

Abstract
Protein splicing is an autocatalytic process involving self-excision of an internal protein domain, the intein, and concomitant ligation of the two flanking sequences, the exteins, with a peptide bond. Protein splicing can also take place in trans by naturally split inteins or artificially split inteins, ligating the exteins on two different polypeptide chains into one polypeptide chain. Protein trans-splicing could work in foreign contexts by replacing the native extein sequences with other protein sequences. Protein ligation using protein trans-splicing increasingly becomes a useful tool for biotechnological applications such as semi-synthesis of proteins, segmental isotopic labeling, and in vivo protein engineering. However, only a few split inteins have been successfully applied for protein ligation. Naturally split inteins have been widely used, but they are cross-reactive to each other, limiting their applications to multiple-fragment ligation. Based on the three-dimensional structures including two newly determined intein structures, we derived 21 new split inteins from four highly efficient cis-splicing inteins, in order to develop novel split inteins suitable for protein ligation. We systematically compared trans-splicing of 24 split inteins and tested the cross-activities among them to identify orthogonal split intein fragments that could be used in chemical biology and biotechnological applications.
Introduction
Protein splicing is a post-translational modification in which an intervening fragment of the precursor protein, termed intein, is cleaved off and the two flanking fragments are simultaneously ligated together by forming a peptide bond.1–3 Inteins have increasingly become valuable tools in diverse biotechnological applications such as protein purification and specific modifications because of their unique chemical reaction.4–8 Fragment assembly of split inteins can catalyze ligation of two separate foreign polypeptide chains into one and has emerged as a powerful protein ligation tool.9–12 Naturally split DnaE inteins from cyanobacteria have been widely used in biotechnological applications because they do not require any refolding for protein trans-splicing (PTS) unlike other artificially split inteins.13–17 However, naturally split DnaE inteins are cross-reactive (non-orthogonal) to each other.15,16 The cross-reactivity among split inteins often restricts their applications in multi-fragment ligation by protein trans-splicing because of undesired combinatorial ligation.18–20 It is of special interest to discover novel orthogonal split inteins that could ligate various target proteins by PTS.21 Several protein engineering approaches exploiting in vitro refolding, reaction kinetics, and different split sites have been employed to circumvent the cross-reactivity among split inteins for multiple-fragment ligation.19,22 However, each strategy has some disadvantages. For ‘one pot’ multiple-fragment ligation, it would be desirable to use two or more robust split inteins. Ideal split inteins for such applications should have the following properties: (1) high ligation efficiency (fast ligation kinetics), (2) high tolerance of junction sequences, (3) high specificity of intein fragments (orthogonality), and (4) non-disturbance to fused targets (high solubility of split fragments and/or shorter fragments). To date, a number of functional split inteins have been reported,18,23–29 although none of artificially or naturally split inteins has all of the ideal properties. In this study, guided by the three-dimensional structures, we created novel split inteins from cis-splicing mini-inteins with a goal to develop new robust split inteins bearing all, or at least some, of the ideal properties for protein ligation of foreign peptides. In total, 21 new split inteins were derived from cis-splicing and natural split inteins. We characterized their PTS activities and their cross-reactivity in order to identify potentially useful split inteins for protein ligation by PTS. This report sheds light on the design-strategy for novel split inteins for protein ligation and the evolutionary origin of naturally split inteins.
Results and discussion
Nomenclature of split inteins
Even though a number of split inteins have been previously reported,23–29 there has been no systematic way to name different split inteins, making their comparison difficult. The lengths of inteins vary from 129 to > 1000 residues due to various insertions and deletions. The variation in length was also observed for mini-inteins that lack typical insertions of homing endonuclease domains.30–32 Because of the various sizes, naming of split sites has been arbitrary.23–29 It is of practical importance to define a nomenclature applicable to different split inteins in order to facilitate comparisons between different split inteins derived from inteins with various sizes. Since the ligation of the two flanking sequences invariably takes place between the preceding (the −1 position) and following (the +1 position) residues of an intein, it is practical to name split inteins based on their lengths from either the N or C terminus of inteins, as depicted in Fig. 1. The distance from the N or C terminus is also useful for designing shorter intein fragments, which can be easily fused to other proteins or chemically synthesized. We introduced a simple naming system where, for example, a split site of “N35” indicates that the split site is located after the first 35 residues from the N terminus of the intein (Fig. 1a). Whereas the N-terminal fragment is called “N35”, the remaining C-terminal intein fragment is termed “delta N35 (ΔN35)”, because it lacks the N-terminal 35 residues. N35/ΔN35 indicates the N- and C-terminal split intein fragments, which is split at the N35 site and used as suffixes in subscript, together with the intein name. NpuDnaEN35/ΔN35 thus indicates a pair of the N-terminal fragment (NpuDnaEN35) and the C-terminal fragment (NpuDnaEΔN35), which are derived by splitting at the N35 site of DnaE intein from Nostoc punctiforme (Npu) (Fig. 1a). There are two possible names for one split intein because of the two termini (Fig. 1a and b). When the split site is located closer to the C terminus, we counted the length from the C terminus and indicated the terminus used for counting by “C” instead of “N” (Fig. 1b). It is noteworthy that we counted the methionine start codon for the C-terminal fragment in some of our previous reports because the naturally split DnaE intein always included such first methionine. In this article, however, the number stricdy indicates the length from the terminus excluding the first residue translated from the start codon and we will consistently use this nomenclature.
Fig. 1.

The systematic nomenclature system for split inteins and engineered mini-inteins. (a) Naming of a split intein if the N-terminal split intein fragment is shorter than the other split intein fragment. (b) Naming of a split intein when the split site is closer to the C terminus. The lengths of IntC and IntN are indicated in subscript together with the name of the original intein. (c) Naming of artificially engineered mini-inteins. The number of residues deleted is indicated in superscript with Δ(delta).
Nomenclature of engineered mini-inteins
Intein structures can be divided into two functional entities, i.e. splicing and endonuclease domains. These two domains are functionally independent, as shown by successful deletion of endonuclease domains without affecting protein-splicing activity.24,33–36 Therefore, it is possible to design smaller functional split inteins by eliminating endonuclease domains. The insertion site of endonuclease domains is highly conserved and found in the loop preceding the block F (Fig. S1, ESI†). However, it is not always obvious how to identify boundaries of the endonuclease domain regions without the three-dimensional structures,34–36 because the size of the endonuclease insertion also varies extensively. It is often necessary to optimize deletions in order to obtain functional engineered mini-inteins.34,35 Thus, it would be practically useful to define a general naming system for engineered mini-inteins with various deletions. We decided to differentiate mini-inteins with different lengths using the number of residues deleted from the original intein as a suffix in superscript together with Δ(delta), which indicates the deletion (Fig. 1c). For example, a mini-intein derived from NpuDnaB intein by deleting 283 residues at the endonuclease insertion site will be called here NpuDnaBΔ283 intein (Fig. 1c). This naming convention will be used here together with the split intein naming system.
A model system for testing novel split inteins
For protein-splicing assays we chose the in vivo system using dual-expression vectors that we previously developed (Fig. 2).12,15,26 This approach allows us to test many split inteins and also simplifies the comparison with previously published data from our laboratory. In this system, an N-terminally hexahistidine (H6) tagged yeast SUMO domain (Smt3) and a C-terminally H6-tagged B1 domain of IgG binding protein G (GB1) are used as N- and C-exteins (target proteins for protein ligation), respectively (Fig. 2a). The expression of two split precursors can be induced under two different promoters (T7 and arabinose) by addition of IPTG and L-arabinose, respectively.12,15,26 We introduced an additional H6-tag at the C terminus permitting quantification of not only the spliced (ligated) product but also the side-products of the N- and C-cleaved products (Fig. 2a).
Fig. 2.

The dual expression system for testing PTS of various new split inteins. (a) Schematic presentation of plasmid design and possible reactions of protein trans-splicing and cleavages. Protein ligation by PTS produces the ligation product of H6-Smt3-GB1-H6. (b) Schematic presentation of the new split inteins tested. Solid lines indicate the new split inteins reported in this study. Previously reported split inteins are marked with broken lines. The total lengths of the inteins are shown on the right side of each intein.
Selection of inteins for engineering novel split inteins
We previously compared cis-splicing of 20 different inteins to identify those with efficient splicing activity in identical extein contexts.33 For designing novel split inteins, we chose four highly efficient cis-splicing mini-inteins lacking endonuclease domains (Fig. S1, ESI†). This is because artificially split inteins with endonuclease domains are often poorly soluble. They were RadA intein from Pyrococcus horikoshii (PhoRadA), NpuDnaE intein, NpuDnaBmin (NpuDnaBΔ283) intein, and VMA intein from Thermoplasma volcanium GSS1 (TvoVMA). High splicing efficiency of these four inteins is also favourable for their potential applications. In addition, we previously elucidated the three-dimensional structures of NpuDnaE intein and PhoRadA intein, facilitating the rational design of new split inteins.36–38 We now also obtained high-resolution crystal stmctures of NpuDnaBΔ290 and TvoVMAΔ21 inteins (see below) and created 21 new split inteins from these four inteins (Fig. 2b). Together with the previously reported split inteins, we investigated protein trans-splicing of 24 different split inteins for the comparison and for testing their cross-reactivity.
Crystal stmctures of NpuDnaBΔ290 and TvoVMAΔ21 inteins
The three-dimensional structures have been useful for creating novel split inteins and mini-inteins25,36,37 and for providing a better understanding of the structure–function relationships of inteins.36–40 To support the design of new split inteins, we additionally determined the crystal structures of two inteins used for this study, i.e. NpuDnaBΔ290 and TvoVMAΔ21 inteins. The initially engineered mini-intein from NpuDnaB intein (NpuDnaBΔ283) is functional and consists of 146 residues, minimized from the original 429 residues33 (Fig. S2a, ESI†). However, we could not obtain well-diffracting crystals of this construct, presumably due to incomplete deletion of the endonuclease insertion region and the remaining disordered region. Further deletion of seven residues allowed growth of crystals that were highly optimized for X-ray analysis, diffracting to the resolution as high as 1.4 Å. Crystal structure of NpuDnaBΔ290 intein was solved by molecular replacement using the coordinates of the DnaB intein from Synechocystis sp. PCC 6803 (PDB, 1MI8)40 as the starting model (Fig. 3b and Table 1). Both inteins share high sequence homology (68% identity) and a very similar three-dimensional structure (r.m.s.d. for backbone atoms of 141 residues is 1.0 Å), although NpuDnaB mini-intein (NpuDnaBΔ283) has considerably superior cis-splicing activity compared to SspDnaB mini-intein (SspDnaBΔ275).33
Fig. 3.
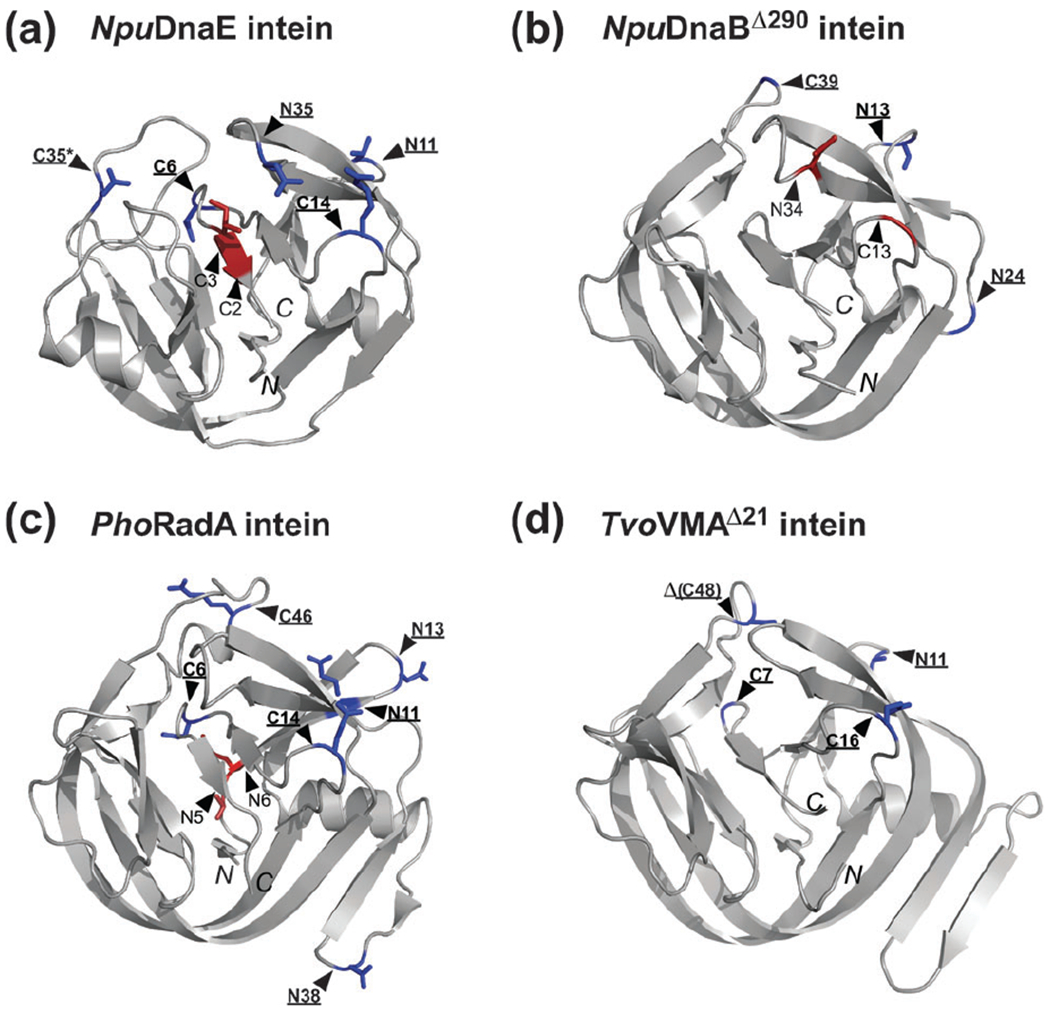
Locations of the split sites presented on the intein structures. (a) The NMR structure of NpuDnaE intein (2KEQ). (b) The crystal structure of NpuDnaBΔ290 intein (4O1R). (c) The NMR structure of PhoRadA intein (2LQM). (d) The crystal structure of TvoVMAΔ21 intein (4O1S). Split sites are indicated by triangles. The residues preceding each split site are shown as stick models in blue for functional split sites or in red for non-functional split sites. C48 split site of TvoVMA intein is located in the deleted region indicated by Δ. Functional split sites are also highlighted in bold and underlined. An asterisk indicates the natural split site of NpuDnaE intein. N and C in italic indicate the N- and C-termini of each intein.
Table 1.
Data collections and structure refinements of NpuDnaBΔ290 intein and TvoVMAΔ21 intein
| NpuDnaBΔ290 intein | TvoVMAΔ21 intein | |
|---|---|---|
| Data collection | ESRF ID14-1 | Diamond 104 |
| Space group | P4322 | P63 |
| Molecules/a.u. | 1 | 2 |
| Unit cell a, b, c (Å); | 61.05, 61.05, 122.2 | 154.4, 154.4, 49.0 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 120 |
| Resolutiona (Å) | 29.6–1.4 (1.45–1.40) | 46.0–2.7 (2.75–2.70) |
| Rmergeb (%) | 8.1 (93.2) | 13.1 (59.8) |
| No. of reflections (measured/unique) | 1298 648/46 443 | 71 837/18 582 |
| ⟨I/σI⟩ | 17.3 (2.5) | 9.8 (1.8) |
| Completeness (%) | 100.0 (100.0) | 99.5 (99.3) |
| Redundancy | 28.0 (28.6) | 3.9 (3.9) |
| Refinement | ||
| Resolution (Å) | 29.6–1.40 | 46.0–2.7 |
| No. of reflections (total/Rfree) | 46 286/948 | 17 824/908 |
| R/Rfreec | 0.161/0.189 | 0.180/0.229 |
| No. of atoms | ||
| Protein | 2354d | 2626 |
| Ligands | 19 | 63 |
| Water | 279 | 125 |
| R.m.s. deviations from ideal targets | ||
| Bond lengths (Å) | 0.009 | 0.011 |
| Bond angles (°) | 1.29 | 1.45 |
| Ramachandran plot (%) | ||
| Favored | 98.6 | 91.2 |
| Allowed | 1.4 | 8.2 |
| Outliers | 0 | 0.7 |
| PDB code | 4O1R | 4O1S |
The highest resolution shell is shown in parentheses.
, where Ii is the observed intensity of the i-th measurement of reflection h, and ⟨I⟩ is the average intensity of that reflection obtained from multiple observations.
, where Fo and Fc are the observed and calculated structure factors, respectively, calculated for all data. Rfree was defined in ref. 56.
Including partially occupied and hydrogen atoms.
TvoVMA intein consists of 186 amino acids (Fig. S2b, ESI†). Initially, we did not succeed in obtaining crystals that would diffract to high resolution. Even though there is no obvious endonuclease insertion, NMR analysis of the TvoVMA intein suggests the presence of disordered regions, as evidenced by the relatively strong signals between 8.0 ppm and 8.5 ppm observed in the [1H, 15N]-HSQC spectrum (Fig. S3, ESI†). We constructed deletion variants of the TvoVMA intein based on the sequence alignments, aiming to trim the sequences around the conserved endonuclease insertion site. We monitored the HSQC spectra and splicing activity in order not to disrupt the entire architecture of the TvoVMA intein (Fig. S3, ESI†). We created two functional deletion variants i.e. TvoVMAΔ13 and TvoVMAΔ21 inteins (Fig. S2b, ESI†). Only TvoVMAΔ21 intein consisting of 165 residues could be crystallized, which diffracts to only 2.7 Å resolution. Its structure was solved by molecular replacement (Fig. 3d and Table 1). The crystal stmcture of the TvoVMAΔ21 intein revealed that the deleted region was indeed located near the crystal contacts with neighbouring molecules. The longer loop in this region probably disturbed the same crystal contacts, preventing growth of well-ordered crystals. The crystal structure of the TvoVMAΔ21 intein shows high similarities with PI-PkoII (2CW7)41 with a Z score of 23.1, PI-PfuI (1DQ3)42 with Z score of 22.4, PhoRadA intein (4E2T)36 with a Z-score of 21.7, and MjaKlbA intein (2JMZ)43 with a Z-score of 20.8, as identified by DALI server 44 These four inteins are from thermophilic archaea and share the same feature of an insertion following the helix after the block A (Fig. S1, ESI†). Such insertion might be important for stabilizing HINT domains as this insertion is observed for all the intein structures from thermophilic organisms, which are currendy available in the Protein Data Bank.
Novel split inteins
In this report we created 21 new split inteins from NpuDnaE intein (4 sites), NpuDnaBΔ283 intein (5 sites), PhoRadA intein (8 sites), and TvoVMA intein (4 sites) (Fig. 2b and Fig. S1, ESI†). Together with the previously reported split sites for NpuDnaE intein (3 sites, dotted lines in Fig. 2b), 24 split inteins were tested and compared with the aim of identifying functional split sites. The comparison could provide a rationale for designing novel split inteins based on the high-resolution three-dimensional structures (Fig. 3). Our primary targets were split inteins with the previously known split sites (N11, C6, C14, and C35) and with even shorter N- or C-terminal fragments (C2, C3, N5, and N6). The identified functional split sites are displayed on the structures by blue stick models (Fig. 3) and indicated in the primary structures by filled rectangles (Fig. S1, ESI†). The efficiencies of protein trans-splicing vary considerably between split inteins (Fig. 4), but a majority of longer split inteins could produce some ligation products.
Fig. 4.

In vivo ligation and side-reactions by the new split inteins. Comparison of PTS by different split inteins derived from (a) NpuDnaE intein, (b) NpuDnaBΔ283 intein, (c) PhoRadA intein, and (d) TvoVMA intein. L, N, and C stand for the percentages of ligation reaction (filled bars), N-cleavage (slashed bars), and C-cleavage (open bars) produced from the coexpression of the two precursors, respectively. The split sites are shown below the graphs. Asterisks indicate that the cleavage reactions could not be analysed due to poor separation between the cleaved product and the precursor in SDS-PAGEs. Data presents mean values ± s.d. (n = 3).
Quantitative comparison of splicing efficiencies of the new split inteins
As we introduced N- and C-terminal H6-tags at both ends (Fig. 2), protein trans-splicing and side-reactions were quantitatively analyzed by SDS-PAGE of the elution fractions from IMAC using Ni-NTA spin columns (Fig. S4, ESI†). Because of the dual His-tags, the elution fractions could contain unreacted N- and C-precursors, the ligated (spliced) product, and N- and C-cleaved products (Fig. S4, ESI†). Adjustment of two precursor proteins at an equimolar ratio with the in vivo system is not trivial and requires careful optimization, because the expression levels deviate between different precursors. Therefore, one of the two precursors can easily be in excess over the other precursor, complicating the quantitative analysis. When one of the precursors was nearly undetectable in the presence of the ligated product and/or the cleaved products, we assumed that such a precursor was completely consumed (reacted), leaving the remaining precursor in excess. In other words, we assumed that the reacted precursors ended up in one of the three reactions (ligation, N- and C-cleavages).
Fig. 4 summarizes protein trans-splicing efficiencies of 24 split inteins, together with two side-reactions of N- and C-cleavages. Nineteen out of 24 split inteins tested could produce ligated products (L). Split inteins with C2, C3, N5 and N6 split sites did not produce any detectable ligation products as judged by SDS-PAGE. Therefore, we did not further investigate these sites for all the four inteins. It is noteworthy that split inteins with the split site at the corresponding endonuclease insertion site, i.e. C35 for NpuDnaE intein, C39 for NpuDnaBΔ283 intein, C46 for PhoRadA intein, and C48 for TvoVMA intein have higher splicing efficiency with fewer side-reactions. This suggests that the highly conserved site for endonuclease domain insertion is probably due to structural requirements for efficient splicing activity. Fig. 4 does not indicate how fast splicing reaction takes place during the expression and purification steps but shows the proportions of side-reactions resulting in undesired products. Interestingly, some split inteins induce cleavages rather than splicing even though their amino acid compositions are nearly identical, suggesting that the protein-folding process upon association of the two precursors is more crucial than the primary structure for productive protein-splicing. In an ideal case with very high splicing kinetics and no side-reaction, we do not expect any remaining precursors and cleaved products in the elution fraction. Fig. 5 presents the normalized yields, which are the ratios between the ligated product and the other remaining proteins in the elution fractions, ignoring the adjustment of unbalanced co-expressions of the two precursors. Naturally split NpuDnaE intein with the C35 split site has the highest yield without any remaining precursors and side-products. It is clearly the best split intein with lowest amounts of side-reactions among the tested split inteins in this article. The previously reported split NpuDnaE intein at the C14 is also comparable to splicing of the wild-type split NpuDnaE intein.26
Fig. 5.

Normalized protein ligation yields from in vivo PTS experiments. Ligation yields were quantified as the percentage of the ligation product against all the proteins in the elution fraction. Names of inteins and split sites are indicated below the graph. Data presents mean values ± s.d. (n = 3).
Practicality of the new split inteins for protein ligation
It is often more critical to produce large amounts of the ligated product than to have higher ligation kinetics of the split intein, although both usually correlate. In particular, yields per litre of growth medium matters for practical applications such as segmental isotopic labeling of proteins.45 However, we found that some split inteins could not be expressed at high levels with high splicing activity. For example, inteins with endonuclease domains could often be expressed well but were insoluble and/or unreactive.23 To take this into account, we compared the yields of the ligated product by 24 pairs of different split inteins under the same conditions from the same volume of the E. coli culture medium. Even though some split inteins derived from PhoRadA and TvoVMA inteins appear to be robust for their protein trans-splicing activity (Fig. 4), the relative ligation yields were much lower compared with other inteins like NpuDnaE and NpuDnaBΔ283 inteins. This is due to the lower expression level of precursor proteins. PhoRadA and TvoVMA inteins were from thermophilic organisms and contain a number of codons that are rare in E. coli, presumably lowering the expression levels. This problem can be alleviated by supplementing tRNAs for rare codons in E. coli (Fig. 6) or might be solved by optimizing the codons used in the intein genes. Among the 24 split inteins, split intein at C14 of NpuDnaE intein and at C39 of NpuDnaBΔ283 intein were the best in terms of the yield per culture volume, although the deviations in individual experiments were large. Considering the lower amounts of the side-reactions (Fig. 4), C14 of NpuDnaE intein is the best split intein with less side-reactions and better final yields under the conditions tested (Fig. S5, ESI†). This is probably because the shorter length of the C-terminal intein fragment split at C14 site is less disturbing the solubility of the fused protein, yet interacting specifically with the N-terminal intein fragment. Moreover, the N-terminal split intein fragment (NpuDnaEΔC14) does not induce any side-reaction without the C-terminal split intein fragment unlike other longer N-terminal split intein fragments that induce cleavage without their counter partners.
Fig. 6.

Relative ligation yields from the different new split inteins. Ligation yields of the in vivo PTS reactions were estimated from SDS-PAGEs based on the intensities of the ligation product bands obtained from independent experiments of different combinations of split inteins. Names of inteins and split sites are indicated below the graph. The data presents mean values ± s.d. (n = 3). Ligation yields of split PhoRadA inteins with and without tRNAs supplementation for rare codons are shown by open and grey bars, respectively.
In vitro protein trans-splicing of the new split inteins
For semi-synthesis,9 it is necessary to perform protein trans-splicing in vitro rather than in vivo. In vitro protein ligation also provides various opportunities to optimize the ligation condition such as pH and additives. Many split intein fragments became insoluble after splitting, even when they are fused to highly soluble proteins like GB1 or SUMO domain. This problem is more frequently observed for artificially split inteins, restricting their use. For example, the NpuDnaBΔ283 intein with C39 split site showed excellent splicing activity in vivo (Fig. 4b, 5, and 6), but the N-terminal intein fragment was mostly insoluble and could not be purified for in vitro experiments when it was expressed as Smt3 fusion. Whereas longer split inteins tend to be less soluble, a shorter fragment (<15 residues) is clearly more soluble when it is fused with soluble proteins as previously shown.26 The short fragments are advantageous for semi-synthetic applications because shorter peptides can be easily synthesized. However, a shorter intein fragment requires longer counter fragment that could already induce undesired cleavages without their partners. This problem was observed particularly for C2, C3, and C6 split sites (Fig. 4). Therefore, the shortest intein fragment is not necessarily the best split intein for in vitro applications. We could test in vitro protein trans-splicing of the two new split inteins. The two pairs of PhoRadAΔC6/C6 and TvoVMAN11/ΔN11 precursors could be purified for in vitro ligation, resulting in acceptable ligation efficiencies (>50%) (Fig. 7). Both new split inteins are comparable to the in vitro ligation by the previously reported NpuDnaEΔC14/C14 (>60%).26 Although these new split inteins do not have remarkable ligation kinetics and lower side-reactions, they might be useful for in vitro protein ligation by PTS.
Fig. 7.

In vitro protein trans-splicing and their ligation kinetics. Kinetic analysis of PTS by (a) PhoRadΔC6/C6 and (b) TvoVMAN11/ΔN11 inteins. SDS-PAGE analysis of the time course of the reaction (left panel) and kinetic analysis (right panel). Time from the start of the reaction is given above lanes in minutes. The bands of precursor proteins and products are indicated. The ligation product is highlighted in bold. The errors were estimated from three independent experiments and the standard deviations are shown. The reaction kinetics were analysed by fitting to first-order kinetics function.
Split inteins derived from NpuDnaE intein
NpuDnaE intein is one of the best natural split inteins with highly efficient splicing activity and has been widely used for protein ligation.15,46,47 We previously created functional split inteins at C14 and C6 sites from the as-splicing single-chain variant of NpuDnaE intein, based on its NMR structure.37 The next question is whether it is possible to make the C-terminal fragment even shorter or to create split intein closer to the N terminus. We tested four new split inteins at N11, N35, C2, and C3 sites in addition to the natural split site (C35) and the previously reported C14 and C6 split sites. The shorter C-terminal intein fragments with less than 6 residues resulted only in N-cleavage reaction. The C-terminal six residues were minimally required for trans-splicing, although the split intein at C6 site also produced large amounts of cleaved products. This observation suggests that N–S acyl shift at the N-terminal junction does not require the last few C-terminal residues and that folding of the three-dimensional structure induces N–S acyl shift without the C-terminal catalytic residues. We assumed that C2 and C3 sites with other inteins could result in similar cleavages.
HINT (Hedgehog/Intein) fold can be divided into two pseudo domains with C2 symmetry, which is most likely to be the result of gene duplication 48 C35 site in the structure of NpuDnaE intein roughly corresponds to N35 site in the other pseudo domain because of this C2 symmetry (Fig. 3a). The N35 site of the NpuDnaE intein has a relatively high ligation efficiency with only small amounts of the side-reactions (Fig. 4). However, an unreactive N-terminal precursor was observed more than that of the C35 site, thereby gready reducing the normalized yield (Fig. 5). This indicates that protein folding of the split NpuDnaE intein at N35 is not as efficient as the C35 site.
N11 site similarly corresponds to the C14 site in terms of the length. This site also corresponds to the previously reported S1 site in SspDnaB intein27 and is found to be functional for the other three inteins. The normalized yields of these split inteins at N11 site are, however, much worse than the C-terminal split sites. These results from split NpuDnaE inteins suggest that there are differences in the folding process of each pseudo domain that are crucial for productive trans-splicing and further understanding of folding process might help the design of better split inteins.
Orthogonality of novel split inteins
One of the limitations of protein trans-splicing is that split inteins can react with each other due to their lower specificity as found among naturally split DnaE inteins.15,16,19,22 It is of considerable interest to identify orthogonal split inteins that are not cross-reactive for multiple-fragment protein ligation.18,19,49 Table 2 summarizes 43 combinations of split inteins tested for cross-reactivity among different split inteins classified by the split sites. Among the five inteins split within the loop after block A (N11 site), only split NpuDnaBΔ283 and SspDnaBΔ275 inteins at this location cross-reacted (Fig. S6, ESI† and Table 2a). This was not surprising because the sequence identities of the first 10–14 residues between the two inteins are about 50–60%. Three functional split inteins at the loop of block F (C14 site) site did not exhibit any cross-activity, confirming their orthogonality (Table 2b and Fig. S7a, ESI†). On the other hand, among the split inteins at block G, PhoRadA split inteins at C7 site and TvoVMA split intein at C6 were cross-reactive (Fig. S7b, ESI†). These two C-terminal fragments have an identical C-terminal sequence with the variation only at the loop region. The shorter fragment of the split inteins at C6 site will probably reduce the probability to be orthogonal and might not be the best site for designing orthogonal split inteins. C14 site seems to be minimally suitable location for creating orthogonal split inteins.
Table 2.
(a) Orthogonality of inteins split at the loop at the end of block A, (b) orthogonality of inteins split at the loop within the block F and (c) orthogonality of inteins split at the loop within the block G
| (a) | |||||
|---|---|---|---|---|---|
| Block A | NpuDnaEN11 | TvoVMAN11 | PhoRadAN11 | ||
| NpuDnaEΔN11 | + | − | − | − | − |
| − | + | − | − | + | |
| TvoVMAΔN11 | − | − | + | − | − |
| PhoRadAΔN11 | − | − | − | + | − |
| − | − | − | − | + | |
| (b) | |||||
| Block F | NpuDnaEC14 | TvoVMAC16 | PhoRadAC14 | ||
| NpuDnaEΔC14 | + | − | − | ||
| TvoVMAΔC16 | − | + | − | ||
| PhoRadAΔC14 | − | − | + | ||
| (c) | |||||
| Block G | NpuDnaEC6 | TvoVMAC7 | PhoRadAC6 | ||
| NpuDnaEΔC6 | + | − | − | ||
| TvoVMAΔC7 | − | + | + | ||
| PhoRadAΔC6 | − | + | + | ||
“+” and “−” indicate “with” and “without” trans-splicing activity, respectively.
Conclusions
In summary, we tested and compared 24 split inteins including the 21 new split inteins designed based on their three-dimensional structures, of which the two new crystal structures of TvoVMAΔ21 and NpuDnaBΔ290 inteins were presented here. We found that the NpuDnaEΔC14/C14 intein was the best split intein for the practical use for protein ligation because of the high yield with fewer side-reactions. Shorter intein fragments with less than 6 residues are generally not suitable as split inteins because they tend to be more cross-reactive and induce more undesired premature cleavages to the N- or C-precursors without their counter partners. Split inteins with very short fragments thus require further engineering to prevent premature N- and C-cleavages. A majority of the new split inteins are orthogonal to each other when the sequence similarities are less than about 40%. Most of functional split inteins identified from the in vivo assay are not always appropriate for in vitro experiments because the split intein precursors are often insoluble or prone to aggregation during purification. The split inteins at the conserved endonuclease insertion site are usually the best split inteins in the in vivo assays, suggesting that this site is less disturbing protein folding of HINT fold and results in more productive protein-splicing. This observation also explains why the endonuclease insertion sites and naturally split sites are highly conserved.
Experimental
The dual vector system for testing protein trans-splicing in vivo
All N-terminal split intein precursors were cloned in pRSF-vectors as H6-tagged yeast ubiquitin-like protein (Smt3) fusion, which carry RSF3010 origin, resistance to kanamycin, and a T7-promoter, whereas all C-terminal split intein precursors were cloned in pBAD-vectors as fusion protein with the C-terminally H6-tagged B1 domain of protein G from Streptococcus spp (GB1), bearing ColE1 origin, ampicillin resistant gene, and arabinose promoter. All plasmids used in this study are summarized in ESI,† Table S1.
Construction of split inteins for in vivo studies
All the plasmids encoding the split inteins (except for H6-Smt3-NpuDnaEΔC6, H6-Smt3-NpuDnaEΔC3, H6-Smt3-NpuDnaEΔC2, , and H6-Smt3-NpuDnaBΔN12) for in vivo experiments were constructed by amplifying the genes of split inteins from previously constructed plasmids as the templates (ESI,† Table S2) by PCR using a pair of two oligonucleotides listed in ESI,† Table S3. The amplified genes were inserted into pHYRSF5350 for the N-terminal split intein precursors or into pMHBAD14C46 for the C-terminal split intein precursors, together with a few native extein residues33,51 (except for the N-junction sequence of NpuDnaE intein).15 H6-Smt3-NpuDnaEΔC6 construct was generated by transferring the gene of NpuDnaEΔC6 from pHYDuet9337 into pHYRSF53. H6-Smt3-NpuDnaEΔC3, H6-Smt3-NpuDnaEΔC2, and were constructed by introducing a stop codon in pSKDuet16 (NpuDnaE)33 or pMMDuet19 (NpuDnaBΔ283)33 using inversion PCR with pairs of two oligonucleotides listed in ESI,† Table S3. was constructed by transferring the gene of intein from pTWIN2 vector (NEB) using the two oligonucleotides of HK039 and HK040 into the pDuet-vector. Two rounds of inversion PCR were performed to mutate the −1 residue to glycine using the two oligonucleotides of HK129 and HK130 and to introduce a stop codon for using the two oligonucleotides of HK142 and HK143. The coding regions of , , NpuDnaEΔC2, NpuDnaEΔC3 were cloned in pHYRSF53.
In vivo protein ligation
Each pair of the two plasmids for the two precursors was co-transformed into E. coli ER2566 strain and grown at 37 °C in 5 mL LB medium supplemented with 25 μg mL−1 kanamycin and 100 μg ml−1 ampicillin. When OD600 reached 0.4–0.6, the C-terminal precursor was induced by addition of a final concentration of 0.08% (w/v) l-arabinose. The C-terminal precursor was expressed for half an hour before the second induction of the N-terminal precursor with a final concentration of 0.5 mM isopropyl β-d-1-thiogalactopyranoside (IPTG). After the second induction the cells were incubated for additional four hours and then harvested by centrifugation. The cell pellet was mixed and lysed with Bacterial protein extraction reagent (B-PER, Thermo Scientific). After centrifugation the supernatant was loaded on Ni-NTA spin columns (Qiagen). The bound proteins were eluted in buffer B (50 mM sodium phosphate, 300 mM NaCl, 250 mM imidazole, pH 8.0) after washing with washing buffer (50 mM sodium phosphate, 300 mM NaCl, 30 mM imidazole, pH 8.0). The elution fractions were analyzed by 18% SDS-PAGE. Protein expressions by supplementing tRNA for rare codons in E. coli were performed by co-transformation of an additional pRARE plasmid, which bears genes for rare tRNAs and growth medium was additionally supplemented with 5 μg mL−1 chloramphenicol.
In vitro protein ligation experiments
The N-terminal precursors of H6-Smt3-TvoVMAN11 (pSARSF53-110) and H6-Smt3-PhoRadAΔC6 (pSARSF53-165) were expressed in E. coli ER2566 cell strain for 3 hours by IPTG induction and purified from 1 liter LB-medium. To supplement for rare codons in E. coli, pSARSF53-165 was co-transformed with the pRARE plasmid. The harvested cell pellets were resuspended in buffer A (50 mM sodium phosphate, 300 mM NaCl, pH 8.0) and stored at −70 °C. The cells were lysed by ultrasonication. The supernatant after centrifugation at 38 360g at 4 °C for 50 minutes was filtered through a 0.45 μm filter and loaded on a 5 mL HisTrapHP (GE Healthcare). H6-tagged proteins were eluted with a linear gradient from 0 to 100% of buffer B. The C-terminal split intein precursor coded in pSABAD14-98 (TvoVMAΔN11-GB1-H6) and pSABAD14-172 (PhoRadAC6-GB1-H6) were expressed and purified by the same IMAC procedure. The cells bearing the plasmid pSABAD14-98 or pSABAD14-172 were grown in 1 or 2 litres LB-medium supplemented with 100 μg ml−1 ampicillin. The protein expression was induced by addition of a final concentration of 0.08% (w/v) l-arabinose. The precursor of TvoVMAΔN11-GB1-H6 was purified by IMAC with Profinia Purification System (BioRad) according to the manufacturers protocol. The elution fractions containing the intein precursors were dialyzed against ligation buffer (0.5 M NaCl, 10 mM Tris-HCl, 1 mM EDTA, pH 7.0) overnight at 10 °C. The in vitro protein ligation was monitored after mixing equimolar amounts (15 μM each) of the N- and C-precursors at 25 °C in the presence of 0.5 mM TCEP (tris(2-carboxyethyl)phosphine). The reaction was followed by taking samples, of which reaction were stopped by mixing with SDS-loading buffer. The samples were analyzed from 18% SDS-PAGEs as described below.
Quantification of the splicing reactions by the image analysis of SDS-PAGE
The ratios of N-, C-cleavages and ligation products were analysed from SDS-PAGEs stained with PhastGel™ Blue R (GE Healthcare) based on band intensities using the software Image J (NIH). The insoluble fraction was not taken into account for the analysis. The percentages of ligation (L), N-, and C-cleavages (N and C) were calculated as the proportion of each reacted product (L, N, and C) against all the reacted products, excluding unreacted precursors. The normalized yields were calculated as the proportion of the ligation product (L) against the sum of all the remaining precursors and ligated and cleaved products present in the elution fraction. The band intensities were normalized according to their molecule sizes. The relative ligation yields were determined by comparing the intensities of ligation product bands from the different protein expressions of different combinations of the split inteins. To reduce artefacts due to staining, the analysis was normalized based on intensities of a standard (bands from the protein weight marker loaded on each gel). Yields were normalized among each other by setting the highest yield ( intein) to one. To analyze the reactions in vitro, intensities of the precursor and product bands at each time point were quantified. Ligation kinetics was determined with SigmaPlot (Systat Software Inc) by fitting the first-order kinetics function to the band intensities. The amount of ligation was calculated as the proportion of the ligation product against the remaining precursors, ligation, and cleavage products. All experiments were performed in triplicates.
Cloning TvoVMA intein, TvoVMAΔ21 intein and NpuDnaBΔ290 intein for the structure determination
H6-Smt3 fusion with TvoVMA intein containing C1A, T+1A mutations, and a stop codon after +1A residue was cloned from pSKDuet2633 by PCR with two oligonucleotides of HK313 and HK264 and inserted into pHYRSF53, resulting in pHYRSF175. TvoVMAΔ21 intein with the deletion of residues 124–144 was constructed from plasmid pSKDuet26 by inversion PCR using two oligonucleotides I423 and I424, resulting in pJODuet72. An inactive variant (C1A, T+1A) of TvoVMAΔ21 intein was cloned as H6-Smt3 fusion in pHYRSF53 using two oligonucleotides of HK370 and HK264 (pJORSF73).
NpuDnaBΔ283 intein (pMMDuet19)33 was further minimized by PCR using the oligonucleotides of HK151, HK506, HK504, and HK505 to construct NpuDnaBΔ290 intein. The nine residues were removed from the loop where the endonuclease domain had been removed in pMMDuet19. The gene of NpuDnaBΔ290 intein was cloned into pSKDuet16, resulting in pALBDuet28 that encodes cis-splicing precursor bearing NpuDnaBΔ290 and two GB1s as exteins. The inactive variant of NpuDnaBΔ290 intein bearing C1A, S+1A mutations and a stop codon after the +1A residue was created as H6-Smt3 fusion by cloning the gene in pHYRSF53 using two oligonucleotides of HK763 and HK764, which resulted in pCARSF02.
Protein expression and purification for structural studies
Protein expression and purification of NpuDnaBΔ290 intein (pCARSF02), TvoVMA intein (pHYRSF175), and TvoVMAΔ21 intein (pJORSF73) were performed in the same way except for PJORSF73, which was additionally co-transformed with pRARE plasmid. pCARSF02 or pHYRSF175 was transformed into E. coli ER2566 strain. The transformed cells were grown at 37 °C in LB-media supplemented with appropriate antibiotics. At OD600 = ~0.6, the cell culture was induced for three hours with a final concentration of 0.5 mM IPTG. For NMR studies, the constructs in pHYRSF175 and pJORSF73 were expressed for 5 hours in stable isotope-labeled medium using M9-medium supplemented with 15NH4Cl as sole nitrogen. The harvested cell pellet was resuspended with buffer A and flash-frozen for storage at −80 °C. The frozen cell pellets were lysed by French Press or ultrasonication and purified as described previously using HisTrapFF column (5 mL) (GE Healthcare).50 After the removal of H6-Smt3 tag, NpuDnaBΔ290 intein, TvoVMA intein, and TvoVMAΔ21 intein were dialyzed against MQ grade water at 10 °C. The proteins were concentrated using Vivaspin 20 centrifugal filter device (GE Healthcare, MWCO 5 000).
NMR spectroscopy
[1H, 15N]-TROSY HSQC spectra of 0.4 mM TvoVMA intein in 10 mM sodium phosphate buffer (pH 6) at 307 K and 0.5 mM TvoVMAΔ21 intein in 20 mM sodium phosphate buffer (pH 7) at 298 K were recorded on a Varian INOVA 600 MHz or 800 MHz spectrometer equipped with a cryogenic probe head.
Protein crystallization
The final protein concentrations of 63 mg mL−1 for NpuDnaBΔ290 intein and 45 mg mL−1 for TvoVMAΔ21 intein were used for protein crystallization. Crystallization conditions were screened at 293 K using Index HT screen (Hampton Research) by sitting drop vapour diffusion in a 96 well plate (Innovadyne SD-2), with a 80 μL reservoir solution and protein drops of 100 nL mixed with 100 nL reservoir solution. Crystallization hit of NpuDnaBΔ290 intein in 0.1 M bis-Tris, pH 5.5, and 3.0 M NaCl was repeated manually. Crystals for diffraction data collection were prepared using sitting drop vapour diffusion with 300 μL reservoir solution and 1 μL protein drops mixed with 1 μL reservoir solution. From the crystallization condition a crystal was picked and cryo-protected with a 20% glycerol solution mixed in mother liquid before vitrification. Crystallization conditions for TvoVMAΔ21 intein were optimized by grid screen from the initial hits and by adjusting the protein concentration to 20 mg mL−1. The best diffraction crystal was collected from a drop grown in 0.1 M HEPES (pH 7.0) and 25% MPD using sitting drop vapour diffusion with 500 μL reservoir solution and 0.5 μL protein drops mixed with 0.5 μL reservoir solution.
Data processing and structure refinement
Diffraction data for the crystal of NpuDnaBΔ290 intein were collected in a single pass on beamline ID14-1 at ESRF/Grenoble and were subsequently indexed, integrated, and scaled to 1.40 Å resolution using the program HKL3000,52 with crystal parameters and data processing statistics listed in Table 1. The estimated Matthews coefficient is 3.64 Å3 Da−1, corresponding to 66.2% solvent content.53 The structure was solved by molecular replacement using SspDnaBΔ275 intein (1MI8) as a starting model, resulting in a fully interpretable electron density map. Further refinement was performed with Refmac554 and Phenix,55 using all data between 29.6 and 1.40 Å, after setting aside 2.1% of randomly selected reflections (~1000 total) for calculation of Rfree.56 Manual corrections were performed with COOT.57 Isotropic individual temperature factors were refined, with the TLS parameters added in the final stages of refinement (Table 1).
Diffraction data for the crystal of TvoVMAΔ21 intein were collected in a single pass on beamline 104 at the Diamond synchrotron and were subsequently indexed, integrated, and scaled to 2.70 Å resolution using the program HKL300052 (Table 1). Although the nominal resolution of measured data was higher, diffraction beyond that limit was not retained due to generally weak scattering from the only suitable crystal. While the asymmetric unit could easily accommodate up to four molecules of TvoVMAΔ21 intein, only two are actually present, explaining the relatively weak diffraction. The estimated Matthews coefficient53 is 4.4 Å3 Da−1, corresponding to 72.1% solvent content. The structure was solved by molecular replacement using as a starting model the coordinates of the Mycobacterium tuberculosis recA mini-intein (2IN0; 20% sequence identity),58 identified with the program BALBES.59 Structure solution utilized the program MOLREP60 implemented in HKL3000. However, solution was possible only after trimming the side chains in the starting model with the program CHAINSAW61 or by using a polyalanine model, whereas no solution could be obtained with either unmodified 2IN0 coordinates, or with any modification of the coordinates of the minimized RadA intein from Pyrococcus horikoshii (4E2U),36 closer in its amino acid sequence (32% identity) to the target intein. The unique solution was automatically rebuilt with BUCCANEER62 and refined with Refmac554 within HKL3000.52 Further refinementwas performed with Phenix,55 using all data between 46.0 and 2.7 Å, after setting aside 5.1% of randomly selected reflections (~900 total) for calculation of Rfree.56 Isotropic individual temperature factors were refined, with the TLS parameters added in the final stages of refinement (Table 1).
Supplementary Material
Acknowledgements
We thank C. Albert and S. Ferkau for technical assistance in the protein and plasmid preparations, R. Kolodziejczyk and K. Kogan for collection of diffraction data, and Ivan Shabalin and Wladek Minor (University of Virginia) for assistance with determining the structure of TvoVMA.Δ21 intein. A.S.A. acknowledges Viikki Doctoral Programme in Molecular Biosciences for financial support. J.S.O. acknowledges the National Doctoral Programme in Informational and Structural Biology for financial support. This work was supported in part by the Academy of Finland (137995), Sigrid Jusélius Foundation, and Biocenter Finland (for H.I., the crystallization, and NMR facilities at the Institute of Biotechnology), and in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Notes and references
- 1.Hirata R, Ohsumk Y, Nakano A, Kawasaki H, Suzuki K and Anraku Y, J. Biol Chem, 1990, 265, 6726–6733. [PubMed] [Google Scholar]
- 2.Kane PM, Yamashiro CT, Wolczyk DF, Neff N, Goebl M and Stevens TH, Science, 1990, 250, 651–657. [DOI] [PubMed] [Google Scholar]
- 3.Paulus H, Annu. Rev. Biochem, 2000, 69, 447–496. [DOI] [PubMed] [Google Scholar]
- 4.Chong S, Mersha FB, Comb DG, Scott ME, Landry D, Vence LM, Perler FB, Benner J, Kucera RB, Hirvonen CA, Pelletier JJ, Paulus H and Xu MQ, Gene, 1997, 192, 271–281. [DOI] [PubMed] [Google Scholar]
- 5.Chong S, Montello GE, Zhang A, Cantor EJ, Liao W, Xu MQ and Benner J, Nucleic Acids Res, 1998, 26, 5109–5115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saleh L and Perler FB, Chem. Rec, 2006, 6, 183–193. [DOI] [PubMed] [Google Scholar]
- 7.Noren CJ, Wang J and Perler FB, Angew. Chem., Int. Ed, 2000, 39, 450–466. [PubMed] [Google Scholar]
- 8.Southworth MW, Amaya K, Evans TC, Xu MQ and Perler FB, BioTechniques, 1999, 27, 110–114, 116,, 118–120. [DOI] [PubMed] [Google Scholar]
- 9.Muir TW, Annu. Rev. Biochem, 2003, 72, 249–289. [DOI] [PubMed] [Google Scholar]
- 10.Ludwig C, Schwarzer D and Mootz HD, J. Biol. Chem, 2008, 283, 25264–25272. [DOI] [PubMed] [Google Scholar]
- 11.Skrisovska L, Schubert M and Allain FHT, J. Biomol. NMR, 2010, 46, 51–65. [DOI] [PubMed] [Google Scholar]
- 12.Züger S and Iwai H, Nat. Biotechnol, 2005, 23, 736–740. [DOI] [PubMed] [Google Scholar]
- 13.Wu H, Hu Z and Liu XQ, Proc. Natl. Acad. Sci. U. S. A, 1998, 95, 9226–9231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martin DD, Xu MQ and Evans TC Jr., Biochemistry, 2001, 40, 1393–1402. [DOI] [PubMed] [Google Scholar]
- 15.Iwai H, Züger S, Jin J and Tam PH, FEES Lett, 2006, 580, 1853–1858. [DOI] [PubMed] [Google Scholar]
- 16.Dassa B, Amitai G, Caspi J, Schueler-Furman O and Pietrokovski S, Biochemistry, 2007, 46, 322–330. [DOI] [PubMed] [Google Scholar]
- 17.Shah NH, Dann GP, Vila-Perello M, Liu Z and Muir TW, J. Am. Chem. Soc, 2012, 134, 11338–11341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shi J and Muir TW, J. Am. Chem. Soc, 2005,127, 6198–6206. [DOI] [PubMed] [Google Scholar]
- 19.Busche AE, Aranko AS, Talebzadeh-Farooji M, Bernhard F, Dotsch V and Iwaï H’, Angew. Chem., Int. Ed, 2009, 48, 6128–6131. [DOI] [PubMed] [Google Scholar]
- 20.Evans TC, Martin D, Kolly R, Panne D, Sun L, Ghosh I, Chen L, Benner J, Liu XQ and Xu MQ, J. Biol. Chem, 2000, 275, 9091–9094. [DOI] [PubMed] [Google Scholar]
- 21.Volkmann G and Iwaï H, Mol. BioSyst, 2010, 6, 2110–2121. [DOI] [PubMed] [Google Scholar]
- 22.Shah NH, Vila-Perello M and Muir TW, Angew. Chem., Int. Ed, 2011, 50, 6511–6515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Otomo T, Teruya K, Uegaki K, Yamazaki T and Kyogoku Y, J. Biomol. NMR, 1999, 14, 105–114. [DOI] [PubMed] [Google Scholar]
- 24.Shingledecker K, Jiang SQ and Paulus H, Gene, 1998, 207, 187–195.9511761 [Google Scholar]
- 25.Brenzel S, Kurpiers T and Mootz HD, Biochemistry, 2006, 45, 1571–1578. [DOI] [PubMed] [Google Scholar]
- 26.Aranko AS, Züger S, Buchinger E and Iwaï H, PLoS One, 2009, 4, e5185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun W, Yang J and Liu XQ, J. Biol. Chem, 2004, 279, 35281–35286. [DOI] [PubMed] [Google Scholar]
- 28.Song H, Meng Q and Liu XQ, PLoS One, 2013, 7, e45355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Appleby JH, Zhou K, Volkmann G and Liu XQ, J. Biol. Chem, 2009, 284, 6194–6199. [DOI] [PubMed] [Google Scholar]
- 30.Perler FB, Nucleic Acids Res, 2002, 30, 383–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gorbalenya AE, Nucleic Acids Res, 1998, 26, 1741–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu H, Xu MQ and Liu XQ, Biochim. Biophys. Acta, 1998, 1387, 422–432. [DOI] [PubMed] [Google Scholar]
- 33.Ellilä S, Jurvansuu JM and Iwaï H, FEES Lett, 2011, 585, 3471–3477. [DOI] [PubMed] [Google Scholar]
- 34.Elleuche S, Döring K and Pöggeler S, Biochem. Biophys. Res. Commun, 2008, 366, 239–243. [DOI] [PubMed] [Google Scholar]
- 35.Hiraga K, Derbyshire V, Dansereau JT, Van Roey P and Belfort M, J. Mol. Biol, 2005, 354, 916–926. [DOI] [PubMed] [Google Scholar]
- 36.Oeemig JS, Zhou D, Kajander T, Wlodawer A and Iwaï H, J. Mol. Biol, 2012, 421, 85–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oeemig JS, Aranko AS, Djupsjöbacka J, Heinämäki K and Iwaï H, FEBS Lett, 2009, 583, 1451–1456. [DOI] [PubMed] [Google Scholar]
- 38.Aranko AS, Oeemig JS, Kajander T and Iwaï H, Nat. Chem. Biol, 2013, 9, 616–622. [DOI] [PubMed] [Google Scholar]
- 39.Klabunde T, Sharma S, Telenti A, Jacobs WR and Sacchettini JC, Nat. Struct. Biol, 1998, 5, 31–36. [DOI] [PubMed] [Google Scholar]
- 40.Ding Y, Xu MQ, Ghosh I, Chen X, Ferrandon S, Lesage G and Rao Z, J. Biol. Chem, 2003, 278, 39133–39142. [DOI] [PubMed] [Google Scholar]
- 41.Matsumura H, Takahashi H, Inoue T, Yamamoto T, Hashimoto H, Nishioka M, Fujiwara S, Takagi M, Imanaka T and Kai Y, Proteins, 2006, 63, 711–715. [DOI] [PubMed] [Google Scholar]
- 42.Ichiyanagi K, Ishino Y, Ariyoshi M, Komori K and Morikawa K, J. Mol. Biol, 2000, 300, 889–901. [DOI] [PubMed] [Google Scholar]
- 43.Johnson MA, Southworth MW, Herrmann T, Brace L, Perler FB and Wüthrich K, Protein Sci, 2007,16, 1316–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Holm L and Rosenstrom P, Nucleic Acids Res, 2010, 38, W545–W549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Minato Y, Ueda T, Machiyama A, Shimada I and Iwaï H, J. Biomol. NMR, 2012, 53, 191–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Buchinger E, Aachmann FL, Aranko AS, Valla S, Skjak-Braek G, Iwaï H and Wimmer R, Protein Sci, 2010, 19, 1534–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Muona M, Aranko AS and Iwaï H, ChemBioChem, 2008, 9, 2958–2961. [DOI] [PubMed] [Google Scholar]
- 48.Hall TM, Porter JA, Young KE, Koonin EV, Beachy PA and Leahy DJ, Cell, 1997, 91, 85–97. [DOI] [PubMed] [Google Scholar]
- 49.Otomo T, Ito N, Kyogoku Y and Yamazaki T, Biochemistry, 1999, 38, 16040–16044. [DOI] [PubMed] [Google Scholar]
- 50.Heinämäki K, Oeemig JS, Djupsjöbacka J and Iwaï H, Biomol. NMR Assignments, 2009, 3, 41–43. [DOI] [PubMed] [Google Scholar]
- 51.Amitai G, Callahan BP, Stanger MJ, Belfort G and Belfort M, Proc. Natl. Acad. Sci. U. S. A, 2009, 106, 11005–11010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Minor W, Cymborowski M, Otwinowski Z and Chruszcz M, Acta Crystallogr., Sect. D: Biol. Crystallogr, 2006, 62, 859–866. [DOI] [PubMed] [Google Scholar]
- 53.Matthews BW, J. Mol. Biol, 1968, 33, 491–497. [DOI] [PubMed] [Google Scholar]
- 54.Murshudov GN, Vagin AA and Dodson EJ, Acta Crystallogr., Sect. D: Biol. Crystallogr, 1997, 53, 240–255. [DOI] [PubMed] [Google Scholar]
- 55.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK and Terwilliger TC, Acta Crystallogr., Sect. D: Biol. Crystallogr, 2002, 58, 1948–1954. [DOI] [PubMed] [Google Scholar]
- 56.Brünger AT, Nature, 1992, 355, 472–475. [DOI] [PubMed] [Google Scholar]
- 57.Emsley P and Cowtan K, Acta Crystallogr., Sect. D: Biol. Crystallogr, 2004, 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- 58.Van Roey P, Pereira B, Li Z, Hiraga K, Belfort M and Derbyshire V, J. Mol. Biol, 2007, 367, 162–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Long F, Vagin AA, Young P and Murshudov GN, Acta Crystallogr., Sect. D: Biol. Crystallogr, 2008, 64, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vagin A and Teplyakov A, J. Appl. Crystallogr, 1997, 30, 1022–1025. [Google Scholar]
- 61.Stein N, J. Appl. Crystallogr, 2008, 41, 641–643. [Google Scholar]
- 62.Cowtan K, Acta Crystallogr., Sect. D: Biol. Crystallogr, 2006, 62, 1002–1011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.