] [Google Scholar]
- 8.Pinkerneil M, Hoffmann MJ, Schulz WA, Niegisch G. HDACs and HDAC inhibitors in urothelial carcinoma-perspectives for an antineoplastic treatment. Curr Med Chem. 2017;24:4151–4165. doi: 10.2174/0929867324666170207142740. [DOI] [PubMed] [Google Scholar]
- 9.Jaenisch R, Bird A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(Suppl):S245–S254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 10.Gaisina IN, Tueckmantel W, Ugolkov A, Shen S, Hoffen J, Dubrovskyi O, Mazar A, Schoon RA, Billadeau D, Kozikowski AP. Identification of HDAC6-selective inhibitors of low cancer cell cytotoxicity. Chem Med Chem. 2016;11:81–92. doi: 10.1002/cmdc.201500456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Renaud JP. Wiley; 2020. Structural biology in drug discovery: Methods, Techniques, and Practices. [DOI] [Google Scholar]
- 12.Glozak MA, Seto E. Histone deacetylases and cancer. Oncogene. 2007;26:5420–5432. doi: 10.1038/sj.onc.1210610. [DOI] [PubMed] [Google Scholar]
- 13.Halsall JA, Turner BM. Histone deacetylase inhibitors for cancer therapy: An evolutionarily ancient resistance response may explain their limited success. Bioessays. 2016;38:1102–1110. doi: 10.1002/bies.201600070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Benedetti R, Conte M, Altucci L. Targeting histone deacetylases in diseases: Where are we? Antioxid Redox Signal. 2015;23:99–126. doi: 10.1089/ars.2013.5776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Subramanian S, Bates SE, Wright JJ, Espinoza-Delgado I, Piekarz RL. Clinical toxicities of histone deacetylase inhibitors. Pharmaceuticals (Basel) 2010;3:2751–2767. doi: 10.3390/ph3092751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang J, Zhong Q. Histone deacetylase inhibitors and cell death. Cell Mol Life Sci. 2014;71:3885–3901. doi: 10.1007/s00018-014-1656-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.https://chemoth.com/
- 18.Rana Z, Diermeier S, Hanif M, Rosengren RJ. Understanding failure and improving treatment using HDAC inhibitors for prostate cancer. Biomedicines. 2020;8:22. doi: 10.3390/biomedicines8020022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cavenagh JD, Popat R. Optimal management of histone deacetylase inhibitor-related adverse events in patients with multiple myeloma: A focus on panobinostat. Clin Lymphoma Myeloma Leuk. 2018;18:501–507. doi: 10.1016/j.clml.2018.05.007. [DOI] [PubMed] [Google Scholar]
- 20.Shah MH, Binkley P, Chan K, Xiao J, Arbogast D, Collamore M, Farra Y, Young D, Grever M. Cardiotoxicity of histone deacetylase inhibitor depsipeptide in patients with metastatic neuroendocrine tumors. Clin Cancer Res. 2006;12:3997–4003. doi: 10.1158/1078-0432.CCR-05-2689. [DOI] [PubMed] [Google Scholar]
- 21.de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem J. 2003;370:737–749. doi: 10.1042/bj20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Y, Shin D, Kwon SH. Histone deacetylase 6 plays a role as a distinct regulator of diverse cellular processes. FEBS J. 2013;280:775–793. doi: 10.1111/febs.12079. [DOI] [PubMed] [Google Scholar]
- 23.Pernet L, Faure V, Gilquin B, Dufour-Guérin S, Khochbin S, Vourc'h C. HDAC6-ubiquitin interaction controls the duration of HSF1 activation after heat shock. Mol Biol Cell. 2014;25:4187–4194. doi: 10.1091/mbc.e14-06-1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boyault C, Sadoul K, Pabion M, Khochbin S. HDAC6, at the crossroads between cytoskeleton and cell signaling by acetylation and ubiquitination. Oncogene. 2007;26:5468–5476. doi: 10.1038/sj.onc.1210614. [DOI] [PubMed] [Google Scholar]
- 25.Valenzuela-Fernández A, Cabrero JR, Serrador JM, Sánchez-Madrid F. HDAC6: A key regulator of cytoskeleton, cell migration and cell-cell interactions. Trends Cell Biol. 2008;18:291–297. doi: 10.1016/j.tcb.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 26.Zuo Q, Wu W, Li X, Zhao L, Chen W. HDAC6 and SIRT2 promote bladder cancer cell migration and invasion by targeting cortactin. Oncol Rep. 2012;27:819–824. doi: 10.3892/or.2011.1553. [DOI] [PubMed] [Google Scholar]
- 27.Cha TL, Chuang MJ, Wu ST, Sun GH, Chang SY, Yu DS, Huang SM, Huan SK, Cheng TC, Chen TT, et al. Dual degradation of Aurora A and B kinases by the histone deacetylase inhibitor LBH589 Induces G2-M arrest and apoptosis of renal cancer cells. Clin Cancer Res. 2009;15:840–850. doi: 10.1158/1078-0432.CCR-08-1918. [DOI] [PubMed] [Google Scholar]
- 28.Gao YS, Hubbert CC, Yao TP. The microtubule-associated histone deacetylase 6 (HDAC6) regulates epidermal growth factor receptor (EGFR) endocytic trafficking and degradation. J Biol Chem. 2010;285:11219–11226. doi: 10.1074/jbc.M109.042754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lienlaf M, Perez-Villarroel P, Knox T, Pabon M, Sahakian E, Powers J, Woan KV, Lee C, Cheng F, Deng S, et al. Essential role of HDAC6 in the regulation of PD-L1 in melanoma. Mol Oncol. 2016;10:735–750. doi: 10.1016/j.molonc.2015.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Niegisch G, Knievel J, Koch A, Hader C, Fischer U, Albers P, Schulz WA. Changes in histone deacetylase (HDAC) expression patterns and activity of HDAC inhibitors in urothelial cancers. Urol Oncol. 2013;31:1770–1779. doi: 10.1016/j.urolonc.2012.06.015. [DOI] [PubMed] [Google Scholar]
- 31.Brierley JD, Gospodarowicz MK, Wittekind C. 8th edition. John Wiley & Sons, Ltd.; 2017. TNM classification of malignant Tumours. [Google Scholar]
- 32.The Japanese Society of Pathology, Japanese Society of Urology (ed.), corp-author Third edition (in Japanese) Kanahara Publishing; Tokyo, Japan: 2001. General Rule for Clinical and Pathological Studies on Bladder Cancer; p. 102. [Google Scholar]
- 33.Bilim V, Yuuki K, Itoi T, Muto A, Kato T, Nagaoka A, Motoyama T, Tomita Y. Double inhibition of XIAP and Bcl-2 axis is beneficial for retrieving sensitivity of renal cell cancer to apoptosis. Br J Cancer. 2008;98:941–949. doi: 10.1038/sj.bjc.6604268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seo J, Min SK, Park HR, Kim DH, Kwon MJ, Kim LS, Ju YS. Expression of Histone Deacetylases HDAC1, HDAC2, HDAC3, and HDAC6 in invasive ductal carcinomas of the breast. J Breast Cancer. 2014;17:323–331. doi: 10.4048/jbc.2014.17.4.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WE, Poddubskaya E, Antonia S, Pluzanski A, Vokes EE, Holgado E, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med. 2015;373:123–135. doi: 10.1056/NEJMoa1504627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bilim V, Kawasaki T, Katagiri A, Wakatsuki S, Takahashi K, Tomita Y. Altered expression of beta-catenin in renal cell cancer and transitional cell cancer with the absence of beta-catenin gene mutations. Clin Cancer Res. 2000;6:460–466. [PubMed] [Google Scholar]
- 37.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 38.Liu Y, Peng L, Seto E, Huang S, Qiu Y. Modulation of histone deacetylase 6 (HDAC6) nuclear import and tubulin deacetylase activity through acetylation. J Biol Chem. 2012;287:29168–29174. doi: 10.1074/jbc.M112.371120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Finzer P, Kuntzen C, Soto U, zur Hausen H, Rösl F. Inhibitors of histone deacetylase arrest cell cycle and induce apoptosis in cervical carcinoma cells circumventing human papillomavirus oncogene expression. Oncogene. 2001;20:4768–4776. doi: 10.1038/sj.onc.1204652. [DOI] [PubMed] [Google Scholar]
- 40.Du Y, Seibenhener ML, Yan J, Jiang J, Wooten MC. aPKC Phosphorylation of HDAC6 results in increased deacetylation activity. PLoS One. 2015;10:e0123191. doi: 10.1371/journal.pone.0123191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rosik L, Niegisch G, Fischer U, Jung M, Schulz WA, Hoffmann MJ. Limited efficacy of specific HDAC6 inhibition in urothelial cancer cells. Cancer Biol Ther. 2014;15:742–757. doi: 10.4161/cbt.28469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Knox T, Sahakian E, Banik D, Hadley M, Palmer E, Noonepalle S, Kim J, Powers J, Gracia-Hernandez M, Oliveira V, et al. Selective HDAC6 inhibitors improve anti-PD-1 immune checkpoint blockade therapy by decreasing the anti-inflammatory phenotype of macrophages and down-regulation of immunosuppressive proteins in tumor cells. Sci Rep. 2019;9:6136. doi: 10.1038/s41598-019-51403-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Patsoukis N, Wang Q, Strauss L, Boussiotis VA. Revisiting the PD-1 pathway. Sci Adv. 2020;6:eabd2712. doi: 10.1126/sciadv.abd2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Keremu A, Aimaiti A, Liang Z, Zou X. Role of the HDAC6/STAT3 pathway in regulating PD-L1 expression in osteosarcoma cell lines. Cancer Chemother Pharmacol. 2019;83:255–264. doi: 10.1007/s00280-018-3721-6. [DOI] [PubMed] [Google Scholar]
- 45.Ota S, Zhou ZQ, Hurlin PJ. Suppression of FGFR3- and MYC-dependent oncogenesis by tubacin: Association with HDAC6-dependent and independent activities. Oncotarget. 2017;9:3172–3187. doi: 10.18632/oncotarget.22816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tavares MT, Shen S, Knox T, Hadley M, Kutil Z, Bařinka C, Villagra A, Kozikowski AP. Synthesis and pharmacological evaluation of selective histone deacetylase 6 inhibitors in melanoma models. ACS Med Chem Lett. 2017;8:1031–1036. doi: 10.1021/acsmedchemlett.7b00223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shen S, Benoy V, Bergman JA, Kalin JH, Frojuello M, Vistoli G, Haeck W, Van Den Bosch L, Kozikowski AP. Bicyclic-capped histone deacetylase 6 inhibitors with improved activity in a model of axonal charcot-marie-tooth disease. ACS Chem Neurosci. 2016;7:240–258. doi: 10.1021/acschemneuro.5b00286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kozikowski AS, Bergman J. Preparation of tetrahydroquinoline substituted hydroxamic acids as selective histone deacetylase 6 inhibitors (ed.) AP (ed.) 2017 [Google Scholar]
- 49.Gaisina IN, Lee SH, Kaidery NA, Ben Aissa M, Ahuja M, Smirnova NN, Wakade S, Gaisin A, Bourassa MW, Ratan RR, et al. Activation of Nrf2 and hypoxic adaptive response contribute to neuroprotection elicited by phenylhydroxamic acid selective HDAC6 inhibitors. ACS Chem Neurosci. 2018;9:894–900. doi: 10.1021/acschemneuro.7b00435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Osipyants AI, Poloznikov AA, Smirnova NA, Hushpulian DM, Khristichenko AY, Chubar TA, Zakhariants AA, Ahuja M, Gaisina IN, Thomas B, et al. L-ascorbic acid: A true substrate for HIF prolyl hydroxylase? Biochimie. 2018;147:46–54. doi: 10.1016/j.biochi.2017.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shen S, Hadley M, Ustinova K, Pavlicek J, Knox T, Noonepalle S, Tavares MT, Zimprich CA, Zhang G, Robers MB, et al. Discovery of a new Isoxazole-3-hydroxamate-Based histone deacetylase 6 inhibitor SS-208 with antitumor activity in syngeneic melanoma mouse models. J Med Chem. 2019;62:8557–8577. doi: 10.1021/acs.jmedchem.9b00946. [DOI] [PubMed] [Google Scholar]
- 52.Segretti MC, Vallerini GP, Brochier C, Langley B, Wang L, Hancock WW, Kozikowski AP. Thiol-based potent and selective HDAC6 inhibitors promote tubulin acetylation and T-regulatory cell suppressive function. ACS Med Chem Lett. 2015;6:1156–1161. doi: 10.1021/acsmedchemlett.5b00303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lv W, Zhang G, Barinka C, Eubanks JH, Kozikowski AP. Design and synthesis of mercaptoacetamides as potent, selective, and brain permeable histone deacetylase 6 inhibitors. ACS Med Chem Lett. 2017;8:510–515. doi: 10.1021/acsmedchemlett.7b00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.