

Abstract
Two new sesquiterpenoid tropolone glycosides, liriosmasides A (1) and B (2), along with two known compounds, secoxyloganin and oplopanpheside C, were isolated from a methanol extract of the roots of Liriosma ovata. The structures of 1 and 2 were elucidated by spectroscopic methods including 1D and 2D NMR and by high-resolution mass spectrometry involving an ultra-high-performance liquid chromatography–quadrupole-orbital ion trap mass spectrometric (UHPLC–Q-Orbitrap MS) method. Compound 1 showed weak inhibitory activity against HIV RNase H.
Graphical Abstract
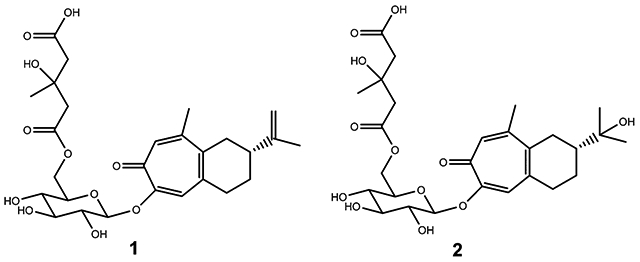
The tropical tree Liriosma ovata Miers [syn. Dulacia inopiflora (Miers) Kuntze, Olacaceae],1 commonly known as “Muira Puama”, is native to Amazonian Brazil, Colombia, Peru, and Venezuela.2,3 In Brazil and France, this plant is used in the form of a fluid extract and other preparations for the treatment of various nervous disorders and as a nerve stimulant and aphrodisiac.2 Ptychopetalum olacoides Benth., from the same family Olacaceae, also known as “Muira Puama”, is used interchangeably with L. ovata.4 In the United States, many dietary supplements labeled as containing L. ovata, P. olacoides, and “Muira Puama” are available from retail stores and Internet suppliers. In addition, since the continuous exploitation and lack of cultivation of P. olacoides have led to significant reduction of its natural populations, Croton echioides Baill., Euphorbiaceae, a small tree growing in Northeastern Brazil, has been used as a substitute for P. olacoides.5 Systematic phytochemical studies are warranted on the authenticated L. ovata plant material to isolate characteristic marker compounds to monitor the authenticity of material source and to determine safety and efficacy of dietary supplements labeled as containing L. ovata. Two phytochemical studies have been conducted on L. ovata, and five compounds were isolated and identified from the roots and three compounds from the bark.6,7 Herein, the isolation and structural elucidation of two new sesquiterpenoid tropolone glycosides, 1 and 2, are described from a methanol extract of the roots of L. ovata. Their structures were determined using spectroscopic methods, including 1D (1H and 13C) and 2D (COSY, HSQC, HMBC, and NOESY) NMR experiments as well as by high-resolution mass spectrometric analysis.
The powdered root material of L. ovata was extracted with MeOH and partitioned sequentially with hexanes, EtOAc, and n-BuOH. The n-BuOH and EtOAc fractions were subjected to extensive fractionation by chromatographic techniques to isolate two new compounds, 1 and 2, and two known compounds, secoxyloganin and oplopanpheside C. Herein are described the structure elucidation and the biological evaluation of 1 and 2.
In this study, an ultra-high-performance liquid chromatography–quadrupole-orbital ion trap mass spectrometer (UHPLC–Q-Orbitrap MS) was used for high-resolution mass spectrometric analyses. Compound 1 was obtained as a pale yellow powder and produced a molecular ion, [M ─ H]−, at m/z 535.2191 using electrospray ionization in the negative mode, consistent with the molecular formula C27H36O11. This suggested 10 degrees of unsaturation. Its UV spectrum showed absorption maxima at 243 and 340 nm, and the IR spectrum supported the presence of hydroxy (3435 cm−1), ester (1731 cm−1), and carbonyl (1645 cm−1) groups. The 13C and HSQC NMR spectra showed the occurrence of three methyls, seven methylenes, eight methines, and nine quaternary carbon atoms. A detailed analysis of 1H and 13C NMR spectra of 1 (Table 1) and the literature suggested the presence of a bicyclic tropolone moiety with an isopropenyl group8 and a 3-hydroxy-3-methylglutaric acid moiety with a C-6 connected glucose.9 This was confirmed by the two-dimensional NMR experiments described below. Closer examination of HMBC connectivities to carbons 5, 9, 10, and 11 of the tropolone group and COSY connections between H-1 and H-2 and also H-3 and H-4 (Table 1, Figure 1) indicated the presence of a fused six-membered ring system, for which the seven-spin system was assigned in the following manner. Two doublets of doublets at δ 2.82 and 2.56 were assigned to H-1a and H-1b, respectively, and a flanking, spin-coupled multiplet (dddd) at δ 2.33 to H-2. Adjacent multiplets (both dddd) at δ 1.98 and 1.66 belonged to H-3a and H-3b, and the multiplets (both ddd) at δ 3.08 and 2.95 to H-4a and H-4b. In addition, singlets at δ 7.20 and 7.24 were assigned by HMBC correlations to respectively H-5 and H-8 in the tropolone ring and singlets at δ 4.83 and 4.81 to H-13a and H-13b in the isopropenyl group, which was placed at C-2 (Table 1). Finally, singlets at δ 1.83 and 2.40 corresponded to two methyl groups, CH3-14 and CH3-15, respectively. The aglycone of compound 1 shares a similar skeleton to manicol, for which the absolute configuration was determined as 2R by X-ray crystallographic analysis.8 Manicol was isolated from a plant in the same genus as compound 1.8 Accordingly, compound 1 was assigned with the same 2R configuration as that of manicol, for biogenetic reasons.
Table 1.
1H and 13C NMR Data for 1 and 2 (600 and 150 MHz, CD3OD)
| position | 1 |
2 |
|||
|---|---|---|---|---|---|
| δC | δH (J in Hz) | HMBC | δC | δH (J in Hz) | |
| 1 | 37.2 | 2.82 (1H, dd, 17.9, 4.6) eq | 3b, 15 (w)a | 33.3 | 2.84 (1H, dd, 17.9, 4.4) |
| 2.56 (1H, dd, 17.9, 10.6) ax | 2.49 (1H, dd, 17.9, 11.7) | ||||
| 2 | 42.1 | 2.33 (1H, dddd, 11.4, 10.6, 4.6, 3.0) ax | 1a, 1b, 3b, 13a, 13b, 14 | 46.1 | 1.67 (1H, dddd, 12.3, 11.7, 4.4, 3.3) |
| 3 | 28.0 | 1.98 (1H, dddd, 11.4, 4.7, 3.0, 2.5) eq | 1a, 1b (w), 2 (w), 4a, 4b (w) | 24.6 | 2.06 (1H, m, 12.3, 3.3)b |
| 1.66 (1H, dddd, 11.4, 11.4, 11.4, 5.3) ax | 1.42 (1H, m, 12.6, S.4)b | ||||
| 4 | 38.0 | 3.08 (1H, ddd, 18.1, 11.4, 4.7) ax | 3b, 5 | 38.9 | 3.04 (1H, m, 17.6, 12.3)b |
| 2.95 (1H, ddd, 18.1, 5.3, 2.5) eq | 2.95 (1H, m, 17.6, S.0)b | ||||
| 5 | 125.7 | 7.20 (1H, s) | 1b, 4a (w), 4b | 125.6 | 7.24 (1H, s) |
| 6 | 159.1 | 5, 8, 1′ | 159.2 | ||
| 7 | 180.1 | 5, 8 | 180.1 | ||
| 8 | 137.4 | 7.24 (1H, s) | 15 | 137.4 | 7.28 (1H, s) |
| 9 | 153.8 | 1a, 1b (w), 8, 15 | 154.1 | ||
| 10 | 142.2 | 1a, 1b, 4a, 5, 8, 15 | 143.0 | ||
| 11 | 143.4 | 1a, 1b, 3b, 4a, 4b (w), 5, 8 (w) | 143.9 | ||
| 12 | 150.0 | 1a, 1b, 3b (w), 13a, 13b, 14 | 73.1 | ||
| 13 | 110.5 | 4.83c (1H, s) | 2, 14 | 27.3 | 1.27 (3H, s) |
| 4.81d (1H, s) | |||||
| 14 | 21.0 | 1.83 (3H, s) | 2, 13a, 13b | 26.8 | 1.25 (3H, s) |
| 15 | 27.4 | 2.40 (3H, s) | 8 | 27.4 | 2.45 (3H, s) |
| glucose moiety | |||||
| 1′ | 101.8 | 5.00 (1H, d, 7.6) | 2′, 3′, 5′ | 101.8 | 5.02 (1H, d, 7.6) |
| 2′ | 74.5 | 3.57 (1H, dd, 9.2, 7.6) | 3′ | 74.5 | 3.57 (1H, dd, 9.2, 7.6) |
| 3′ | 77.1 | 3.51 (1H, t, 9.2) | V, 2′, 4′, 5′ | 77.1 | 3.51 (1H, t, 9.2) |
| 4′ | 71.6 | 3.38 (1H, t, 9.2) | 2′, 3′, 5′, 6′a, 6′b (w) | 71.6 | 3.39 (1H, t, 9.2) |
| 5′ | 75.9 | 3.75 (1H, ddd, 9.2, 6.7, 1.8) | 1′, 4′, 6′a (w), 6′b | 75.9 | 3.75 (1H, ddd, 9.2, 6.6, 1.6) |
| 6′ | 64.9 | 4.55 (1H, dd, 12.0, 1.8) | 4′, 5′ | 64.9 | 4.53 (1H, dd, 12.0, 1.6) |
| 4.15 (1H, dd, 12.0, 6.7) | 4.15 (1H, dd, 12.0, 6.6) | ||||
| glutaryl group | |||||
| 1″ | 172.5 | 6′a, 6′b, 2″a, 2″b | 172.6 | ||
| 2″ | 46.3 | 2.74 (1H, d, 14.8) | 4″, 6″ | 46.9 | 2.68 (1H, d, 14.5) |
| 2.68 (1H, d, 14.8) | 2.62 (1H, d, 14.5) | ||||
| 3″ | 70.8 | 2″a, 2″b, 4″, 6″ | 71.0 | ||
| 4″ | 46.0 | 2.63 (2H, brs) | 2″a, 2″b, 6″ | 46.9 | 2.61 (2H, brs) |
| 5″ | 175.0 | 4″ | 175.0 | ||
| 6″ | 27.9 | 1.34 (3H, s) | 2″a, 2″b, 4″ | 28.0 | 1.31 (3H, s) |
w: weak.
Only identifiable couplings.
cis to Me-14.
trans to Me-14.
Figure 1.
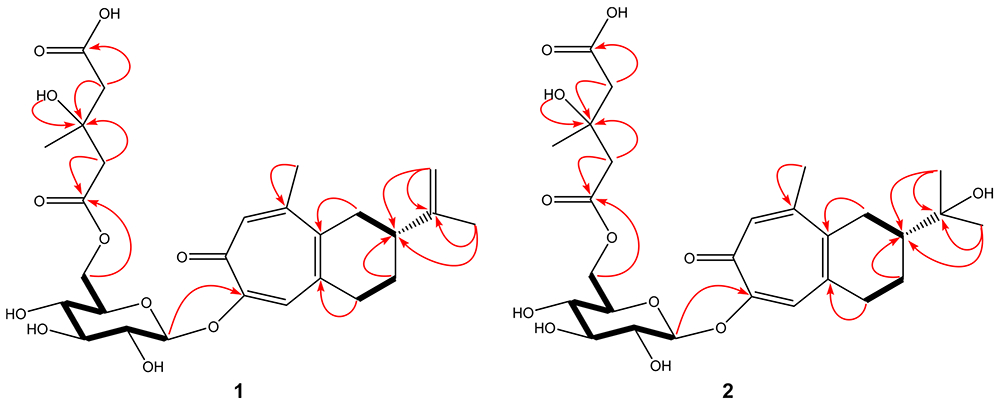
Key 1H─1H COSY (bold ─) and HMBC (red →) correlations of compounds 1 and 2.
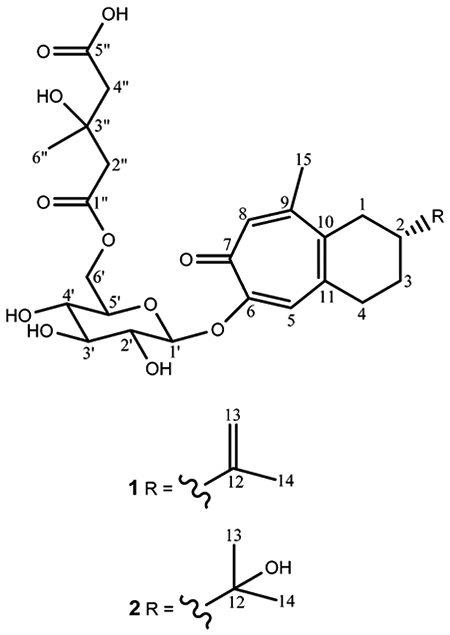
A glucose moiety in 1 was determined by the 1H NMR signals at δ 5.00, 4.55, 4.15, 3.75, 3.57, 3.51, and 3.38 and the 13C NMR resonances at δ 101.8, 77.1, 75.9, 74.5, 71.6, and 64.9.9 The 7.6 Hz coupling constant for the anomeric proton H-1′ at δ 5.00 confirmed its axial orientation.10 Acid hydrolysis and GC-MS analysis of the thiazolidine derivatives substantiated the sugar unit as being β-D-glucose. The 1H NMR resonances at δ 2.74, 2.68, 2.63, and 1.34 and the 13C NMR signals at δ 175.0, 172.5, 70.8, 46.3, 46.0, and 27.9 revealed the existence of a 3-hydroxy-3-methylglutaric acid moiety.9,11
The HMBC spectrum (Table 1, Figure 1) was then employed to determine how the isopropenyl, fused cyclohexene-tropolone, glucose, and 3-hydroxy-3-methylglutaric acid groups were connected. Correlations from H-13a, H-13b, and CH3-14 to C-12 and C-2 confirmed that the isopropenyl group is linked to C-2 of the fused cyclohexene-tropolone unit. The HMBC correlation from CH3-15 to C-9 required that the 15-methyl group be attached to C-9 of the same tropolone moiety. The HMBC correlation from the anomeric proton (H-1′) of glucose to C-6 of the sesquiterpenoid tropolone necessitated that the anomeric carbon (C-1′) was attached to C-6 by an ether bond. Finally, the correlations from H-6′a and H-6′b to C-1″ suggested that C-1″ of the 3-hydroxy-3-methylglutaric acid group is attached to C-6′ of glucose by an ester linkage. Lastly, strong NOESY correlations between H-13a (δ 4.83) and CH3-14 (δ 1.83) and between H-13b (δ 4.81) and H-1a (δ 2.82), H-1b (δ 2.56), H-2 (δ 2.33), H-3a (δ 1.98), and H-3b (δ 1.66) demonstrated that H-13a is cis and H-13b is trans to CH3-14. The foregoing data demonstrated that compound 1 is a new sesquiterpenoid tropolone glycoside, identified as 2-isopropenyl-9-methyl-1,2,3,4-tetrahydrobenzocyclohepten-7-one-6-O-[6′-(3″-hydroxy-3″-methylglutaryl)]-β-D-glucopyranoside and given the trivial name liriosmaside A.
Compound 2 was isolated as a pale yellow powder and using high-resolution mass spectrometry produced a molecular ion, [M ─ H]−, at m/z 553.2302 using electrospray ionization in the negative mode, consistent with the molecular formula, C27H38O12. This suggested nine degrees of unsaturation. Its UV spectrum showed absorption maxima at 243 and 335 nm. The 1H and 13C NMR spectra of 2 were similar to those of 1 (Table 1), indicating that they share the same skeleton: a sesquiterpenoid tropolone glycoside with an attached 3-hydroxy-3-methylglutaric acid group. Similarly, the sugar unit was confirmed as β-D-glucose using the same method as for compound 1. The only difference observed between 1 and 2 was that C-2 was found to be substituted with an isopropanol group in compound 2 instead of an isopropenyl group in compound 1. The absolute configuration of C-2 also followed the absolute configuration of C-2 in manicol.8 The HMBC spectrum (Figure 1) showed correlations from CH3-13 and CH3-14 to C-12 and C-2, requiring that the isopropanol group be joined to C-2. The HMBC correlation from CH3-15 to C-9 necessitated that the 15-methyl group is attached at C-9. The HMBC correlation from H-1′ to C-6 confirmed that C-1′ in glucose is linked to C-6 in the sesquiterpenoid tropolone moiety by an ether bond, and the correlations from H-6′a and H-6′b to C-1″ established that C-1″ in the 3-hydroxy-3-methylglutaric acid group is joined to C-6′ in glucose by an ester linkage. The foregoing data suggested that compound 2 is also a new sesquiterpenoid tropolone glycoside, identified as 2-isopropanol-9-methyl-1,2,3,4-tetrahydrobenzocyclohepten-7-one-6-O-[6′-(3″-hydroxy-3″-methylglutaryl)]-β-D-glucopyranoside and was assigned the trivial name liriosmaside B.
Two additional known compounds, secoxyloganin and oplopanpheside C, were isolated from the methanol extract of the roots of L. ovata. Their structures were confirmed by comparison to published spectroscopic data.9,12 In 1980, the sesquiterpene manicol was isolated from the root bark of the Guyanan tree Dulacia guianensis (Olacaceae).13 Originally a eudesmane-type structure was assigned to manicol, but on further degradation and derivatization experiments it was found to be inconsistent with the eudesmane skeleton. X-ray crystallographic analysis of the compound revealed the skeleton of manicol as a hydroxytropolone compound.8 Recently, manicol was reported as a potent and specific inhibitor of ribonuclease H (RNase H) activity of human immunodeficiency virus (HIV) reverse transcriptase (RT) in vitro.14
In an in vitro study, compound 1 weakly inhibited RNase H enzymatic activity, with an IC50 value of 34 μM, with compound 2 inactive at the dose tested (Figure 2 and Table 2). In addition, tropolones previously tested in which the extra hydroxy group was missing have shown only weak inhibitory activity in this assay at best.14 Neither 1 nor 2 was active in the HIV cytopathicity assay up to a high concentration of 50 μM, and neither compound was cytotoxic to CEM-SS cells up to that concentration, which is in accordance with other cell growth inhibition data (Table 2).
Figure 2.
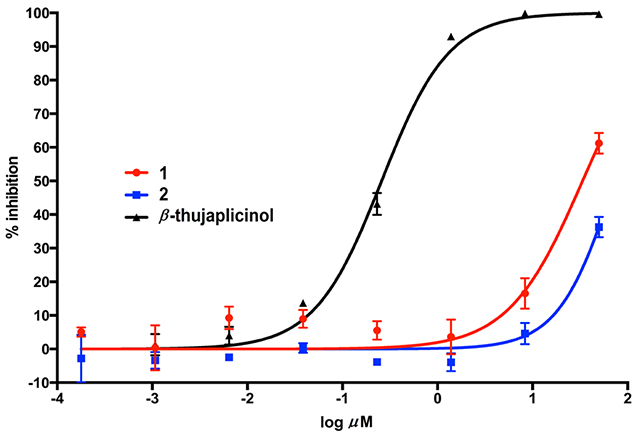
RNase H inhibitory evaluation of compounds 1 and 2 and β-thujaplicinol.
Table 2.
HIV RNase H Inhibitory and Cytopathicity Assay Dataa
| compound | IC50 HIV RNase H (μM) |
EC50 HIV cytopathicity (μM) |
IC50 HIV cytopathicity (μM) |
|---|---|---|---|
| 1 | 34 | no protection | >50 |
| 2 | >50 | no protection | >50 |
| β-thujaplicinol | 0.26 | no protection | 2.3 |
| dideoxycytidine | not tested | 0.079 | 61 |
IC50 values are the average of triplicate analyses.
EXPERIMENTAL SECTION
General Experimental Procedures.
Optical rotations were measured on an Autopol II automatic polarimeter (Rudolph Research Analytical, Hackettstown, NJ, USA). UV spectra were measured on an Agilent 8453 UV–visible spectroscopy system (Agilent Technologies, Santa Clara, CA, USA). Infrared spectra were measured on an Agilent FTS 7000e IR spectrometer. NMR spectra were recorded on an Agilent MR600 with a 13C/1H optimized OneProbe with compounds dissolved in CD3OD. A Exactive quadrupole–Orbitrap mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) coupled to an Accela UHPLC system (Thermo Fisher Scientific, San Jose, CA, USA) via a HESI-II electrospray ionization (ESI) source was used in this study for high-resolution mass spectrometry. The chromatographic separation was carried out using a 150 mm × 2.1 mm i.d., 1.7 μm particle size, Acquity UPLC BEH C18 reversed-phase analytical column (Waters, Milford, MA, USA) maintained at 35 °C. The injection volume was 2.0 μL. The autosampler temperature was maintained at 6 °C. The mobile phase consisted of 0.1% formic acid in water (eluent A) and 0.1% formic acid in acetonitrile (eluent B). The gradient elution was performed as follows: 0–1 min eluent B 5% (flow 200 μL/min); 1–10 min eluent B 5–60% (flow 200 μL/min); 10–12 min eluent B 60% (flow 200 μL/mm); column equilibration 12–15 min eluent B 5% (flow 200 μL/min). The mobile phase flow was diverted to waste between 3.0 and 10.0 min of the run. The mass spectrometer was operated in negative ESI full MS acquisition mode with the use of the following parameter settings: spray voltage, 3.5 kV; sheath gas, 35 arbitrary units; auxiliary gas, 10 arbitrary units; sweep gas, 3 arbitrary units; capillary temperature, 350 °C; S lens RF level, 50; heater temperature, 400 °C. Full mass spectra were recorded at a mass resolving power of 140 000 full width at half-maximum (fwhm, calculated for m/z 200) in the range m/z 100–800. TLC analysis was performed on silica gel 60 F254 plates (Merck KGaA, Darmstadt, Germany) using 0.1% formic acid in a CHCl3─MeOH solvent system with compounds visualized by spraying with anisaldehyde solution (Sigma-Aldrich, St. Louis, MO, USA) and heating at 120 °C. Sephadex LH-20 (25–100 μm) was purchased from Sigma-Aldrich. Flash chromatography was performed using a Biotage Isolera One flash purification system with silica gel and C18 SNAP flash cartridges (Biotage, Charlotte, NC, USA). GC-MS analysis was carried out on an Agilent 6890N GC system and 5973 mass selective detector [column: Restek Rtx-5MS, 0.25 μm, 0.25 mm × 30 m; carrier gas He; injection temperature 280 °C; detection temperature 280 °C; column temperature 150 °C (1 min), 10 °C/min to 250 °C (20 min)]. Sugar standards were purchased from Sigma-Aldrich.
Plant Material.
The roots of Liriosma ovata were collected in Nina Rumi, Maynas Province, District of San Juan, Peru (S 3°50′32.9″; W 73°22′39.7″) in January 2012 following international conventions in regard to plant collections and authenticated by Mr. Cesar Grandez and Mr. Victor Pinedo, Faculty of Biological Sciences, National University of the Peruvian Amazon, Iquitos, Peru. A voucher specimen (reference: 19478) was deposited at Botanical Liaisons, LLC, Boulder, CO, USA.
Extraction and Isolation.
The powdered root material of L. ovata (1 kg) was extracted exhaustively with MeOH at room temperature (4 L × 10, 3 h each time) and concentrated in vacuo to yield 159.0 g of MeOH extract. A portion of the MeOH extract (156.4 g) was suspended in water (1 L), then partitioned sequentially with the same volumes of hexanes, EtOAc, and n-BuOH, three times for each solvent, to afford hexanes (16.0 g), EtOAc (61.1 g), and n-BuOH (28.9 g) fractions, respectively. The EtOAc fraction (61.1 g) was defatted with hexanes (1 L × 3) to yield 3.3 g of defatted EtOAc fraction (E). Fraction E (3.3 g) was fractionated on a Sephadex LH-20 column (4.1 cm × 50 cm) eluting with MeOH with a flow rate of 1 mL/min (25 mL/fraction) to produce the combined fraction E3 (650 mg). Fraction E3 (650 mg) underwent RP-C18 flash chromatography (120 g, 50 μm, 39 × 157 mm, H2O─MeOH, from 80:20 to 10:90, 25 mL/min flow rate, monitored at 244 and 341 nm, 22 mL/fraction) to afford the combined subfraction E3-2 (58 mg). The n-BuOH fraction (B, 28.9 g) was fractionated on a Sephadex LH-20 column using the same conditions described previously to produce the combined fraction B2 (20.0 g). Fraction B2 (20.0 g) underwent RP-C18 flash chromatography (400 g, 50 μm, 71 × 168 mm, H2O─MeOH, from 90:10 to 50:50, 50 mL/min flow rate, monitored at 254 nm, 120 mL/fraction) to give 10 combined subfractions, B2-1 to B2-10. Subfraction B2-10 (6.7 g) was subjected to RP-C18 flash chromatography (400 g, 50 μm, 71 × 168 mm, H2O─MeOH, from 70:30 to 20:80, 50 mL/min flow rate, monitored at 254 nm, 120 mL/fraction) to produce the combined subfraction B2-10-1 (166 mg). On the basis of their similar TLC profiles, subfractions B2-10-1 and E3-2 were combined and further purified by silica gel flash chromatography (50 g, 25 μm, 39 × 81 mm, 0.1% formic acid in CHCl3─MeOH, from 90:10 to 70:30, 30 mL/min flow rate, monitored at 244 and 341 nm, 9 mL/fraction) to yield compound 1 (30 mg). Subfraction B2-7 (775 mg) was subjected to multiple RP-C18 flash chromatography separations (0.1% formic acid in H2O─MeOH, monitored at 245 and 345 nm: 120 g, 50 μm, 39 × 157 mm, from 80:20 to 40:60, 25 mL/min flow rate, 22 mL/fraction; 30 g, 50 μm, 30 × 72 mm, from 70:30 to 50:50, 15 mL/min flow rate, 9 mL/fraction; and 12 g, 50 μm, 21 × 55 mm, from 58:42 to 53:47, 10 mL/min flow rate, 5 mL/fraction), then further purified by silica gel flash chromatography (10 g, 25 μm, 21 × 55 mm, 0.1% formic acid in CHCl3─MeOH, from 90:10 to 85:15, 10 mL/min flow rate, monitored at 245 and 345 nm, 5 mL/fraction) to yield compound 2 (3 mg). Subfraction B2-2 (197 mg) was subjected to RP-C18 flash chromatography (30 g, 50 μm, 30 × 72 mm, H2O─MeOH, from 95:5 to 75:25, 15 mL/min flow rate, monitored at 243 nm, 9 mL/fraction), silica gel flash chromatography (25 g, 25 μm, 30 × 72 mm, 0.1% formic acid in CHCl3─MeOH, from 90:10 to 70:30, 15 mL/min, monitored at 236 nm, 15 mL/fraction), and RP-C18 flash chromatography (12 g, 50 μm, 21 × 55 mm, 0.1% formic acid in H2O─MeOH, 80:20 to 75:25, 10 mL/min flow rate, monitored 220 and 236 nm, 5 mL/fraction) to yield secoxyloganin (7 mg). Subfraction B2-3 (358 mg) was subjected to silica gel flash chromatography (25 g, 25 μm, 30 × 72 mm, 0.1% formic acid in CHCl3─MeOH, from 90:10 to 90:20, 15 mL/min, monitored at 236 nm, 15 mL/fraction) and RP-C18 flash chromatography (12 g, 50 μm, 21 × 55 mm, 0.1% formic acid in H2O─MeOH, 80:20 to 75:25, 10 mL/min flow rate, monitored 223 and 265 nm, 5 mL/fraction) to yield oplopanpheside C (6 mg).
Liriosmaside A (1): pale yellow powder; [α]25D −16.9 (c 0.21, MeOH); UV (MeOH) λmax (log ε) 243 (4.43), 340 (3.90) nm; IR (KBr) νmax 3435, 2926, 2855, 1731, 1645, 1595, 1541, 1457, 1384, 1248, 1215, 1067, 1026, 892, 719 cm−1; 1H and 13C NMR data, see Table 1; HRESIMS m/z 535.2191 [M ─ H]− (calcd for C27H35O11, 535.2185).
Liriosmaside B (2): pale yellow powder; [α] 25D −2.6 (c 0.27, MeOH); UV (MeOH) λmax (log ε) 243 (3.86), 335 (3.27) nm; IR (KBr) υmax 3432, 2926, 2854, 2826, 2740, 1730, 1652, 1607, 1386, 1353, 1069, 792 cm−1; 1H and 13C NMR data, see Table 1; HRESIMS m/z 553.2302 [M – H]− (calcd for C27H37O12, 553.2290).
Determination of Absolute Configuration of Sugars.
The absolute configurations of the sugars were determined by comparing the GC-MS retention times of the acetylated thiazolidine derivative of the standard sugar and the sugar obtained by hydrolysis of the compound as described previously.15 The retention times of standards D-glucose and L-glucose were 22.9 and 23.4 min, respectively.
HIV RNase H Inhibitory and Cytopathicity Assay.
Inhibitory activity against HIV RT-associated ribonuclease H was determined as previously described,16 and β-thujaplicinol was used as a positive control. The HIV cytopathicity assay was conducted as previously reported,17 and dideoxycytidine was used as a positive control.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Dr. J. I. Rader (U.S. FDA) for her scientific advice, Dr. L. Vaclavik (U.S. FDA) for the HRESIMS measurements, Dr. A. R Fardin-Kia (U.S. FDA) for the UV spectroscopic measurements, and Dr. M. M. Mossoba (U.S. FDA) for the IR spectroscopic measurements. We thank Mr. C. Grandez and Mr. V. Pinedo for the collection and authentication of plant material.
Footnotes
Supporting Information
1H NMR, 13C NMR, 1H─1H COSY, HSQC, and HMBC spectra of compounds 1 and 2 are available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- (1).McGuffin M; Kartesz JT; Leung AY; Tucker AQ Herbs of Commerce, 2nd ed.; American Herbal Products Association: Silver Spring, MD, 2000; p 59. [Google Scholar]
- (2).Youngken HW In American Druggist and Pharmaceutical Record; American Druggist Publishing Co.: New York, 1921; Vol. 69, September, p 31. [Google Scholar]
- (3).Sleumer HO Flora Neotropica, New York Botanical Garden: Bronx, NY, 1984; Monograph 38, pp 12, 127. [Google Scholar]
- (4).Gladstar R Rosemary Gladstar’s Herbal Recipes for Vibrant Health: 175 Teas, Tonics, Oils, Salves, Tinctures, and Other Natural Remedies for the Entire Family, Storey Publishing: North Adams, MA, 2008; p 351. [Google Scholar]
- (5).Novello CR; Marques LC; Miyazaki CR; Milaneze-Gutierre MA; Carneiro-Torres DS; Sarragiotto MH; de Mello JCP Braz. J. Pharmacogn 2012, 22, 946–956. [Google Scholar]
- (6).Iwasa J; Kimura Y Yakugaku Zasshi 1969, 89, 1172–1174. [DOI] [PubMed] [Google Scholar]
- (7).Picerno P; Mencherini T; Rastrelli L; Piccinelli A; Aquino R J. Nat Prod 2008, 71, 265–268. [DOI] [PubMed] [Google Scholar]
- (8).Polonsky J; Beloeil JC; Prange T; Pascard C; Jacquemin H; Donnelly DMX; Kenny PTM Tetrahedron 1983, 39, 2647–2655. [Google Scholar]
- (9).Huang WH; Zhang QW; Meng LZ; Yuan CS; Wang CZ; Li SP Chem. Pharm. Bull 2011, 59, 676–679. [DOI] [PubMed] [Google Scholar]
- (10).Smite E; Lundgren LN; Andersson R Phytochemistry 1993, 32, 365–369. [Google Scholar]
- (11).Dubois MA; Wierer M; Wagner H Phytochemistry 1990, 29, 3369–3371. [Google Scholar]
- (12).Calis I; Sticher O Phytochemistry 1984, 23, 2539–2540. [Google Scholar]
- (13).Polonsky J; Varon Z; Jacquemin H; Donnelly DMX; Meegan MJ J. Chem. Soc. Perkin Trans 1 1980, 2065–2069. [Google Scholar]
- (14).Budihas SR; Gorshkova I; Gaidamakov S; Wamiru A; Bona MK; Parniak MA; Crouch RJ; McMahon JB; Beutler JA; Le Grice SF Nucleic Acids Res. 2005, 33, 1249–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Ma J; Whittaker P; Keller AC; Mazzola EP; Pawar RS; White KD; Callahan JH; Kennelly EJ; Krynitsky AJ; Rader JI Planta Med. 2010, 76, 1758–1761. [DOI] [PubMed] [Google Scholar]
- (16).Parniak MA; Min KL; Budihas SR; Le Grice SF; Beutler JA Anal. Biochem 2003, 322, 33–39. [DOI] [PubMed] [Google Scholar]
- (17).Weislow OS; Kiser R; Fine DL; Bader J; Shoemaker RH; Boyd MR J. Natl. Cancer Inst 1989, 81, 577–586. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.