

Abstract
Two new compounds tryptoquivalines W (1) and X (2) were isolated from a Hawaiian soil fungal strain Aspergillus terreus FS107. The soil sample was collected on the top of Mauna Kea, the tallest mountain in Hawaii. The structures of compounds 1 and 2 were determined on the basis of MS spectroscopic and NMR analysis, and NMR calculation. The absolute configuration (AC) was determined by ECD calculations. Compounds 4 and 5 showed inhibition against NF-κB with IC50 values of 3.45 and 6.76 μM, respectively.
Keywords: Fungus, Tryptoquivaline, NMR, NF-κB
Fungi, including unicellular yeast, multicellular mold, mildews, rusts, smuts, and mushrooms, are widely distributed all over the world. It was suggested that there may be as many as 120,000 species of microfungi within the United States of America and 1.5 million worldwide. Fungi in the genus Aspergillus are well-known as a source of biologically active secondary metabolites [1]. The Aspergillus Secondary Metabolites Database (A2MDB) provides a phylogenetic representation of over 2500 strains, catalogs 807 secondary metabolites from 675 Aspergillus species, and presents a detailed chemical information of secondary metabolites [2]. Among the widely distributing Aspergillus strains are A. flavus, A. fumigatus, A. niger, A. tubingensis, A. oryzae, A. versicolor, and A. terreus etc [2]. The common producers of secondary metabolites in the genus Aspergillus include A. niger, A. terreus, A. versicolor, A. flavus, A. tubingensis, and A. fumigatus, etc [2]. Aspergillus species produce not just mycotoxins, but also biosynthesize diverse types of secondary metabolites. A. terreus is a saprotrophic fungal species found in soil sources throughout the world. They are prevalent in tropical and subtropical regions, but they are also found in very harsh environmental conditions. In our continuous search of new and biologically active compounds from Hawaiian fungi [3–16], we studied a fungal strain, Aspergillus terreus FS107 that was isolated from a soil sample collected on the top of the Mauna Kea mountain, the highest mountain in the State of Hawaii, and isolated six secondary metabolites, including two new compounds (1 and 2) and four known molecules (3–6) (Fig. 1). Herein, we present the isolation, structural elucidation, plausible biosynthesis and biological evaluation of these metabolites.
Fig. 1.
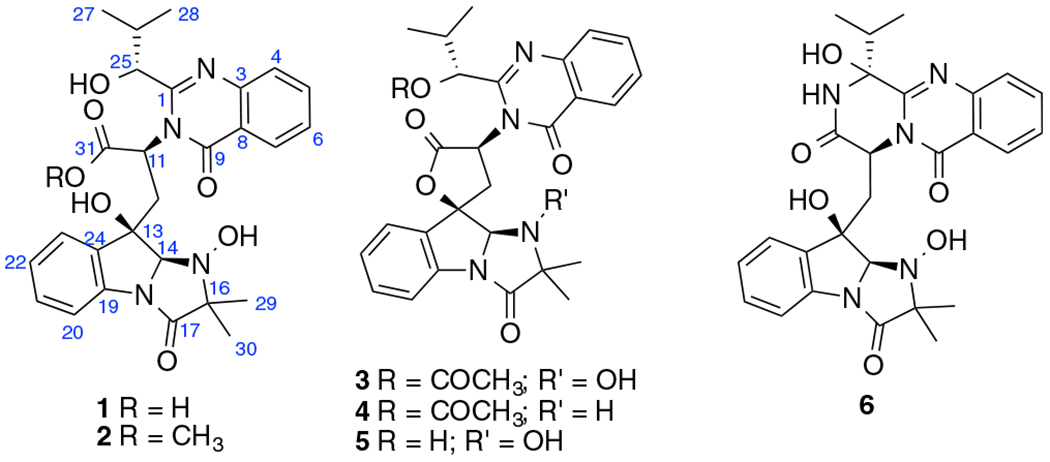
Structures of compounds 1–6.
Compound 1 [17] was isolated as a colorless powder. Its molecular formula was determined to be C27H30N4O7 by HR-ESIMS (m/z 523.2194, calcd for [M + H]+ 523.2193), with fifteen degrees of unsaturation. The 1H NMR and HSQC spectra (Table 1) of 1 exhibited the presence of eight aromatic protons in the low field, four methyl groups in the high field, and six protons between them including four methines and one methylene. In the 1H–1H COSY spectrum of 1 (Fig. 2), four spin systems were identified including two AA’BB’ spin systems, one 1-oxygenated 2-methylpropyl group [(CH3)2CH-CH(O-], and a –(COO)CH(N)–CH2-spin system that accounted for the α- and β-protons of an amino acid. In the HMBC spectrum, the two methyl singlets showed correlations to a carbonyl carbon at δC 173.1 and a nirogenated tertiary carbon at δC 70.8 indicating a 2-methyl alanine (MeAla) or 2-amino-isobutanoic acid (AIBA) unit in the molecule. Further HMBC analysis indicated the presence of two 1,2-disubstituted aromatic rings, one of which should be derived from tryptophan with the indole being reduced to form an indoline moiety. Based on the four fragments identified (Fig. 2), a search on the “Dictionary of Natural Products” with the molecular weight defined from 500 to 550 Dalton revealed that 1 was very similar to tryptoquivaline A (3) [18], N-dehydroxy tryptoquivaline A (deoxytryptoquivaline) (4) [19], and especially O-deacetyl-tryptoquivaline A (5) [18], which were also isolated in this study. In the HMBC of 3, H-7 and H-23 showed correlations to C-9 and C-13, respectively, which further confirmed the presence of anthranilic acid and tryptophan moieties in these analogs. Since the molecular weight of 1 was 522 Dalton, which is 18 units more than that of 5, it was readily concluded that 1 was an acid with a carboxyl and a hydroxyl group at 13-position instead of a γ-lactone, which was supported by an IR band in the region of 1600–1700 cm−1. From the biosynthetic point of view, we argued that compound 1 should have the same configuration [11(S), 13(R), 14 (R), and 25(R] as that of the known compound, tryptoquivaline A (3). To verify our hypothesis, we carried out NMR calculations of 1 [11(S), 13(R), 14(R), and 25(R)] and its 25-epimer [1-epimer: 11(S), 13(R), 14(R), and 25(S)] [20]. Each configuration was submitted for the configurational search by using the OPLS_2005 force field in MacroModel with an energy window of 5.02 kcal/mol. Geometry optimization and frequency calculation were performed at the B3LYP/6–31 + G(d,p) level in gas phase. NMR shielding tensors were computed with the GIAO method at the B3LYP/6–311 + G (2d,p) level in methanol. For the ECD calculation, both geometry optimization and TDDFT calculation were performed at the APFD/6–311 + G(2d,p) level with methanol as solvent. Results showed that the calculated NMR data of 1 matched the experimental NMR data of 1 better than 1-epimer (Table 2). To confirm our hypothesis, we carried out ECD calculations for 1, 1-epimer and 1e (enantiomer of 1) [21,22]. The experimental ECD spectrum of compound 1 showed a strong negative Cotton effect at 220250 nm. The calculated weighted ECD spectrum of the stereoisomer 1 coincides well with the recorded ECD spectrum of 1 rather than 1-epimer and 1e (Fig. 3). Hence, the structure of 1 was determined as shown.
Table 1.
1H and 13C NMR data of 1 and 2.
| 1 |
2 |
|||
|---|---|---|---|---|
| no. | δH, J (Hz)a | δCb | δH, J (Hz)a | δCb |
| 1 | 168.9 | 168.9 | ||
| 3 | ||||
| 4 | 7.83 d (7.8) | 127.9 | 7.83 d (7.8) | 127.9 |
| 5 | 7.49 overlapped | 131.4 | 7.54 overlapped | 131.6 |
| 6 | 7.21 t (7.8) | 125.1 | 7.22 overlapped | 125 |
| 7 | 8.45 d (7.8) | 120.8 | 8.50 d (7.8) | 120.8 |
| 8 | ||||
| 9 | 175.4 | 174.4 | ||
| 10 | ||||
| 11 | 5.04 dd (3.1, 8.7) | 50.6 | 5.04 dd (3.1, 8.70) | 50.1 |
| 12 | 2.51 dd (3.1, 15.0) | 39.8 | 2.50 dd (3.1, 15.0) | 38.5 |
| 2.77 dd (8.7, 15.0) | 2.80 dd (8.7, 15.0) | |||
| 13 | 76.6 | 76.5 | ||
| 14 | 5.26 s | 85.1 | 5.13 s | 84.2 |
| 15 | ||||
| 16 | 70.8 | 70.5 | ||
| 17 | 173.1 | 173 | ||
| 18 | ||||
| 19 | 137.3 | 137.3 | ||
| 20 | 7.48 d (7.8) | 115.1 | 7.50 overlapped | 115.1 |
| 21 | 7.36 t (7.8) | 129.4 | 7.39 t (7.8) | 129.5 |
| 22 | 7.20 t (7.8) | 123.3 | 7.21 overlapped | 123.1 |
| 23 | 7.53 d (7.8) | 124.5 | 7.52 overlapped | 124.3 |
| 24 | 137.3 | 137.3 | ||
| 25 | 3.97 d (3.4) | 76.2 | 3.98 d (3.4) | 76.3 |
| 26 | 2.18 m | 31.5 | 2.14 m | 32 |
| 27 | 0.85 d (7.0) | 14.8 | 0.89 d (7.0) | 15.22 |
| 28 | 1.05 d (7.0) | 18.2 | 1.04 d (7.0) | 18.4 |
| 29 | 1.38 s | 21.6 | 1.34 s | 21.8 |
| 30 | 1.45 s | 15.9 | 1.40 s | 15.9 |
| 31 | 172.6 | 172.6 | ||
| 32 | 3.77 s | 51.3 | ||
Spectra recorded at 400 MHz.
Spectra recorded at 100 MHz. Data based on 1H, 13C, HSQC, and HMBC experiments.
Fig. 2.
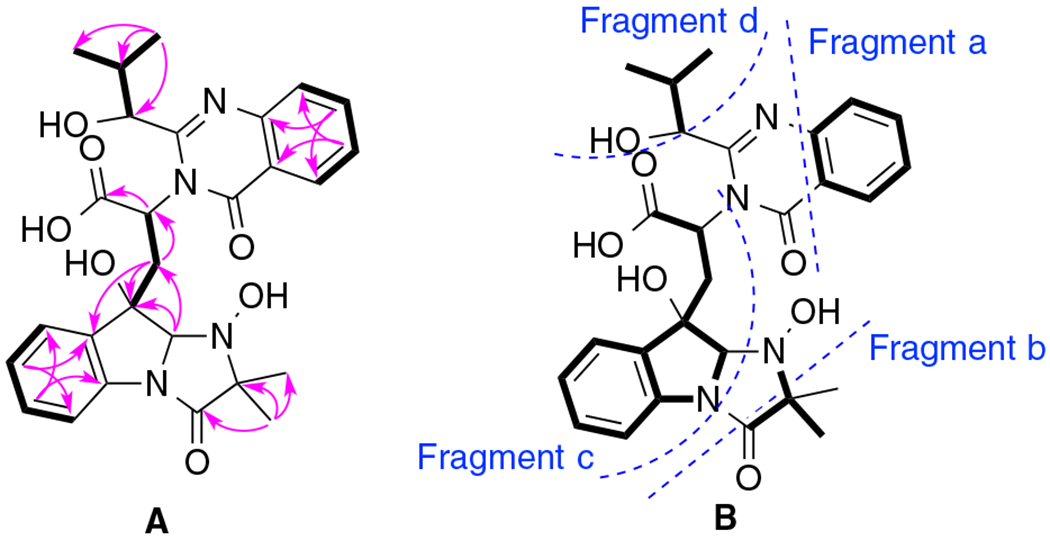
A. Key COSY (Bold) and HMBC (Single headed) correlations of 1; B. Four fragments identified in 1.
Table 2.
Calculated 1H and 13C NMR data of 1 and 1-epimer.
| 1 |
1-epimer |
|||
|---|---|---|---|---|
| no. | δH, J (Hz) | δδC | δH, J (Hz) | δC |
| 1 | 160.91 | 157.69 | ||
| 3 | 147.30 | 147.93 | ||
| 4 | 7.67 | 127.05 | 7.72 | 127.91 |
| 5 | 7.80 | 135.99 | 7.86 | 136.05 |
| 6 | 7.53 | 127.59 | 7.55 | 127.86 |
| 7 | 8.27 | 127.52 | 8.29 | 128.07 |
| 8 | 121.37 | 120.75 | ||
| 9 | 163.19 | 165.03 | ||
| 10 | ||||
| 11 | 5.34 | 58.51 | 6.63 | 56.39 |
| 12 | 2.63/3.50 | 37.39 | 3.00/3.64 | 38.30 |
| 13 | 82.86 | 80.08 | ||
| 14 | 5.39 | 84.25 | 5.34 | 88.87 |
| 15 | ||||
| 16 | 80.55 | 76.04 | ||
| 17 | 173.83 | 173.10 | ||
| 18 | ||||
| 19 | 136.94 | 138.31 | ||
| 20 | 7.51 | 113.87 | 7.41 | 115.87 |
| 21 | 7.32 | 130.64 | 7.34 | 130.31 |
| 22 | 7.08 | 125.10 | 7.14 | 125.52 |
| 23 | 7.23 | 124.68 | 7.43 | 124.65 |
| 24 | 141.21 | 140.31 | ||
| 25 | 5.40 | 75.94 | 4.50 | 77.01 |
| 26 | 2.20 | 39.32 | 2.54 | 37.77 |
| 27 | 1.08 | 15.52 | 1.23 | 17.23 |
| 28 | 1.12 | 19.36 | 1.09 | 17.23 |
| 29 | 1.49 | 21.28 | 1.51 | 18.04 |
| 30 | 1.44 | 19.44 | 1.36 | 22.37 |
| 31 | 172.60 | 173.90 | ||
| 32 | 3.75 | 51.0 | ||
Fig. 3.
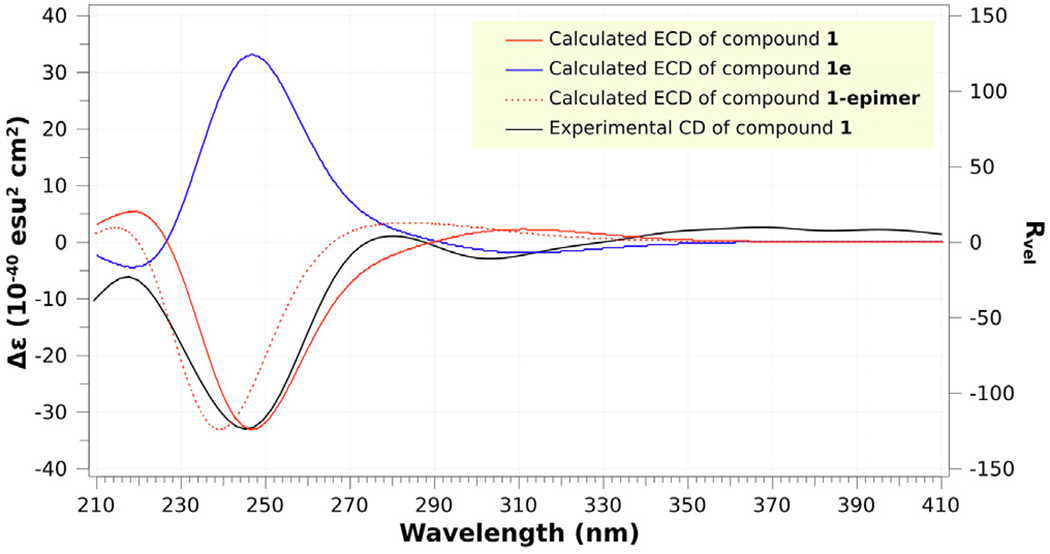
ECD spectra of experimental and calculated 1, 1e, and 1-epimer.
Compound 2 [17] was isolated as a colorless solid. Its molecular formula was determined to be C28H32N4O7 by HR-ESIMS (m/z 537.1501, calcd for [M + H]+ 537.2349), which was 14 units more than that of 1. Analysis of NMR data (Table 1) indicated the presence of an extra methoxy group when compared with that of compound 1. Hence, the structure of compound 2 was readily determined as the methyl ester of compound 1, and it was named as tryptoquivalines X.
Besides the new compounds 1 and 2, four known compounds tryptoquivaline A (3) [18], deoxytryptoquivaline (4) [19], O-deacetyl-tryptoquivaline A (5) [18], and epifiscalin E (6) [23] were also purified. The structures of these known compounds were determined based on comparisons of NMR and HRESIMS data with previously reported data.
Biogenetically, all the compounds isolated in this study could be derived from a cyclic tripeptide-like precursor (I, valine-tryptophan-anthranilic acid) [24]. Nucleophilic attack from the nitrogen at the α-position of tryptophan on the carbonyl carbon of valine would yield intermediate II. Hydrolysis of the amide bond in the lactam ring with an isopropyl group could produce intermediate III. Oxidation followed by coupling between IVb and 2-methyl alanine would generate V. Hydroxylation at 15-position of V followed by oxidative deamination would produce the new compound 1, which could be esterified to compound 2. Cyclization between 13-hydroxy group and the carboxyl group at 11-position would generate compound 5, which would yield compound 3 via acetoxylation of the 25-hydroxy group. Compound 4 could be produced from V after oxidative deamination, acetoxylation of the 25-hydroxy group and cyclization between 13-hydroxyl and the carboxyl group at 11-position. Coupling of the oxidative product of II with MaAla followed by hydroxylation 25-position would generate compound 6 (Fig. 4).
Fig. 4.
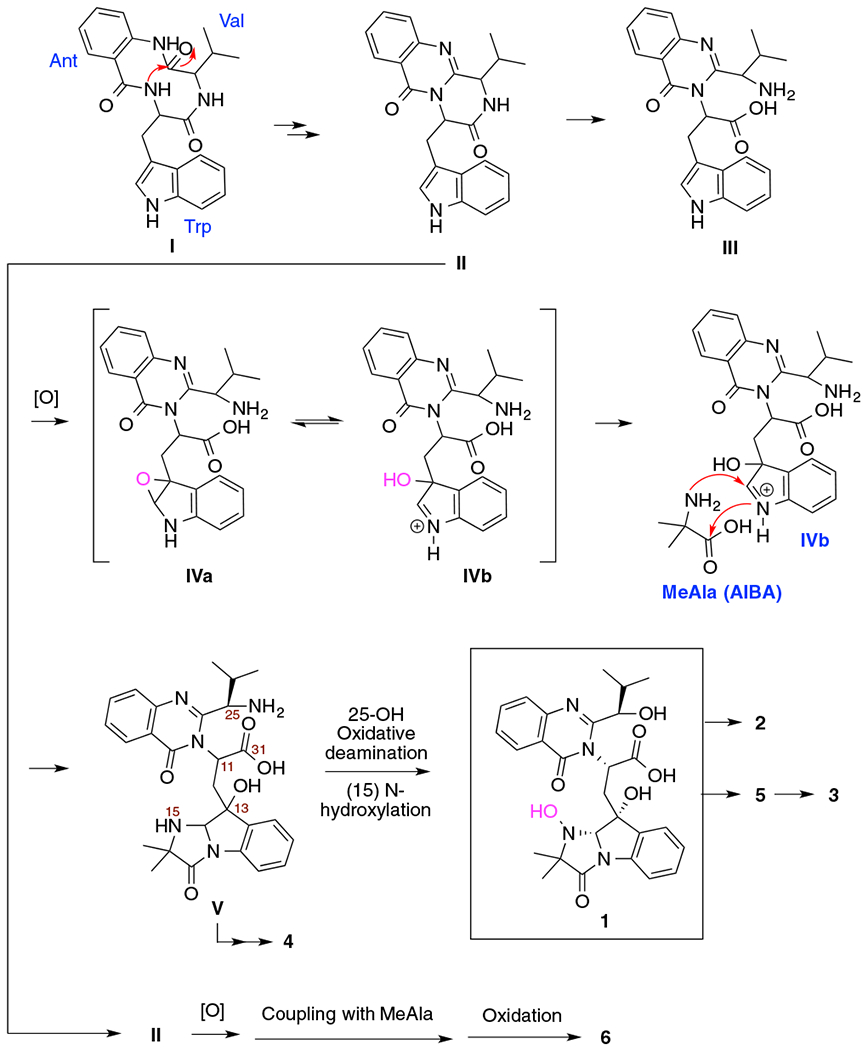
Proposed biosynthetic pathway for compounds 1–6.
Tryptoquivalines are widely distributed in nature. So far, twenty two tryptoquivalines (A-V) [18,25–34] have been isolated. These tryptoquivalines were isolated from two fungal genera. Tryptoquivalines A-O were obtained from Aspergillus clavatus (or Aspergillus fumigatus), while tryptoquivalines P-V from Neosartorya species (N. laciniosa, N. takakii, and N. pseudofischeri). It was reported that tryptoquivalines A and B showed tremorgenic property, and tryptoquivaline O demonstrated antifungal activity. Besides the two subclasses (1–5, and 6) as identified in this study, some tri- or tetra-peptide precursors formed different or even more complicated molecules with diverse ring systems, for examples, fumiquinazoline K (7) [27], fumiquinazoline D (8) [29,35], quinadoline B (9), [36] fumiquinazoline C (10) [35], fumiquinazoline J (11) [35,37], and isochaetominine C (12) [34] (Fig. 5).
Fig. 5.
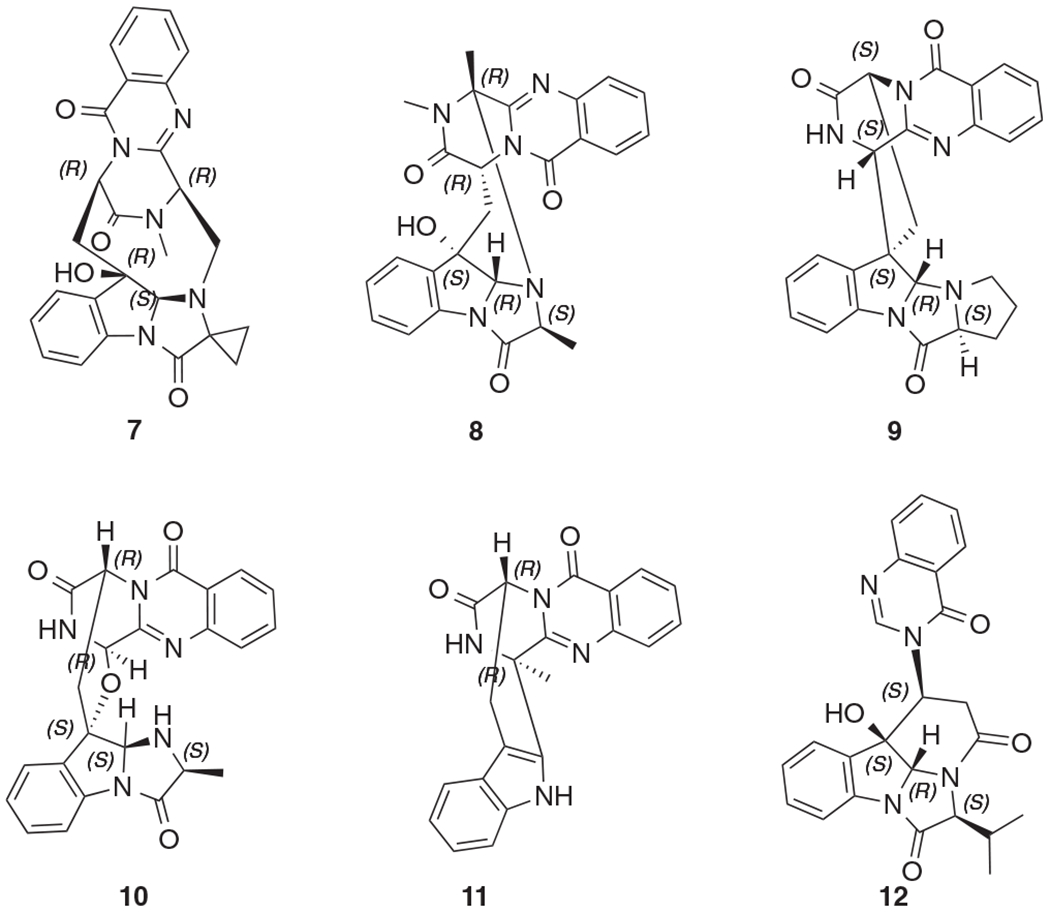
Some analogs (7–12) of compounds 1–6.
Compounds 1–6, which were pure enough for NMR analysis and bioassays, were tested for their anti-proliferative activity against A2780 human ovarian cancer cells [38] and antibacterial activity against S. aureus and E. coli [39], but none was active at 40 μM, and 100 μM, respectively. When evaluated in a mammalian cell-based assay designed to monitor TNF-α-induced NF-κB activity [40], compounds 4 and 5 showed NF-κB inhibition with IC50 values of 3.45 and 6.76 μM, respectively. Compounds 1–6 were also evaluated for their cytotoxicity against the human embryonic kidney cells 293 (HEK 293) using the same conditions as the NF-κB assay, and no cytotoxicity was observed at a concentration of 50 μM. Hence, in the absence of cytotoxicity, inhibition of NF-κB activity suggests the potential of mediating a cancer chemopreventive response.
Supplementary Material
Acknowledgments
This work was financially supported mainly by a start-up funding from Daniel K. Inouye College of Pharmacy (DKICP), Seed Grants from University of Hawaii at Hilo (UHH), and the Victoria S. and Bradley L. Geist Foundation (15ADVC-74420 and 17CON-86295) (to SC). Funding for this work was also supported by Hawaii IDeA Network for Biomedical Research Excellence III and IV (INBRE-III and INBRE-IV) project: NIGMS Grant 5P20GM103466. We would like to thank Dr. Ruisheng Peng, Institute of Astronomy, University of Hawaii, for the sample collection. We would also like to express our gratitude to Mr. Justin Reinicke for his help with NMR and for his kind assistance with optical rotation and ECD data collection.
Footnotes
Declaration of Competing Interest Statement
The authors declare no competing financial interest.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.tetlet.2020.151730.
References
- [1].Zhang HW, Song YC, Tan RX, Nat. Prod. Rep 23 (2006) 753. [DOI] [PubMed] [Google Scholar]
- [2].Vadlapudi V, Borah N, Yellusani KR, Gade S, Reddy P, Rajamanikyam M, Vempati LNS, Gubbala SP, Chopra P, Upadhyayula SM, Amanchy R, Sci. Rep 7 (2017) 7325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Li C, Yang B, Fenstemacher R, Turkson J, Cao S, Tetrahedron Lett. 56 (2015) 1724. [Google Scholar]
- [4].Li C, Ding Y, Yang B, Miklossy G, Yin H, Walker LA, Turkson J, Cao S, Org. Lett 17 (2015)3556. [DOI] [PubMed] [Google Scholar]
- [5].Li C, Ding Y, Yang B, Yin GH-Q, Turkson J, Cao S, Phytochemistry. 126 (2016) 41. [DOI] [PubMed] [Google Scholar]
- [6].Li C, Ren G, Yang B, Miklossy G, Turkson J, Fei P, Ding Y, Walker LA, Cao S, Org. Lett 18 (2016) 2335. [DOI] [PubMed] [Google Scholar]
- [7].Fei-Zhang DJ, Li C, Cao S, Cancer Biol. Ther 17 (2016) 709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Li C, Yang B, Turkson J, Cao S, Phytochemistry 140 (2017) 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Li C, Sarotti AM, Turkson J, Cao S, Tetrahedron Lett. 58 (2017) 2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Huang P, Li C, Sarotti AM, Turkson J, Cao S, Tetrahedron Lett. 58 (2017) 1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Li C, Sarotti AM, Yang B, Turkson J, Cao S, Molecules 22 (2017) pii:E1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Li C, Sarotti AM, Huang P, Dang UT, Hurdle JG, Kondratyuk TP, Pezzuto JM, Turkson J, Cao S, Sci. Rep 7 (2017) 10424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Li C, Sarotti AM, Yoshida W, Cao S, Tetrahedron Lett. 58 (2018) 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Li C, Hu Z, Liu Q, Wu X, Cao S, Tetrahedron Lett. 59 (2018) 3381. [Google Scholar]
- [15].Li C, Sarotti AM, Wu X, Yang B, Turkson J, Chen Y, Liu Q, Cao S, Molecules 24 (2019) pii:E196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wang F, Li C, Hu Z, Wu X, Cao S, Tetrahedron Lett. 59 (2019) 42. [Google Scholar]
- [17].(a) General experimental procedures9(b) Strain isolation and fermentation: The strain Aspergillus terreus was isolated from a soil sample collected from 14,048.1 feet altitude of Mauna Kea Mountain (latitude: 19.822932° N; Longitude: 155.470194° W), Hawaii, in September 2018, Hawaii. The rDNA ITS1-4 region sequence offungus has been submitted to GenBank (Accession number MN749355) and was deposited in an −80 °C freezer at Daniel K. Inouye College of Pharmacy, University of Hawaii at Hilo, HI, USA. After activating on PDA plates at 28 °C for 5 days, it was cut into small pieces and inoculated into 10 L autoclaved sterilized liquid medium [mannitol 20 g, glucose 10g, monosodium glutamate 5 g, KH2PO4 (0.5 g), MgSO4·7H22O 0.3 g and yeast extract 3 g for 1 L distilled water; pH 6.5 prior sterilization] for fermentation at 24 °C for 28 days.(c) Extraction and compound isolation: The mycelia of FS107 were filtered and extracted with acetone under ultrasonic (1 L × 3 times), followed by removal of acetone under reduced pressure to afford an aqueous solution. After combining the aqueous mycelia extraction and supernatant solution, it was subjected to HP-20 column eluted with MeOH-H2O (10, 50, 90 and 100%) to afford four fractions (Fr.1-4). Fraction 3 (2.4 g) was separated by prep-HPLC (Phenyl-Hexyl, 5 μ, 100 × 21.2 mm; 8 mL/min) eluted with 65-90% MeOH-H2O in 25 min to yield sub-fractions (SFr. 1-30). SFr 6 was purified by semi-preparative HPLC (45% isocratic of MeOH-H2O with 0.1% formic acid for 20 min; 3 mL/min) to afford compound 1 (1.2 mg, tR 13 min). Compound 2 (1.36 mg, tR 23.5 min). Compound 4 (0.92 mg, tR 15 min), 5 (1.3 mg, tR 12 min) and 6 (1.4 mg, tR 8 min) were separated from SFr 12 by using the same HPLC (60% isocratic of MeOH-H2O with 0.1% formic acid for 20 min). And finally compound 3 (2.5 mg, tR 14 min) was isolated from SFr17 by using 70% isocratic of MeOH-H2O with 0.1% formic acid for 20 min (3 mL/min). Tryptoquivaline W (1): white powder; [α]D −16 (c 0.2, MeOH); UV (MeOH) λmax (log ε) 210 (3.56), 303 (2.56) & 349 (2.78) nm; IR(KBr) νmax 3326, 2944, 2832, 1657, 1449, 1406, 1114, 1021 cm−1; 1H and 13C NMR data, Table 1; HRESIMS m/z 523.2194 [M + H]+ (calcd for C27H30N4O7, 523.2193). Tryptoquivaline X (2): white powder; [α]D −28 (c 0.2, MeOH); UV (MeOH) λmax (log ε) 208 (3.07), 288 (2.64) & 349 (2.59) nm; IR (KBr) νmax 3308, 2950, 2836, 1652, 1449, 1404, 1105, 1012 cm−1; 1H and 13C NMR data, Table 1; HRESIMS m/z 537.1501 [M + H]+ (calcd for C28H32N4O7, 537.2349).
- [18].Clardy J, Springer JP, Buchi G, Matsuo K, Wightman R, J. Am. Chem. Soc 1975 (97) (2017) 663. [DOI] [PubMed] [Google Scholar]
- [19].Buechi G, Luk KC, Kobbe B, Townsend JM, J. Org. Chem 42 (1977) 244. [DOI] [PubMed] [Google Scholar]
- [20].Kim KH, Ramadhar TR, Beemelmanns C, Cao S, Poulsen M, Currie CR, Clardy J, Chem. Sci 5 (2014) 4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Wang Q, Hu Z, Luo X, Liu J, Li G, Cao S, Liu Q, J. Nat. Prod 82 (2019) 1331. [DOI] [PubMed] [Google Scholar]
- [22].Wang Q, Hu Z, Li X, Wang A, Wu H, Liu J, Cao S, Liu Q, J. Nat. Prod 81 (2018) 2531. [DOI] [PubMed] [Google Scholar]
- [23].Qian SY, Yang C-L, Khan A, Chen R-X, Wu M-S, Tuo L, Liu Q, Wang J-G, Cheng G-G, Nat. Prod. Res 33 (2018) 1387. [DOI] [PubMed] [Google Scholar]
- [24].Gao X, Chooi Y-H, Ames BD, Wang P, Walsh CT, Tang Y, J. Am. Chem. Soc 133 (2011) 2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Yamazaki M, Fujimoto H, Okuyama E, Chem. Pharm. Bull 25 (1977) 2554. [DOI] [PubMed] [Google Scholar]
- [26].Yamazaki M, Fujimoto H, Okuyama E, Chem. Pharm. Bull 26 (1977) 111. [DOI] [PubMed] [Google Scholar]
- [27].Zhou Y, Debbab A, Mándi A, Wray V, Schulz B, Müller WEG, Kassack M, Lin W-H, Kurtán T, Proksch P, Aly AH, Eur. J. Org. Chem 5 (2013) 894. [Google Scholar]
- [28].Yamazaki M, Okuyama E, Maebayashi Y, Chem. Pharm. Bull 27 (1979) 1611. [DOI] [PubMed] [Google Scholar]
- [29].Li X, Zhang Q, Zhang AL, Gao JM, J. Agric. Food Chem. 60 (2012) 3424. [DOI] [PubMed] [Google Scholar]
- [30].Xu N, Cao Y, Wang L, Chen G, Pei YH, J. Asian Nat. Prod. Res 14 (2012)1109. [DOI] [PubMed] [Google Scholar]
- [31].Xu N, Cao Y, Wang L, Chen G, Pei YH, J. Asian Nat. Prod. Res 15 (2013) 731. [DOI] [PubMed] [Google Scholar]
- [32].Gomes NM, Bessa LJ, Buttachon S, Costa PM, Buaruang J, Dethoup T, Silva AM, Kijjoa A, Mar. Drugs. 12 (2014) 822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zin WW, Buttachon S, Buaruang J, Gales L, Pereira JA, Pinto MM, Silva AM, Kijjoa A, Mar. Drugs. 13 (2015) 3776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Paluka J, Kanokmedhakul K, Soytong M, Soytong K, Kanokmedhakul S, Fitoterapia 137 (2019) 104257. [DOI] [PubMed] [Google Scholar]
- [35].Yan D, Chen Q, Gao J, Bai J, Liu B, Zhang Y, Zhang L, Zhang C, Zou Y, Hu Y, Org Lett. 21 (2019) 1475. [DOI] [PubMed] [Google Scholar]
- [36].Koyama N, Inoue Y, Sekine M, Hayakawa Y, Homma H, Omura S, Tomoda H, Org Lett. 10 (2008) 5273. [DOI] [PubMed] [Google Scholar]
- [37].Huang LH, Xu MY, Li HJ, Li JQ, Chen YX, Ma WZ, Li YP, Xu J, Yang DP, Lan WJ, Org. Lett 19 (2017) 4888. [DOI] [PubMed] [Google Scholar]
- [38].Anti-proliferative Assays9
- [39].Antibacterial assay: Bacteria were grown on agar plates [Tryptic Soy Agar (TSA) or Luria–Bertani Agar (LBA)] for 1 day at 37 °C and then added to a liquid medium (TSB for S. aureus and LB for E. coli). After incubation at 37 °C for 20 h, initially sensitivity assay was done by disk diffusion method, followed by broth microdilution method to determine MIC values of the sensitive compounds. For broth microdilution method the overnight incubated cultures were diluted with TSB or LB media to obtain an OD600 value of approx. 0.1. The bacterium-containing media (50 μL) were then added to each well of 96-well plates, containing a DMSO [10%] solution (50 μL) of each compound. The 96-well plates were placed in an incubator at 37 °C for 24 h. A resazurin dye (30 μL, 0.015%) was then added to each well. After letting the mixtures incubated at 37 °C for 2 h, a color change was observed. DMSO [10%] was used as a negative control whereas chloramphenicol was used as a positive control, which was active against S. aureus and E. coli with MIC values of 19.35 and 9.66 μM, respectively. The maximum concentration of the used compounds was 100 μM. All experiments were performed in triplicate.
- [40].NF-κB assay: We employed HEK 293 from Panomics for monitoring changes occurring along the NF-κB pathway. Stable constructed cells were seeded into 96-well plates at 20 × 103 cells per well. Cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen Co.), supplemented with 10% FBS, 100 units/mL penicillin, 100 μg/mL streptomycin, and 2 mM l-glutamine. After 48 h incubation, the medium was replaced and the cells were treated with various concentrations of test substances. TNF-α (human, recombinant, E. coli, Calbiochem) was used as an activator at a concentration of 2 ng/mL (0.14 nM). The plate was incubated for 6 h. Spent medium was discarded, and the cells were washed once with PBS. Cells were lysed using 50 μL (for 96-well plate) of reporter lysis buffer from Promega, by incubating for 5 min on a shaker, and stored at −80 °C. The luciferase assay was performed using the Luc assay system from Promega. The gene product, luciferase enzyme, reacts with luciferase substrate, emitting light, which was detected using a luminometer (LUMIstar Galaxy BMG). Data for NF-κB inhibition are expressed as IC50 values (i.e., concentration required to inhibit TNF-α-induced NF-κB activity by 50%). As positive controls, two known NF-κB inhibitors were used, TPCK (Nκ-tosyl-l-phenylalanine chloromethyl ketone) and BAY-11-7082 (which selectively and irreversibly inhibits NF-κB activation by blocking TNF-α-induced phosphorylation of IκB-α without affecting constitutive IκB-α phosphorylation), yielding IC50 values of 5.32±0.9 and 11±1.8 μM, respectively. All experiments were performed in triplicate.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.