

Abstract
Bacterial flagellin can elicit production of TLR5-mediated IL-22 and NLRC4-mediated IL-18, cytokines that act in concert to cure and prevent rotavirus (RV) infection. This study investigated the mechanism by which these cytokines act to impede RV. Although IL-18 and IL-22 induce each other’s expression, we found that IL-18 and IL-22 both impeded RV independently of one another and did so by distinct mechanisms that involved activation of their cognate receptors in intestinal epithelial cells (IEC). IL-22 drove IEC proliferation and migration toward villus tips, which resulted in increased extrusion of highly differentiated IEC that serve as the site of RV replication. In contrast, IL-18 induced cell death of RV-infected IEC thus directly interrupting the RV replication cycle, resulting in spewing of incompetent virus into the intestinal lumen and causing a rapid drop in the number of RV-infected IEC. Together, these actions resulted in rapid and complete expulsion of RV, even in hosts with severely compromised immune systems. These results suggest that a cocktail of IL-18 and IL-22 might be a means of treating viral infections that preferentially target short-lived epithelial cells.
Keywords: Cell proliferation, anoikis, cell shedding, pyroptosis
Introduction
Rotavirus (RV) remains a scourge to humanity, causing severe distress (“morbidity”) to many children and contributes to thousands of childhood deaths annually, particularly in developing countries wherein RV vaccines have only moderate efficacy (1). RV is a double-stranded RNA virus that primarily infects intestinal epithelial cells (IEC) that line the villus tips of the ileum, resulting in severe life-threatening diarrhea in young children and moderate gastrointestinal distress in adults (2–4). Such tropism and pathogenesis is faithfully recapitulated in RV-infected mice, making the mouse model of RV useful for studying basic aspects of RV immunity and disease pathophysiology. Further, the RV mouse model may prove a useful platform for discovery of novel means to treat and prevent RV infection, especially in scenarios when adaptive immunity, which normally plays an essential role in clearing RV, is not functioning adequately. Toward this end, we previously reported that administration of bacterial flagellin rapidly cured, and/or protected against, RV infection. Such protection was independent of interferon and adaptive immunity and dependent upon generation of both toll-like receptor 5 (TLR5)-mediated IL-22 and NOD-like receptor C4 (NLRC4)-mediated IL-18, which together resulted in prevention and/or cure of RV infection, and its associated diarrhea (5). However, the mechanisms by which these cytokines impede RV infection remained unknown. Herein, we report that IL-22 acts upon IEC to drive proliferation, migration, and ultimately extrusion of infected IEC into the intestinal lumen, whereas IL-18 drives rapid death of RV-infected IEC. The combined actions of IL-22 and IL-18 eliminate RV from the intestine independent of adaptive immunity.
Results
IL-22 and IL-18 activate their receptors on epithelial cells to protect against rotavirus
We previously reported that systemic administration of bacterial flagellin elicits TLR5-mediated production of IL-22 and NLRC4-mediated generation of IL-18 that can act in concert to prevent or treat rotavirus (RV) and some other enteric viral infections (5). Specifically, as shown in Figure S1 and our previous work, chronic RV infections that developed in RV-inoculated immune-deficient C57BL/6 Rag-1−/− mice were cured by combined systemic treatment with IL-18 and IL-22, whereas injection of either cytokine alone reduced RV loads but did not clear the virus, regardless of cytokine dose and duration of administration. In these particular experiments, RV infection was assayed by measuring fecal RV antigens by ELISA, but measurement of RV genomes in the intestine yields similar results (5). In WT mice, a sufficiently high doses of recombinant IL-22 can, by itself, fully prevent RV infection while lower doses of exogenously administered IL-22 and IL-18 dramatically reduced the extent of RV infection, while the combination of these cytokines eliminated evidence of infection (Figure 1A). The central goal of this study was to elucidate mechanisms by which these cytokines act in concert to control and prevent RV infection.
Fig. 1. IL-22 and IL-18 elicit distinct antiviral activities against mRV invasion.
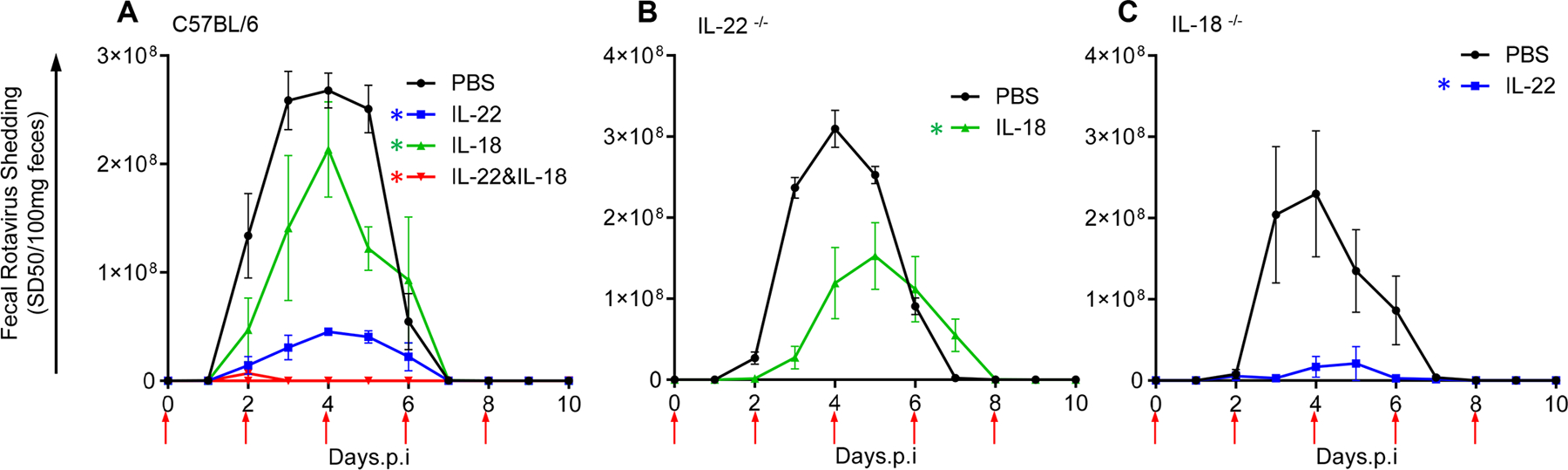
Mice were administered PBS, IL-22 (2 μg) and/or IL-18 (1 μg) via intraperitoneal injection, 2 hours prior to, or 2, 4, 6, or 8 days after (indicated by arrows) oral inoculation with mRV. Fecal RV levels were measured over time by ELISA. (A) C57BL/6 mice n=4. (B) IL-22−/− mice, n=5 and 7 for PBS and IL-18, respectively. (C) IL-18−/− mice, n=5. * indicates significantly different from PBS by two-way ANOVA, P < 0.0001.
In the context of parasitic infection, both IL-18 and IL-22 promote expression of each other, and loss of either impairs immunity to Toxoplasma. gondii (6). We thus hypothesized that administration of IL-18 might impede RV as a result of its ability to induce IL-22 expression. This hypothesis predicted that ability of IL-18 to protect against RV infection would be largely absent in IL-22−/− mice. However, administration of IL-18 upon RV inoculation clearly reduced the extent of RV infection in IL-22−/− mice, which argued strongly against this hypothesis (Figure 1B). We considered the converse hypothesis, namely that IL-22 might impede RV infection by elicitation of IL-18, but we observed that recombinant IL-22 markedly prevented RV infection in IL-18−/− mice (Figure 1C). Although IL-18 and IL-22 may play important roles in inducing each other’s expression, our results indicate that they each activate distinct signaling pathways that cooperate to impede RV infection.
Next, we examined the extent by which IL-18 and IL-22 acted upon the hematopoietic or non-hematopoietic compartment to impede RV infection. We used WT, IL-18-R−/−, and IL-22-R−/− mice to generate irradiated bone marrow chimeric mice that expressed the receptors for IL-22 or IL-18 in only bone marrow-derived or radioresistant cells. Such mice were inoculated with RV, treated with recombinant IL-22 or IL-18, and RV infection was monitored via measuring fecal RV antigens by ELISA. Figure 1 used a relatively low dose of cytokine that highlighted the cooperativity of IL-18 and IL-22, but successive experiments used 5-fold higher doses to enable a robust effect that could be dissected via bone marrow chimeric mice. Mice that expressed the IL-22 receptor only in bone marrow-derived cells were not protected from RV infection by treatment with IL-22 (Figure 2A), whereas mice with IL-22 receptor only in radioresistant cells were almost completely protected by this cytokine (Figure 2B). These results suggest that IL-22 protects mice from RV infection by acting on IEC, which are known to be populated from radioresistant stem cells and responsive to IL-22 (7). In accord with this notion, we observed that multiple IEC cell lines are responsive to IL-22 in vitro via STAT3 phosphorylation although IL-22, like flagellin and IL-18, did not impact RV infection in vitro (Figure S2). Studies with IL-18-R chimeric mice similarly revealed that expression of this receptor in only bone marrow-derived cells conferred only a modest non-significant reduction (12 ± 3.8 %) in the extent of RV infection upon IL-18 administration (Figure 2C). In contrast, in mice that expressed IL-18-R in only radioresistant cells, IL-18 reduced extent of RV infection by 76 ± 8.7 % (Figure 2D). Together, these results suggest that agonizing IL-18 and IL-22 receptors on IEC result in generation of signals that impede RV in vivo but not in vitro.
Fig. 2. Both IL-22 and IL-18-mediated antiviral pathway utilize their cognate non-hematopoietic cell receptors.
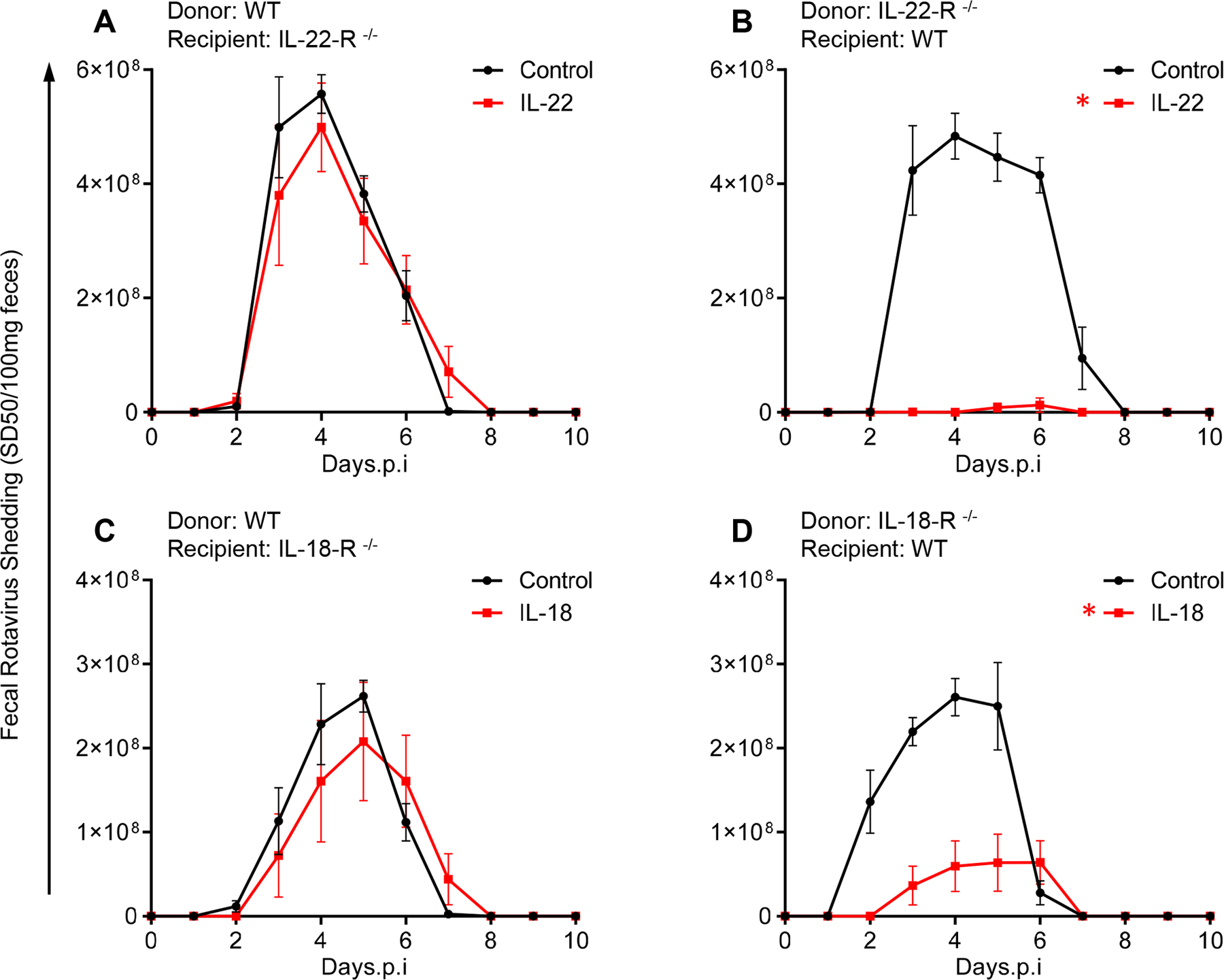
Indicated bone marrow-irradiated chimeric mice were administered PBS (control), IL-22 (10 μg), or IL-18 (2 μg) via intraperitoneal injection, 2 hours prior to, or 2, 4, 6, or 8 days after oral inoculation with mRV. Fecal RV levels were measured over time by ELISA. Differences between control and cytokine groups for each chimera/panel was analyzed by two-way ANOVA. (A) n = 7, P = 0.7715). (B) n=4 and 7 for PBS and IL-22, respectively. (C) n=7 and 6 for PBS and IL-18, respectively. (D) n=4 and 6 for PBS and IL-18, respectively. * indicates significantly different from PBS by two-way ANOVA, P < 0.0001.
IL-22 and IL-18 promote IEC proliferation/migration
In cell culture and organoid models, IL-22 promotes IEC proliferation, migration, and stem cell regeneration (8–10), which together are thought to contribute to ability of IL-22 to promote healing in response to an array of insults, including exposure to radiation and dextran sodium sulfate (DSS) in vivo (11–14). In contrast to such severe injuries, RV infection is generally characterized by a lack of overt intestinal inflammation (15, 16). We hypothesized that IL-22 may promote IEC proliferation and/or migration that might reduce the extent of RV infection by increasing the rate of IEC turnover, especially near villus tips, which is the predominant site of RV infection (2–4). We further reasoned that IL-18 might trigger the same kind of response and further increase IEC proliferation and turnover. Mice were administered BrdU and treated with IL-22 and/or IL-18. Sixteen hours later, mice were euthanized and intestines were subjected to fluorescence microscopy to measure rates at which IEC migrated toward villus tips (17). In accord with our hypothesis, administration of IL-22 approximately doubled the rate at which IEC migrated toward villus tips (Figure 3A, B). IL-18 administration also increased rate of IEC migration to a lesser extent. The combination of these cytokines did not result in a faster rate of IEC migration relative to IL-22 alone. Epidermal growth factor (EGF) is known to promote IEC proliferation and migration (18, 19), so we tested whether this cytokine might protect against RV infection. In accord with EGF promoting proliferation in a variety of tissues, EGF treatment induced IEC migration up the crypt villus axis (Figure 3C, D) albeit not quite as robustly as IL-22 (1.43 vs.1.95 fold increase respectively). Moreover, EGF had the ability to reduce the extent of RV infection (Figure 3E) but not as completely as IL-22. Together these results support the hypothesis that IL-22 and IL-18 promote IEC replication and migration, which contributes to protection against RV infection.
Fig. 3. Accelerated proliferation rate and migration levels of IEC are correlated with debilitation of mRV infectivity.
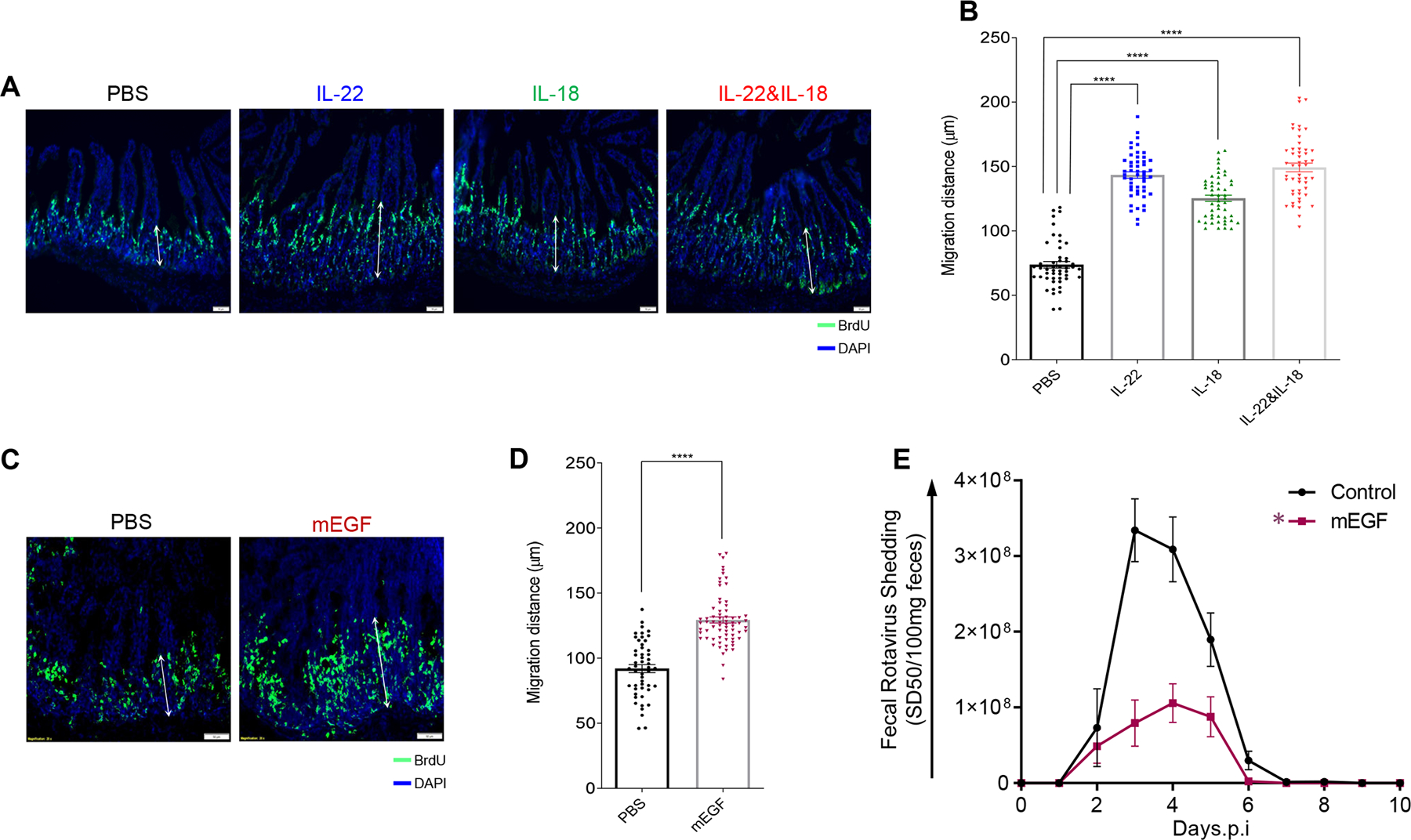
Mice were i.p injected with PBS, IL-22, (10 μg) IL-18 (2 μg), both cytokines, or murine epidermal growth factor (mEGF). 1h later, mice were administered BrdU. Mice were euthanized 16-hour post-BrdU administration, and BrDU was visualized (A and C) and migration was measured (B and D) by microscopy and image analysis, respectively. Images shown in A and C are representative. For B and D, sections were scored at least from 50 villus per group of mice (n = 5). Distance of the foremost migrating cells along the crypt-villus axis were measured with ImageJ software. Results are presented as mean ± SEM. Statistical significance was evaluated by Student’s t-test (**** denotes P < 0.0001). (E) Mice were i.p injected with PBS or EGF (10 μg) murine EGF two hours prior to, or 2, 4, 6, or 8 days after oral inoculation with mRV. Fecal RV levels were measured over time by ELISA. Data are mean ± SEM, n=5 * indicates significantly different from PBS by two-way ANOVA, P < 0.0001.
IL-22 promotes extrusion of IEC into small intestinal lumen
We next considered how promoting IEC proliferation might impede RV infection. Increased extrusion of IEC into the lumen is a likely consequence of increased IEC proliferation/migration, which is thought to occur such that cells remain alive until extrusion is completed in order to preserve the gut barrier (20). We hypothesized that increased proliferation/migration induced by IL-22 and/or IL-18 treatments might result in increased extrusion of villus tip cells, which are the site of RV infection. We investigated this hypothesis using a previously described method (21) in which cross sections of hematoxylin and eosin stained pieces of ileum are examined for visual evidence of cell shedding. We were unable to consistently distinguish IEC from other luminal contents, so we visualized cells using the DNA stain. This approach suggested a greater presence of IEC in the lumen of mice treated with cytokines, particularly IL-22 (Figure 4A), but it was difficult to quantitate such a difference via cell counting so we sought to evaluate levels of host cells via qPCR of 18S DNA in the ileum. The highly degradative environment of the intestine would likely degrade IEC shed into the lumen, but since such cells are extruded in a relatively intact state, their DNA might survive long enough to enable quantitation by qPCR. Small intestinal contents were extracted, and 18S DNA quantitated and expressed as number of cells per 100 mg of luminal content using known numbers of mouse epithelial cells to generate a standard curve. This approach indicated that IL-22 treatment markedly increased the level of IEC present in the lumen (Figure 4B) suggesting increased IEC shedding. IL-18 induced only a modest level of IEC shedding that appeared to be additive to the shedding induced by IL-22. A generally similar pattern was observed in the cecum (Figure 4C). In contrast, these cytokines did not impact levels of 18S DNA present in the lumen of the colon (Figure 4D), perhaps reflecting that the impact of these cytokines on IEC shedding is specific to the ileum/cecum and/or that the DNA of shed IEC is quickly degraded in the bacterial-dense colon. An even greater amount of shedding of IEC into the ileum was induced by treating mice with flagellin, although two treatments of IL-18/22 could match this level, which suggested that production of these cytokines might be sufficient to recapitulate the IEC shedding induced by flagellin (Figure 4E). The greater potency of flagellin may reflect ability of IL-18 and IL-22 to promote each other’s expression. Use of IL-22−/− and IL-18−/− mice revealed that these cytokines, both of which are necessary for flagellin’s anti-RV action (5), were both necessary for flagellin-induced cell shedding (Figure 4F). Collectively, these results support the notion that increased extrusion of IEC, particularly in response to IL-22, might be central to this cytokine’s ability to impede RV infection, but this data did not offer insight into how IL-22 and IL-18 cooperate to offer stronger protection against this virus.
Fig. 4. IL-22 promotes cell extrusion into intestinal lumen.
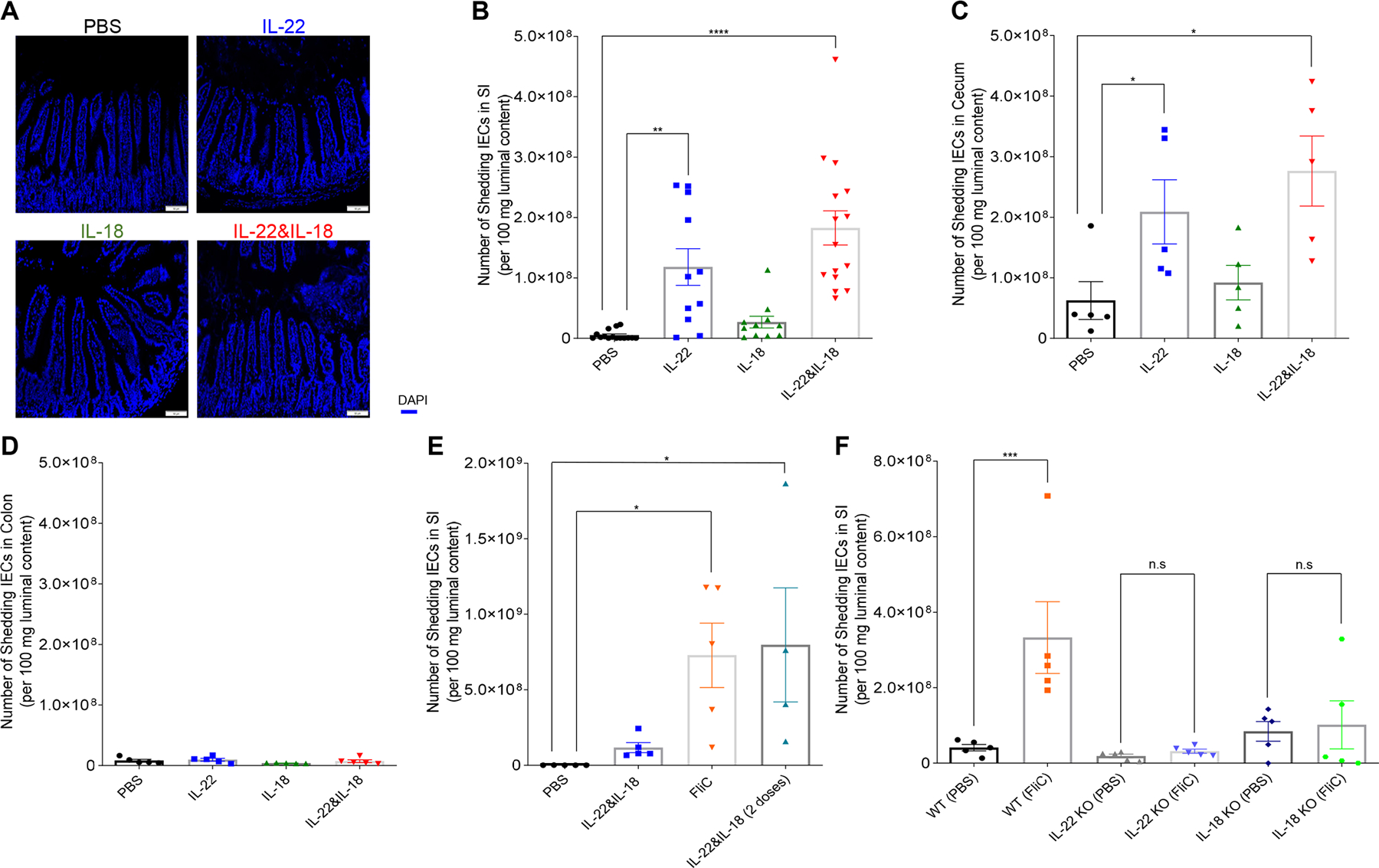
Mice (WT or indicated KO strain) received a single (except where indicated otherwise) i.p injection of PBS, IL-22, (10 μg) IL-18 (2 μg), both cytokines, or bacterial flagellin, FliC (15 μg). 8h later, mice were euthanized and intestine was isolated and luminal content were collected. (A) Microscopic appearance of DAPI-stained section to visualize shed cells in lumen. (B to F) Measurements of shed cells in different regions of the gastrointestinal (GI) tract via 18s by q-PCR (B, E and F) small intestine, (C) cecum, (D) colon (double doses of IL-22 plus IL-18 in E were 12 hours apart). Data in B-F are mean ± SEM (B), with significance assessed by Student’s t test, n = 5–15 mice as indicated by number of data points; *represents P < 0.05, ** represents P < 0.01, **** represents P < 0.0001, n.s., not significant.
IL-18 induces death of RV-infected IEC
Next, we examined how IL-22 and IL-18 might impact IEC in the presence of an active RV infection. We utilized WT mice 3 days following inoculation with RV, a time approaching peak levels of RV shedding (Figure 1A). RV-infected and uninfected mice were administered IL-22 and/or IL-18, euthanized 6 h later, and small intestinal content was isolated. Like IL-18/22 administration, RV infection upregulated IEC extrusion, with a marked further increase in IEC extrusion being observed by administration of IL-18/22 to RV-infected mice (Figure 5A). This suggests that increased IEC extrusion may normally contribute to innate defense against RV (2) and that exogenously administered IL-18/22 (or flagellin) may enhance this protective mechanism. Yet, like the case in uninfected mice, the promotion of IEC extrusion appeared to be driven by IL-22 and not IL-18 (Figure 5B).
Fig. 5. IL-18 induced TUNEL-positive cell death in villus tips of RV-infected mice.
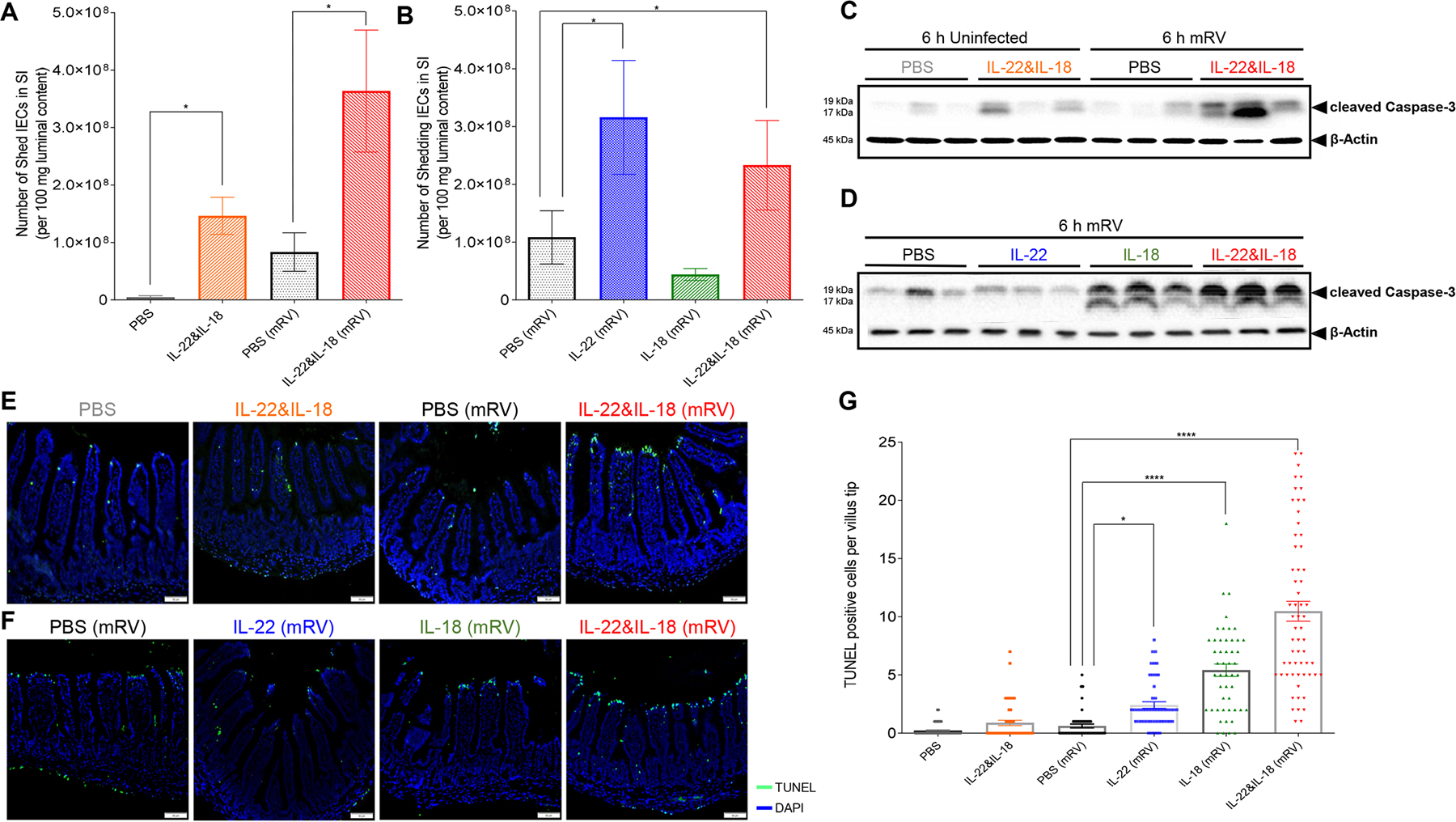
Mice were orally inoculated with mRV, or not(sham?) and were i.p injected at 3 dpi with PBS, IL-22, (10 μg) IL-18 (2 μg), or both cytokines. Mice were euthanized 6 h later and following assays were carried out.
(A-B) Assay of cell extrusion (i.e. measure of cells in lumen) as performed in response to cytokines in Fig 4. (C and D) Assay cleaved caspase-3 in IEC was assayed by SDS-PAGE immunoblotting. (E and F) Visualization of cell death by TUNEL staining, counterstained with DAPI. (G) Quantitation of TUNEL-positive cells at villus tip region based on visual counts. Data in panels A, B, and G are mean ± SEM. Panes A and B used 5 mice per condition to generate one value per mouse. Panel G used 5 mice per condition and assayed 6–10 villi per mouse, which are indicated by data points. Significance was determined by Student’s t test, * represents P < 0.05, **** represents P < 0.0001).
Next, we sought to investigate events in IEC that remained part of the small intestine at the time of increased IEC extrusion. Specifically, we examined if IL-18 and/or IL-22 might impact cell death. We observed that IL-18/22 or RV induced modest and variable induction of cleaved caspase-3. In contrast, administration of these cytokines to RV-infected mice induced marked elevations in cleaved caspase-3 (Figure 5C). Caspase-3 cleavage was also observed in response to IL-18 but not IL-22 (Figure 5D). Quantitation of cell death by TUNEL yielded a similar pattern of results. Specifically, both IL-18/22 and RV by themselves resulted in a modest increase in TUNEL-positive cells, which appeared sporadically throughout the villi (Figure 5E and Figure S3A, B). In contrast, treating RV-infected mice with IL-18 or the combination of IL-18 and IL-22, but not IL-22 itself, resulted in striking TUNEL-positivity at the villus tips (Figure 5 E, F, G), known sites of RV infection. Cytokine-induced TUNEL-positivity, which did not occur in the absence of RV, appeared to localize in the villus tip, where RV was localized prior to cytokine treatment, thus suggesting that IL-18 was promoting cell death in RV-infected cells (Figure S3C).
Cell death can occur via numerous pathways, so we hypothesized that IL-18-induced cell death might occur via pyroptosis, which appears to be a frequent form of cell death for infected cells (22). In accord with this possibility, IL-18 administration to RV-infected mice result in cleaved gasdermin D (Figure 6A), whose activity is essential for pyroptosis. To test the role of gasdermin D activation in IL-18 induced cell death, we performed experiments in mice lacking gasdermin D and gasdermin E, the latter of which is thought to compensate for lack of gasdermin D in some scenarios. Interestingly, our initial experiments found that gasdermin-deficient mice were highly resistant to RV infection (Fig S4). However, such resistance was associated with high levels of segmented filamentous bacteria (SFB), which we have recently shown drives spontaneous resistance to RV in Rag1−/− mice (23). Cross-fostering on gasdermin-deficient mice removed SFB and restored susceptibility to RV infection, thus extending our recent findings to mice with functional adaptive immunity. This model could also address if the IL-18-induced cell death that associates with clearance of RV is mediated by pyroptosis. IL-18 administration did not induce cleaved gasdermin D in mice lacking this gene (Figure 6A), thus verifying the specificity of the antibody we utilized, IL-18 induced cell death of RV-infected mice proceeded at least as robustly as had been observed in WT mice (Figure 6B). Specifically, although gasdermin-deficient mice had mild elevations in basal caspase 3, they still upregulated this caspase in response to IL-18, albeit at markedly lower levels compared with WT mice. IL-18 induced marked TUNEL-positivity in these mice (Figure 6C, D) and fully protected gasdermin-deficient mice against RV infection (Figure 6E). These results argue that IL-18-induced cell death and associated clearance of RV is not mediated by pyroptosis.
Fig. 6. Pyroptosis mediator Gasdermin is not required for IL-18-induced cell death or its protection against mRV infection.
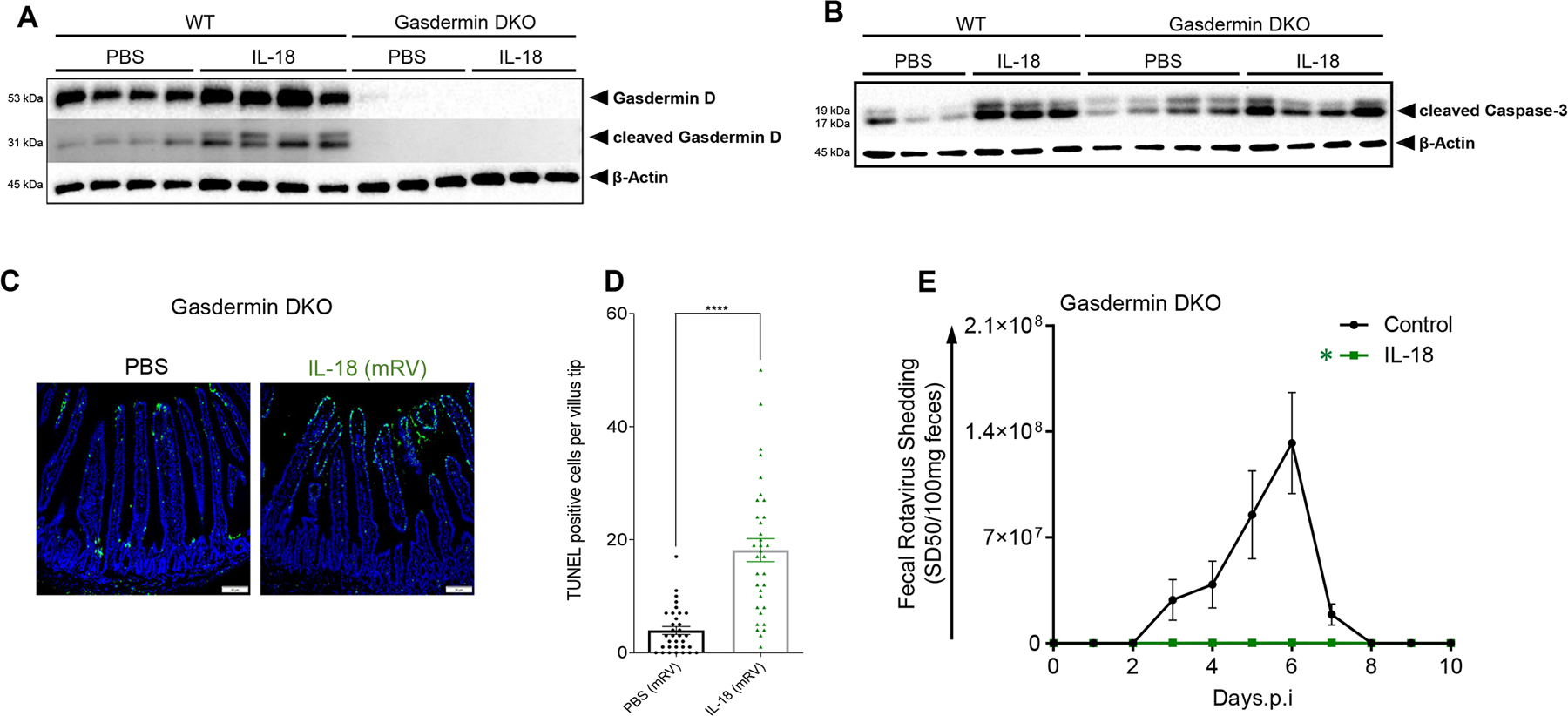
(A-D) Gasdermin-deficient, or WT, mice were administered PBS or IL-18 (2 μg) three days post-mRV inoculation. Mice were euthanized 6 h later and jejunums were analyzed. (A and B) IEC were analyzed by SDS-PAGE immunoblotting for detection of gasdermin-D, cleaved gasdermin D and cleaved caspase 3, respectively. (C) Cell death by TUNEL, counterstained with DAPI. (D) Quantitation of TUNEL-positive cells at villus tip region based on visual counts. Experiments included 5 mice per condition. Data in panel G was based on assay 6–8 villi per mouse, which are indicated by data points **** indicates P < 0.0001 by Student’s t test. (E) Gasdermin-deficient mice were administered PBS or IL-18 (2 μg) via intraperitoneal injection, 2 hours prior to, or 2, 4, 6 or 8 days after (indicated by arrows), oral inoculation with mRV. Fecal RV levels were measured over time by ELISA. . Data are mean ± SEM, n=5 * indicates significantly different from control by two-way ANOVA, P < 0.0001.
IL-18 interrupts viral replication
We examined the extent by which IL-22-induced IEC extrusion and IL-18-induced IEC death was associated with RV reduction in the ileum at 6h and 24h following administration of these cytokines. We measured the levels of RV genomes and the ratio of positive to negative (+/−) RV strands in both the lumen and IEC, which reflects levels of active replication since most positive strands encode RV proteins and do not get incorporated into virions (24). In accord with our previous work, we observed that in the epithelium, both IL-22 and IL-18 led to a clear reduction in both the level of RV genomes and RV replication by 6h (Figure 7 A, B). In contrast, the small intestinal lumen had a dramatic but variable increase in the level of RV genomes and a stark increase in RV +/− strand ratios 6h following administration of IL-18 with the combination of IL-18 and IL-22 but not IL-22 alone (Figure 7C, D). By 24h, levels of RV in the lumen had dropped dramatically whereas the miniscule levels of remaining virus appeared to not be actively replicating (Figure 7E, F). Collectively, these results support a model wherein IL-18-induced cell death interrupts active RV replication, spewing incompletely replicated virus into the lumen while IL-22 induces IEC migration and subsequent extrusion of the mature IEC that RV targets, thus together working in concert to resolve RV infection.
Fig. 7. Administration of IL-18 rapidly releases the replicating virus into the luminal side.
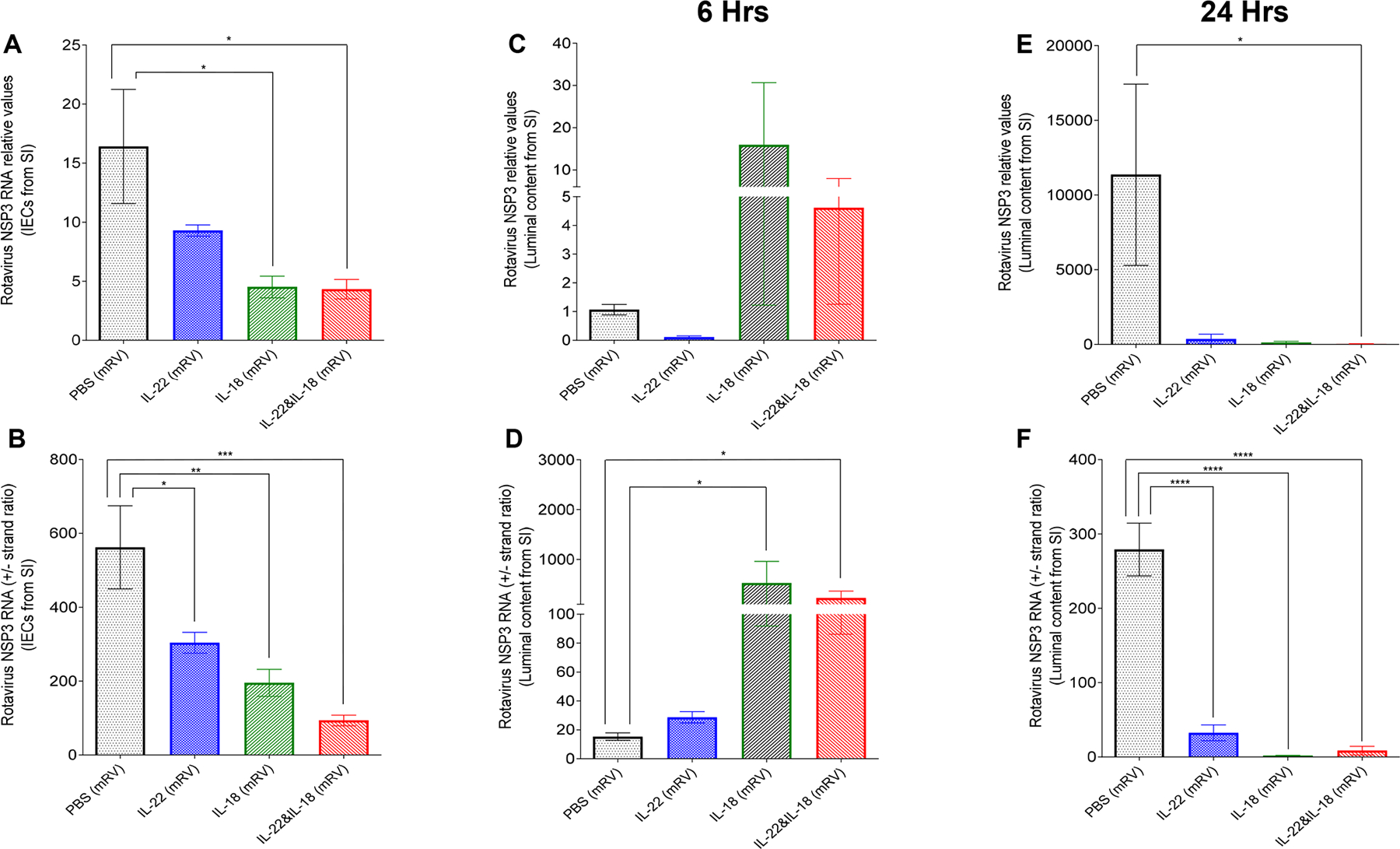
mRV-infected mice were i.p injected with PBS, IL-22 (10μg), IL-18 (2 μg) or both cytokines on day 3 post-mRV inoculation. 6 or 24 h, later, mice were euthanized and contents of jejunums were isolated. RNA was extracted and used to measure of mRV genomes and replication status as reflected by NSP3 RNA levels and the ratio of NSP3 (+) RNA strand to complimentary NSP3 (−) RNA strand. (A and B) The overall mRV genome and efficacy of virus replication in small intestinal epithelial cells. (C to F) The overall mRV genome and efficacy of virus replication in luminal content from small intestine (one-way ANOVA, n = 5–10, * represents P < 0.05, ** represents P < 0.01, *** represents P < 0.001, **** represents P < 0.0001).
Discussion
The central focus of this study was to determine the mechanisms by which IL-18 and IL-22, which are elicited by bacterial flagellin, contribute to preventing or curing RV infection. We initially considered that the ability of IL-18 and IL-22 to promote each other’s expression allowed them to use a shared mechanism to promote RV clearance. We found that irrespective of such mutual promotion, IL-18 and IL-22 both impeded RV independent of each other and did so by distinct mechanisms, which is illustrated in Figure 8. Specifically, IL-22 drove IEC proliferation and migration toward villus tips, thus accelerating the ongoing process of extrusion of highly differentiated IEC at the major site of RV replication. In contrast, administration of IL-18 to RV-infected mice induced rapid cell death, as defined by TUNEL, at villus tips where RV is localized. Such induction of TUNEL positivity, which is not typically seen at significant levels in the intestine, was associated with rapid abortion of the RV replication cycle followed by a marked reduction of RV antigens in the intestinal tract. These actions of IL-22 and IL-18 together resulted in rapid and complete expulsion of RV, thus providing a mechanism that explains how this combination of cytokines prevents and cures RV infection.
Fig. 8. Proposed mechanism by which IL-22 and IL-18 prevent and cure RV infection. This will need a more descriptive caption.
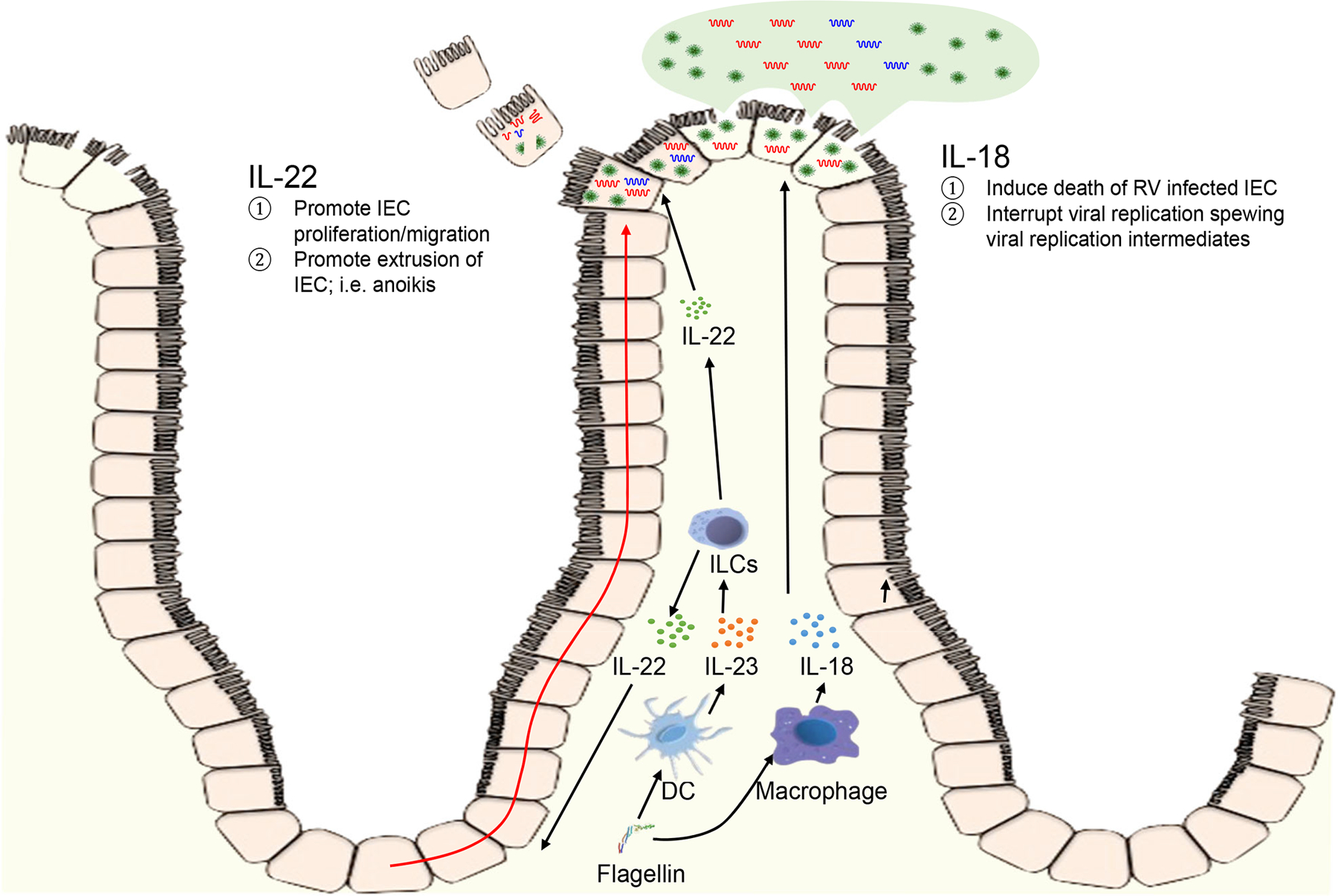
IL-22 increases epithelial proliferation thus increasing extrusion of epithelial cells, including RV-infected cells. into lumen the intestinal lumen; i.e. anoikis. IL-18 induces rapid cell death, associated with loss of cell rupturing of RV-infected cells.
RV does not induce detectable increases in IL-22 expression nor does genetic deletion of IL-22 appear to markedly augment RV infection (5), thus arguing that IL-22 does not normally play a major role in clearance of this pathogen. The known cooperation of IL-22 and interferon-λ in activating antiviral gene expression [3] suggests the possibility that RV may have evolved strategies to deliberately avoid or block IL-22 induction. Nonetheless, the downstream action of IL-22, particularly its promotion of IEC turnover may be shared by endogenous anti-RV host defense mechanisms. The role of adaptive immune-independent host defense against RV is most easily appreciated in immune compromised mice wherein RV loads decline markedly from their peak levels, but it may also play a role in protecting against RV even in immune competent mice. Innate host defense against RV is likely multifactorial, and may involve type III interferon (3), particularly in neonate mice. Our observations in adult mice indicate that RV infection increases IEC extrusion, and this mechanism combined with previous observations that RV infection activates intestinal stem cell proliferation suggests that increased IEC turnover may limit RV infection (2). We do not think such a mechanism is necessarily unique to IL-22 as EGF has ability to drive similar events. Moreover, we recently showed that SFB also drives enterocyte proliferation independent of IL-22 and is not required for adaptive immunity (23). Hence, we presume that IL-22 can activate a primitive mechanism of host defense against a variety of challenges, especially those impacting IEC.
IEC are rapidly proliferating cells with average lifetimes of about 3 days (24), which means that the intestine must eliminate vast numbers of cells continuously. The overwhelming majority of IEC are eliminated via cell extrusion at villus tips through a process termed anoikis. A central tenet of anoikis is that cells remain alive at the time of extrusion followed by the lack of attachment to other cells resulting in induction of a programmed death process (25). A key aspect of this process is that cells can be eliminated without comprising gut barrier function, thus avoiding infection and inflammation that might otherwise occur. Accordingly, administration of IL-22 is associated with few adverse effects and has been shown to resolve inflammation in several different scenarios. (26). Moreover, IL-22 plays a broad role of maintaining gut health in the intestinal tract, including mediating microbiota-dependent impacts of dietary fiber (27). It is possible that increasing anoikis via IL-22 results in extrusion of RV-containing cells in a manner that prevents viral escape and, consequent infection of other IEC. However, inability of IL-22 to induce detectable increases in luminal RV argues against this possibility. Rather, we envisage that the cell death process following IEC extrusion might result in destruction of RV in these cells. We also hypothesize that the accelerated IEC turnover induced by IL-22 may result in villus IEC being less differentiated and less susceptible to RV infection. In accord with this possibility, we observed that that flagellin administration resulted in an IL-22-dependent increase in CD44+26− IEC (Figure. S5), which are known to be RV-resistant (28). It is difficult to discern the relative importance of IL-22 in the induction of IEC extrusion versus its impact on differentiation state of villus IEC. IL-22-induced reduction in RV levels in chronically infected Rag-1−/− mice occurs over a course of several days supports a role for the latter mechanism. Use of IL-22 receptor bone marrow chimera mice demonstrated that IL-22 acts directly on IEC to affect RV infection. (7). IL-22 induced signaling is generally thought to be mediated by STAT3 (5, 10), and IL-22 induced phosphorylation of STAT3 in IEC in vivo. However, we observed that IEC-specific STAT3-KO mice could still be protected against RV by IL-22, suggesting that this mechanism of action may not proceed by a characterized signaling mechanism (Fig. S6). Thus, how the IL-22 receptor signals to impact IEC phenotype remains incompletely understood.
In contrast to IL-22, recent work indicates induction of IL-18 plays a role in endogenous immunity against RV, wherein caspase 1-mediated IL-18 production results from activation of the NLR9pb inflammasome. Such IL-18 induction paralleled gasdermin-dependent cell death, the absence of which resulted in delayed clearance of RV (29, 30). Based on this work, we hypothesized that exogenously administered IL-18 might enhance RV-induced death of RV-infected cells and/or increase IEC turnover analogous to IL-22. Administration of IL-18 in the absence of RV elicited a modest increase in the number of TUNEL- positive cells as well as a modest increase in IEC proliferation/migration that was not accompanied by increased IEC extrusion, suggesting the increased proliferation compensated for cell death. However, TUNEL positive cells were scattered along the villus. In RV infected mice, IL-18 led to TUNEL-positive cells at the villus tips, which is also the primary site of RV-infection. It is tempting to envisage localized impacts of IL-18 reflect the pattern of expression of the IL-18 receptor, including localization to villus tips and/or induced by RV, but limited knowledge of the determinants of its expression and lack of available reagents to study it render these ideas as speculative.
The manner of IL-18-induced cell death, namely its striking TUNEL induction, which was associated with spewing of RV replication intermediates, suggested pyroptotic cell death. However, we found that lack of gasdermin D and E, which are thought to be essential for pyroptosis, did not impede IL-18-induced cell death in RV-infected cells thus arguing such cell death does not fit perfectly into any known cell death pathways. Induction of IL-18 receptor-mediated signaling by itself is not sufficient to induce cell death in villus tip epithelial cells but triggers death in cells primed as a result of RV infection. The nature of such priming is not understood but may involve IEC signaling pathways, including NLR9pb, TLR3, and PKR, which are capable of recognizing RV components and/or responding to intracellular stress in general (30–32). In this context, the ability of IL-22 to enhance IL-18-induced TUNEL positivity in RV-infected cells may reflect an intersection of IL-22-R and IL-18-R signaling or be a manifestation of these cytokines to promote each other’s expression.
The central limitation in our study was that our approaches were largely correlative. Specifically, we lacked modalities to specifically block IEC migration or cell death in response to IL-22 and IL-18 respectively. Another limitation is that we were not able to demonstrate that the TUNEL-positive cells actually contained RV. Indeed, our attempts to do so via double-staining were not successful, possibly reflecting that the disappearance of RV after cytokine treatment likely occurs early in the cell death process while the DNA fragmentation that underlies TUNEL positivity is considered a late event in the cell death process. Thus, more specific identification of processes that mediate cell death of RV-infected IEC in response to IL-18 is an important target of future studies.
The improved understanding of the mechanism by which IL-18/22 controls RV infection reported herein should inform use of these cytokines to treat viral infection in humans. Chronic RV infections can occur in immune compromised humans, suggesting that IL-18/22 may be explored as a possible treatment for this and other chronic viral infections. Our results suggest that this cytokine treatment may be effective for viruses that preferentially infect villus epithelial cells and possibly other epithelia that have high cell turnover rates. In contrast, this combination of cytokines seems unlikely to impact viruses that inhabit more long-lived cells, including hematopoietic cells that are generally not responsive to IL-22. We observed that flagellin and IL-18/22 has some efficacy against reovirus, particularly early in infection when it infects gut epithelial cells, as well as some efficacy against influenza, which initially infects lung epithelial cells, but did not show any impact on hepatitis C virus as assayed in mice engrafted with human hepatocytes, which are thought to be long lived cells. IL-18/22 can protect mice against norovirus infection, which infects B cells and tuft cells (33, 34), but human norovirus is thought to primarily infect epithelial cells, particularly in immunocompromised persons who develop chronic norovirus infections (35). SARS-CoV2, the causative agent of COVID-19 has also been observed to replicate in intestinal epithelial cells (36), and like RV, appears to replicate in mature IEC, which express the SARS-CoV2 receptor ACE2. Intestinal replication of SARS-CoV-2 is thought to contribute to extra-respiratory pathologies associated with COVID-19 (37). As such, the use of IL-18/22-based therapy may be a potential strategy to treat chronic rotavirus and/or norovirus infections in person with immune dysfunction and, moreover, might serve to mitigate severe cases of COVID-19.
Materials and Methods
Study Design
This study sought to ascertain the mechanism by which IL-22 and IL-18 prevent and cure RV infection. Mice were orally administered RV. Extent of infection was assayed my measuring viral genomes and proteins in the intestine. IL18 and or IL-22 were administered to mice with various genetic deficiencies. Cell extrusion and cell death were measured. All procedures were Rotavirus-infected mice were approved by GSU’s animal care and use committee (IACUC # 17047).
Mice
All mice used herein were adults (i.e. 4–8 week-old) on a C57BL/6 background bred at Georgia State University (Atlanta, GA). Rotavirus-infected mice were housed in an animal biosafety level 2 facility. WT, Rag-1−/−, IL-18−/−, IL-18-R−/−, Stat3flox, and Villin-cre were purchased from Jackson Laboratories (Bar Harbor, ME, USA). NLRC4−/−, IL-22−/−, and IL-22-R−/− mice were provided by Genentech (South San Francisco, CA, USA). TLR5−/− and TLR5−/−/NLRC4−/− and WT littermates were maintained as previously described (5). Gasdermin D/E−/− mice, whose generation and initial characterization was previously described (22) were shipped to GSU and studied in original and cross-fostered state as indicated in results.
Materials
Murine Fc-IL-22 was provided by Genentech, Inc. Murine IL-18 was purchased from Sino Biological Inc (Beijing, China). Procedures for isolation of flagellin and verification of purity, were described previously (5). Recombinant murine epidermal growth factor (mEGF) was purchased from PEPROTECH.
Rotavirus infection
Acute Models:
Age- and sex-matched adult mice (8–12 weeks of age) were orally administrated 100 μl 1.33% sodium bicarbonate (Sigma) and then inoculated with 105 SD50 of murine rotavirus EC strain. Approach used to determine SD50 has been described previously (5). Chronic model: 5-week-old Rag-1−/− mice were infected with mRV (same infection procedure as described in Acute Models). Feces were collected 3-week post rotavirus inoculation to confirm the establishment of chronic infection.
In vitro model:
Cell culture-adapted rhesus RV was trypsin-activated (10 μg/ml trypsin in serum-free RPMI-1640 (Cellgro) at 37°C for 30 min. The basolateral side of the polarized Caco-2 cells were stimulated with cytokines, 1.5 hours prior to expose to rhesus RV infection as previously described (5). The upper chamber of transwells were infected with trypsin-pretreated RRV and allowed to adsorb at 37°C for 40 min before being washed with serum-free medium (SFM). The presence of cytokines was maintained at a constant level throughout the experiment.
Fecal Rotavirus Antigen Detection
Fecal pellets were collected daily from individual mouse on days 0–10 post rotavirus inoculation. Samples were suspended in PBS (10% wt./vol.) and, after centrifugation, supernatants of fecal homogenates were analyzed by enzyme-linked immunosorbent assay (ELISA) after multiple serial dilutions, more detailed descriptions of experimental procedures are previously described (5).
Generation of Bone Marrow Chimeric mice
Mice were subjected to X-ray irradiation using an 8.5 Gy equivalent followed by injection of 2×107 bone marrow cells administered intravenously as previously described (5). All mice were afforded an 8-week recovery period before experimental use. For the first two weeks post-transfer, mice were maintained in sterile cages, and supplied with drinking water containing 2 mg/ml neomycin (Mediatech/Corning).
Visual assessment of IEC shed into small intestinal lumen
Intestinal sections were fixed in methanol-Carnoy’s fixative solution (60% methanol, 30% chloroform, 10% glacial acetic acid) for 48 hours at 4°C. Fixed tissues were washed two times in dry methanol for 30 min each, followed by two times in absolute ethanol for 20 min each, then incubated in two baths of xylene before proceeding to paraffin embedding. 4 μm-thin sections were sliced from paraffin-embedded tissues, and placed on glass slides after floating on a water bath. The sections were dewaxed by initial incubation at 60°C for 20 min, followed by two baths in prewarmed xylene substitute solution for 10 min each. Deparaffinized sections were then hydrated in solutions with decreasing concentration of ethanol (100, 95, 70, 50, and 30%) every 5 min in each bath. Last, slides allowed to dry almost completely and were then mounted with Prolong antifade mounting media containing DAPI before analysis by fluorescence microscopy.
Immunohistochemistry for TUNEL staining
Intestinal sections were fixed in 10% buffered formalin at room temperature for 48 hours, and then embedded in paraffin. Tissues were sectioned at 4 μm thickness, and IEC death was detected by TUNEL assay using the In Situ Cell Death Detection Kit, Fluorescein (Roche) according to the manufacturer’s instructions.
Immunoblot Analysis for assay of cleaved Caspase-3, Phospho-STAT3 and cleaved Gasdermin D
Intestinal epithelial cells lysate (20 μg per lane) were separated by SDS-PAGE through 4%−20% Mini-PROTEAN® TGX™ gel (Bio-rad, USA), transferred to nitrocellulose membranes, and analyzed by immunoblot, as previously described (5). Briefly, isolated IEC were incubated with RIPA lysis buffer (Santa Cruz Biotechnology, USA) for 30 min at room temperature. Subsequently, cell lysates were homogenized by pipette and subjected to full-speed centrifugation. Protein bands were detected for cleaved caspase 3, phosphor-STAT3 and anti-β-actin (Cell Signaling), and incubated with horseradish peroxidase-conjugated anti-rabbit secondary antibody. Immunoblotted proteins were visualized with Western blotting detection Reagents (GE Healthcare) and then imaged using the ChemiDoc XRS+ system (Bio-RAD).
Isolation of intestinal epithelial cells
The entire small intestine was harvested from mice according to indicated experimental design, and sliced longitudinally before being washed gently in PBS to remove the luminal content. Tissues were processed and maintained at 4°C throughout. Cleaned tissue samples were further minced into 1–2-mm3 pieces, and shaken in 20 ml HBSS containing 2mM EDTA and 10 mM HEPES for 30 min. An additional step of vigorous vortexing in fresh HBSS (10 mM HEPES) after EDTA incubation facilitated cell disaggregation. Intestinal epithelial cells (IEC) were then filtered through 70-μm nylon mesh strainer (BD Biosciences), centrifuged, and resuspended in PBS.
Antibody Staining and Flow Cytometry Analysis
Bulk leukocytes and intestinal epithelial cells isolated above were incubated with succinimidyl esters (NHS ester)-Alexa Fluor 430, which permitted determination of cell viability. Cells were then blocked by incubation with 10 μg/ml anti-CD16/anti-CD-32 (clone 2.4G2 ATCC). 20 min later, cells were stained with fluorescently conjugated antibodies: CD26-PE (clone: H194–112, eBioscience), CD44-PECy7 (clone: IM7, eBioscience), CD45-FITC (clone: 30-F11, eBioscience), CD326-APC (clone: G8.8, eBioscience). Finally, stained cells were fixed with 4% formaldehyde for 10 mins before whole cell population was analyzed on a BD LSR II flow cytometer. Collected data was analyzed using FlowJo.
Quantification of IEC shedding from luminal content
Host DNA was quantitated from 100 mg of luminal content (100 mg) from small intestine by using QIAamp DNA Stool Mini kit (Qiagen), and subjected to quantitative PCR using QuantiFast SYBR Green PCR kit (Bio-Rad) in a CDX96 apparatus (Bio-Rad) with specific mouse 18S oligonucleotides primers. The sense and antisense oligonucleotides primers used were: 18s-1F: 5’-GTAACCCGTTGAACCCCATT-3’ and 18s-1R: 5’-CCATCCAATCGGTAGTAGCG-3’. PCR results were expressed as actual numbers of IEC shedding per 100 mg of luminal content, calculated using a standard curve, which was generated using two-fold serial dilutions of mouse colon carcinoma cell line MC26. DNA was extracted from each vial with known number of MC26 cells after serial dilutions, and then real-time quantitative PCR was performed. The cycle quantification (Cq) values (X-axis) are inversely proportional to the amount of target genes (18S) (Y-axis), and a standard curve is applied to estimate the numbers of cell shedding from luminal content based on the quantity of target copies (18S) from each sample.
Quantification of RV genomes and replication in IEC and luminal content
To extract RNA, cell pellets were homogenized with TRIzol™ (Invitrogen), and chloroform was added to the homogenate to separate RNA (an upper aqueous layer) from DNA and proteins (a red lower organic layer). Isopropanol facilitated the precipitation of RNA out of solution, and after centrifugation, the impurities were removed by washing with 75% ethanol. RNA pellets were resuspended in RNase-free water and underwent quantitative qRT-PCR. Total RNA from luminal content was purified from RNeasy PowerMicrobiome Kit according to the manufacturer’s instructions. Primers that target NSP3 region: EC.C (+) (5’-GTTCGTTGTGCCTCATTCG-3’ and EC.C (−) (5’-TCGGAACGTACTTCTGGAC-3’) were applied to quantify viral genomes from IEC and luminal content. RV replication was quantitated as previously described (38).
BrdU pulse-chase labeling analysis of intestinal enterocyte migration
A pulse-chase experimental strategy was used to label intestinal enterocytes with 5-bromo-2-deoxyuridine (BrdU) to estimate the IEC migration rate along the crypt-villus axis over a defined period of time. Briefly, 8-week-old mice were intraperitoneally injected with either PBS or cytokine(s) (IL-22 and/or IL-18) one hour prior to BrdU treatment i.p. (50 μg per mg of mice body weight). After 16 hours, mice were euthanized, and a segment of the jejunum were resected, immediately embedded in OCT (expand) and then underwent tissue sectioning. 4 μm tissue sections were firstly fixed in 4% formaldehyde for 30 min at room temperature, and then washed three times in PBS. DNA denaturation was performed by incubating the sections in prewarmed 1.5 N HCl for 30 min at 37 °C, and then acid was neutralized by rinsing sections three times in PBS. Before BrdU immunostaining, sections were blocked with rabbit serum (BioGenex, Fremont, CA) for 1 h at room temperature, then incubated with anti-BrdU (Abcam) 2 hours at 37 °C, and counterstained with 4’, 6-diamidino-2-phenylindole (DAPI). The BrdU-labeled cells were visualized by fluorescence microscopy.
mRV antigen detection by fluorescence immunohistochemistry
The proximal jejunum was imbedded into O.C.T. compound, and then sliced into 6 μm thin sections. Tissue slides were incubated in 4% paraformaldehyde for 15 min, followed by 5 min washing of PBS twice. Autofluorescence caused by free aldehydes was quenched by incubating slides in 50 mM NH4Cl in PBS or 0.1 M Glycine in OBS for 14 min at room temperature (RT), followed by 5 min PBS washing 3 times. 3% BSA-PBS was used to block the tissue samples for 1 hour at RT. The slides were then washed with PBS for 5 min, followed by incubation with primary antibody (hyperimmune guinea pig anti-RRV serum, 1:100) in blocking buffer overnight at 4°C. After slides were washed three times with PBS, secondary antibody (donkey anti-guinea pig IgG, Jackson Immuno Research: 706-586-148) was applied to the sample slides for 1–2 hours at RT. The fluorescence emission of mRV antigen was detected by fluorescence microscopy.
Quantification and statistical analysis
Significance was determined using the one-way analysis of variance (ANOVA), or student’s t-test (GraphPad Prism software, version 6.04). Differences were noted as significant *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
Supplementary Material
Fig S1. Impact of IL-22 and IL-18 on RV infection in immune deficient, Rag-1−/− mice.
Fig S2. Lack of effect of IL-22 and IL-18 on RV infection in cultured IECs.
Fig. S3. Impact of IL-18 on IEC death.
Fig. S4. Cross-fostering on Gasdermin-deficient mice conferred susceptibility to RV infection.
Fig S5. Flagellin-mediated changes of cell subpopulations along intestinal villus-crypt axis.
Fig S6. Impact of IL-22 and flagellin on RV infection in mice lacking STAT3 in IEC.
Acknowledgements
Funding: This work was supported by NIH grants DK083890 and DK099071 (to ATG). JZ is supported by career development award from American Diabetes Association. B.C. is supported by a Starting Grant from the European Research Council, an Innovator Award from the Kenneth Rainin Foundation, and a Chaire d’Excellence from Paris University.
Abbreviations:
- RV
Rotavirus
- IEC
intestinal epithelial cells
- SI
small intestine
Footnotes
Competing Interests: Andrew T. Gewirtz and Benyue Zhang are inventors on patent application (WO2015054386A1 WIPO) held by Georgia State University that covers “Prevention and Treatment of Rotavirus Infection Using IL-18 and IL-22”.
Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. All mice are either commercially available or available under a material transfer agreement (MTA).
References
- 1.Tate JE, Burton AH, Boschi-Pinto C, Parashar UD, N. World Health Organization-Coordinated Global Rotavirus Surveillance, Global, Regional, and National Estimates of Rotavirus Mortality in Children <5 Years of Age, 2000–2013. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America 62 Suppl 2, S96–S105 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zou WY, Blutt SE, Zeng XL, Chen MS, Lo YH, Castillo-Azofeifa D, Klein OD, Shroyer NF, Donowitz M, Estes MK, Epithelial WNT Ligands Are Essential Drivers of Intestinal Stem Cell Activation. Cell reports 22, 1003–1015 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hernandez PP, Mahlakoiv T, Yang I, Schwierzeck V, Nguyen N, Guendel F, Gronke K, Ryffel B, Hoelscher C, Dumoutier L, Renauld JC, Suerbaum S, Staeheli P, Diefenbach A, Interferon-lambda and interleukin 22 act synergistically for the induction of interferon-stimulated genes and control of rotavirus infection. Nature immunology 16, 698–707 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kapikian AZ, Shope RE, “Rotaviruses, Reoviruses, Coltiviruses, and Orbiviruses” in Medical Microbiology, th, Baron S, Eds. (Galveston (TX), 1996). [PubMed] [Google Scholar]
- 5.Zhang B, Chassaing B, Shi Z, Uchiyama R, Zhang Z, Denning TL, Crawford SE, Pruijssers AJ, Iskarpatyoti JA, Estes MK, Dermody TS, Ouyang W, Williams IR, Vijay-Kumar M, Gewirtz AT, Viral infection. Prevention and cure of rotavirus infection via TLR5/NLRC4-mediated production of IL-22 and IL-18. Science 346, 861–865 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Munoz M, Eidenschenk C, Ota N, Wong K, Lohmann U, Kuhl AA, Wang X, Manzanillo P, Li Y, Rutz S, Zheng Y, Diehl L, Kayagaki N, van Lookeren-Campagne M, Liesenfeld O, Heimesaat M, Ouyang W, Interleukin-22 induces interleukin-18 expression from epithelial cells during intestinal infection. Immunity 42, 321–331 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Wolk K, Kunz S, Witte E, Friedrich M, Asadullah K, Sabat R, IL-22 increases the innate immunity of tissues. Immunity 21, 241–254 (2004). [DOI] [PubMed] [Google Scholar]
- 8.Lindemans CA, Calafiore M, Mertelsmann AM, O’Connor MH, Dudakov JA, Jenq RR, Velardi E, Young LF, Smith OM, Lawrence G, Ivanov JA, Fu YY, Takashima S, Hua G, Martin ML, O’Rourke KP, Lo YH, Mokry M, Romera-Hernandez M, Cupedo T, Dow L, Nieuwenhuis EE, Shroyer NF, Liu C, Kolesnick R, van den Brink MRM, Hanash AM, Interleukin-22 promotes intestinal-stem-cell-mediated epithelial regeneration. Nature 528, 560–564 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pickert G, Neufert C, Leppkes M, Zheng Y, Wittkopf N, Warntjen M, Lehr HA, Hirth S, Weigmann B, Wirtz S, Ouyang W, Neurath MF, Becker C, STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. The Journal of experimental medicine 206, 1465–1472 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nagalakshmi ML, Rascle A, Zurawski S, Menon S, de Waal Malefyt R, Interleukin-22 activates STAT3 and induces IL-10 by colon epithelial cells. International immunopharmacology 4, 679–691 (2004). [DOI] [PubMed] [Google Scholar]
- 11.Bishop JL, Roberts ME, Beer JL, Huang M, Chehal MK, Fan X, Fouser LA, Ma HL, Bacani JT, Harder KW, Lyn activity protects mice from DSS colitis and regulates the production of IL-22 from innate lymphoid cells. Mucosal immunology 7, 405–416 (2014). [DOI] [PubMed] [Google Scholar]
- 12.Dudakov JA, Hanash AM, van den Brink MR, Interleukin-22: immunobiology and pathology. Annual review of immunology 33, 747–785 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hanash AM, Dudakov JA, Hua G, O’Connor MH, Young LF, Singer NV, West ML, Jenq RR, Holland AM, Kappel LW, Ghosh A, Tsai JJ, Rao UK, Yim NL, Smith OM, Velardi E, Hawryluk EB, Murphy GF, Liu C, Fouser LA, Kolesnick R, Blazar BR, van den Brink MR, Interleukin-22 protects intestinal stem cells from immune-mediated tissue damage and regulates sensitivity to graft versus host disease. Immunity 37, 339–350 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, Abbas AR, Modrusan Z, Ghilardi N, de Sauvage FJ, Ouyang W, Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nature medicine 14, 282–289 (2008). [DOI] [PubMed] [Google Scholar]
- 15.Lundgren O, Svensson L, Pathogenesis of rotavirus diarrhea. Microbes and infection 3, 1145–1156 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morris AP, Estes MK, Microbes and microbial toxins: paradigms for microbial-mucosal interactions. VIII. Pathological consequences of rotavirus infection and its enterotoxin. American journal of physiology. Gastrointestinal and liver physiology 281, G303–310 (2001). [DOI] [PubMed] [Google Scholar]
- 17.Peterson LW, Artis D, Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nature reviews. Immunology 14, 141–153 (2014). [DOI] [PubMed] [Google Scholar]
- 18.Tang X, Liu H, Yang S, Li Z, Zhong J, Fang R, Epidermal Growth Factor and Intestinal Barrier Function. Mediators of inflammation 2016, 1927348 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frey MR, Golovin A, Polk DB, Epidermal growth factor-stimulated intestinal epithelial cell migration requires Src family kinase-dependent p38 MAPK signaling. The Journal of biological chemistry 279, 44513–44521 (2004). [DOI] [PubMed] [Google Scholar]
- 20.Eisenhoffer GT, Loftus PD, Yoshigi M, Otsuna H, Chien CB, Morcos PA, Rosenblatt J, Crowding induces live cell extrusion to maintain homeostatic cell numbers in epithelia. Nature 484, 546–549 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Williams JM, Duckworth CA, Watson AJ, Frey MR, Miguel JC, Burkitt MD, Sutton R, Hughes KR, Hall LJ, Caamano JH, Campbell BJ, Pritchard DM, A mouse model of pathological small intestinal epithelial cell apoptosis and shedding induced by systemic administration of lipopolysaccharide. Disease models & mechanisms 6, 1388–1399 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sarhan J, Liu BC, Muendlein HI, Li P, Nilson R, Tang AY, Rongvaux A, Bunnell SC, Shao F, Green DR, Poltorak A, Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc Natl Acad Sci U S A 115, E10888–E10897 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi Z, Zou J, Zhang Z, Zhao X, Noriega J, Zhang B, Zhao C, Ingle H, Bittinger K, Mattei LM, Pruijssers AJ, Plemper RK, Nice TJ, Baldridge MT, Dermody TS, Chassaing B, Gewirtz AT, Segmented Filamentous Bacteria Prevent and Cure Rotavirus Infection. Cell 179, 644–658 e613 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park JH, Kotani T, Konno T, Setiawan J, Kitamura Y, Imada S, Usui Y, Hatano N, Shinohara M, Saito Y, Murata Y, Matozaki T, Promotion of Intestinal Epithelial Cell Turnover by Commensal Bacteria: Role of Short-Chain Fatty Acids. PloS one 11, e0156334 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gilmore AP, Anoikis. Cell death and differentiation 12 Suppl 2, 1473–1477 (2005). [DOI] [PubMed] [Google Scholar]
- 26.Alabbas SY, Begun J, Florin TH, Oancea I, The role of IL-22 in the resolution of sterile and nonsterile inflammation. Clinical & translational immunology 7, e1017 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zou J, Chassaing B, Singh V, Pellizzon M, Ricci M, Fythe MD, Kumar MV, Gewirtz AT, Fiber-Mediated Nourishment of Gut Microbiota Protects against Diet-Induced Obesity by Restoring IL-22-Mediated Colonic Health. Cell Host Microbe 23, 41–53 e44 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sen A, Rothenberg ME, Mukherjee G, Feng N, Kalisky T, Nair N, Johnstone IM, Clarke MF, Greenberg HB, Innate immune response to homologous rotavirus infection in the small intestinal villous epithelium at single-cell resolution. Proceedings of the National Academy of Sciences of the United States of America 109, 20667–20672 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, Yang ZH, Zhong CQ, Han J, Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell research 25, 1285–1298 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu S, Ding S, Wang P, Wei Z, Pan W, Palm NW, Yang Y, Yu H, Li HB, Wang G, Lei X, de Zoete MR, Zhao J, Zheng Y, Chen H, Zhao Y, Jurado KA, Feng N, Shan L, Kluger Y, Lu J, Abraham C, Fikrig E, Greenberg HB, Flavell RA, Nlrp9b inflammasome restricts rotavirus infection in intestinal epithelial cells. Nature 546, 667–670 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garcia MA, Gil J, Ventoso I, Guerra S, Domingo E, Rivas C, Esteban M, Impact of protein kinase PKR in cell biology: from antiviral to antiproliferative action. Microbiology and molecular biology reviews : MMBR 70, 1032–1060 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vercammen E, Staal J, Beyaert R, Sensing of viral infection and activation of innate immunity by toll-like receptor 3. Clinical microbiology reviews 21, 13–25 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilen CB, Lee S, Hsieh LL, Orchard RC, Desai C, Hykes BL Jr., McAllaster MR, Balce DR, Feehley T, Brestoff JR, Hickey CA, Yokoyama CC, Wang YT, MacDuff DA, Kreamalmayer D, Howitt MR, Neil JA, Cadwell K, Allen PM, Handley SA, van Lookeren Campagne M, Baldridge MT, Virgin HW, Tropism for tuft cells determines immune promotion of norovirus pathogenesis. Science 360, 204–208 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jones MK, Watanabe M, Zhu S, Graves CL, Keyes LR, Grau KR, Gonzalez-Hernandez MB, Iovine NM, Wobus CE, Vinje J, Tibbetts SA, Wallet SM, Karst SM, Enteric bacteria promote human and mouse norovirus infection of B cells. Science 346, 755–759 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Karandikar UC, Crawford SE, Ajami NJ, Murakami K, Kou B, Ettayebi K, Papanicolaou GA, Jongwutiwes U, Perales MA, Shia J, Mercer D, Finegold MJ, Vinje J, Atmar RL, Estes MK, Detection of human norovirus in intestinal biopsies from immunocompromised transplant patients. The Journal of general virology 97, 2291–2300 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zang R, Gomez Castro MF, McCune BT, Zeng Q, Rothlauf PW, Sonnek NM, Liu Z, Brulois KF, Wang X, Greenberg HB, Diamond MS, Ciorba MA, Whelan SPJ, Ding S, TMPRSS2 and TMPRSS4 promote SARS-CoV-2 infection of human small intestinal enterocytes. Sci Immunol 5, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pan L, Mu M, Yang P, Sun Y, Wang R, Yan J, Li P, Hu B, Wang J, Hu C, Jin Y, Niu X, Ping R, Du Y, Li T, Xu G, Hu Q, Tu L, Clinical Characteristics of COVID-19 Patients With Digestive Symptoms in Hubei, China: A Descriptive, Cross-Sectional, Multicenter Study. Am J Gastroenterol 115, 766–773 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fenaux M, Cuadras MA, Feng N, Jaimes M, Greenberg HB, Extraintestinal spread and replication of a homologous EC rotavirus strain and a heterologous rhesus rotavirus in BALB/c mice. J Virol 80, 5219–5232 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1. Impact of IL-22 and IL-18 on RV infection in immune deficient, Rag-1−/− mice.
Fig S2. Lack of effect of IL-22 and IL-18 on RV infection in cultured IECs.
Fig. S3. Impact of IL-18 on IEC death.
Fig. S4. Cross-fostering on Gasdermin-deficient mice conferred susceptibility to RV infection.
Fig S5. Flagellin-mediated changes of cell subpopulations along intestinal villus-crypt axis.
Fig S6. Impact of IL-22 and flagellin on RV infection in mice lacking STAT3 in IEC.