

Abstract
ABCB1 (P-glycoprotein/P-gp) is an ATP-binding cassette transporter well known for its association with multidrug resistance in cancer cells. Powered by the hydrolysis of ATP, it effluxes structurally diverse compounds. In this chapter, we discuss current views on the molecular basis of the substrate polyspecificity of P-gp. One of the features that accounts for this property is the structural flexibility observed in P-gp. Several X-ray crystal structures of mouse P-gp have been published recently in the absence of nucleotide, with and without bound inhibitors. All the structures are in an inward-facing conformation exhibiting different degrees of domain separation, thus revealing a highly flexible protein. Biochemical and biophysical studies also demonstrate this flexibility in mouse as well as human P-gp. Site-directed mutagenesis has revealed the existence of multiple transport-active binding sites in P-gp for a single substrate. Thus, drugs can bind at either primary or secondary sites. Biochemical, molecular modeling, and structure-activity relationship studies suggest a large, common drug-binding pocket with overlapping sites for different substrates. We propose that in addition to the structural flexibility, the molecular or chemical flexibility also contributes to the binding of substrates to multiple sites forming the basis of polyspecificity.
1. INTRODUCTION
ATP-binding cassette (ABC) transporters such as ABCB1 (P-glycoprotein/P-gp), ABCG2, and ABCC1 are well known for their association with multidrug resistance (MDR), effluxing structurally diverse compounds, powered by the hydrolysis of ATP (Ambudkar et al., 1999). P-gp also plays an important role in the pharmacokinetics of many drugs, altering their absorption, distribution, and excretion. P-gp has been extensively studied since 1976, when it was identified as the multidrug efflux pump in Chinese hamster ovary cells that had been selected for resistance to colchicine (Juliano & Ling, 1976). It is a 170 kDa single polypeptide chain consisting of two transmembrane domains (TMDs) and two nucleotide-binding domains (NBDs). It is believed that this transporter functions through an alternate access mechanism involving two different conformations (Hollenstein, Dawson, & Locher, 2007; Jardetzk, 1966; Senior, al-Shawi, & Urbatsch, 1995; van Wonderen et al., 2014). Drug binding occurs when the protein adopts an inward-facing conformation (inverted V appearance) observed in P-gps such as those found in mouse and Caenorhabditis elegans P-gp X-ray structures obtained in the absence of nucleotide (Aller et al., 2009; Jin, Oldham, Zhang, & Chen, 2012). This is followed by a significant structural change to an outward-facing conformation (V-shaped in appearance) such as the one exhibited by the X-ray structure of SAV1866 with bound nucleotide (ADP) (Dawson & Locher, 2006), when drug release takes place. Hydrolysis of ATP is believed to reset the protein to the inward-facing form to begin a new cycle of drug binding and release (Callaghan, Ford, & Kerr, 2006; Sauna & Ambudkar, 2000). The switch from inward to outward form certainly requires a highly flexible structure.
Substrate “promiscuity” or polyspecificity is a well-known characteristic of P-gp and the subject of much research. Attempts have been made to understand the ability of P-gp to recognize various chemically and structurally diverse substrates through biochemical investigations and structural studies. Despite all these studies, the molecular basis of this unusual property still remains poorly understood and is a matter of intense debate. Other review articles have been published recently on the subject (Gutmann, Ward, Urbatsch, Chang, & van Veen, 2010; Sharom, 2014; Wong, Ma, Rothnie, Biggin, & Kerr, 2014). In this review, we will focus on recent biochemical and structural studies of P-gp, with discussion primarily on its substrate polyspecificity.
2. MOLECULAR BASIS OF POLYSPECIFICITY
2.1. Structural flexibility revealed by X-ray crystallography
In 2009, Aller et al. (2009) were the first to report an X-ray structure of a mammalian ABC transporter. They solved the structure of mouse P-gp at a resolution of 3.8–4.4 Å, in the absence of nucleotide, with and without bound inhibitors. In the absence of nucleotide or drug-substrate, the mouse P-gp structure (3G5U.pdb) was observed to adopt an inward-facing conformation. It has a large internal cavity of about 6000 Å3 open to both the cytoplasm and the membrane inner leaflet, with a wide separation between the two NBDs. In order to compare the separation of the NBDs in different X-ray structures with biochemical and biophysical data available in the literature that report on the separation between these domains, distances between the domains are measured between the cysteine residues of the Walker A motif. Thus, the distance between the α-carbons of the Walker A cysteines C427 and C1070 in the mouse apo-form P-gp X-ray structure (4M1M.pdb) is 38 Å (see Fig. 1). The distances between these residues in another mouse P-gp structure (4KSC.pdb) and C. elegans P-gp (4F4C. pdb) are 44 and 53 Å, respectively (Jin et al., 2012; Ward et al., 2013).
Figure 1.
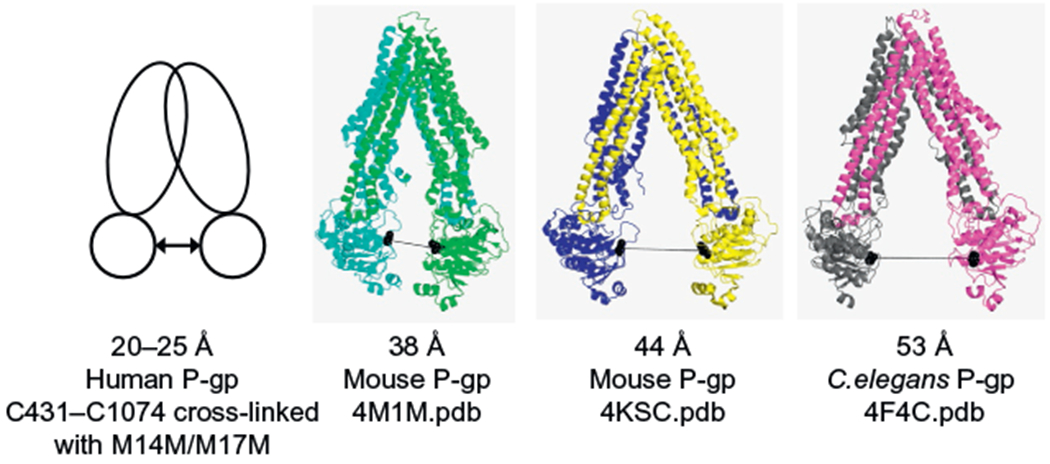
Comparison of the separation of the two nucleotide-binding domains (NBDs) in human, mouse, and C. elegans P-gps based on cross-linking studies and X-ray crystallography. The first panel to the left shows a schematic representation of the human P-gp structure based on cross-linking studies (Sim, Bhatnagar, Chufan, Kapoor, & Ambudkar, 2013), while the rest of the panels gather the crystal structures of mouse and C. elegans P-gps as cartoon models (4F4C.pdb, Jin et al., 2012; 4M1M.pdb, Li, Jaimes, & Aller, 2014; 4KSC.pdb, Ward et al., 2013). The figure is ordered from the data that show the NBD domains least separated to the most separated. The distance in Angstroms indicates the separation between the cysteine residues of the Walker A motif (431-1074 in human; 427-1070 in mouse; 455-1116 in C. elegans). The double arrow symbol denotes the distance between residues C431-C1074 in human P-gp. The X-ray structures are colored in the following manner: green and cyan, yellow and blue, and magenta and gray for the N-terminal and C-terminal halves of mouse P-gp 4M1M. pdb, 4KSC.pdb and C. elegans P-gp 4F4C.pdb, respectively. Both Walker A cysteine residues are shown as black balls in the X-ray crystal structures, and the black line represents the distance between them.
Mouse P-gp was also co-crystallized with two stereo-isomers of cyclic hexapeptide inhibitors, cyclic-tris-(R)-valineselenazole (QZ59-RRR) and cyclic-tris-(S)-valineselenazole (QZ59-SSS) (Aller et al., 2009; Tao et al., 2011). While one molecule of QZ59-RRR binds one molecule of P-gp, two molecules of QZ59-SSS were found in the central cavity of P-gp (Fig. 2A). This is consistent with biochemical studies demonstrating binding of two different compounds in the drug-binding pocket (Ambudkar, Kim, & Sauna, 2006). Also, this clearly shows that the internal cavity is able to accommodate at least two molecules of the same compound at the same time. The biochemical results also showed that small differences in the physicochemical features of a compound such as the optical property (R-enantiomer vs. the S-enantiomer) are sufficient to be differentiated in terms of binding sites. On the other hand, the fact that there are minimal structural differences between the apo-form and the inhibitor-bound form has raised some concerns (Gottesman, Ambudkar, & Xia, 2009). Biochemical evidence suggests that drug binding proceeds through an induced-fit mechanism (Loo, Bartlett, & Clarke, 2003b) and hence one would expect at least some conformational changes upon inhibitor binding.
Figure 2.
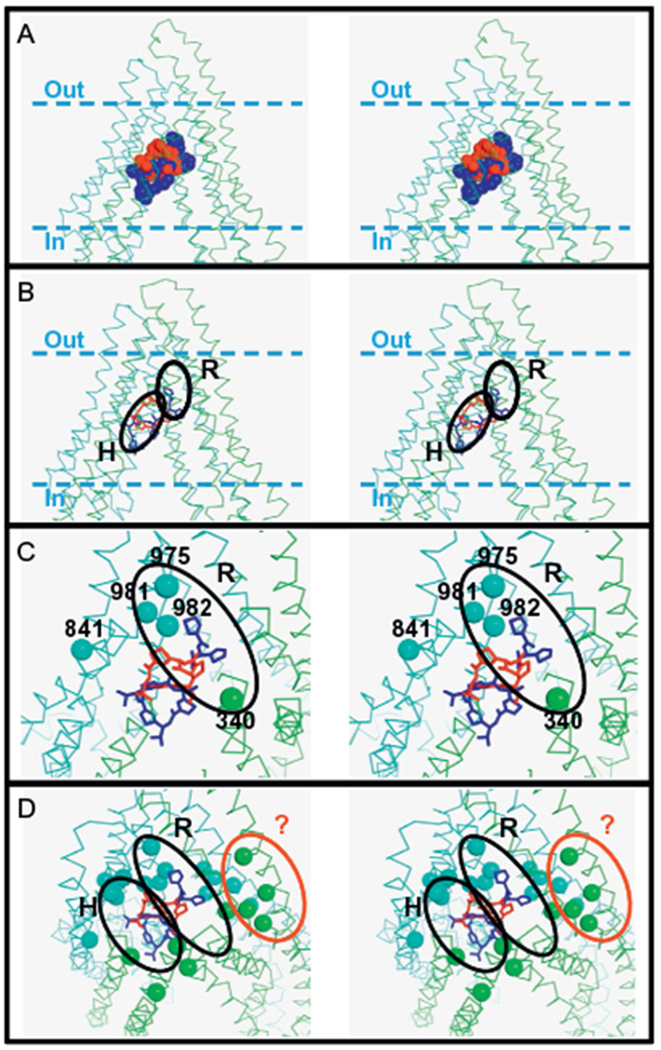
The proposed drug-binding sites on P-gp. Panel A: The sites where the cyclic peptides QZ59-RRR (red) and QZ59-SSS (blue) bind mouse P-gp as determined by X-ray crystallography (4M2S.pdb and 4M2T.pdb, respectively) are shown in ball models. Panel B: The proposed location of the R and H sites are shown in the homology model of human P-gp based on the mouse P-gp structure (Martinez et al., 2014; Pajeva et al., 2013). Panel C: The residues of the rhodamine B binding site determined by cysteine-scanning mutagenesis (Loo & Clarke, 2002) are shown as balls (at the α-carbon position) and compared with the proposed location of the R site. Panel D: The residues of the verapamil-binding site determined by cysteine-scanning mutagenesis are shown as balls (at the α-carbon position) and compared with the R and H sites. A group of residues (61-64-65, 118-125, 942-945, 868-871-872) suggest the existence of a verapamil-binding site different from the proposed R and H; this site is demarcated as a red oval and indicated with a question mark. In all the panels, the structure of P-gp is shown as a ribbon model in green (N-terminal) and cyan (C-terminal). The approximate location of the plasma membrane is demarcated with dashed lines in panels A and B. Stick models of the QZ59 molecules (RRR-isomer in red and SSS-isomer in blue) are shown in panels B, C, and D for reference. The four panels are shown as stereo images. The figures were prepared with PyMOL 1.5.0.5.
Very recently, Aller and coworkers published refined X-ray structures of mouse P-gp (Li et al., 2014). In these improved models, major registry shift corrections were made to six transmembrane helices in the drug-binding pocket as well as three of the four intracellular helices, the “elbow helices” which are important for communication between the TMDs and NBDs and minor corrections were made to the extracellular loops. The new structures are certainly better models than the original ones, as reflected by their Ramachandran plots. The new structures contain 95% of residues in the favorable Ramachandran region compared to 57% for the previous models.
Shortly after the publication of the first mouse P-gp structures, the crystal structure of the P-gp of another species, C. elegans was reported at 3.4 Å resolution (Jin et al., 2012). C. elegans P-gp (gene P-gp-1, GenBank accession code AB01232.1) is only 46% identical to human P-gp, whereas mouse P-gp is 87% identical. The C. elegans P-gp similarity is even lower if the sequence is compared at the drug-binding pocket (20% identical, considering the residues that were found to interact with the QZ59 cyclic peptides in mouse P-gp X-ray structures). Sixty ABC transporter genes from the C. elegans genome have been annotated in GenBank and fifteen were assigned as P-gp genes (P-gp-1 to P-gp-15) (Sheps, Ralph, Zhao, Baillie, & Ling, 2004). The C. elegans P-gp-3 gene confers resistance to colchicine and chloroquine in nematodes as determined by transposon-mediated deletion mutagenesis studies, suggesting that soil nematodes may express P-gp to protect themselves against environmental toxins (Broeks, Janssen, Calafat, & Plasterk, 1995). Jin et al. (2012) showed that cells infected by recombinant baculovirus carrying the C. elegans P-gp-1 gene acquire resistance to actinomycin D and paclitaxel, two anticancer drugs which are also well-known substrates of human P-gp. Nonetheless, important differences are expected at least in terms of substrate specificity between human and C. elegans P-gps, based on the low sequence similarity at the drug-binding pocket.
The structure of C. elegans P-gp also shows an inward-facing conformation, similar to that seen in mouse P-gp as reported by Aller and coworkers. The drug translocation pathway is open to the cytoplasmic surface and also continuous with the membrane inner leaflet. The degree of separation between the two NBDs in C. elegans is found to be much larger (53 Å) compared to that of the mouse P-gp, as measured by the distance that separates the Walker A cysteine residues (Fig. 1).
More recently, Ward et al. (2013) reported three new mouse P-gp X-ray structures, all in the inward-facing conformation. The crystals were obtained in the absence of nucleotides and inhibitors, but one of them was prepared in the presence of a nanobody that binds P-gp at the C-terminal side of the first NBD. In the apo-X-ray structures, the NBDs are more separated than in the first reported mouse P-gp structures (Aller et al., 2009; Li et al., 2014). The distances between the NBDs, as measured by the alpha carbon atom of Walker A cysteine residues 427 (NBD1) and 1070 (NBD2), are 42 and 44 Å in both apo-P-gp structures solved at 3.8 and 4.2 Å resolution, respectively. The separation is wider than that observed in the first-reported mouse P-gp crystal structure (38 Å, see Fig. 1) (Aller et al., 2009), but shorter than the distance observed in C. elegans P-gp (53 Å). When mouse P-gp is co-crystallized with a nanobody, the inward-facing conformation is the narrowest, with a distance of about 35 Å between the two Walker A cysteine residues.
2.2. Structural flexibility probed with disulfide cross-linking and biophysical methods
As the molecular structure of human P-gp has not yet been determined, the distance between the NBDs of human P-gp has only been investigated biochemically using disulfide cross-linking studies between cysteine residues in the NBDs. In a recent study, Sim et al. (2013) found that the native cysteines (C431 and C1074) in the Walker A domain of the NBDs (in a cysless background P-gp) can be successfully cross-linked with homo-bifunctional thiol-reactive cross-linkers. These cross-linkers have methane thiol sulfonate (MTS) functional groups on both ends, which react specifically with cysteines. As such, these MTS cross-linkers act as molecular rulers, allowing the determination of distances between the reporter cysteines at the NBDs. With cross-linkers M14M and M17M of spacer arm lengths defined to be 20 and 25 Å, respectively, it was found that C431 and C1074 can be cross-linked via disulfide cross-linking to form a cross-linked product that travels with slower mobility and so appears as a separate band at a higher molecular weight from the uncross-linked P-gp (Sim et al., 2013). This report showed that the distance between the NBDs of human P-gp is the closest (20–25 Å), when compared to the mouse and the C. elegans P-gp crystal structures (see Fig. 1). It was not possible to determine if the NBDs could be cross-linked beyond 25 Å due to the lack of availability of longer cross-linkers. Nonetheless, these cross-linking results show that the NBDs of human P-gp can still vary in distance by approximately 5 Å. Thus, the biochemical studies also support the structural work on the flexibility of P-gp.
The structural flexibility of P-gp has also been investigated using biophysical methods such as electron paramagnetic resonance (EPR) (Wen, Verhalen, Wilkens, Mchaourab, & Tajkhorshid, 2013) and fluorescence resonance energy transfer (FRET) (Verhalen, Ernst, Borsch, & Wilkens, 2012). In the report by Wen et al., the EPR signals obtained from the spin-labeled NBD mutants of mouse P-gp show wide distributions covering both longer and shorter distances than those observed in the crystal structures. The broad EPR signals of the three double-Cys pairs used in this study indicates a heterogeneous population of molecules with different conformations. For one pair of mutations (613C and 1258C in the NBDs), the distance distribution was centered at about 58 Å and had a width over 20 Å. Also, using molecular dynamics, the distance between the two NBDs in equilibrium is at least 20 Å in the apo state. These examples illustrate once again the structural flexibility of mouse P-gp, which appears to be even more flexible than human P-gp. In this study, the authors suggested that the TMDs of P-gp might be more flexible than in other ABC exporters or in P-gp bacterial homologs. Their structural explanation was that there is more helical kinking and/or unwinding within the TMDs as a result of a higher number of helix-breaking residues such as glycine or proline. These glycine/proline residues may provide the extra flexibility needed to accommodate substrates of various shapes and sizes. Interestingly, in P-gp, there are significantly more glycine residues in the TMDs than in other ABC exporters (Wen et al., 2013).
Using the single-molecule FRET technique, Verhalen and coworkers (Verhalen et al., 2012) tested four conditions. Those conditions were (i) apo; (ii) Mg-ATP and verapamil; (iii) vanadate-trapped (Mg-ATP plus verapamil plus orthovanadate); and (iv) Mg-ATP and cyclosporine A. It was observed that in apo-P-gp, there is low FRET efficiency and the authors proposed that this is likely to be due to the large distance between the dye molecules that are conjugated to the NBDs. In all cases, there was a broad distribution in the FRET efficiency profiles due to the flexibility of the dynamic protein that is constantly sampling a wide range of NBD conformations.
Even though many biochemical and biophysical studies done with P-gp in the inward-facing conformation have provided convincing results indicating that P-gp is a highly dynamic protein with NBDs covering a wide range of distances, the inward-facing conformation, as seen in the mouse and C. elegans crystal structure, is still regarded by some as a conformation that may not be physiologically relevant. The main reason for such concerns is that it is unlikely that P-gp would have no bound ATP, considering the affinity for ATP is around 0.5 mM and the ATP concentration in the cell is much higher than that (3–5 mM) (Ambudkar, Cardarelli, Pashinsky, & Stein, 1997; Gottesman et al., 2009). Further, disulfide cross-linking data (Loo, Bartlett, & Clarke, 2010; Verhalen & Wilkens, 2011) showed that TMDs and NBDs can be cross-linked and still have drug-stimulated ATPase activity, suggesting these domains can be close together and the wide separation between the NBDs as seen in the crystal structure may not be present during the steady state turnover. A human ABC transporter found in the inner membrane of mitochondria, ABCB10, has been recently crystallized in the presence of nucleotide (Shintre et al., 2013). Interestingly, three structures with bound ATP analogs (one with AMPPNP and two with AMPPCP) show the protein in the inward-facing conformation. Although the nucleotide-bound NBDs are not close enough to effectively produce ATP hydrolysis, these structures show that the inward-facing conformation may exist in the presence of ATP. Also, they show a great degree of plasticity in transmembrane α-helices, further supporting the observation that in general ABC transporters are highly flexible proteins.
In an early paper by Loo and Clarke (2000b), using disulfide cross-linking in double cysteine mutants engineered between transmembrane segments 4, 5, 6, 10, 11, and 12, disulfide cross-linking was seen using an oxidant that spontaneously cross-linked these cysteine residues between TM6 and TM10, and TM6 and TM11. Disulfide cross-linking was also seen between TM4 and TM12, and TM5 and TM12. In the presence of drug substrates such as colchicine, verapamil, cyclosporine A, or vinblastine, this cross-linking effect was altered, suggesting the TM segments allow conformational changes to accommodate the binding of these structurally diverse substrates. Therefore in hindsight, the concept of structural flexibility was already suggested many years ago based on biochemical studies (Loo & Clarke, 2000b).
The range of distances between the NBDs of P-gp found in various X-ray structures, in biochemical determinations using cross-linkers of different lengths, EPR analyses as well as FRET experiments, clearly illustrate that human and mouse P-gp are highly flexible in the absence of nucleotide and that P-gp can sample a wide range of domain separations in inward-facing conformations. One of the remarkable features of P-gp is its ability to recognize substrates of very different molecular weights. It can transport small molecules such as cimetidine (MW 252) (Pan, Dutt, & Nelson, 1994), small peptides such as N-acetyl-leucyl-leucyl-norleucinal (ALLN, MW 383) (Sharma, Inoue, Roitelman, Schimke, & Simoni, 1992), as well as large molecules such as cyclosporine A (MW 1203) (Kerr, Sauna, & Ambudkar, 2001; Miller et al., 2000) and gramicidin D (MW 1882) (Schinkel, 1999). In order to transport molecules of very different sizes, it appears that P-gp must have a certain amount of structural flexibility.
2.3. Substrate polyspecificity and ligand-based studies
One of the most remarkable features of P-gp is its ability to bind and transport hundreds of structurally and functionally diverse substrates. A substrate is a molecule that binds to the protein and is transported outside the cell. In terms of cell biology, a substrate is better defined as a chemical compound to which P-gp-expressing cells exhibit resistance in cytotoxicity assays. Long lists of P-gp substrates have been reported in previous reviews [see for example, Eckford & Sharom, 2009; Kapoor, Sim, & Ambudkar, 2013].
P-gp substrates are mainly hydrophobic and amphipathic compounds. They comprise a great variety of substances that include anticancer drugs (e.g., doxorubicin, paclitaxel, vinblastine); tyrosine kinase inhibitors (e.g., nilotinib, imatinib); calcium channel blockers (e.g., verapamil) and antibiotics (e.g., gramicidin D); HIV protease inhibitors (ritonavir, saquinavir); fluorescent dyes (e.g., rhodamine 123, daunorubicin), among many others. Surprisingly, this large group of substrates includes compounds of very different molecular weights, compounds composed of different chemical groups (e.g., aromatic; methoxy; amide linkages), and compounds exhibiting different topologies (e.g., organic molecules; linear and cyclic peptides, conjugate structures). It is thus difficult to describe what chemical features a compound must exhibit in order to be a P-gp substrate. In other words, it seems difficult to predict if a chemical compound is a P-gp substrate based solely on an analysis of its molecular structure. However, a number of attempts have been reported and some of them are described below.
In silico methods for predicting if a molecular entity is a P-gp substrate, such as measuring quantitative structure–activity relationships or the use of support vector machine method, are based on similarity to chemical structures and physicochemical properties of known substrates. The broad substrate specificity of P-gp makes it clear that there is no unique pharmacophore to describe the molecular features of a chemical entity to be recognized by P-gp. Therefore, studies have reported multiple pharmacophores for P-gp even though they have in common a certain degree of hydrophobicity and the presence of hydrogen bond acceptors (ether, carbonyl, hydroxyl, tertiary amino groups) (Demel et al., 2008). The presence of many nonpolar residues facing the central cavity of P-gp, such as phenylalanine, valine, leucine, and isoleucine, as well as of residues able to act as hydrogen bond donors, such as tyrosine and glutamine (see Fig. 3), are consistent with the common features of multiple P-gp pharmacophores. Donmez Cakil et al. (2014) recently showed that, for example, Y953 (TM11) forms a hydrogen bond with rhodamine 123 and propafenone analogs.
Figure 3.

Sequence alignment of transmembrane domains 1 and 2 of human P-gp. Residues 52–390 of domain 1 and 712–1033 of domain 2 were selected for alignment. Using the homology model of human P-gp based on mouse P-gp X-ray structure 4M1M.pdb, the residues facing the central cavity were selected and are shown in red color. The symbol “+” denotes similarity between residues.
Anna Seelig reported two P-gp pharmacophores based on a comparison of 100 chemical compounds recognized as P-gp substrates (Seelig, 1998). One pharmacophore has two hydrogen bond acceptors separated by a distance of 2.5 ± 0.3 Å and the second one has two or three hydrogen bond acceptors separated by 4.6 ± 0.6 Å (in the case of three acceptors, the 4.6 Å distance is between the two outer groups) (Seelig, 1998). Recently, the pharmacophoric features of nilotinib were investigated in relation to its ability to interact not only with P-gp but also with another ABC transporter (ABCG2) and the BCR-ABL kinase (Shukla, Kouanda, Silverton, Talele, & Ambudkar, 2014). Nilotinib is a second-generation tyrosine kinase inhibitor used in the treatment of chronic myeloid leukemia. The results showed seven pharmacophoric features for P-gp inhibitors, AADDRRR (A = hydrogen bond acceptor; D = hydrogen bond donor, and R = aromatic ring), with the hydrogen bond acceptors separated by 6 Å. Research on ligand–base predictions has been productive, and many articles and reviews on the topic have been published (Cramer, Kopp, Bates, Chiba, & Ecker, 2007; Demel et al., 2008; Ferreira, dos Santos, Ferreira, & Guedes, 2011; Li, Li, Eksterowicz, Ling, & Cardozo, 2007; Pajeva, Globisch, & Wiese, 2009; Pajeva & Wiese, 2002; Penzotti, Lamb, Evensen, & Grootenhuis, 2002).
2.4. P-glycoprotein portals
All X-ray structures of mouse P-gp show two portals open to the inner leaflet of the membrane, delineated by the α-helices TMs 3 and 4 on one side, and TMs 9 and 10 on the other side (Li et al., 2014; Ward et al., 2013). These portals are created upon TM4 and 5 (and TM10 and 11) crossovers to make extensive contacts with the α-helical bundle of the opposite domain. The arrangement suggests the drugs can enter the central cavity through these particular portals or gates, although no experimental data is available to rule out the possibility drugs can enter through other ways.
This structural motif is also conserved in C. elegans P-gp (Jin et al., 2012), in the bacterial exporter MsbA (Ward, Reyes, Yu, Roth, & Chang, 2007) and in the mitochondrial ABCB10 (Shintre et al., 2013). In the case of the structure of C. elegans P-gp, an N-terminal helical hairpin seems to block one of these portals. Nonetheless, a truncation mutant that lacks this helical hairpin and the full length protein behaves essentially the same based on the stimulation of the ATPase activity by drugs (actinomycin D and paclitaxel) and a cytotoxicity assay (Jin et al., 2012). The structural role of this helical hairpin therefore remains unknown.
2.5. Drug-binding sites
In enzymes, the substrate-binding site is defined as the site or pocket where the chemical reaction takes place. In transporters, as there is no chemical reaction, the definition of drug-binding site is different. Furthermore, the substrate must interact with many residues from the moment it enters the central cavity of the transporter until the moment when it is released to the extracellular space. However, the conformational change from inward-opening (competent for drug binding) to outward-opening (competent for drug release) must be triggered for an interaction between the substrate and specific residues. Although the situation could be more complex than the one we are describing here, the specific residues that trigger important protein conformational changes can define the drug-binding site(s) in transporters. One of the salient features of P-gp and other MDR-linked ABC drug-transporters is that there are no positively or negatively charged residues in the drug-binding pocket. Therefore, the major interactions between substrates and the protein residues are hydrogen-bonding, van der Waals, and hydrophobic interactions.
The identification of the drug-binding site(s) in P-gp has been the goal of many investigations and it is currently the subject of extensive studies and debate. Many years ago, Loo and Clarke demonstrated that the drug-binding sites are within the TMDs of P-gp, because deletion of the NBD does not prevent drug binding (Loo & Clarke, 1999). Nonetheless, the TMD is large enough to hold many sites and attempts to identify those sites have been made. One of the methods used to identify drug-binding residues is cysteine-scanning mutagenesis and reaction with thiol-reactive agents. This method is feasible in P-gp because replacing all seven cysteines with alanines yields an active transporter (cysless P-gp) (Loo & Clarke, 1995). Residues from all 12 α-helices were mutated to cysteine in a cysless background and thiol-reactive substrates such as dibromobimane, MTS-verapamil, and MTS-rhodamine were tested for their reactivity against single cysteine mutants. The MTS is very reactive toward accessible cysteines in the protein. The approach consists in testing whether drugs (verapamil, rhodamine B) can protect the cysteine mutants from inhibition by MTS drugs. Those studies showed that many residues from different helices contribute to the drug-binding sites of verapamil and rhodamine B.
More than 20 residues from almost all α-helices were found to be part of the verapamil-binding site of P-gp [61, 64, 65 (TM1); 118, 125 (TM2); 222 (TM4); 306 (TM5); 339, 342 (TM6); 725, 728, 729 (TM7); 766 (TM8); 841, 842 (TM9); 868, 871, 872 (TM10); 942, 945 (TM11); 975, 982, 984, 985 (TM12)] (Loo, Bartlett, & Clarke, 2003a, 2006; Loo & Clarke, 1997, 2000a, 2001). Intriguingly, the residues cannot be clustered in a single binding site because some of them are separated by large distances (example, A841-H61 = 35 Å). Since the publication of these data, not much elaboration has been reported on the binding site(s) of verapamil. Using this approach and other approaches, residues of the binding site for rhodamine dyes (see below), propafenones, vinblastine, and colchicine (among others) have been reported, although they usually are not sufficient to define the drug-binding site for them.
2.6. The proposed R, H, and P sites
Approximately 15 years ago, Shapiro et al. proposed three drug-binding sites for P-glycoprotein, designated as the H (Hoechst), R (rhodamine), and P (prazosin and progesterone) sites, based on kinetics studies of drugs transported in isolated P-gp-rich plasma membrane vesicles from Chinese hamster ovary CHRB30 cells (Shapiro, Fox, Lam, & Ling, 1999; Shapiro & Ling, 1997). Both the H and R sites are proposed to be active for transport while the P site is essentially an allosteric site. The R site preferentially binds rhodamine 123 and anthracyclines (daunorubicin, doxorubicin); the H site preferentially binds Hoechst 33342, Hoechst 33258, quercetin, and colchicine; while the P site binds preferentially prazosin and progesterone. Interestingly, a positive cooperative effect between the R and H sites was observed. In other words, substrate binding to the R site stimulates transport of the substrate binding to the H site, and vice versa. The P site has different characteristics (see below). The authors also reported that the R and H sites have similar affinities for vinblastine, etoposide, and actinomycin D. At that time, not much structural information about the location of these three drug-binding sites was available.
Loo and Clarke found that residues I340 (TM6), A841 (TM9), L975 (TM12), V981 (TM12), and V982 (TM12) are part of the rhodamine B binding site, because this dye significantly protects the ATPase activity of mutants I340C, A841C, L975C, V981C, and V982C from inhibition by MTS-rhodamine (Loo & Clarke, 2002). In other words, these observations suggest that these residues are part of the R site originally proposed by Shapiro and coworkers. The homology model of human P-gp based on the refined X-ray structures of mouse P-gp shows residues I340, L975, V981, and V982 close enough to envision a drug-binding site for rhodamine dyes (Fig. 2B and C). Residues L975, V981, and V982 are all in helix 12, and the side chain of I340 (helix 6) is 11 Å away from the side chain of V982. The space between these residues is enough to accommodate a substrate and it seems is preserved in the closed conformation, after ATP hydrolysis, as suggested for the 13 Å separation these residues exhibit in the homology model of human P-gp based on the X-ray structure of SAV1866 (O’Mara & Tieleman, 2007). Residue A841 (TM9) is distant from this site (A841-I340 = 23 and 26 Å in the open and closed conformations, respectively). However, it is located at one of the portals and could be relevant for rhodamine entrance to the central cavity of P-gp (Fig. 2C).
Recently, Martinez et al. (2014) proposed the location of the R and H sites, taking advantage of the known position of the two cyclic peptides, QZ59-RRR and QZ59-SSS, at the central cavity of the mouse P-gp X-ray structure (see above) (Aller et al., 2009). The authors measured the effects of both QZ59 compounds on the transport of Hoechst 33342 (for the H site) and daunorubicin (for the R site) using a NIH3T3 cell line transfected with human wild-type MDR1 (ABCB1). And based on docking simulations (AutoDock Vina, using the 4LSG.pdb mouse X-ray sructure), they proposed the location of the H site along the central cavity, with residues F303, Y307, and Y310 as part of this site, with the R site at a deeper position in the cavity, overlapping the location of the QZ59-SSS molecule most embedded in the structure (Martinez et al., 2014) (Fig. 2B). The authors also made clear that the R site only slightly overlapped with the location of QZ59-RRR. This location of the R site coincides with previous modeling studies suggesting that rhodamine B and other molecules bind deeper in the cavity, although these simulations are somewhat in doubt because they were carried out with the unrefined mouse structure 3G60.pdb (Dolghih, Bryant, Renslo, & Jacobson, 2011). It is important to note that these docking simulations were performed with a flexible receptor, because dissimilar results were obtained using a rigid receptor. Modeling of rhodamine B in a rigid homology model of human P-gp based on 3G60.pdb show the molecule in approximately the same site as QZ59-RRR, below the proposed R site obtained through simulations using a flexible receptor (Bikadi et al., 2011). Although the use of rigid versus flexible receptor may be a matter of debate, evidence that drug binding proceeds through an induced-fit mechanism in P-gp (Loo et al., 2003b) plus the low resolution of the available X-ray structures of P-gp support the use of a flexible receptor for docking simulations.
In another recent study, Pajeva, Sterz, et al. (2013) proposed approximately the same location for both R and H sites, as described above (Fig. 2B). The authors also proposed that the potent inhibitors tariquidar and elacridar bind to P-gp sites that coincide or overlap with the putative R and H sites, based on the effects of these inhibitors on the accumulation of rhodamine 123 and Hoechst 33342 in adriamycin-resistant human ovarian carcinoma cells (Pajeva, Sterz, et al., 2013).
As mentioned above, the residues that form part of the verapamil-binding sites as determined by cysteine-scanning mutagenesis cannot be clustered in a single site, but several of them are within the proposed R and H sites (Fig. 2D). Interestingly, a group of residues (61-64-65, 118–125, 942–945, 868-871-872) suggests the existence of another binding site different from the R and H sites; this site is delineated in the homology model of human P-gp based on the mouse P-gp X-ray structure, in Fig. 2D ((dark gray in the print version) oval with a question mark).
2.7. Primary and secondary sites
Chufan et al. (2013) have recently reported that residues I306-Y307 (TM5), F343 (TM6), Q725-F728 (TM7), and F978-V982 (TM12) are part of a common drug-binding pocket for QZ59-S-SSS, cyclosporine A, tariquidar, valinomycin, and FSBA. These studies included QZ59-S-SSS, which is an analog of QZ59-SSS used for crystallographic studies, containing sulfur instead of selenium. It is expected that QZ59-S-SSS binds human P-gp at the same sites QZ59-SSS binds mouse P-gp, which was determined by X-ray crystallography (Li et al., 2014). In accord with the above-described location for the R and H sites, residues I306-Y307-F343-Q725-F728 are part of the H site, while F978-V982 are common to both the R and H sites. These data suggested that cyclosporine A, tariquidar, valinomycin, and FSBA bind to sites that overlap both the R and H sites, reinforcing the idea of overlapping binding sites for different drugs. The most important finding from that work was the observation that drugs bind to secondary sites when residues of the primary site are mutated (Chufan et al., 2013). The primary binding site of P-gp was defined as the site where drugs bind and inhibit the labeling with [125I]-iodoarylazidoprazosin (IAAP), which is a prazosin derivative that photo-cross-links P-gp when exposed to UV light (365 nm). Most substrates and modulators of P-gp inhibit the photo-crosslinking of P-gp in a concentration-dependent manner. Site-directed mutagenesis studies showed that several drugs lose this ability of inhibiting IAAP-photo-cross-linking upon mutation of residues of the common drug-binding pocket. These experiments thus define residues of the primary binding site. Importantly, upon mutation of residues of the primary site, substrates still bind P-gp and are transported outside the cell, revealing the existence of secondary binding sites (Chufan et al., 2013). These observations demonstrate that each drug-substrate can bind to more than one site and all sites (or most of them) are capable of transport function. The existence of multiple similar sites generates several possibilities including molecular or chemical flexibility, in addition to the structural flexibility that can also contribute to the broad substrate specificity of P-gp. As described above, the X-ray structure of mouse P-gp with bound QZ59-SSS (4M2T.pdb) showed two sites for the cyclic peptide (Fig. 2A) (Aller et al., 2009). The Atm1-family ABC exporter from Novosphingobium aromaticivorans (NaAtm1, with approximately 45% sequence identify with the human ABCB7) was recently co-crystallized with two molecules of oxidized glutathione (GSSG), bound to the protein at primary and secondary (lower occupancy) sites (Lee, Yang, Zhitnitsky, Lewinson, & Rees, 2014). Both the QZ59 cyclic peptides and the GSSG molecules bind at the central cavity of the exporter in the TMD, although the GSSG molecules bind closer to the cytoplasmic surface than the QZ59 compounds. Interestingly, the majority of the ligand-protein interactions involve TM5–TM6 and TM11–TM12 in both the mouse P-gp and the NaAtm1 exporter (Lee et al., 2014). Using mass spectrometry, Marcoux et al. (2013) showed that cyclosporine A binds to two sites on P-gp. Clearly, studies carried out with different techniques reveal that P-gp has more than one binding site even for the same substrate.
In an attempt to reconcile the definition of R and H sites with the concepts of primary and secondary sites, it seems appropriate to consider that P-gp has more than one R site (primary and secondary R sites) for rhodamine dyes and also has more than one H site (primary and secondary H sites) for Hoechst 33342. Therefore, upon mutation of residues of these primary R and H sites, it is expected that the respective substrates bind at secondary R and H sites. Further support for the presence of multiple R sites on P-gp comes from previous observations that rhodamine 123 and LDS-751 (that also binds to an R site) (Shapiro & Ling, 1998) bind simultaneously to P-gp, determined by fluorescence titrations using purified Chinese hamster P-gp (Lugo & Sharom, 2005a).
Using the “Site Finder” program in the MOE software package, Pajeva, Hanl, and Wiese (2013) found several pockets at the interdomain space between the NBDs and the ICLs (intracellular loops) that the authors explore as possible drug-binding sites of P-gp. As rhodamine 123 is a small molecule, the authors successfully docked the compound at one of the pockets in this location and proposed this site as another R site for P-gp. Although no experimental data was presented, the authors support this proposal with previous FRET studies by Lugo and Sharom that mapped the location of the putative R site to 18–25 Å from the line connecting the Walker A cysteines of the NBDs (Lugo & Sharom, 2005b). However, it is important to note that the proposed location in the NBD-ICL interdomain region has not been validated with mutagenesis studies.
Localization for the P site, the site that binds preferentially prazosin and progesterone, has also been proposed. The characteristics of this site seem to be different from those of the R and H sites. Substrate binding to the P site stimulates the transport of both rhodamine 123 and Hoechst 33342. Substrate binding to the P and R sites stimulates the transport of Hoechst 33342. In contrast, substrate binding to both the P and H sites stimulates the transport of rhodamine 123 less than that of binding to each individually (Shapiro et al., 1999). As progesterone was found not to be transported by P-gp (Ueda et al., 1992) and prazosin had been reported as a poor substrate (Greenberger, 1993), the authors proposed that the P site is essentially an allosteric site (Shapiro et al., 1999). However, the bodipy-FL prazosin is a very good substrate (Gribar, Ramachandra, Hrycyna, Dey, & Ambudkar, 2000). Thus, the P site seems to be a transport compatible site instead of an allosteric site as proposed earlier (Shapiro et al., 1999). The P site is likely the place where the photoaffinity-labeled substrate IAAP (see above) binds and cross-links P-gp. Dey, Ramachandra, Pastan, Gottesman, and Ambudkar (1997) found that P-gp contains two nonidentical IAAP binding sites, one at the N-terminal and the other one at the C-terminal half, suggesting the existence of two P sites, or a single P site formed by residues of both the N- and C-terminal halves of P-gp.
2.8. Pseudo-symmetric sites
Peter Chiba and coworkers showed that photo-activated propafenone derivatives label residues in pseudo-symmetric positions including TM segments 3, 5, 8, and 11 (Pleban et al., 2005). These results suggested the existence of two substrate–protein interaction modes. These ideas were inspired by the assumption that P-gp evolved from a half-transporter by gene duplication (Gottesman & Pastran, 1993). Indeed, several bacterial ABC exporters are half-transporters that require homo- or hetero-dimerization for function; however P-gp is a full transporter in which gene duplication would have led to the formation of a single polypeptide chain. While homo-dimeric half-transporters have the TM helices with their respective amino acid sequence in duplicate, P-gp exhibits helices with indeed similar amino acid sequences (50% similarity, 29% identity; see Fig. 3). To prove that P-gp has two solute translocation pathways, Parveen et al. (2011) introduced positively charged arginines in symmetric positions in P-gp (Q132R-TM2 and Q773R-TM8) to prevent protonatable compounds such as rhodamine dyes from entering either of the substrate translocation paths. The presence of charge residues decreases the transport of substrates, but the effect was not of the same magnitude for all substrates. Rhodamine 123 transport was more affected in mutant Q773C, while the transport for verapamil, vinblastine, and propafenone analogs was more affected in the Q132R mutant. These observations seem to indicate that substrates can use both translocation paths, although they show preference for one over the other one (Parveen et al., 2011).
The alignment of the sequences of the TMDs 1 and 2 was carried out using BLAST (Altschul et al., 1997) and is shown in Fig. 3. The residues that face the central cavity in the X-ray structures of P-gp were selected and are underlined in Fig. 3. It is clear that some residues of the drug-binding pocket of P-gp are identical in symmetric positions of the α-helices. For example, 310 (TM5) and 953 (TM11) are tyrosines, and 332 (TM6) and 975 (TM12) are leucines. But more important is the fact that several residues are slightly different [339 (TM6) and 982 (TM12) are leucine and valine] or drastically different [65 (TM1) and 725 (TM7) are leucine and glutamine]. These differences reveal different specificity in the two symmetric positions of the cavity.
3. MOLECULAR MODELING STUDIES
Since the publication of the first X-ray crystal structures of mouse P-gp in 2009 (Aller et al., 2009), a number of laboratories have employed molecular modeling studies trying to identify the drug-binding sites of P-gp. The high similarity between mouse (mdr1a) and human P-gp sequences (87% identity, 94% similarity) has encouraged many research groups to build homology models of human P-gp based on the structures of mouse P-gp and many modeling studies have been published. In 2012, the publication of the X-ray structure of C. elegans P-gp (Jin et al., 2012) revealed some errors in model building of the first mouse P-gp structures. The use of a template with errors certainly raises questions concerning the results and conclusions of those modeling studies. Finally, in 2013, refined structures of mouse P-gp were deposited in the pdb Bank, using a new electron density map generated from a single-wavelength anomalous dispersion phasing (SAD) of an original dataset, one that exhibited less radiation damage (Li et al., 2014). At approximately the same time, two new X-ray structures of mouse P-gp in the apo form were reported at the same resolution (Ward et al., 2013). The accuracy of the new structures was validated by generating 17 point mutations to cysteine and labeling them with mercury(II).
Although a discontinuity in helix 12 (i.e., the presence of a loop) of the refined structures of mouse P-gp (4M1M.pdb, 4M2S.pdb, and 4M2T.pdb) makes difficult its comparison with the same helix in the new X-ray structures of mouse P-gp (4KSB.pdb and 4KSC.pdb), it seems there is a register shift at TM helix 12 between these structures. The possibility that the helix adopts different conformations when the protein is crystallized in different conditions is a hypothesis to be considered. However, Li et al. (2014) pointed out that registry errors remain for TM12 residues 968–987 in the new mouse structures reported by Ward et al. (4KSB.pdb and 4KSC.pdb) and in the new model deposited in the pdb Bank (4LSG.pdb). This discrepancy is relevant for modeling studies. For example, while a phenylalanine (F979-mouse; F983-human) interacts with the QZ59 cyclic peptides and faces the central cavity in the refined structures of mouse P-gp, a valine (V978-mouse; V982-human) faces the central cavity in the new structures (4KSB.pdb and 4KSC.pdb) (see Fig. 4). Certainly, docking analysis will yield different results depending on the model selected for the studies. This difference in TM12 has to be sorted out in order for research groups to use these structures to carry out reliable modeling studies.
Figure 4.
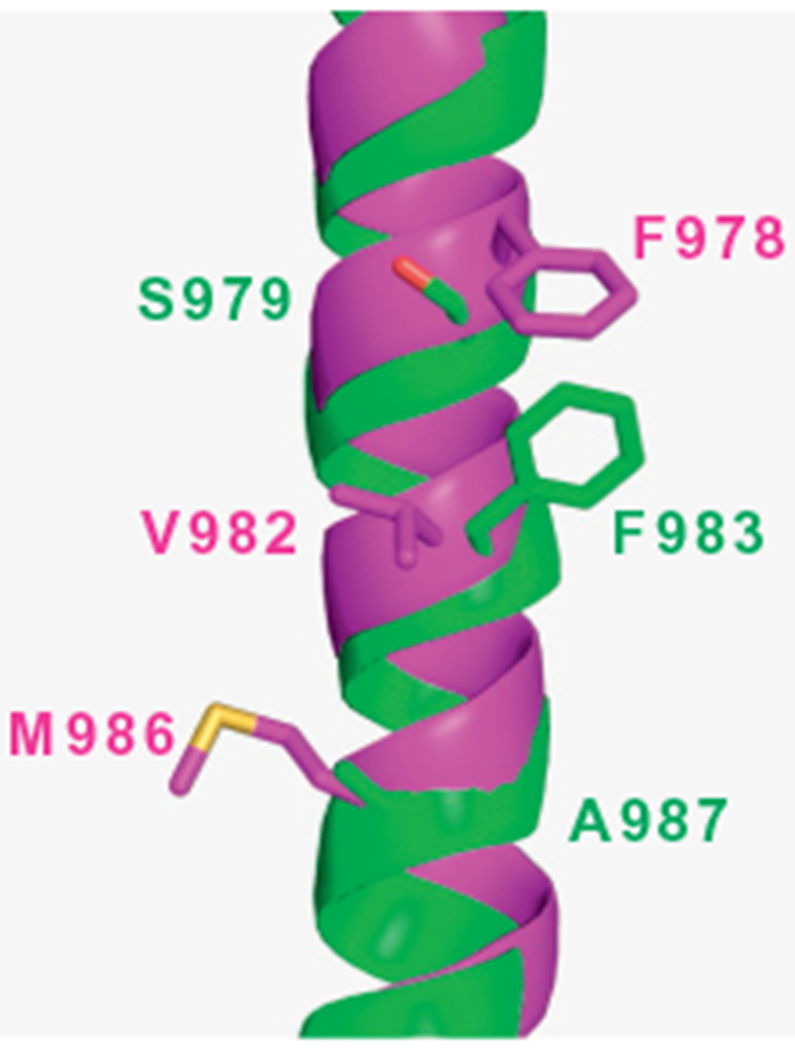
Register shift at transmembrane helix 12 between the refined structure of mouse P-gp published by Li et al. (2014) (4M1M.pdb) and the new crystal structure of mouse P-gp published by Ward et al. (2013) (4KSB.pdb). The structures were aligned in PyMOL 1.5.0.5 and only the TM12s are shown for clarity. The helices are shown as cartoon models in green (4M1M.pdb) and magenta (4KSB.pdb). Residues facing the central cavity are shown as stick models: S979, F983, and A987 of 4M1M.pdb and F978, V982, and M986 of 4KSB.pdb (residue numbers denote the human P-gp sequence).
4. CONCLUSIONS AND PERSPECTIVES
The ability of P-gp to recognize a wide variety of substrates is remarkable and thus it has been the subject of intense research for almost 40 years. The field has certainly shown advances, as we know today (i) the overall molecular structure of P-gp, (ii) the structure is highly flexible, (iii) the substrates are transported through a central cavity, and (iv) the alternate access mechanism of transport that enjoys a significant consensus in the ABC transporter community. It seems evident that P-gp has a large drug-binding pocket with different overlapping sites for different drugs. It is also evident that each drug has multiple transport-active binding sites. Nonetheless, the identification of the particular sites for various drugs remains elusive. Perhaps the only case with experimental support is the drug-binding sites of the cyclic peptides QZ59-RRR and QZ59-SSS, determined by X-ray crystallography. In a collective effort that required studies carried out by different groups at different times, the R, H, and P sites have been proposed, which include the relative positions of these sites in the common drug-binding pocket of P-gp. The concepts of primary and secondary sites as well as the idea of pseudo-symmetric sites (as part of the pseudo-symmetric translocation pathways) have also been introduced.
As discussed in this review, an appreciable amount of biochemical and structural data has been generated and progress has been achieved in the field. However, we are still far from understanding the mechanism of polyspecificity of P-gp. Since 2009, no more X-ray structures of P-gp with bound substrates or modulators have been reported. Site-directed mutagenesis studies are very laborious and the data sometimes are not conclusive. The fact that P-gp is a highly flexible membrane protein poses a great challenge for biophysical studies using EPR (DEER) and FRET techniques. And molecular modeling studies such as substrate-docking still cannot offer reliable results because the drug-binding pocket in the available structures (inward-opening conformation) is large and open. Therefore, high-resolution structures of ABC transporters obtained in the presence of substrates and inhibitors are required to further our understanding of the molecular basis of the polyspecificity of ABC drug transporters.
ACKNOWLEDGMENTS
We thank George Leiman for editorial assistance in the preparation of the chapter and the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research, for financial support. H.-M. S. was supported in part by a NUS-OPF fellowship from the National University of Singapore. A critical reading of the chapter by Drs. John Golin and Atish Patel is also gratefully acknowledged.
REFERENCES
- Aller SG, Yu J, Ward A, Weng Y, Chittaboina S, Zhuo RP, et al. (2009). Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science, 323, 1718–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang JH, Zhang Z, Miller W, et al. (1997). Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Research, 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambudkar SV, Cardarelli CO, Pashinsky I, & Stein WD (1997). Relation between the turnover number for vinblastine transport and for vinblastine-stimulated ATP hydrolysis by human P-glycoprotein. Journal of Biological Chemistry, 272, 21160–21166. [DOI] [PubMed] [Google Scholar]
- Ambudkar SV, Dey S, Hrycyna CA, Ramachandra M, Pastan I, & Gottesman MM (1999). Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annual Review of Pharmacology and Toxicology, 39, 361–398. [DOI] [PubMed] [Google Scholar]
- Ambudkar SV, Kim IW, & Sauna ZE (2006). The power of the pump: Mechanisms of action of P-glycoprotein (ABCB1). European Journal of Pharmaceutical Sciences, 27, 392–400. [DOI] [PubMed] [Google Scholar]
- Bikadi Z, Hazai I, Malik D, Jemnitz K, Veres Z, Hari P, et al. (2011). Predicting P-glycoprotein-mediated drug transport based on support vector machine and three-dimensional crystal structure of P-glycoprotein. PLoS One, 6, e25815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broeks A, Janssen HW, Calafat J, & Plasterk RH (1995). A P-glycoprotein protects Caenorhabditis elegans against natural toxins. The EMBO Journal, 14, 1858–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callaghan R, Ford RC, & Kerr ID (2006). The translocation mechanism of P-glycoprotein. FEBS Letters, 580, 1056–1063. [DOI] [PubMed] [Google Scholar]
- Chufan EE, Kapoor K, Sim HM, Singh S, Talele TT, Durell SR, et al. (2013). Multiple transport-active binding sites are available for a single substrate on human P-glycoprotein (ABCB1). PLoS One, 8, e82463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer J, Kopp S, Bates SE, Chiba P, &Ecker GF. (2007). Multispecificity of drug transporters: Probing inhibitor selectivity for the human drug efflux transporters ABCB1 and ABCG2. ChemMedChem, 2, 1783–1788. [DOI] [PubMed] [Google Scholar]
- Dawson RJP, & Locher KP (2006). Structure of a bacterial multidrug ABC transporter. Nature, 443, 180–185. [DOI] [PubMed] [Google Scholar]
- Demel MA, Schwaha R, Kramer O, Ettmayer P, Haaksma EEJ, & Ecker GF (2008). In silico prediction of substrate properties for ABC-multidrug transporters. Expert Opinion on Drug Metabolism & Toxicology, 4, 1167–1180. [DOI] [PubMed] [Google Scholar]
- Dey S, Ramachandra M, Pastan I, Gottesman MM, & Ambudkar SV (1997). Evidence for two nonidentical drug-interaction sites in the human P-glycoprotein. Proceedings of the National Academy of Sciences of the United States of America, 94, 10594–10599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolghih E, Bryant C, Renslo AR, & Jacobson MP (2011). Predicting binding to P-glycoprotein by flexible receptor docking. PLoS Computational Biology, 7, e1002083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donmez Cakil Y, Khunweeraphong N, Parveen Z, Schmid D, Artaker M, Ecker GF, et al. (2014). Pore-exposed tyrosine residues of P-glycoprotein are important hydrogen-bonding partners for drugs. Molecular Pharmacology, 85, 420–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckford PDW, & Sharom FJ (2009). ABC efflux pump-based resistance to chemotherapy drugs. Chemical Reviews, 109, 2989–3011. [DOI] [PubMed] [Google Scholar]
- Ferreira RJ, dos Santos DJVA, Ferreira MJU, & Guedes RC (2011). Toward a better pharmacophore description of P-glycoprotein modulators, based on macrocyclic diterpenes from euphorbia species. Journal of Chemical Information and Modeling, 51, 1315–1324. [DOI] [PubMed] [Google Scholar]
- Gottesman MM, Ambudkar SV, & Xia D (2009). Structure of a multidrug transporter. Nature Biotechnology, 27, 546–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesman MM, & Pastan I (1993). Biochemistry of multidrug resistance mediatedby the multidrug transporter. Annual Review of Biochemistry, 62, 385–427. [DOI] [PubMed] [Google Scholar]
- Greenberger LM (1993). Major photoaffinity drug labeling sites for iodoaryl azidoprazosin in P-glycoprotein are within, or immediately C-terminal to, transmembrane domains 6 and 12. Journal of Biological Chemistry, 268, 11417–11425. [PubMed] [Google Scholar]
- Gribar JJ, Ramachandra M, Hrycyna CA, Dey S, & Ambudkar SV (2000). Functional characterization of glycosylation-deficient human P-glycoprotein using a vaccinia virus expression system. Journal of Membrane Biology, 173, 203–214. [DOI] [PubMed] [Google Scholar]
- Gutmann DAP, Ward A, Urbatsch IL, Chang G, & van Veen HW (2010). Understanding polyspecificity of multidrug ABC transporters: Closing in on the gaps in ABCB1. Trends in Biochemical Sciences, 35, 36–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenstein K, Dawson RJP, & Locher KP (2007). Structure and mechanism of ABC transporter proteins. Current Opinion in Structural Biology, 17(4), 412–418. [DOI] [PubMed] [Google Scholar]
- Jardetzk O (1966). Simple allosteric model for membrane pumps. Nature, 211, 969–973. [DOI] [PubMed] [Google Scholar]
- Jin MS, Oldham ML, Zhang QJ, & Chen J (2012). Crystal structure of the multidrug transporter P-glycoprotein from Caenorhabditis elegans. Nature, 490, 566–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juliano RL, & Ling V (1976). A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochimica et Biophysica Acta, 455, 152–162. [DOI] [PubMed] [Google Scholar]
- Kapoor K, Sim HM, & Ambudkar SV (2013). Multidrug resistance in cancer: A tale of ABC drug transporters Molecular mechanisms of tumor cell resistance to chemotherapy: Vol. 1 (pp. 1–34). New York: Springer. [Google Scholar]
- Kerr KM, Sauna ZE, & Ambudkar SV (2001). Correlation between steady-state ATP hydrolysis and vanadate-induced ADP trapping in human P-glycoprotein. Evidence for ADP release as the rate-limiting step in the catalytic cycle and its modulation by substrates. Journal of Biological Chemistry, 276, 8657–8664. [DOI] [PubMed] [Google Scholar]
- Lee JY, Yang JG, Zhitnitsky D, Lewinson O, & Rees DC (2014). Structural basis for heavy metal detoxification by an Atm1-type ABC exporter. Science, 343, 1133–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JZ, Jaimes KF, & Aller SG (2014). Refined structures of mouse P-glycoprotein. Protein Science, 23, 34–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li WX, Li LP, Eksterowicz J, Ling XFB, & Cardozo M (2007). Significance analysis and multiple pharmacophore models for differentiating P-glycoprotein substrates. Journal of Chemical Information and Modeling, 47, 2429–2438. [DOI] [PubMed] [Google Scholar]
- Loo TW, Bartlett MC, & Clarke DM (2003a). Permanent activation of the human P-glycoprotein by covalent modification of a residue in the drug-binding site. Journal of Biological Chemistry, 278, 20449–20452. [DOI] [PubMed] [Google Scholar]
- Loo TW, Bartlett MC, & Clarke DM (2003b). Substrate-induced conformational, changes in the transmembrane segments of human P-glycoprotein—Direct evidence for the substrate-induced fit mechanism for drug binding. Journal of Biological Chemistry, 278, 13603–13606. [DOI] [PubMed] [Google Scholar]
- Loo TW, Bartlett MC, & Clarke DM (2006). Transmembrane segment 7 of human P-glycoprotein forms part of the drug-binding pocket. Biochemical Journal, 399, 351–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo TW, Bartlett MC, & Clarke DM (2010). Human P-glycoprotein is active when the two halves are clamped together in the closed conformation. Biochemical and Biophysical Research Communications, 395, 436–440. [DOI] [PubMed] [Google Scholar]
- Loo TW, Bartlettt MC, & Clarke DM (2006). Transmembrane segment 1 of human P-glycoprotein contributes to the drug-binding pocket. Biochemical Journal, 396, 537–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo TW, & Clarke DM (1995). Membrane topology of a cysteine-less mutant of human P-glycoprotein. Journal of Biological Chemistry, 270, 843–848. [DOI] [PubMed] [Google Scholar]
- Loo TW, & Clarke DM (1997). Identification of residues in the drug-binding site of human P-glycoprotein using a thiol-reactive substrate. Journal of Biological Chemistry, 272, 31945–31948. [DOI] [PubMed] [Google Scholar]
- Loo TW, & Clarke DM (1999). The transmembrane domains of the human multidrug resistance P-glycoprotein are sufficient to mediate drug binding and trafficking to the cell surface. Journal of Biological Chemistry, 274, 24759–24765. [DOI] [PubMed] [Google Scholar]
- Loo TW, & Clarke DM (2000a). Identification of residues within the drug-binding domain of the human multidrug resistance P-glycoproteinby cysteine-scanning-mutagenesis and reaction with dibromobimane. Journal of Biological Chemistry, 275, 39272–39278. [DOI] [PubMed] [Google Scholar]
- Loo TW, & Clarke DM (2000b). The packing of the transmembrane segments of human multidrug resistance P-glycoprotein is revealed by disulfide cross-linking analysis. Journal of Biological Chemistry, 275, 5253–5256. [DOI] [PubMed] [Google Scholar]
- Loo TW, & Clarke DM (2001). Defining the drug-binding site in the human multidrug resistance P-glycoprotein using a methanethiosulfonate analog of verapamil, MTS-verapamil. Journal of Biological Chemistry, 276, 14972–14979. [DOI] [PubMed] [Google Scholar]
- Loo TW, & Clarke DM (2002). Location of the rhodamine-binding site in the human multidrug resistance P-glycoprotein. Journal of Biological Chemistry, 277, 44332–44338. [DOI] [PubMed] [Google Scholar]
- Lugo MR, & Sharom FJ (2005a). Interaction of LDS-751 and rhodamine 123 with P-glycoprotein: Evidence for simultaneous binding of both drugs. Biochemistry, 44, 14020–14029. [DOI] [PubMed] [Google Scholar]
- Lugo MR, & Sharom FJ (2005b). Interaction of LDS-751 with P-glycoprotein and mapping of the location of the R drug binding site. Biochemistry, 44, 643–655. [DOI] [PubMed] [Google Scholar]
- Marcoux J, Wang SC, Politis A, Reading E, Ma J, Biggin PC, et al. (2013). Mass spectrometry reveals synergistic effects of nucleotides, lipids, and drugs binding to a multidrug resistance efflux pump. Proceedings of the National Academy of Sciences of the United States of America, 110, 9704–9709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez L, Arnaud O, Henin E, Tao HC, Chaptal V, Doshi R, et al. (2014). Understanding polyspecificity within the substrate-binding cavity of the human multidrug resistance P-glycoprotein. The FEBS Journal, 281, 673–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DS, Nobmann SN, Gutmann H, Toeroek M, Drewe J, & Fricker G (2000). Xenobiotic transport across isolated brain microvessels studied by confocal microscopy. Molecular Pharmacology, 58, 1357–1367. [DOI] [PubMed] [Google Scholar]
- O’Mara ML, & Tieleman DP (2007). P-glycoprotein models of the apo and ATP-bound states based on homology with Sav 1866 and MalK. FEBS Letters, 581, 4217–4222. [DOI] [PubMed] [Google Scholar]
- Pajeva IK, Globisch C, &Wiese M (2009). Combined pharmacophore modeling, docking, and 3D QSAR studies of ABCB1 and ABCC1 transporter inhibitors. ChemMedChem, 4, 1883–1896. [DOI] [PubMed] [Google Scholar]
- Pajeva IK, Hanl M, & Wiese M (2013). Protein contacts and ligand binding in the inward-facing model of human P-glycoprotein. ChemMedChem, 8, 748–762. [DOI] [PubMed] [Google Scholar]
- Pajeva IK, Sterz K, Christlieb M, Steggemann K, Marighetti F, & Wiese M (2013). Interactions of the multidrug resistance modulators tariquidar and elacridar and their analogues with P-glycoprotein. ChemMedChem, 8, 1701–1713. [DOI] [PubMed] [Google Scholar]
- Pajeva IK, & Wiese M (2002). Pharmacophore model of drugs involved in P-glycoprotein multidrug resistance: Explanation of structural variety (hypothesis). Journal of Medicinal Chemistry, 45, 5671–5686. [DOI] [PubMed] [Google Scholar]
- Pan BF, Dutt A, & Nelson JA (1994). Enhanced transepithelial flux of cimetidine by madin-darby canine kidney-cells overexpressing human P-glycoprotein. Journal of Pharmacology and Experimental Therapeutics, 270, 1–7. [PubMed] [Google Scholar]
- Parveen Z, Stockner T, Bentele C, Pferschy S, Kraupp M, Freissmuth M, et al. (2011). Molecular dissection of dual pseudosymmetric solute translocation pathways in human P-glycoprotein. Molecular Pharmacology, 79, 443–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzotti JE, Lamb ML, Evensen E, & Grootenhuis PDJ (2002). A computational ensemble pharmacophore model for identifying substrates of P-glycoprotein. Journal of Medicinal Chemistry, 45, 1737–1740. [DOI] [PubMed] [Google Scholar]
- Pleban K, Kopp S, Csaszar E, Peer M, Hrebicek T, Rizzi A, et al. (2005). P-glycoprotein substrate binding domains are located at the transmembrane domain/transmembrane domain interfaces: A combined photoaffinity labeling-protein homology modeling approach. Molecular Pharmacology, 67, 365–374. [DOI] [PubMed] [Google Scholar]
- Sauna ZE, & Ambudkar SV (2000). Evidence for a requirement for ATP hydrolysis at two distinct steps during a single turnover of the catalytic cycle of human P-glycoprotein. Proceedings of the National Academy of Sciences of the United States of America, 97, 2515–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schinkel AH (1999). P-glycoprotein, a gatekeeper in the blood-brain barrier. Advanced Drug Delivery Reviews, 36, 179–194. [DOI] [PubMed] [Google Scholar]
- Seelig A (1998). A general pattern for substrate recognition by P-glycoprotein. European Journal of Biochemistry, 251, 252–261. [DOI] [PubMed] [Google Scholar]
- Senior AE, al-Shawi MK, & Urbatsch IL (1995). The catalytic cycle of P-glycoprotein. FEBS Letters, 377, 285–289. [DOI] [PubMed] [Google Scholar]
- Shapiro AB, Fox K, Lam P,& Ling V (1999). Stimulation of P-glycoprotein-mediated drug transport by prazosin and progesterone. Evidence for a third drug-binding site. European Journal of Biochemistry, 259, 841–850. [DOI] [PubMed] [Google Scholar]
- Shapiro AB, & Ling V (1997). Positively cooperative sites for drug transport by P-glycoprotein with distinct drug specificities. European Journal of Biochemistry, 250, 130–137. [DOI] [PubMed] [Google Scholar]
- Shapiro AB, & Ling V (1998). Transport of LDS-751 from the cytoplasmic leaflet of the plasma membrane by the rhodamine-123-selective site of P-glycoprotein. European Journal of Biochemistry, 254, 181–188. [DOI] [PubMed] [Google Scholar]
- Sharma RC, Inoue S, Roitelman J, Schimke RT, & Simoni RD (1992). Peptide-transport by the multidrug resistance pump. Journal of Biological Chemistry, 267, 5731–5734. [PubMed] [Google Scholar]
- Sharom FJ (2014). Complex interplay between the P-glycoprotein multidrug efflux pump and the membrane: Its role in modulating protein function. Frontiers in Oncology, 4, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheps JA, Ralph S, Zhao ZY, Baillie DL, & Ling V (2004). The ABC transporter gene family of Caenorhabditis elegans has implications for the evolutionary dynamics of multidrug resistance in eukaryotes. Genome Biology, 5, R15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintre CA, Pike ACW, Li Q, Kim JI, Barr AJ, Goubin S, et al. (2013). Structures of ABCB10, a human ATP-binding cassette transporter in apo- and nucleotide-bound states. Proceedings of the National Academy of Sciences of the United States of America, 110, 9710–9715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla S, Kouanda A, Silverton L, Talele TT, & Ambudkar SV (2014). Pharmacophore modeling of nilotinib as an inhibitor of ATP-binding cassette drug transporters and BCR-ABL kinase using a three-dimensional quantitative structure-activity relationship approach. Molecular Pharmaceutics, 11, 2313–2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim HM, Bhatnagar J, Chufan EE, Kapoor K, &Ambudkar SV. (2013). Conserved walker a cysteines 431 and 1074 in human P-glycoprotein are accessible to thiol-specific agents in the Apo and ADP-vanadate trapped conformations. Biochemistry, 52, 7327–7338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao HC, Weng Y, Zhuo RP, Chang G, Urbatsch IL, & Zhang QH (2011). Design and synthesis of selenazole-containing peptides for cocrystallization with P-glycoprotein. Chembiochem, 12, 868–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda K, Okamura N, Hirai M, Tanigawara Y, Saeki T, Kioka N, et al. (1992). Human P-glycoprotein transports cortisol, aldosterone, and dexamethasone, but not progesterone. Journal of Biological Chemistry, 267, 24248–24252. [PubMed] [Google Scholar]
- van Wonderen JH, McMahon RM, O’Mara ML, McDevitt CA, Thomson AJ, Kerr ID, et al. (2014). The central cavity of ABCB1 undergoes alternating access during ATP hydrolysis. The FEBS Journal, 281, 2190–2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhalen B, Ernst S, Borsch M, & Wilkens S (2012). Dynamic ligand-induced conformational rearrangements in P-glycoprotein as probed by fluorescence resonance energy transfer spectroscopy. Journal of Biological Chemistry, 287, 1112–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhalen B, & Wilkens S (2011). P-glycoprotein retains drug-stimulated ATPase activity upon covalent linkage of the two nucleotide binding domains at their C-terminal ends. Journal of Biological Chemistry, 286, 10476–10482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward A, Reyes CL, Yu J, Roth CB, & Chang G (2007). Flexibility in the ABC transporter MsbA: Alternating access with a twist. Proceedings of the National Academy of Sciences of the United States of America, 104, 19005–19010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward AB, Szewczyk P, Grimard V, Lee CW, Martinez L, Doshi R, et al. (2013). Structures of P-glycoprotein reveal its conformational flexibility and an epitope on the nucleotide-binding domain. Proceedings of the National Academy of Sciences of the United States of America, 110, 13386–13391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen PC, Verhalen B, Wilkens S, Mchaourab HS, & Tajkhorshid E (2013). On the origin of large flexibility of P-glycoprotein in the inward-facing state. Journal of Biological Chemistry, 288, 19211–19220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong K, Ma J, Rothnie A, Biggin PC, & Kerr ID (2014). Towards understanding promiscuity in multidrug efflux pumps. Trends in Biochemical Sciences, 39, 8–16. [DOI] [PubMed] [Google Scholar]