

Summary
Purpose
PI3K/AKT/mTOR and RAS/RAF/MEK pathways are frequently dysregulated in colorectal cancer (CRC). We conducted a biomarker-driven trial of the combination of MK-2206, an allosteric AKT 1/2/3 inhibitor, and selumetinib, a MEK 1/2 inhibitor, in patients with CRC to evaluate inhibition of phosphorylated ERK (pERK) and AKT (pAKT) in paired tumor biopsies.
Patients and Methods
Adult patients with advanced CRC were enrolled in successive cohorts stratified by KRAS mutation status. Initially, 12 patients received oral MK-2206 90 mg weekly with oral selumetinib 75 mg daily in 28-day cycles. Following an interim analysis, the doses of MK-2206 and selumetinib were increased to 135 mg weekly and 100 mg daily, respectively. Paired tumor biopsies were evaluated for target modulation.
Results
Common toxicities were gastrointestinal, hepatic, dermatologic, and hematologic. Of 21 patients enrolled, there were no objective responses. Target modulation did not achieve the pre-specified criteria of dual 70 % inhibition of pERK and pAKT levels in paired tumor biopsies.
Conclusion
Despite strong scientific rationale and preclinical data, clinical activity was not observed. The desired level of target inhibition was not achieved. Overlapping toxicities limited the ability to dose escalate to achieve exposures likely needed for clinical activity, highlighting the challenges in developing optimal combinations of targeted agents.
Keywords: MK-2206, Selumetinib, pAKT, pERK, Colorectal carcinoma
Introduction
Activating mutations in the KRAS gene, most commonly involving codons 12 or 13, are detected in approximately 30– 40 % of patients with colorectal cancer (CRC) and predict for resistance to EGFR inhibitors [1, 2]. AKT upregulation occurs in approximately 60 % of CRC cases [3]. Activating mutations in the PI3K-AKT pathway and increased phosphorylation of AKT through a MEK-EGFR-PI3K feedback loop can result in resistance to MEK inhibitors [4, 5]. Therefore, dual inhibition of both pathways may represent an effective therapeutic strategy to provide clinical benefit to patients with refractory CRC.
MK-2206, an allosteric inhibitor of AKT, has been shown to potently inhibit the three human isoforms of AKT (AKT1, AKT2, and AKT3) with IC50’s in the nanomolar range [6]. Two different drug administration schedules of MK-2206 were initially evaluated in early phase trials: an every other day schedule and a once a week schedule [6, 7]. Based on pharmacokinetic data and toxicities, the once weekly dosing schedule was pursued for further development [6]. Administration of single doses of MK-2206 in A2780 ovarian tumor xenografts showed greater than 80 % inhibition of AKT1 and AKT2 kinase activity in peripheral blood1h after dosing, and ~70 % inhibition of pSer473AKT in tumor tissue at 6 h after dosing, a level of inhibition that was maintained for at least 24 h at doses higher than 30 mg/kg (personal communication, MH Lam).
Selumetinib is a potent, oral, non-ATP competitive, small molecule inhibitor of the mitogen-activated protein (MAP) kinase kinase, MEK-1/2 (IC50 10–14 nM) [8]. In preclinical models of CRC, > 70 % inhibition of nuclear pERK in tumor tissue was achieved 4 h after a single 25 mg/kg dose in both Colo-205 and SW-620 xenografts, with concurrent evidence of growth inhibition [8]. Additional work demonstrated statistically significant inhibition of tumor growth in HCT116 xenografts, with pharmacodynamic assessments following single doses of study drugs (alone and in combination) showing >70 % inhibition of pSer473AKT and pERK levels at 3 h post-administration of the combination, that persisted up to 5 h (Fig. 1; personal communication, MH Lam).
Fig. 1.
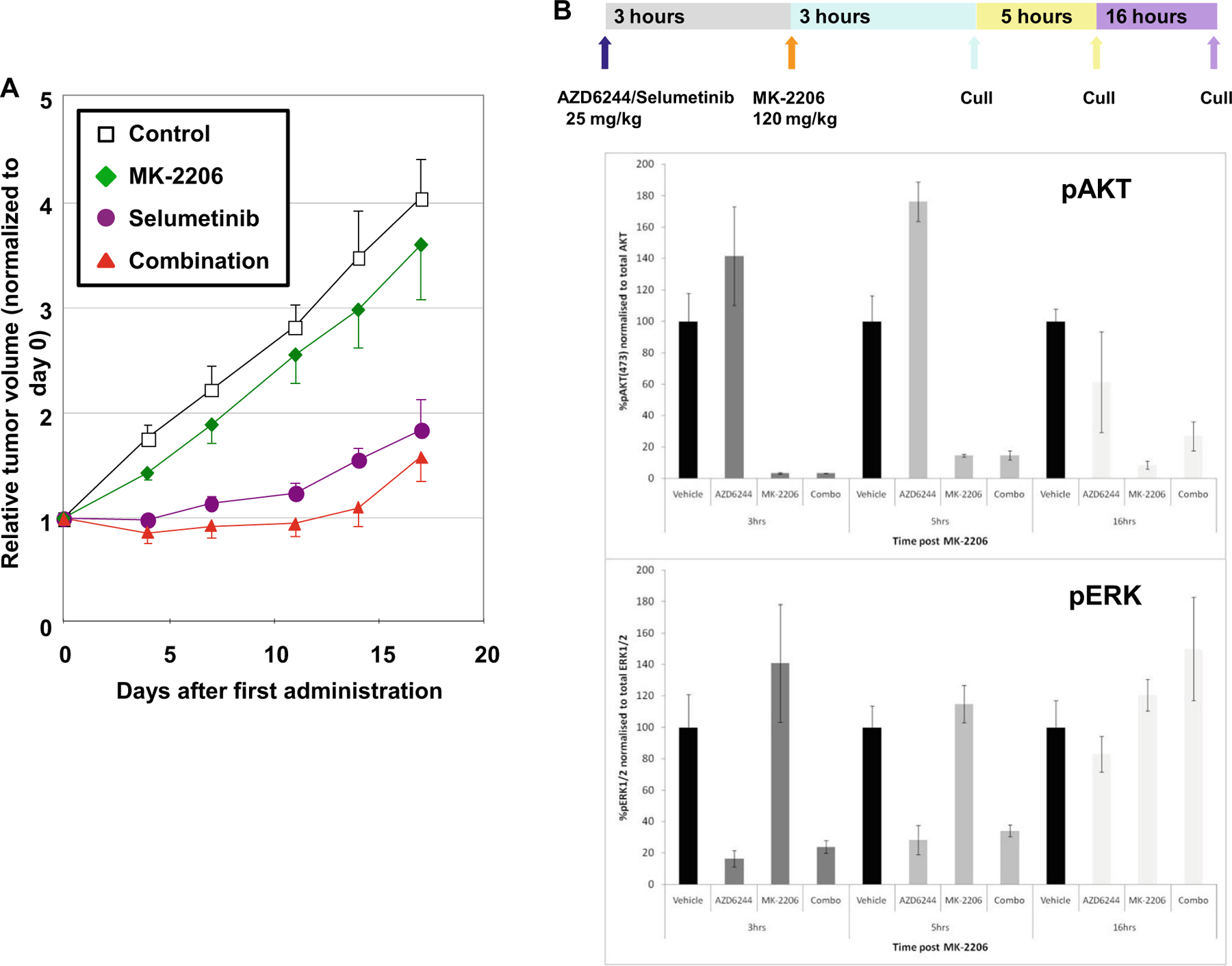
a Efficacy of MK-2206 and selumetinib in HCT116 tumor xenografts (n = 5). Mice were treated with MK-2206 monotherapy (120 mg/kg PO QOD for 2 weeks), selumetinib monotherapy (25 mg/kg PO BID 5 days per week for 2 weeks), or the combination using monotherapy doses. The combination showed a statistically significant decrease in tumor growth inhibition as compared to MK-2206 monotherapy (p<0.05). Error bars represent standard error of the mean. b In vivo pharmacodynamic effect of MK-2206 and selumetinib on pAKT and pERK in HCT116 tumor models. Selumetinib was administered at a dose of 25 mg/kg, and MK-2206 was administered at a dose of 120 mg/kg 3 h post-selumetinib dosing. Tumor tissue was harvested at 3, 5, and 16 h after administration of MK-2206 in combination with selumetinib. Mesoscale analysis of pharmacodynamic target engagement was carried out for total AKT, pSer427AKT, total ERK, and pThr202/Tyr204ERK expression. Greater than 70 % inhibition of both pAKT and pERK was observed with combination treatment persisting out to 5 h. Error bars reflect the standard deviation and were based on the mean values of 4 mice per timepoint. [Courtesy of Merck; adapted with permission.]
Based on doses established in an initial phase 1 trial [9] and interest in evaluating this combination in advanced CRC, we conducted a biomarker-driven, phase 2 study of the combination of MK-2206 and selumetinib in patients with refractory CRC, with the objectives of demonstrating modulation of phosphorylated ERK (pERK) and AKT (pAKT) in tumor biopsies following the administration of study drugs, and determining the antitumor activity of the combination. Dual pAKT and pERK inhibition of at least 70 % from baseline levels in tumor biopsies 3–6 h post drug administration was chosen as the objective of the current trial to correspond to statistically significant tumor growth in single agent experiments utilizing selumetinib or MK-2206, respectively [8, 10].
Patients and methods
Patient population
This open-label, single arm, biomarker-driven, phase 2 study evaluated levels of pERK and pAKT in tumor biopsies prior to and following administration of the combination of MK-2206 and selumetinib; patients were ≥ 18 years old with histologically-confirmed, metastatic CRC that had progressed or recurred following oxaliplatin- and irinotecan-based chemotherapy administered for metastatic disease. Patients were stratified based on their KRAS mutation status (defined by presence of codon 12/13 mutation on archival tissue) within each cohort (Fig. 2). Patients were required to have disease amenable to biopsy, be willing to undergo biopsies, and have an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1. Additionally, adequate organ function was required, defined as absolute neutrophil count ≥ 1500/µL, platelet count ≥ 100,000/µL, total bilirubin ≤ 1.5X institutional upper limit of normal (ULN), AST and ALT ≤ 3 x ULN (up to 5 X ULN was allowed in the setting of liver metastases), and creatinine < 1.5 X ULN. Exclusion criteria included poorly controlled diabetes (defined as fasting blood glucose > 160 mg/dL or HgbA1C > 8 %), or prolonged QTc (defined as QTc >450 msec for men and 470 msec for women by the Bazett’s formula).
Fig. 2.

Enrollment schema. Patients with histologically confirmed CRC were stratified by KRAS mutational analysis and enrolled in successive cohorts of 6 patients each (3 KRAS wild-type and 3 KRAS-mutation positive disease) based on timing of biopsies: post-biopsy C1D1 (Cohort A), post-biopsy C1D22 (Cohort B), or post-biopsy C1 D4 (Cohort C). If no patient satisfied dual inhibition of pERK and pAKT of ≥ 70 % among the patients accrued to either KRAS mutation status subset in a specific cohort, no further patients were to be accrued to that cohort subset
Study design
Patients received MK-2206 orally once a week in combination with selumetinib orally once a day in 28-day cycles. Successive cohorts of 6 patients each (3 with wild-type KRAS and 3 with KRAS-mutation positive disease) were enrolled with different post-biopsy timings: post-biopsy on cycle 1, day 1 (C1D1; cohort A), post-biopsy C1D22 (cohort B), and if significant combined inhibition of pAKT and pERK were observed in cohorts A and B, then post-biopsy on C1D4, 72 h after the first dose of MK-2206 to evaluate potential recovery of AKT inhibition (cohort C). Tumor biopsies (18-gauge core needle) were obtained at baseline (prior to drug administration) and then 3 to 6 h after dosing of study drugs on the day of the post-drug biopsy. A predetermined target inhibition of 70 % of both pERK and pAKT, by quantitative chemiluminescence immunoassay, was defined as significant based on the levels observed in preclinical models that were associated with antitumor activity [8, 10]. If no patient satisfied this criterion in a specific cohort, no further patients were to be accrued to that cohort. If 1 patient satisfied this criterion, 2 additional patients were to be accrued to that specific KRAS subset. If at least 2 of 5 patients in the subset demonstrated at least 70 % inhibition of both pERK and pAKT, the combination would be declared promising for that KRAS subset. This variation of a Simon 2-stage design had an 89 % power to detect a 60 % rate of combined pERK/pAKT inhibition across patients in each KRAS subset, with a 0.92 probability of rejecting for a 10 % rate across patients (potential of false positive), and a 0.73 probability of terminating the cohort subset early [11]. This design yielded a false positive rate (i.e., declaring the drug combination promising when the rate of combined inhibition was only 10 %) of 0.08 in each KRAS subset (of each cohort), and an overall false positive rate of 0.15 in each cohort.
Six patients each, in cohorts A and B, received MK-2206 orally at 90 mg once a week in combination with selumetinib orally at 75 mg once daily throughout the 28-day cycle and underwent paired tumor sampling. Since dual target inhibition of > 70 % was not observed in tumor biopsies obtained on days 1 and 22 compared to baseline, patients were not enrolled on cohort C to evaluate target recovery. Concurrently, the ongoing phase 1 trial of the combination in solid tumors established the safety of higher doses of both agents (ClinicalTrials.gov identifier: NCT01021748); therefore, a decision was made to amend the study and enroll six additional patients (3 KRAS-mutation positive and 3 KRAS wild-type) at a higher dose of MK-2206 (135 mg once a week) in combination with 100 mg selumetinib daily. The objective was to compare the observed target modulation between the 2 dose levels evaluated in this study. Because repeated dosing would likely demonstrate cumulative effects on target inhibition, tumor biopsies in these 6 patients were performed post-dose on C1D22. If significant dual target inhibition were observed in these 6 patients, then additional patients were to be enrolled to evaluate target recovery. Adverse events were graded according to National Cancer Institute-Common Toxicity Criteria (NCI-CTCAE version 4.0). Dose adjustments and treatment delays of up to 2 weeks were permitted for resolution of toxicities. Patients were considered evaluable if they received study treatment and underwent paired tumor biopsies; patients not meeting these criteria were evaluable for toxicity and eligible to continue study treatment per study criteria but were replaced for the purposes of the final analyses.
This trial was conducted under a National Cancer Institute-sponsored IND with institutional review board approval. Protocol design and conduct followed all applicable regulations, guidances, and local policies (ClinicalTrials.gov identifier: NCT01333475).
Safety assessments
Patients were monitored weekly during the first 2 cycles and then every 2 weeks for subsequent cycles with history, physical examination, and laboratory evaluations. Hemoglobin A1c levels were obtained at baseline and at the start of each cycle, urinalysis at baseline and every 2 cycles, and electrocardiogram at baseline and 4 to 10 h following the dose of MK-2206 on days 1, 8, 15, and 22 of the first cycle to monitor QTc interval at the time of peak concentration of the study drug in plasma. Ophthalmologic examinations were performed at baseline, at the start of the second cycle, and as clinically indicated, and included visual acuity, intraocular pressure, and dilated retinal exams and ocular coherence tomography (OCT). Radiologic response assessments by computerized tomography (CT) scans were performed every 2 cycles using the Response Evaluation Criteria in Solid Tumors (RECIST) (version 1.1) [12].
Pharmacodynamics
Tumor biopsy collection and preparation
Tumor biopsies were immediately dry flash frozen and stored in liquid nitrogen or −80 °C until processing for measurement of pERK and pAKT levels. At the time of processing, biopsies were lysed by mincing and sonication (3×1 min at 2–8 °C) in 150 µL of lysate buffer containing 1 mM EDTA, 0.5 % TritonX-100, 5 mM sodium fluoride (Sigma-Aldrich), 6 M urea (Sigma-Aldrich), 1 mM activated sodium orthovanadate (Sigma-Aldrich), 2.5 mM sodium pyrophosphate (Sigma-Aldrich), 1X protease inhibitors (Roche Applied Science), and 100 µM phenylmethylsulfonyl fluoride (Sigma-Aldrich) in 1x PBS pH 7.2–7.4. Specimens were then supplemented with 1 % SDS and boiled for 5 min to stop all enzymatic catalysis, snap-cooled in an ice bath, clarified by centrifugation (5 min of 2000×g at 2–8 °C), aliquoted, and stored at −80 °C. Specimen extraction was optimized for use with the chemiluminescence immunoassay.
Chemiluminescence immunoassay
R&D Systems Phospho-ERK 1 (Thr202/Tyr204)/ERK2 (Thr185/Tyr187) DuoSet IC and Phospho-AKT (Ser473) Pan Specific DuoSet IC ELISA kits (Cat# DYC1018B and DYC887B, respectively) were adapted to chemiluminescence readout and validated to quantitatively measure pERK and pAKT levels in human tissue (dilution linearity R2=0.98 pERK, R2= 0.99 pAKT; %CV at 0 < 6). pERK and pAKT standards were serially diluted in Reagent Diluent (1 M urea and 5 mM NaF) to a range of 39 to 2500 pg phosphoprotein/mL and served as standard controls. All samples were loaded in duplicate or triplicate with 100 µL total volume per well: clinical samples (0.05 to 0.1 µg total protein/µL), pERK and pAKT standards, and high and low controls (generated from Jurkat and MCF-7 cells obtained from the Division of Cancer Treatment & Diagnosis Repository, NCI at Frederick, authenticated by DNA short tandem repeat screening using AmpFLSTR® Identifiler® methodology [Applied Biosystems], held for no more than 20 passages, and processed and extracted as for patient specimens). Extract protein loads were selected to provide pAKT and pERK readouts that spanned the range of the standard curve. Final readout for each sample was reported as pg pERK or pAKT per 1 µg total protein. Mean recovery for pERK1/2 was 102 %. Recovery for pAKT was 100 %. Dilution linearity was established for a tumor extract protein load of 1.5 to 5 µg per well.
Results
Patient population and disposition
A total of 21 patients were enrolled between June 2011 and October 2013; 11 patients had KRAS-mutation positive CRC. Median age was 52 years (range 19–77), M/F ratio was 14/7, and median number of prior regimens was 3 (range 2–9). Sixteen patients were considered evaluable (10 in the initial dosing part of the study and 6 at the higher doses). Paired tumor biopsies were not available for 5 patients. Four patients experienced toxicities during the first cycle requiring treatment interruption; therefore, post-treatment biopsies were not performed in these patients. One patient withdrew from the study after 16 days due to the need for emergent radiation therapy to the spine.
Efficacy
Three patients demonstrated clinical progression of disease prior to the first restaging, and 6 patients progressed at restaging after 2 cycles (Fig. 3). The average length of time on study was 56 days (2 cycles). One patient remained on study with stable disease for 182 days; this patient had a mixed response with some decrease in liver and lung metastatic lesions but with eventual progression of an adrenal lesion that was confirmed to be metastatic CRC following an excisional biopsy.
Fig. 3.
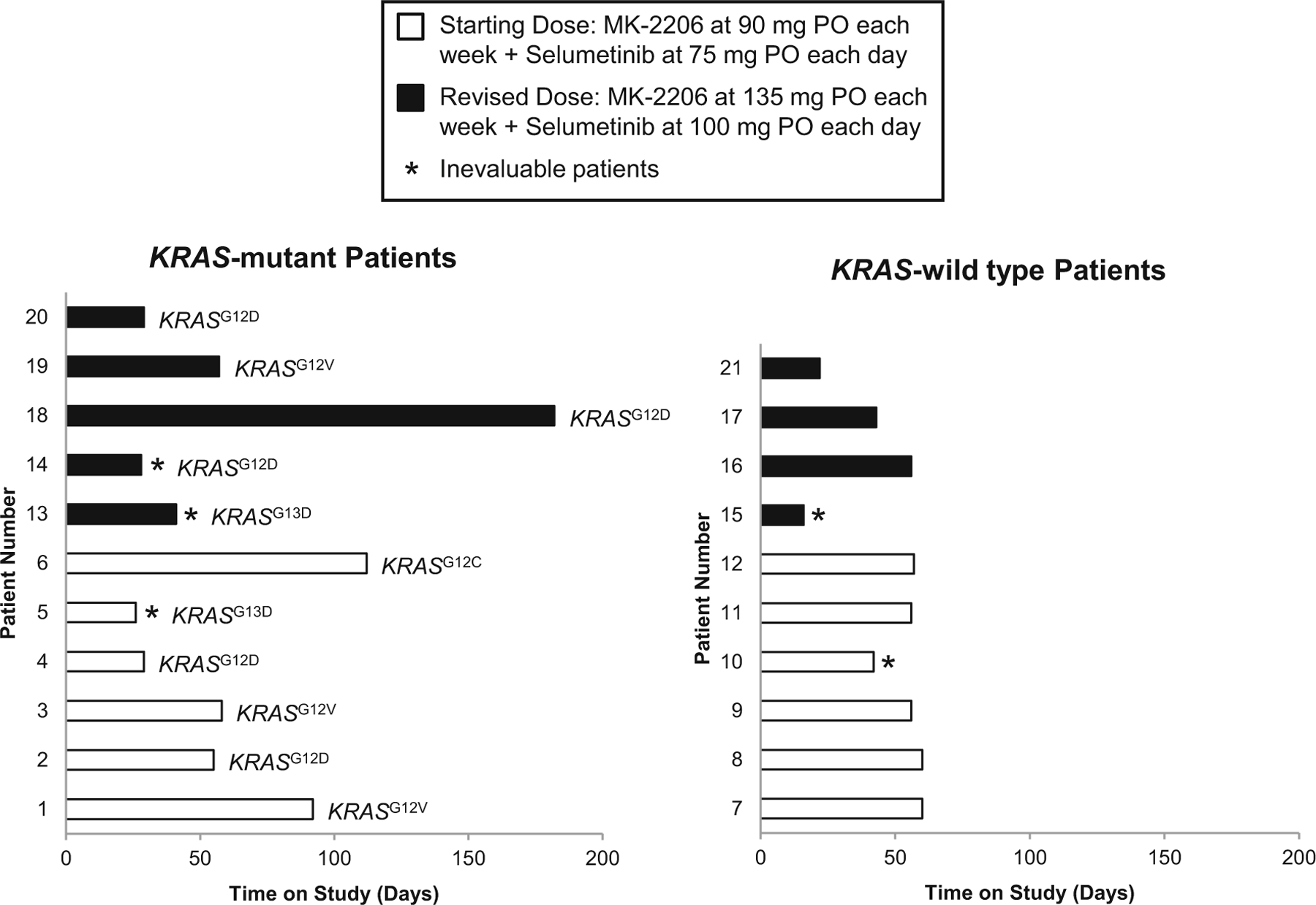
Length of time on study for each patient, separated by KRAS mutation status. KRAS-specific mutations are listed next to each patient (e.g., G12D = glycine (G) to aspartic acid (D) substitution at position 12). The first 12 patients received the initial dose of 90 mg oral MK-2206 each week + 75 mg oral selumetinib daily for a 28-day cycle. The remaining 9 patients received the higher dose of 135 mg oral MK-2206 each week + 100 mg oral selumetinib daily. Mean length of time for all patients on study was 56 days. Five patients were inevaluable for pharmacodynamic endpoints due to the lack of paired tumor biopsies (*)
Adverse events
Three patients were taken off-study due to toxicities. One patient developed grade 3 transaminitis in the setting of extensive liver involvement but in the absence of documented progression of disease at the time of data capture, and in spite of dose reduction of both agents. This patient was subsequently found to have tumor infiltration of the bile ducts within 2 weeks after discontinuation of treatment on study. One patient developed grade 3 rash during the first cycle which did not resolve within 2 weeks despite treatment interruption and palliative measures. Another patient developed recurrence of grade 2 subretinal fluid accumulation with retinal detachment detected in both eyes on OCT at the start of the second cycle despite treatment interruption and dose reduction of both agents following the first occurrence on cycle 1 day 8 of treatment (Fig. 4). Two additional patients had grade 1 subretinal detachments during the first cycle without associated symptoms at the time of the exam, which resolved with treatment interruption of both drugs. Both these patients had reported intermittent blurry vision at cycle 1 day 15 evaluation, thus prompting ophthalmic exams, but otherwise reported no symptoms nor visual acuity deficits at the day of the exam. These patients were able to continue on treatment at the same dose without recurrence of symptoms or objective findings on ophthalmologic exam. Even though retinal detachments were previously documented with MEK inhibition, the doses of both agents were reduced due to concerns for possible increase in toxicities with the combination. No QTc interval prolongations were documented (Table 1).
Fig. 4.
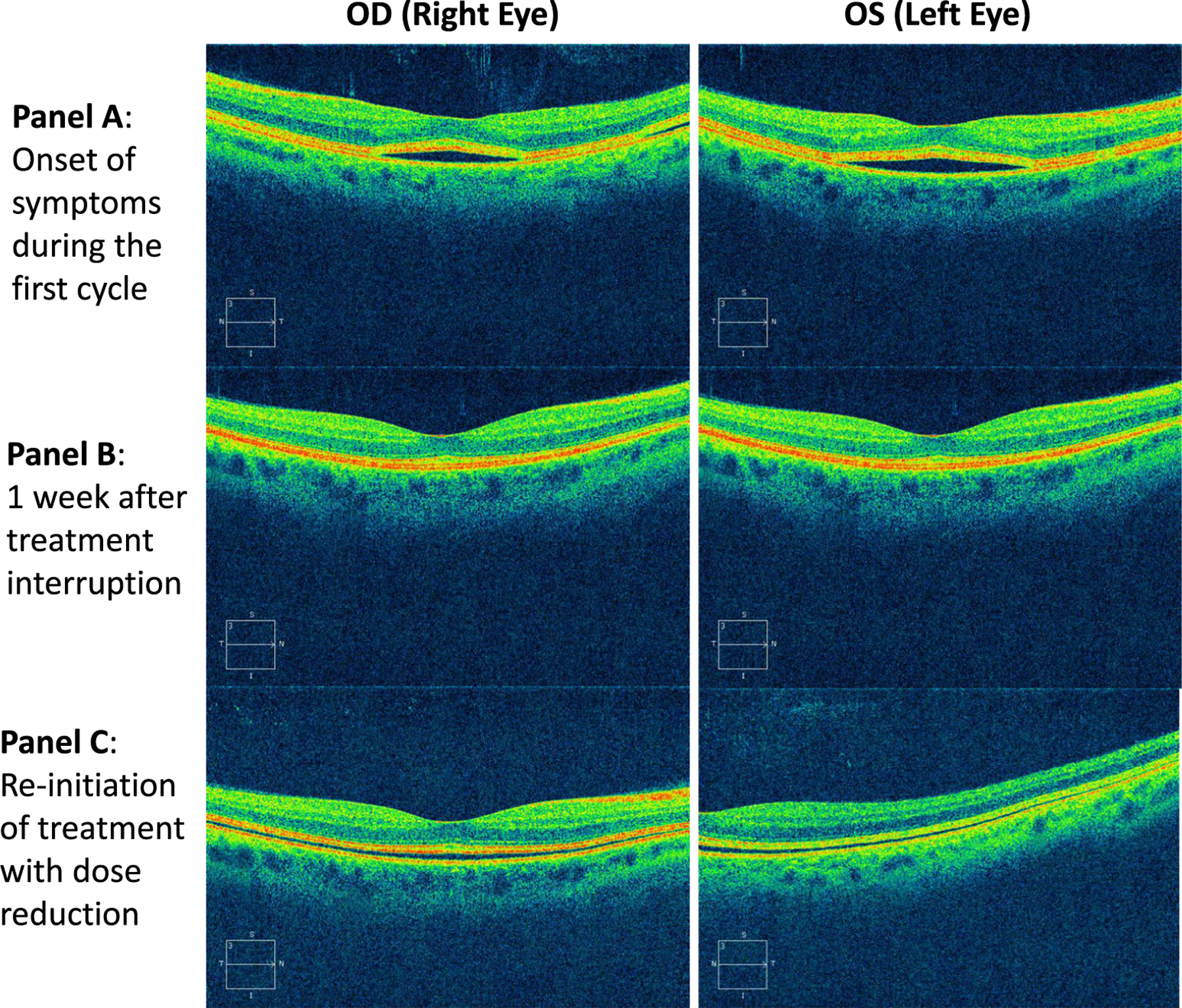
Ocular coherence tomography images for one patient who developed grade 2 bilateral subretinal fluid accumulations and detachment on study. The patient reported blurry vision within 2 h after taking medication at the start of cycle 1 day 8. At presentation of symptoms, vision was reduced slightly to 20/32 OD (right eye), and 20/25 OS (left eye). Pinhole vision, which stimulates optimal spectacle correction, was 20/20 in each eye at presentation. Subsequent examination 1 week later with treatment interruption of both agents revealed resolution of detachments. Within 1 week of reinitiating treatment with dose reduction of both agents, examination revealed more shallow bilateral detachments. On all subsequent exams, vision measured 20/20 in each eye in the patient’s standard spectacle correction
Table 1.
Summary of treatment-related adverse events (AEs)
| Adverse eventc | Starting dosea (n=12) |
Revised doseb (n=9) |
||||
|---|---|---|---|---|---|---|
| Grade 1 | Grade 2 | Grade 3 | Grade 1 | Grade 2 | Grade 3 | |
| Increase in AST | 5 | 3 | – | 1 | 1 | 1 |
| Increase in ALT | 3 | 1 | 1 | 1 | 2 | – |
| Hyperbilirubinemia | 3 | 1 | – | 1 | – | – |
| Alkaline phosphatase | 2 | 2 | – | – | 3 | – |
| Hypoalbuminemia | 1 | 2 | – | 1 | – | – |
| Neutropenia | – | – | – | – | 1 | – |
| Lymphopenia | 4 | 5 | – | 1 | 2 | – |
| Anemia | 3 | – | – | 1 | 1 | – |
| Thrombocytopenia | 3 | – | – | – | – | – |
| Acneiform rash | 5 | 1 | – | 3 | 2 | 1 |
| Maculopapular rash | 2 | 2 | – | 2 | – | 1 |
| Papulopustular rash | 2 | – | – | – | – | – |
| Dry skin | 1 | – | – | – | – | – |
| Pruritis | – | – | – | 2 | 1 | – |
| Oral mucositis | 2 | – | – | – | – | – |
| Dysgeusia | – | – | – | 1 | – | – |
| Nausea | 6 | 1 | – | 4 | – | – |
| Vomiting | 4 | – | – | 1 | – | – |
| Diarrhea | 4 | – | – | 2 | – | – |
| Abdominal pain | 2 | – | – | – | 1 | – |
| Abdominal bloating | 1 | – | – | 1 | – | – |
| Dyspepsia | – | 1 | – | – | – | – |
| Anorexia | 2 | – | – | 1 | – | – |
| Dehydration | – | 2 | – | – | – | – |
| Fatigue | 2 | – | – | 1 | 1 | – |
| Hyperglycemia | 2 | 1 | – | – | 2 | – |
| Hypercalcemia | 2 | – | – | – | 1 | – |
| Increased CPK | 2 | – | – | 1 | – | – |
| Dizziness | 2 | – | – | 1 | – | – |
| Edema | 3 | – | – | 1 | – | – |
| Hypertension | – | – | – | – | 1 | – |
| Palpitations | 1 | – | – | – | – | – |
| Sinus bradycardia | 2 | – | – | 1 | – | – |
| Retinal detachment | 2 | 1 | – | – | – | – |
| Blurred vision | 2 | 1 | – | – | – | – |
| Floaters | 1 | – | – | – | – | – |
Starting dose: MK-2206 90 mg each week plus AZD6244 75 mg each day; n=12
Revised dose: MK-2206 135 mg each week plus AZD6244 100 mg each day; n=9
All adverse events represent the worse grade per patient. No Grade 4 or 5 events were seen at either dose
Pharmacodynamic studies
Dual target inhibition of > 70 % in pERK and pAKT levels in tumor biopsies compared to baseline was not observed in any tumor sample. Half of the patients at both dose levels showed at least a 56 % decrease in pAKT levels, whereas only a quarter of all the patients had ≥ 53 % decreases in pERK levels (Fig. 5). Three patients demonstrated a trend toward biomarker response with both pERK and pAKT levels approaching 60 % inhibition, two at the initial dose and one at the higher dose. Additionally, it is interesting to note that tumor samples from the patient with KRAS-mutated CRC who remained on study for the longest duration did demonstrate inhibition of pAKT levels (56 %), but had an increase in pERK level (49 %) in post-dose biopsies. Due to inconsistent inhibition of pERK and pAKT on C1D1 and C1D22, we did not pursue enrollment to cohort C to evaluate target recovery by C1D4 at either dose level.
Fig. 5.
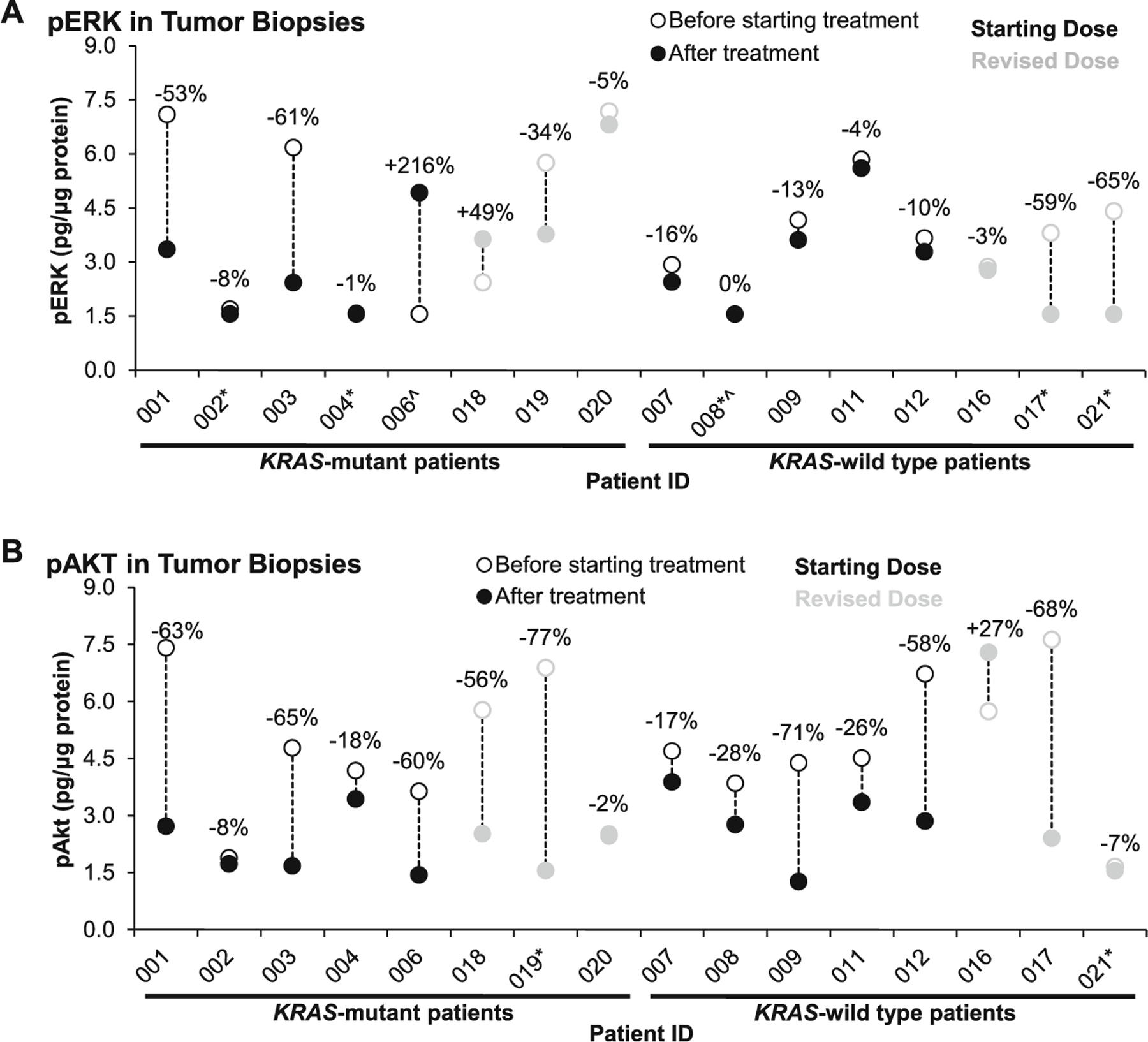
Modulation of pERK and pAKT in paired tumor biopsies, separated by KRAS mutation status. Three patients demonstrated a trend toward biomarker response with dual inhibition of pERK and pAKT levels approaching 60 % inhibition (patients 001, 003, and 017); however, no patient met the pre-specified criteria of ≥ 70 % inhibition of both biomarkers. Patients 1–3 and 7–9 had post-treatment biopsies taken 3–6 h after their cycle 1 day 1 doses, and patients 4, 6, 11, 12, and 16–21 had post-treatment biopsies taken 3–6 h after their cycle 1 day 22 doses. a Two patients had pERK levels below the lower limit of quantitation (LLQ) before treatment (^), and five had pERK levels below LLQ after treatment (*). The pERK LLQ was 0.78 pg/µg protein (for patients 1–9) and 1.56 pg/µg protein (for patients 10–21). b Two patients had pAKT levels below the LLQ after treatment (*). The pAKT LLQ was 0.78 pg/µg protein (for patients 1–9) and 1.56 pg/µg protein (for patients 10–21)
Discussion
Significant dual target inhibition, defined from preclinical data as 70 % inhibition of both pERK and pAKT, was not reached in this study, potentially due to the chosen dosing schedule. In the first-in-human study of MK-2206 administered at the single-agent MTD of 60 mg every other day, paired tumor biopsy samples from 9 patients were evaluated for target modulation. pSer473AKT was decreased in all patients (range, 44.9 to 95.6 %; p=0.015) with greater than 70 % inhibition observed in 7 of 9 patients [7]; average steady-state trough concentrations were higher than the concentration (56.8 nM) required for 70 % inhibition of pSer473AKT in whole blood associated with antitumor activity in preclinical models. Due to the long half-life of MK-2206 (T1/2 60–80 h) and evidence of inhibition of AKT Ser473 phosphorylation in platelet-rich plasma up to 96 h after dosing at the 200 mg weekly dose, the study sponsor chose to pursue the once a week dosing schedule for further clinical development. It is worth noting that, despite steady state concentrations exceeding 56.8 nM, >70 % inhibition of pSer473AKT levels was demonstrated in only 1 of the 6 pairs of tumor biopsies and did not show adequate target inhibition in the majority of samples on this dosing schedule [6].
Single agent studies of selumetinib, evaluating an earlier powder formulation, established the MTD at 100 mg orally BID, and demonstrated inhibition of ERK phosphorylation levels in paired tumor biopsies (mean 79 % inhibition) [13]. A subsequent single-agent study of the hydrogen sulfate capsule formulation, which was also used in the current study, established the MTD at 75 mg orally BID. In this trial, significant inhibition of ERK phosphorylation was seen in PBMCs (91 % inhibition at a mean IC50 352 ng/mL) [14]; however, inhibition of target was not evaluated in tumor samples. Since target modulation was not consistently performed across dose levels in single agent trials of either agent, a dose-PD response relationship for the single-agents could not be established to guide dose adjustments for the combination. Additionally, correlation of target modulation with plasma exposures was not performed in the phase 1 trial of this combination [15]. In our study, no promising clinical activity was observed at the doses evaluated, and overlapping toxicities limited dose escalation and attempts to achieve higher levels of target inhibition to optimize efficacy.
Selectively targeting multiple intracellular pathways in an effort to optimize antitumor efficacy and prevent the development of resistance requires an understanding of the respective pathways, their interactions, the degree and duration of inhibition required for optimal antitumor effect, and the consequences of inhibition on both tumor cells and normal tissues. Normal tissue vulnerability can limit the ability to administer sufficient doses of study drugs for optimal target inhibition and, ultimately, antitumor efficacy, as was our experience in this trial. In spite of the lower doses of study drugs, reduced due to overlapping skin and GI toxicities in the phase 1 trial, we observed acneiform rash in 58.3 % of patients at the initial dose level, and 55.6 % of patients at the revised, higher dose level.
The degree of dual target inhibition required for clinical benefit is not known for this study combination. We utilized observations from preclinical models and single-agent trials to guide our criterion for target modulation. The lack of clinical benefit could be due to inadequate target inhibition, or alternatively, may be attributed to additional genetic aberrations such as PTEN loss, activation of the NF-κB pathway, or cross-talk with other signaling pathways such as the TGF-β or Wnt signaling pathways that have previously been implicated in refractory CRC [16]. In the absence of consistent target modulation and availability of a multiplex assay that could measure the activity of compensatory pathways from a small biopsy sample, no definitive conclusion regarding lack of response can be drawn.
Our trial highlights the need to define target modulation in the tissue of interest very early in clinical development. While PBMCs and platelet rich plasma allow for ease of longitudinal sampling, these and other normal tissues do not necessarily reflect the molecular changes occurring within tumor tissue. Additionally, the plasma milieu of PBMCs may allow for higher drug exposure than that of tumor tissue and, as such, optimal timing of sampling of PBMCs and maximal effect on target may be different than for tumor tissue [17].
For future optimization of targeted agent combinations, the relationship of antitumor activity to target modulation must be established first in preclinical models. Early clinical trials can then be designed to demonstrate target modulation in tumor tissues at clinically achievable drug concentrations, including establishing the dose range that would result in the desired level of target inhibition. Information regarding dose range is essential when considering drug combinations that necessitate dose de-escalation for safety. Due to overlapping toxicities and lack of clinical benefit in this study, we would not advocate further investigation of this combination in this patient population.
Acknowledgments
The authors thank the patients and their families for their contributions to this study. Additionally, we thank Dr. Andrea Regier Voth, Leidos Biomedical Research, Inc., for medical writing support, and Dr. Kate Ferry-Galow, Leidos Biomedical Research, Inc., for quality assurance auditing of the pharmacodynamic data presented in this manuscript.
This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under Contracts No. HHSN261200800001E and N01-CM-2011–00032. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. MK-2206 and selumetinib were generously provided by Merck and AstraZeneca, respectively, under a collaborative agreement with the Division of Cancer Treatment and Diagnosis, National Cancer Institute, NIH.
Footnotes
Conflict of interest Michael H. Lam is an employee of Merck and Company. No potential conflicts of interest were disclosed by the other authors.
Contributor Information
Khanh Do, Division of Cancer Treatment and Diagnosis, National Cancer Institute, 31 Center Drive, Bldg 31, Rm 3A44, Bethesda, MD 20892, USA.
Giovanna Speranza, Division of Cancer Treatment and Diagnosis, National Cancer Institute, 31 Center Drive, Bldg 31, Rm 3A44, Bethesda, MD 20892, USA.
Rachel Bishop, National Eye Institute, Bethesda, USA.
Sonny Khin, Frederick National Laboratory for Cancer Research, Leidos Biomedical Research, Inc., Frederick, USA.
Larry Rubinstein, Division of Cancer Treatment and Diagnosis, National Cancer Institute, 31 Center Drive, Bldg 31, Rm 3A44, Bethesda, MD 20892, USA.
Robert J. Kinders, Frederick National Laboratory for Cancer Research, Leidos Biomedical Research, Inc., Frederick, USA
Manuel Datiles, National Eye Institute, Bethesda, USA.
Michelle Eugeni, Division of Cancer Treatment and Diagnosis, National Cancer Institute, 31 Center Drive, Bldg 31, Rm 3A44, Bethesda, MD 20892, USA.
Michael H. Lam, Merck Research Laboratories – Oncology, Boston, USA
L. Austin Doyle, Division of Cancer Treatment and Diagnosis, National Cancer Institute, 31 Center Drive, Bldg 31, Rm 3A44, Bethesda, MD 20892, USA.
James H. Doroshow, Division of Cancer Treatment and Diagnosis, National Cancer Institute, 31 Center Drive, Bldg 31, Rm 3A44, Bethesda, MD 20892, USA
Shivaani Kummar, Division of Cancer Treatment and Diagnosis, National Cancer Institute, 31 Center Drive, Bldg 31, Rm 3A44, Bethesda, MD 20892, USA.
References
- 1.Vaughn CP, Zobell SD, Furtado LV, Baker CL, Samowitz WS (2011) Frequency of KRAS, BRAF, and NRAS mutations in colorectal cancer. Genes Chromosomes Cancer 50(5):307–312. doi: 10.1002/gcc.20854 [DOI] [PubMed] [Google Scholar]
- 2.Van Cutsem E, Kohne CH, Lang I, Folprecht G, Nowacki MP, Cascinu S, Shchepotin I, Maurel J, Cunningham D, Tejpar S, Schlichting M, Zubel A, Celik I, Rougier P, Ciardiello F (2011) Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J Clin Oncol 29(15):2011–2019. doi: 10.1200/JCO.2010.33.5091 [DOI] [PubMed] [Google Scholar]
- 3.Roy HK, Olusola BF, Clemens DL, Karolski WJ, Ratashak A, Lynch HT, Smyrk TC (2002) AKT proto-oncogene overexpression is an early event during sporadic colon carcinogenesis. Carcinogenesis 23(1):201–205 [DOI] [PubMed] [Google Scholar]
- 4.Turke AB, Song Y, Costa C, Cook R, Arteaga CL, Asara JM, Engelman JA (2012) MEK inhibition leads to PI3K/AKT activation by relieving a negative feedback on ERBB receptors. Cancer Res 72(13):3228–3237. doi: 10.1158/0008-5472.CAN-11-3747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Halilovic E, She QB, Ye Q, Pagliarini R, Sellers WR, Solit DB, Rosen N (2010) PIK3CA mutation uncouples tumor growth and cyclin D1 regulation from MEK/ERK and mutant KRAS signaling. Cancer Res 70(17):6804–6814. doi: 10.1158/0008-5472.CAN-10-0409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Biondo A, Yap TA, Yan L, Patnaik A, Fearen I, Baird RD, Papadopoulos KP, Delgado LM, Taylor A, Lupinacci L, Blackman SC, Decordova S, Tall M, Heaton S, Garrett MD, Sullivan D, De Bono JS, Tolcher AW (2011) Phase I clinical trial of an allosteric AKT inhibitor, MK-2206, using a once weekly (QW) dose regimen in patients with advanced solid tumors. J Clin Oncol 29(suppl):3037, abstract [DOI] [PubMed] [Google Scholar]
- 7.Yap TA, Yan L, Patnaik A, Fearen I, Olmos D, Papadopoulos K, Baird RD, Delgado L, Taylor A, Lupinacci L, Riisnaes R, Pope LL, Heaton SP, Thomas G, Garrett MD, Sullivan DM, de Bono JS, Tolcher AW (2011) First-in-man clinical trial of the oral pan-AKT inhibitor MK-2206 in patients with advanced solid tumors. J Clin Oncol 29(35):4688–4695. doi: 10.1200/JCO.2011.35.5263 [DOI] [PubMed] [Google Scholar]
- 8.Davies BR, Logie A, McKay JS, Martin P, Steele S, Jenkins R, Cockerill M, Cartlidge S, Smith PD (2007) AZD6244 (ARRY-142886), a potent inhibitor of mitogen-activated protein kinase/ extracellular signal-regulated kinase kinase 1/2 kinases: mechanism of action in vivo, pharmacokinetic/pharmacodynamic relationship, and potential for combination in preclinical models. Mol Cancer Ther 6(8):2209–2219. doi: 10.1158/1535-7163.MCT-07-0231 [DOI] [PubMed] [Google Scholar]
- 9.Tolcher AW, Khan K, Ong M, Banerji U, Papadimitrakopoulou V, Gandara DR, Patnaik A, Baird RD, Olmos D, Garrett CR, Skolnik JM, Rubin EH, Smith PD, Huang P, Learoyd M, Shannon KA, Morosky A, Tetteh E, Jou YM, Papadopoulos KP, Moreno V, Kaiser B, Yap TA, Yan L, de Bono JS (2014) Anti-tumour activity in RAS-driven tumours by blocking AKT and MEK. Clin Cancer Res. doi: 10.1158/1078-0432.CCR-14-1901 [DOI] [PMC free article] [PubMed]
- 10.Hirai H, Sootome H, Nakatsuru Y, Miyama K, Taguchi S, Tsujioka K, Ueno Y, Hatch H, Majumder PK, Pan BS, Kotani H (2010) MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol Cancer Ther 9(7):1956–1967. doi: 10.1158/1535-7163.MCT-09-1012 [DOI] [PubMed] [Google Scholar]
- 11.Rubinstein LV, Steinberg SM, Kummar S, Kinders R, Parchment RE, Murgo AJ, Tomaszewski JE, Doroshow JH (2010) The statistics of phase 0 trials. Stat Med 29(10):1072–1076. doi: 10.1002/sim.3840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J (2009) New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45(2):228–247. doi: 10.1016/j.ejca.2008.10.026 [DOI] [PubMed] [Google Scholar]
- 13.Adjei AA, Cohen RB, Franklin W, Morris C, Wilson D, Molina JR, Hanson LJ, Gore L, Chow L, Leong S, Maloney L, Gordon G, Simmons H, Marlow A, Litwiler K, Brown S, Poch G, Kane K, Haney J, Eckhardt SG (2008) Phase I pharmacokinetic and pharmacodynamic study of the oral, small-molecule mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244 (ARRY-142886) in patients with advanced cancers. J Clin Oncol 26(13):2139–2146. doi: 10.1200/JCO.2007.14.4956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Banerji U, Camidge DR, Verheul HM, Agarwal R, Sarker D, Kaye SB, Desar IM, Timmer-Bonte JN, Eckhardt SG, Lewis KD, Brown KH, Cantarini MV, Morris C, George SM, Smith PD, van Herpen CM (2010) The first-in-human study of the hydrogen sulfate (Hyd-sulfate) capsule of the MEK1/2 inhibitor AZD6244 (ARRY-142886): a phase I open-label multicenter trial in patients with advanced cancer. Clin Cancer Res 16(5):1613–1623. doi: 10.1158/1078-0432.CCR-09-2483 [DOI] [PubMed] [Google Scholar]
- 15.Khan KH, Yan L, Mezynski J, Patnaik A, Moreno V, Papadopoulos KP, Garrett CR, Ong M, Shannon KA, Morosky A, Rubin EH, Tetteh E, Skolnik J, Smith IC, Smith PD, De Bono JS, Tolcher AW (2012) A phase I dose escalation study of oral MK-2206 (allosteric Akt inhibitor) with oral selumetinib (AZD6244; ARRY-142866) (MEK 1/2 inhibitor) in patients with advanced or metastatic solid tumors. J Clin Oncol 30(suppl):e13599, abstract [Google Scholar]
- 16.Watanabe T, Kobunai T, Yamamoto Y, Matsuda K, Ishihara S, Nozawa K, Iinuma H, Ikeuchi H, Eshima K (2011) Differential gene expression signatures between colorectal cancers with and without KRAS mutations: crosstalk between the KRAS pathway and other signalling pathways. Eur J Cancer 47(13):1946–1954. doi: 10.1016/j.ejca.2011.03.029 [DOI] [PubMed] [Google Scholar]
- 17.Ang JE, Kaye S, Banerji U (2012) Tissue-based approaches to study pharmacodynamic endpoints in early phase oncology clinical trials. Curr Drug Targets 13(12):1525–1534 [DOI] [PMC free article] [PubMed] [Google Scholar]