

SUMMARY
The NLRP3 inflammasome, a critical component of the innate immune system, induces caspase-1 activation and interleukin (IL)-1β maturation in response to microbial infection and cellular damage. However, aberrant activation of the NLRP3 inflammasome contributes to the pathogenesis of several inflammatory disorders, including cryopyrin-associated periodic syndromes, Alzheimer’s disease, type 2 diabetes, and atherosclerosis. Here, we identify the receptor for activated protein C kinase 1 (RACK1) as a component of the NLRP3 complexes in macrophages. RACK1 interacts with NLRP3 and NEK7 but not ASC. Suppression of RACK1 expression abrogates caspase-1 activation and IL-1β release in response to NLRP3- but not NLRC4- or AIM2-activating stimuli. This RACK1 function is independent of its ribosomal binding activity. Mechanistically, RACK1 promotes the active conformation of NLRP3 induced by activating stimuli and subsequent inflammasome assembly. These results demonstrate that RACK1 is a critical mediator for NLRP3 inflammasome activation.
Graphical Abstract
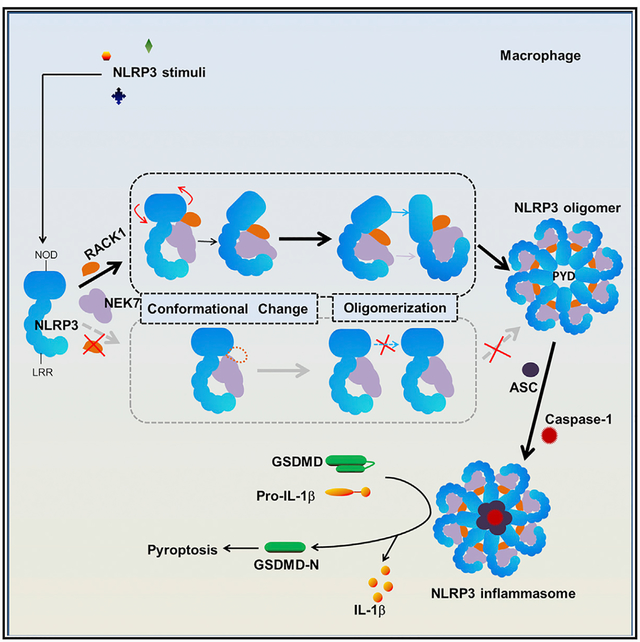
In Brief
The NLRP3 inflammasome is a critical component of the innate immune system. Duan et al. show that the receptor for activated protein C kinase 1 (RACK1) mediates NLRP3 inflammasome activation by promoting NLRP3 active conformation and inflammasome assembly, thereby establishing RACK1 as an important regulator of the NLRP3 inflammasome.
INTRODUCTION
Inflammasomes are intracellular, multimeric protein complexes, which have critical roles in host defense against infection and various non-infectious stresses. Inflammasome assembly is initiated by a pattern recognition receptor (PRR), which in most cases, recruits the adaptor protein, apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) leading to caspase-1 activation (Schroder and Tschopp, 2010). Activated caspase-1 induces the maturation and secretion of proinflammatory cytokines, including interleukin (IL)-1β and IL-18, as well as gasdermin D (GSDMD)-mediated cell death (Martinon et al., 2002; Shi et al., 2015). To date, there are six PRRs that have been well documented to form inflammasomes: the nucleotide-binding oligomerization domain (NOD); the leucine-rich repeat (LRR)-containing protein (NLR) family members NLRP1, NLRP3, NLRP6, and NLRC4; and absent-in-melanoma 2 (AIM2) and pyrin (Broz and Dixit, 2016; Sharma and Kanneganti, 2016). Among these inflammasomes, the NLRP3 inflammasome is most extensively studied because of its activation by a diverse range of stimuli as well as its contribution to the pathogenesis of several inflammatory diseases, including cryopyrin-associated periodic syndromes, Alzheimer’s disease, type 2 diabetes, gout, and atherosclerosis (Kelley et al., 2019; Swanson et al., 2019).
Despite the contribution of NLRP3 to multiple diseases, the mechanism leading to NLRP3 inflammasome activation is not fully understood. Currently, a two-signal model has been proposed for NLRP3 inflammasome activation in macrophages (He et al., 2016a). In that model, a priming signal (signal 1), induced by ligands for toll-like receptors (TLRs), NLRs, or cytokine receptors, upregulates the expression of NLRP3 and pro-IL-1β via the activation of transcription factor nuclear factor kB (NF-kB); an activation signal (signal 2), provided by stimuli such as ATP, pore-forming toxins, crystalline substances, nucleic acids, hyaluronan, and fungal, bacterial, or viral pathogens, triggers NLRP3 inflammasome assembly and activation. Alteration of intracellular ion homeostasis, mitochondrial dysfunction, reactive oxygen species (ROS) production, or lysosomal damage has been proposed as the intracellular event or signal for NLRP3 inflammasome activation (Kelley et al., 2019; Swanson et al., 2019). In addition, a number of NLRP3-interacting proteins, such as SGT1, HSP90, TXNIP, MAVS, MARK4, MIF, NEK7, and DDX3X, have been shown to promote NLRP3 inflammasome activation (He et al., 2016b; Lang et al., 2018; Li et al., 2017; Mayor et al., 2007; Samir et al., 2019; Shi et al., 2016; Subramanian et al., 2013; Zhou et al., 2010). However, how those identified NLRP3-interacting partners orchestrate NLRP3 inflammasome activation remains unclear.
We previously identified NEK7 as an NLRP3-interacting partner critical for NLRP3 inflammasome activation by using a combinatorial approach of affinity purification and mass spectrometry (He et al., 2016b). In our search for other NLRP3-interacting proteins that might be involved in NLRP3 inflammasome activation, we found the receptor for activated protein C kinase 1 (RACK1), which was originally discovered as an intracellular receptor for the activated protein C kinase (PKC) isoform βII (Ron et al., 1994; Stebbins and Mochly-Rosen, 2001). As a highly conserved member of the tryptophan-aspartic acid (WD) repeat protein family, RACK1 is now recognized as a multifunctional scaffold protein involved in many biological processes, such as proliferation, cell migration, apoptosis, and development (Adams et al., 2011). In addition, RACK1 is an integral component of the small ribosomal 40S subunit and acts as a signaling platform to regulate translational activity and selectivity (Rabl et al., 2011; Sengupta et al., 2004). Notably, RACK1 is also found in plant innate immune complexes that have a pivotal role in host defense against infection (Cheng et al., 2015; Nakashima et al., 2008). The role of RACK1 in the NLRP3 inflammasome pathway of mammalian innate immunity has not yet, to our knowledge, been explored. In this study, we analyzed the role of RACK1 in NLRP3 inflammasome activation and provide evidence that non-ribosomal RACK1 serves as a critical NLRP3-interacting protein that mediates NLRP3 inflammasome activation by promoting NLRP3 active conformation and inflammasome assembly.
RESULTS
RACK1 Interacts with NLRP3 and NEK7
To identify NLRP3-interacting proteins that are involved in NLRP3 inflammasome activation in macrophages, we used immortalized bone marrow-derived macrophages (iBMDM) derived from Nlrp3−/− mice and reconstituted these macrophages with a triple-tagged NLRP3 (NLRP3-SFP; S-tag, FLAG and streptavidin-binding tag at the carboxyl terminus). This system allowed us to isolate NLRP3 complexes from macrophages by a two-step affinity purification method without the usage of an antibody and competition from endogenous NLRP3. Using this system, we previously identified NEK7 as an NLRP3-interacting protein critical for NLRP3 inflammasome activation (He et al., 2016b). Among other NLRP3-interacting proteins in NLRP3 complexes, RACK1 is particularly notable as it is involved in the assembly of intracellular signaling complexes and plant innate immunity (Adams et al., 2011; Cheng et al., 2015; Nakashima et al., 2008). Compared with NEK7, RACK1 had a lower peptide coverage in our mass spectrometry analysis and was identified in NLRP3 protein complexes from macrophages treated with lipopolysaccharide (LPS) alone or after LPS plus ATP stimulation (Figure 1A). To validate those data, we immunoprecipitated NLRP3 complexes from cell lysates of macrophages with an anti-FLAG antibody instead of the above-mentioned antibody-independent pull-down method. The parental Nlrp3−/− iBMDM, transduced with an empty vector, was included as a control in our experimental settings. Consistent with our mass spectrometry data, RACK1 was co-immuno-precipitated with NLRP3 and NEK7 from NLRP3-reconstituted macrophages but not from control cells under both conditions of stimulation (Figure 1B). Likewise, we immunoprecipitated NEK7 from NEK7-deficient macrophages reconstituted with tagged NEK7 (NEK7-SFP) and found that RACK1 was also co-immunoprecipitated with NEK7 and NLRP3 from reconstituted macrophages stimulated with LPS plus ATP (Figure 1C). Furthermore, co-immunoprecipitation experiments also revealed that RACK1 interacted with NLRP3 in primary macrophages (Figure 1D).
Figure 1. RACK1 Interacts with NLRP3 and NEK7.
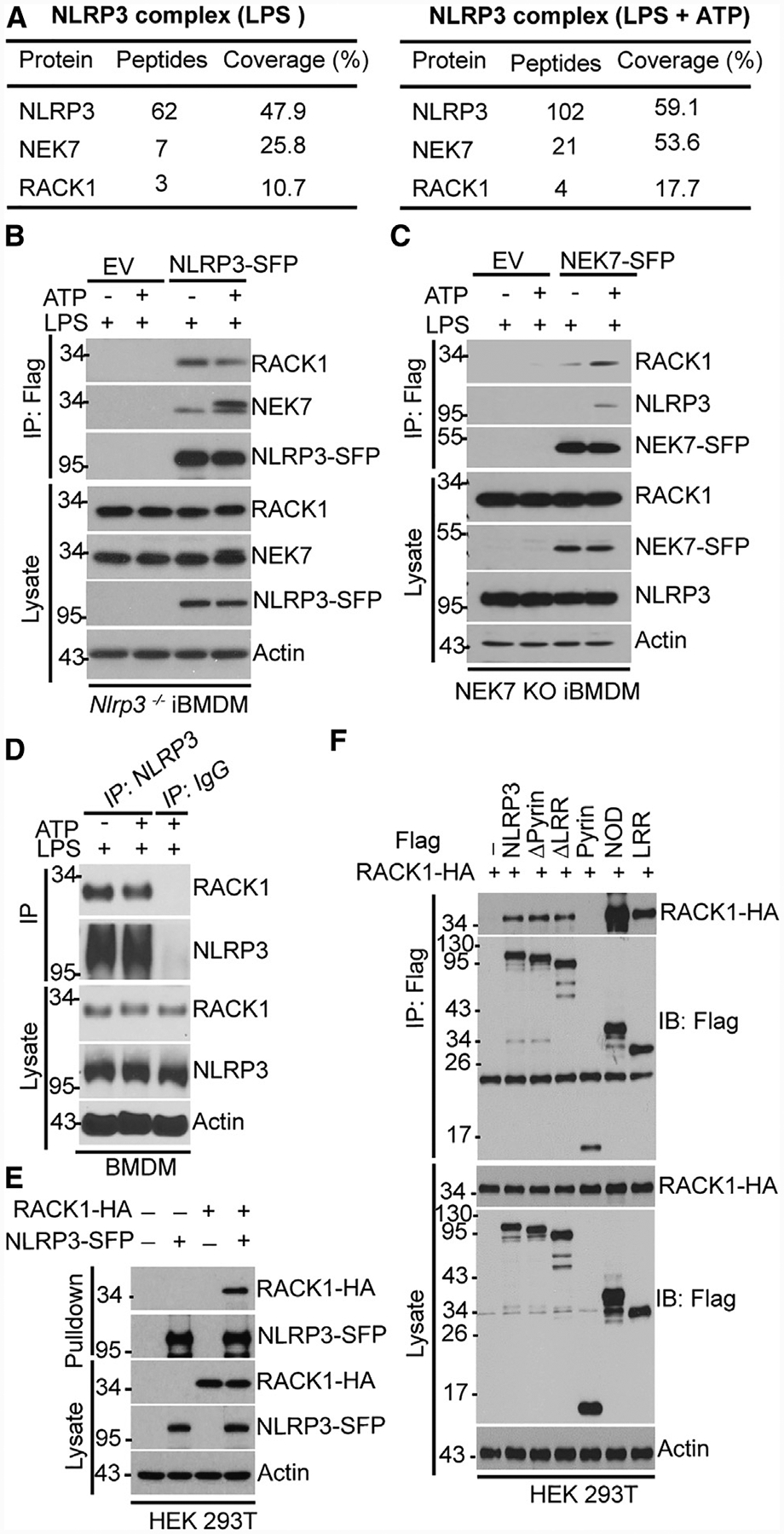
(A) Mass spectrometry analysis of NLRP3, RACK1, and NEK7 peptides after purification of NLRP3 complexes.
(B and C) Tagged NLRP3 (NLRP3-SFP) or NEK7 (NEK7-SFP) was immunoprecipitated (IP) with anti-FLAG antibody from iBMDM treated with LPS (200 ng Ml−1, 4 h) alone or with LPS plus ATP (5 mM, 30 min) and was immunoblotted with the indicated antibodies. EV, empty vector.
(D) BMDM was stimulated with LPS (200 ng mL−1, 4 h) alone or with LPS plus ATP (5 mM, 30 min). Cell lysates were immunoprecipitated and immunoblotted with the indicated antibodies.
(E) NLRP3-SFP was co-expressed with HA-tagged RACK1 in HEK293T cells, pulled down, and analyzed by immunoblotting. HA, hemagglutinin.
(F) FLAG-tagged, full-length or truncated NLRP3 was co-expressed with HA-tagged RACK1 in HEK293T cells, immunoprecipitated, and analyzed by immunoblotting. (Δpyrin, pyrin domain deleted; ΔLRR, leucine-rich repeats deleted; pyrin, pyrin domain only; NOD, NOD domain only; LRR, leucine-rich repeat only). Results are representative of three independent experiments.
We next investigated the interaction between RACK1 and components of the NLRP3 inflammasome. We co-transfected HA-tagged RACK1 with SFP-tagged NLRP3, NEK7, or ASC into human embryonic kidney (HEK) 293T cells, which do not express endogenous NLRP3 and ASC. RACK1 was pulled down with NLRP3 and NEK7 but not with ASC (Figures 1E, S1A, and S1B). We further found that RACK1 interacted with NLRP3 in NEK7-deficient macrophages and with NEK7 in NLRP3-deficient macrophages (Figures S1C and S1D). NLRP3 contains three domains: an N-terminal pyrin domain, a centrally located nucleotide-binding domain (NOD), and a C-terminal leucine-rich repeat (LRR). To determine which domain or domains of NLRP3 interact with RACK1, FLAG-tagged wild-type NLRP3 or various NLRP3-deletion mutants were co-expressed with hem-agglutinin (HA)-tagged RACK1 in HEK293T cells. RACK1 interacted with full-length NLRP3 or NLRP3 lacking the pyrin or LRR domain (Figure 1F). Furthermore, RACK1 interacted with both the NOD and LRR domains of NLRP3, but not with the pyrin domain of NLRP3 alone (Figure 1F). Together, these data indicate that RACK1 interacts with the NOD and LRR domains of NLRP3 and its critical regulator NEK7.
RACK1 Is Required for NLRP3 Inflammasome Activation in Macrophages and In Vivo
To determine the role of RACK1 in NLRP3 inflammasome activation, we suppressed RACK1 expression with RACK1-specific small-interfering RNA (siRNA) and assessed NLRP3 inflammasome activation by ATP in macrophages. RACK1 knockdown did not affect the expression levels of NLRP3 inflammasome components NLRP3, pro-caspase-1, or ASC as compared with non-targeting siRNA (Figure 2A). Additionally, RACK1 depletion had no effect on the induction of NLRP3 by LPS (Figure 2A). However, RACK1 knockdown remarkably attenuated caspase-1 activation and IL-1β secretion induced by ATP in LPS-primed macrophages, whereas the production of tumor necrosis factor alpha (TNF-α), an inflammasome-independent cytokine, was comparable between macrophages treated with RACK1 siRNA and control siRNA (Figures 2A–2C). To determine whether the effect of RACK1 knockdown on NLRP3 inflammasome activation is specific to ATP or common to other NLRP3 activators, we stimulated siRNA-treated macrophages with nigericin, gramicidin, silica, or lysosome membrane damaging agent Leu-Leu-OMe (LLOMe) after LPS priming. RACK1 knockdown markedly reduced caspase-1 activation and IL-1β release without inhibiting TNF-α release (Figures 2D–2F). RACK1 knockdown inhibited NLRP3 inflammasome activation, but did not affect the induction of NLRP3 by LPS, suggesting that RACK1 might be primarily involved in the activation pathway of the NLRP3 inflammasome. To test that, we silenced RACK1 expression in macrophages, which constitutively expressed NLRP3 after lentiviral transduction to induce NLRP3 inflammasome activity in the absence of signal 1. RACK1 knockdown inhibited NLRP3 inflammasome activation induced by nigericin, gramicidin, or LLOMe in those macrophages (Figure S2A). Furthermore, RACK1 knockdown had no effect on caspase-1 activation induced by poly (deoxyadenylic-deoxythymidylic) acid (poly(dA:dT)), which activates the AIM2 inflammasome, or Salmonella, which activates the NLRC4 inflammasome (Figure 2G). We were unsuccessful in generating RACK1 knockout (KO) macrophages by the CRISPR/Cas9 system, which is consistent with the previous observation that complete deletion of mouse RACK1 is lethal for early embryonic development (Volta et al., 2013). To further confirm the role of RACK1 in NLRP3 inflammasome activation, we used two different short hairpin RNAs (shRNAs) to silence RACK1 expression in macrophages. In agreement with our siRNA experiments, suppression of RACK1 expression by shRNA in BMDM or immortalized macrophages remarkably inhibited caspase-1 activation and IL-1β release in response to ATP, nigericin, or silica (Figures S2B–S2F). Inflammasome activation induces GSDMD-mediated cell death (Shi et al., 2015). We found that RACK1 knockdown inhibited GSDMD cleavage and cell death induced by the NLRP3 inflammasome but not by the NLRC4 or AIM2 inflammasome (Figures S3A and S3B). Furthermore, RACK1 knockdown inhibited caspase-1 activation and IL-1β maturation induced by the noncanonical NLRP3 inflammasome after the cytosolic delivery of LPS (Figure S4). Collectively, these data demonstrate that RACK1 is specifically required for NLRP3 inflammasome activation in macrophages.
Figure 2. RACK1 Knockdown Impairs NLRP3 Inflammasome Activation in Macrophages and In Vivo.
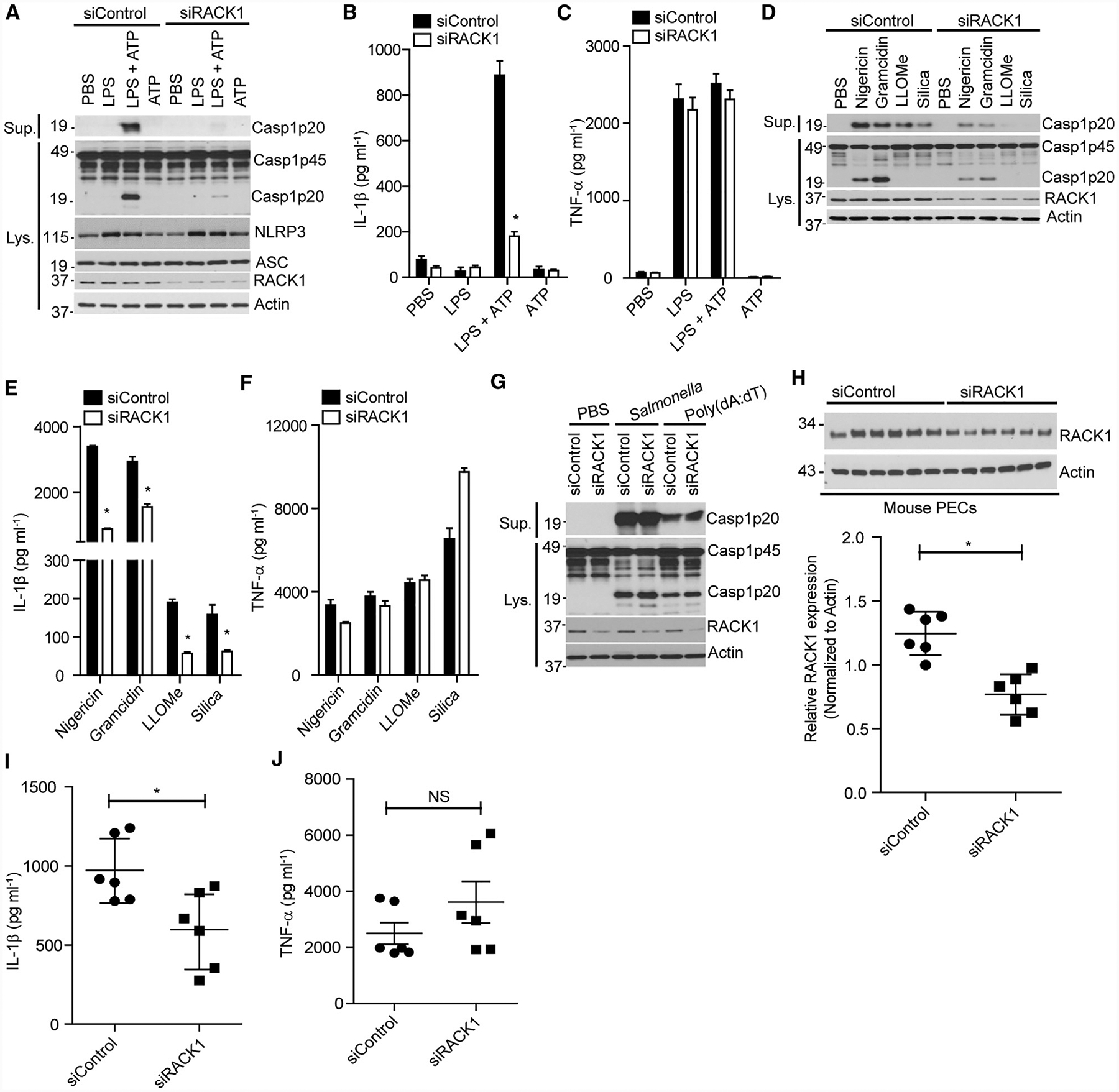
(A–C) iBMDM was treated with control siRNA (siControl) or RACK1 siRNA (siRACK1). The cells were left unstimulated (PBS) or stimulated with LPS (200 ng mL−1, 4 h), ATP (5 mM, 30 min), or LPS (200 ng mL−1, 4 h) plus ATP (5 mM, 30 min). (A) Supernatants (Sup.) and cell lysates (Lys.) were analyzed by immunoblotting with the indicated antibodies. (B and C) IL-1β and TNF-α release in supernatants was analyzed by ELISA.
(D–G) iBMDM were treated with siControl or siRACK1 and then primed by LPS (200 ng mL−1, 4 h). (B–G) Cells were left unstimulated (PBS) or stimulated by nigericin (5 μM, 1 h), gramicidin (0.5 μM, 1 h), LLOMe (2 μM, 4 h), silica (500 μg Ml−1, 4 h), poly(dA:dT) (2 μg mL−1, 4 h), or Salmonella (MOI = 10, 1 h). Caspase-1 (Casp1) in the supernatant (Sup.) and cell lysate (Lys.) were analyzed by immunoblotting (D and G); IL-1β and TNF-α release in the supernatants of (D) was analyzed by ELISA (E and F).
(H–J) Mice were intraperitoneally injected with in vivo-jetPEI-encapsulated siRNA (100 μg per mouse) and then with LPS (25 mg/kg of body weight). Mouse serum was collected after intraperitoneal injection of LPS and analyzed for cytokines IL-1β (I) and TNF-α (J). Peritoneal exudate cells (PECs) from each mouse were collected for assessing RACK1 expression by western blot (H). Actin was immunoblotted as a loading control. Each symbol represents one mouse.
For ELISA data of in vitro experiments (B, C, E, and F), error bars denote SD of triplicate wells. Results (A–G) are representative of three independent experiments. For in vivo experiments (H–J), mean values are indicated by a horizontal bar. Results are representative of two independent experiments. NS, not significant. Unpaired two-tailed Student’s t test, *p < 0.05.
We next assessed the role of RACK1 in NLRP3 inflammasome activation in vivo. To that end, we used an LPS-induced sepsis model in which the NLRP3 inflammasome is required for the production of IL-1β in serum (He et al., 2013; Mariathasan et al., 2006; Sutterwala et al., 2006). We delivered a mixture of siRNA, using the jetPET system, into mice through intraperitoneal injection. Peritoneal exudate cells from mice treated with siRACK1 had reduced expression of RACK1 as compared with those from control siRNA-treated mice (Figure 2H). After intraperitoneal injection with LPS, IL-1β was detected in the sera of siRNA-treated mice. Importantly, mice treated with RACK1 siRNA produced less IL-1β in the serum when compared with controls. In contrast, the production of serum TNF-α was comparable across all groups (Figures 2I and 2J). These data suggest that RACK1 is also required for LPS-induced NLRP3 inflammasome activation in vivo.
RACK1 Is Required for Activating NLRP3 Mutation (R258W)-Induced Inflammasome Activation in Macrophages
NLRP3 gain-of-function mutations that commonly associate with cryopyrin-associated periodic syndrome (CAPS) can trigger inflammasome activation in the absence of NLRP3 activators (Brydges et al., 2009; Hoffman et al., 2001; Meng et al., 2009). To examine whether RACK1 is required for NLRP3 inflammasome activation induced by a gain-of-function mutation of NLRP3, we depleted RACK1 by siRNA in mouse macrophages containing the activating NLRP3 (R258W) mutation, which corresponds to the human NLRP3 (R260W) mutation that causes Muckle-Wells syndrome (Meng et al., 2009). In agreement with previous studies, treatment of NLRP3 (R258W) macrophages with LPS alone was sufficient to activate caspase-1 and IL-1β release (Meng et al., 2009). Importantly, RACK1 knockdown had no effect on TNF-α secretion, but diminished caspase-1 activation and IL-1β release elicited by LPS in NLRP3 (R258W) macrophages, as compared with the control (Figures 3A–3C). Furthermore, RACK1 knockdown also inhibited GSDMD cleavage and cell death induced by this activating mutation of NLRP3 (Figures 3D and 3E). Taken together, these data indicate that RACK1 acts on, or just downstream of, both wild-type and cryopyrin-associated periodic syndrome (CAPS)-associated NLRP3 to mediate NLRP3 inflammasome activation in macrophages.
Figure 3. RACK1 Knockdown Suppresses Mutation-Induced NLRP3 Inflammasome Activation in Macrophages.
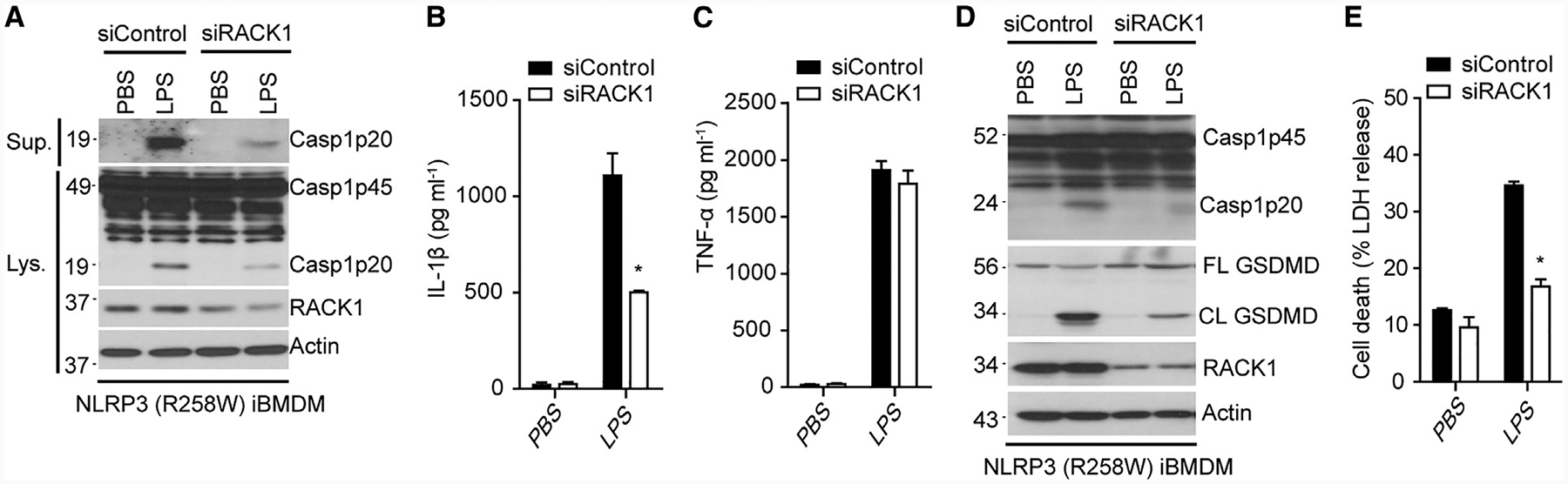
(A–E) Macrophages (NLRP3 R258W) were treated with control siRNA (siControl) or RACK1 siRNA (siRACK1). Cells were left unstimulated (PBS) or stimulated with LPS (200 ng mL−1, 4 h). Cell lysates (Lys.) and culture supernatants (Sup.) were immunoblotted with indicated antibodies (A and D). IL-1β (B), TNF-α (C), and LDH(E) in culture supernatants of macrophages were analyzed. Error bars denote SD of triplicate wells. Results are representative of three independent experiments. Unpaired two-tailed Student’s t test, *p < 0.05.
RACK1 Is Critical for ASC Oligomerization Induced by NLRP3 Activation
We next sought to determine the mechanism by which RACK1 mediates NLRP3 inflammasome activation. RACK1 interacted with NLRP3 and was required for inflammasome activation induced by NLRP3 activators and the activating mutation of NLRP3. Therefore, we hypothesized that RACK1 might control the proximal signaling event of NLRP3. Activation of NLRP3 or other inflammasome sensors triggers the formation of a supra-molecular structure termed ASC speck, which is composed of ASC oligomers present in the detergent-insoluble cellular fraction (Fernandes-Alnemri et al., 2007). As expected, stimulation of macrophages by the NLRP3 activators ATP or nigericin or infection of macrophages by Salmonella (NLRC4 activator) induced the formation of ASC specks in the cytosol. RACK1 knockdown reduced the number of cells that contained ASC specks after stimulation with ATP or nigericin, whereas it did not affect ASC speck formation induced by Salmonella infection (Figures 4A and 4B). Furthermore, analysis of ASC oligomers in the detergent-insoluble fraction revealed that RACK1 knockdown inhibited ATP or nigericin, but not Salmonella, induced ASC oligomerization (Figure 4C). Taken together, these results indicate that RACK1 is specifically required for the NLRP3-dependent ASC oligomerization and speck formation.
Figure 4. RACK1 Is Required for ASC Speck Formation Induced by NLRP3.
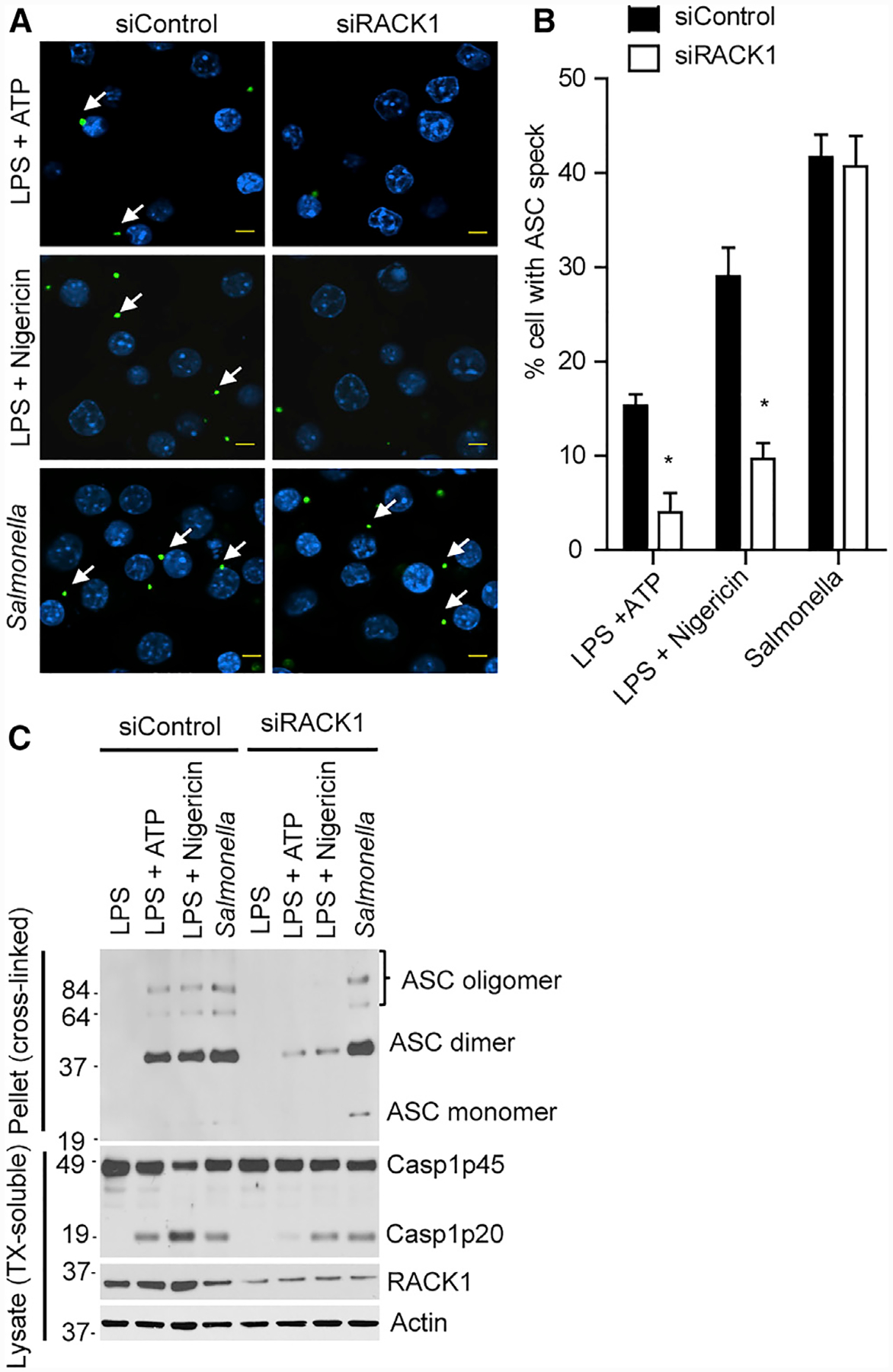
(A) iBMDM was treated with control siRNA (siControl) or RACK1 siRNA (siRACK1). Cells were primed with LPS (200 ng mL−1, 4 h) and then stimulated with ATP (5 mM, 30 min), nigericin (5 μM, 1 h), or Salmonella (MOI = 10, 1 h). Representative images of ASC specks in macrophages treated with indicated stimuli. Scale bars, 5 μm.
(B) Quantification of endogenous ASC specks (arrows) in (A). Data show representative results from three combined independent experiments. Error bars indicate SD. Unpaired two-tailed Student’s t test, *p < 0.05.
(C) ASC oligomerization in Triton X-100 (TX)-insoluble fractions of macrophages was analyzed by immunoblotting after cross-linking. TX-soluble fractions were also immunoblotted with indicated antibodies. Results are representative of three independent experiments.
RACK1 Mediates NLRP3 Inflammasome Activation Independent of Its Ribosomal Binding Activity
RACK1 was originally identified as the receptor for activated protein kinase C (PKC) (Ron et al., 1994). However, inhibition of PKC by chemicals C-1, GF 109203X, or Go 6983 had no effect on caspase-1 activation induced by ATP in LPS-primed BMDM, suggesting a PKC-independent mechanism for RACK1 to regulate NLRP3 inflammasome activation (Figure S5A). Potassium efflux is a key signaling event for the activation of NLRP3, which is required for the NLRP3-NEK7 interaction (He et al., 2016b; Muñoz-Planillo et al., 2013). We found that high concentration of extracellular potassium (50 mM) did not affect the NLRP3-RACK1 interaction, whereas it did block the NLRP3-NEK7 association and caspase-1 activation induced by ATP stimulation (Figure S5B). Furthermore, suppression of RACK1 expression by siRNA did not affect the NLRP3-NEK7 interaction in macrophages (Figure S5C). RACK1 has been reported to regulate the translational process by acting as a component of the ribosomal 40S subunit (Kuroha et al., 2010; Rabl et al., 2011). Furthermore, a previous study showed that inhibition of the translational machinery activates the NLRP3 inflammasome (Vyleta et al., 2012). Therefore, we sought to determine whether the association of RACK1 with ribosomes is required for NLRP3 inflammasome activation. We first assessed whether the NLRP3-RACK1 interaction was dependent on ribosomal binding activity of RACK1 using a mutant RACK1 (R36D/K38E) that cannot interact with ribosomes (Coyle et al., 2009; Sengupta et al., 2004) (Figure S6). Wild-type RACK1 or mutant RACK1 (R36D/K38E) was co-transfected with NLRP3 into HEK293T, and NLRP3 was immunoprecipitated from cell lysates after transfection. Immunoblotting analysis revealed that RACK1 (R36D/K38E) interacted more strongly with NLRP3 than wild-type RACK1 did, suggesting that the ribosomal association of RACK1 is dispensable for the interaction of NLRP3 with RACK1 (Figure 5A). To further assess which form of RACK1, ribosomal or non-ribosomal, is required for NLRP3 inflammasome activation, we expressed HA-tagged siRNA-resistant wild-type RACK1 or its mutant RACK1 (R36D/K38E) in macrophages pre-treated with RACK1 siRNA. Exogenously expressed RACK1 rescued ATP or nigericin-induced caspase-1 activation and IL-1β release, without affecting TNF-α secretion, in macrophages (Figures 5B–5D). Although we consistently had much less expression of RACK1 (R36D/K38E) than wild-type RACK1 produced in our culture system, exogenously expressed RACK1 (R36D/K38E) also restored ATP or nigericin-induced caspase-1 activation and IL-1β release in macrophages treated with RACK1 siRNA (Figures 5B–5D). Taken together, these data suggest that RACK1 interacts with NLRP3 and promotes NLRP3 inflammasome activation independently of its ribosomal-binding activity.
Figure 5. RACK1 Mediates NLRP3 Inflammasome Activation Independent of Its Ribosomal Binding Activity.
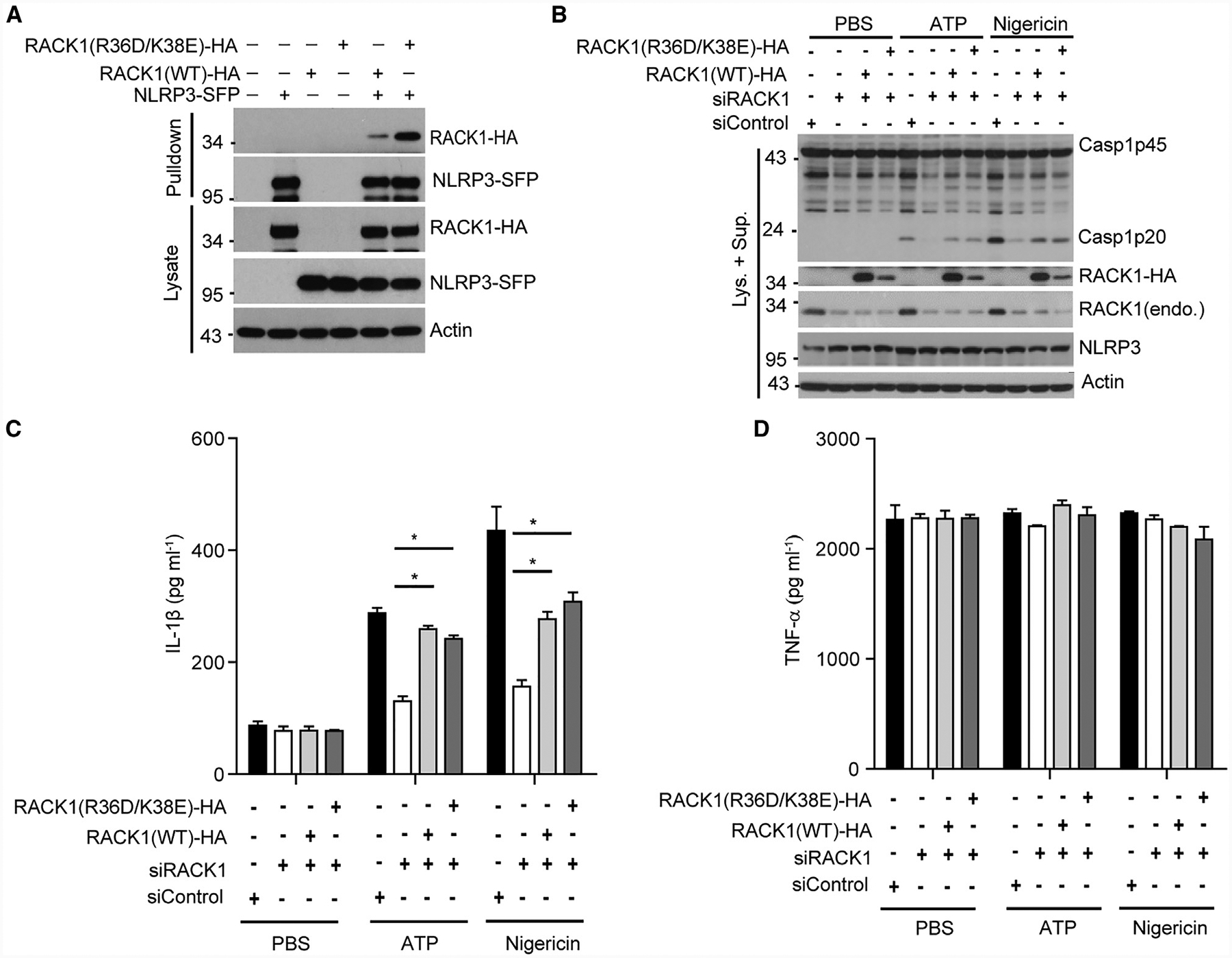
(A) Tagged NLRP3 (NLRP3-SFP) was co-expressed with HA-tagged wild-type RACK1 or mutant RACK1 (R36D/K38E, defective in ribosome binding) in HEK293T cells. Cell lysates were pulled down with streptavidin beads and analyzed by immunoblotting.
(B–D) Mouse macrophages were treated with RACK1 siRNA (siRACK1), followed by transduction with lentivirus expressing HA-tagged, siRNA-resistant, wild-type RACK1 or mutant RACK1 (R36D/K38E). Macrophages were primed with LPS (200 ng mL−1, 4 h) and then stimulated with PBS (control), ATP (5 mM, 30 min), or nigericin (5 μM, 1 h). Mixtures of cell lysates and culture supernatants were analyzed by immunoblotting with the indicated antibodies (B). IL-1β (C) and TNF-α (D) in culture supernatants were analyzed by ELISA. Error bars denote SD of triplicate wells. Results are representative of three independent experiments. Unpaired two-tailed Student’s t test, *p < 0.05.
RACK1 Promotes NLRP3 Active Conformation and Inflammasome Assembly
The centrally located NOD domain of NLRP3 is critical for its oligomerization and inflammasome activation (Duncan et al., 2007). Furthermore, disease-associated mutations in this domain or NLRP3 stimuli induce the active “open” conformation of NLRP3 that coincides with its oligomerization (Tapia-Abellán et al., 2019). Because RACK1 strongly binds to the NOD domain of NLRP3 (Figure 1F), we hypothesized that RACK1 might be required for stimulus-induced NLRP3 conformational changes and inflammasome complex formation. To test that hypothesis, we first analyzed the formation of NLRP3 complexes in response to activating stimuli. Macrophages were stimulated with ATP or nigericin, and then, digitonin-solubilized cell lysates were separated in the first dimension by blue native PAGE and then in a second dimension by SDS–PAGE. In agreement with our previous findings, the formation of high-molecule-weight complexes (>1,200 kDa) containing NLRP3 and NEK7 was observed in macrophages treated with LPS plus ATP or nigericin, but not LPS alone (Figures 6A and 6B) (He et al., 2016b). However, RACK1 knockdown significantly reduced the formation of high-weight protein complexes of NLRP3 and NEK7 in macrophages as compared with cells treated with non-targeting control (Figures 6A and 6B). Next, we examined whether RACK1 was required for stimuli-induced conformational change of NLRP3. To that end, we used a bioluminescence resonance energy transfer (BRET) assay to monitor the conformational change of NLRP3 upon activation, as previously reported (Tapia-Abellán et al., 2019). The luciferase (donor)/YFP (acceptor)-tagged NLRP3 was expressed in HEK293T cells treated with control or RACK1 siRNA (Figure 6C). The BRET signals (“close” conformation) were comparable at the resting state for both siRNA-treated cells; a characteristic drop of the BRET signal was observed in cells after the addition of nigericin, which indicated an “open” conformation for NLRP3 (Figure 6D). However, RACK1 knockdown attenuated this nigericin-induced drop of the BRET signal, suggesting the formation of a partially active conformation of NLRP3 (Figure 6D). Collectively, these results suggest that RACK1 is crucial for the formation of the fully active NLRP3 conformation and the NLRP3-NEK7 inflammasome complex in response to NLRP3 stimuli.
Figure 6. RACK1 Promotes NLRP3 Active Conformation and Inflammasome Assembly.
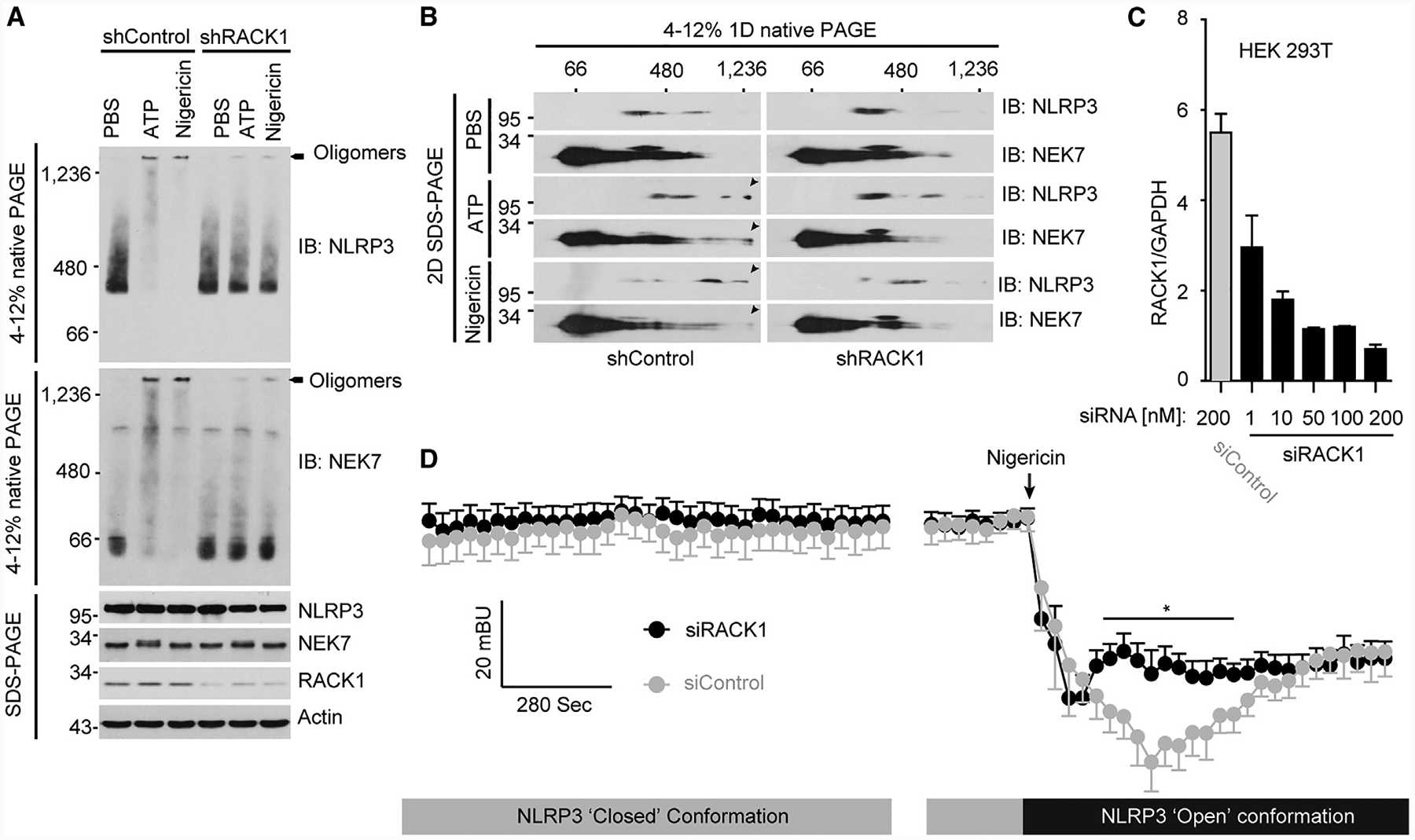
(A) Macrophages were infected with lentivirus expressing control shRNA (shControl) or RACK1 shRNA (shRACK1). After puromycin selection, transduced macrophages were primed with LPS (200 ng mL−1, 4 h) and then stimulated with PBS (control), ATP (5 mM, 30 min), or nigericin (5 μM, 1 h). NLRP3 inflammasome assembly was analyzed by blue native PAGE and immunoblotting. Cell lysates were also analyzed by SDS–PAGE and immunoblotting.
(B) Cell lysates were separated by a first dimension of blue native PAGE, followed by a second dimension of SDS–PAGE.
(C) HEK293T cells stably expressing YFP-NLRP3-Luc were transfected with 200 nM of control siRNA (siControl) or the indicated concentrations of RACK1 siRNA (siRACK1). The relative levels of RACK1 gene expression were analyzed by quantitative reverse transcriptase-PCR and normalized to the level of endogenous glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression.
(D) Intramolecular BRET signal of NLRP3 was measured before and after nigericin (10 μM) treatment. The arrow indicates the time for nigericin addition. Mann-Whitney test, two-tailed for each comparison of siControl (200 nM) with siRACK1 (200 nM). *p < 0.05. Results are representative of three independent experiments.
DISCUSSION
The NLRP3 inflammasome acts as a critical component of innate immunity to promote host defense, yet its aberrant activation contributes to the pathogenesis of several inflammatory diseases (Guo et al., 2015). Although intensively investigated over the past decade, the molecular mechanism of NLRP3 inflammasome activation remains poorly understood. We previously identified NEK7 as a critical NLRP3-interacting partner with a combinatorial approach of affinity tag-based tandem protein purification and mass spectrometry (He et al., 2016b). In this study, we used same approach to identify RACK1 as another critical NLRP3-binding partner. We found that RACK1 interacted with both NLRP3 and NEK7 but not the downstream adaptor ASC, and suppression of RACK1 expression specifically abrogated NLRP3 inflammasome activation induced by a range of NLRP3 stimuli or an activating mutation of NLRP3. Rescue experiments with wild-type RACK1 or its ribosome-binding-deficient mutant showed that non-ribosomal RACK1 contributed to its regulatory activity in NLRP3 inflammasome activation. Furthermore, we found that RACK1 promoted stimuli-induced active conformation of NLRP3 and the formation of large NLRP3-NEK7 complexes. Collectively, our results suggest that RACK1 has a pivotal role in NLRP3 inflammasome assembly and activation.
Although a number of proteins have been shown to regulate NLRP3 inflammasome activation, genetic and biochemical data from different studies have indicated that NEK7 is required for NLRP3 inflammasome activation induced by various NLRP3 activators (Deng et al., 2019; Groß et al., 2016; He et al., 2016b; Schmid-Burgk et al., 2016; Shi et al., 2016). A recent study reveals a cryo-electron microscopy structure of NLRP3 in complex with NEK7 (Sharif et al., 2019). However, NLRP3 in that complex was inactive and failed to form oligomers, suggesting that other NLRP3-interacting partners might be required for NLRP3 activation under the reported in vitro conditions. In this study, we identified RACK1 as a protein that interacts with both NLRP3 and NEK7 and showed its requirement for NLRP3 inflammasome activation. Therefore, RACK1 might be required for the oligomerization of NLRP3 in the aforementioned in vitro NLRP3-NEK7 complexes. Supporting that notion, RACK1 has been shown to induce oligomerization of Bax and promote intrinsic apoptosis (Mamidipudi and Cartwright, 2009). In addition, our findings module critical for NLRP3 self-association (Duncan et al., module critical for NLRP3 self-association (Duncan et al., 2007), suggesting that RACK1 might directly regulate NLRP3 oligomerization through its interaction with this domain. Furthermore, our data show that RACK1 knockdown suppressed the conformational transition of NLRP3 induced by nigericin and the formation of high-molecular-weight complexes composed RACK1-mediated NLRP3 inflammasome activation, in which RACK1-mediated NLRP3 inflammasome activation, in which RACK1 promotes NLRP3 oligomerization and inflammasome assembly through its interaction with the NOD domain of NLRP3. How the interaction of RACK1 with NEK7 or other partners in the inflammasome complex contributes to this function of RACK1 remains to be investigated in future studies.
RACK1 was originally discovered as a binding partner for the activated protein kinase C (Ron et al., 1994). In agreement with previous studies (Zhang et al., 2017), our data showed that NLRP3 inflammasome-dependent caspase-1 activation in response to ATP was not affected by the presence of PKC inhibitors, suggesting that RACK1 mediates NLRP3 inflammasome activation independent of PKC kinase activity. RACK1 is now recognized as a scaffold protein for kinases and receptors and has an important role in a wide range of biological processes, including cell motility, survival, and death; tumorigenesis; and immune signaling (McCahill et al., 2002). RACK1 exists in both ribosome- and non-ribosome-bound forms, and the ribosomal RACK1 promotes translational initiation (Coyle et al., 2009; Sengupta et al., 2004). Previous studies reported that age-associated reduction of RACK1 expression in rat macrophages impairs TNF-α production in response to LPS (Corsini et al., 1999). In contrast, our experiments with macrophages treated with control siRNA or RACK1 siRNA showed that the induction of NLRP3, as well as TNF-α production, were only marginally altered after RACK1 depletion, suggesting that the inhibition of the NLRP3 inflammasome caused by RACK1 knockdown may not be due to the global inhibition of the translation in macrophages under our culture conditions. One possibility is that the non-ribosomal RACK1 form involved in NLRP3 inflammasome activation might disappear earlier than ribosomal RACK1 after RACK1 siRNA treatment. In support of that possibility, our data show that RACK1 ribosome-binding defective mutant rescued NLRP3 inflammasome activation in macrophages with the knockdown of endogenous RACK1. Interestingly, the RACK1 mutant that cannot interact with ribosomes exhibited enhanced ability to associate with NLRP3. Because RACK1 exists in both ribosomal and non-ribosomal forms in cells, one possible explanation is that the expression of the RACK1 mutant increased the availability of the non-ribosomal form of RACK1, which might be responsible for its interaction with NLRP3 and subsequent NLRP3 inflammasome activation. However, other possibilities such as direct enhancement of NLRP3/RACK1 interaction by the mutation may also account for these observations. Collectively, these results are consistent with previous studies showing the differential functions for ribosome- and non-ribosome-bound forms of RACK1 (Kim et al., 2017; Ruan et al., 2012). Although further work is needed, our studies suggest that non-ribosomal RACK1 mediates NLRP3 inflammasome activation in macrophages.
RACK1 is highly conserved among all eukaryotic species and is reported to have a pivotal role in plant innate immune responses; for example, RACK1 is a component of the Rac1 innate immune complex and participates in host immune defense against rice blast fungus infection (Nakashima et al., 2008). In Arabidopsis thaliana, RACK1 is involved in a novel pathogen-secreted protease sensing pathway by serving as a scaffold protein for the G-protein-MAPK signaling cascade (Cheng et al., 2015). Our findings show that mammalian RACK1 has a function in the innate immune response by mediating the NLRP3 inflammasome pathway, implying that RACK1 has a conserved innate immune function in both plants and mammals. Furthermore, RACK1 has been identified as a cytosolic interacting protein for the Yersinia effector protein YopK, and that interaction has been proposed as a requirement for Yersinia pathogenesis (Thorslund et al., 2011). Notably, a later study reported that YopK prevents type-III secretion-system-induced NLRP3 inflammasome activation in response to Yersinia infection (Brodsky et al., 2010). Future studies will determine whether Yersinia Yopk also inhibits the NLRP3 inflammasome by interfering with the interactions of RACK1 with NLRP3 and NEK7 by targeting RACK1.
In summary, our findings reveal that RACK1 is a critical regulator of the NLRP3 inflammasome and mediates NLRP3 inflammasome activation by promoting the active conformation of NLRP3, suggesting conserved RACK1-mediated innate immune signaling for host defense in both plants and mammals. Further biochemical and structural characterizations of the NLRP3 inflammasome are needed to determine how NLRP3, NEK7, RACK1, and other regulators orchestrate inflammasome assembly and activation.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for reagents should be directed to and will be fulfilled by the Lead Contact, Yuan He (yhe@med.wayne.edu).
Materials availability
All unique reagents generated in this study are available from the Lead Contact with a completed Material Transfer Agreement.
Data and code availability
This study did not generate any unique datasets or code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
C57BL/6 mice were originally purchased from Jackson Laboratories and maintained in our specific pathogen-free facility. Both male and female mice were used for this study. Age- and sex-matched 6- to 8- week-old mice after co-housing were used for in vivo studies, and 6- to 12- week-old mice were used for in vitro studies. All animal studies were approved by Wayne State University or the University of Michigan Committee for the Care and Use of Laboratory Animals.
RACK1 knockdown in vivo and LPS treatment
In in vivo studies, RNA interference of RACK1 was performed as previously described (Fry et al., 2012). The siRNAs (siControl: 5´-CUUGCUUCAAUUCAUCUAC-3´; siRACK1: 5´-GUAGAUGAAUUGAAGCAAG-3´) were synthesized and processed for in vivo-ready grade (Biosynthesis). Age- and sex-matched, 6- to 8-week-old wild-type C57BL/6 mice were intraperitoneally injected with 100μg siRNA per mouse using in vivo-jetPEI solution as transfer reagent (Polyplus Transfection) according to the manufacturer’s guidelines. After 48 h of injection, mice were intraperitoneally injected once with LPS (25 mg kg−1 of body weight) and blood was collected from mice after 3 h for the measurement of serum IL-1β and TNF-α by ELISA. Mice were euthanized and peritoneal exudate cells (PECs) were collected for assessing RACK1 knockdown by western blot.
Cell culture
Bone-marrow derived macrophages (BMDM) were prepared by differentiating bone marrow cells from the tibia and femur bones of C57BL/6 for 7 days in 10% FBS IMDM (GIBCO) supplemented with 30% L292-cell supernatant, non-essential amino acids, sodium pyruvate and antibiotics (penicillin/streptomycin). L292-cell and immortalized BMDM (iBMDM) were cultured in 10% FBS IMDM (GIBCO) supplemented with sodium pyruvate and antibiotics (penicillin/streptomycin). HEK293T cells (ATCC) were cultured on DMEM (GIBCO) containing 10% FBS and antibiotics (penicillin/streptomycin).
Bacterial Culture
Salmonella Typhimurium SL1344 was cultured in Luria-Bertani media at 32°C for 16 h.
METHOD DETAILS
NLRP3-interacting proteins purification and mass spectrometry
NLRP3-interacting proteins were purified as previously described (He et al., 2016b). In brief, LPS-primed Nlrp3−/− iBMDM, which were reconstituted with empty lentiviral vector (pHIV-EGFP) or a vector expressing a triple-tagged NLRP3 (NLRP3-SFP; SFP for S-tag, Flag and a streptavidin-binding tag at the carboxyl terminus), were stimulated with 5 mM ATP for 30 min and lysed in ice-cold lysis buffer. The NLRP3-interacting proteins were isolated from the soluble fraction of the cell lysates by a two-step immuno-precipitation method with streptavidin agarose beads followed by S-protein agarose beads. The purified protein complexes were separated by SDS-PAGE and analyzed by liquid chromatography–mass spectrometry.
Plasmids and HEK293T cell transfection
HEK293T cells were plated into 6-well tissue culture plates (6.0 × 105 cells per well in 2 mL complete DMEM) overnight. Cells were single- or co-transfected for 16 h with plasmids expressing HA-tagged RACK1 (0.5 μg), SFP-tagged NLRP3 (0.5 μg), NEK7 (1.0 μg), RACK1 (0.5 μg), triple Flag-tagged full-length NLRP3 (0.5 μg), pyrin domain deletion mutant (Δpyrin; amino acids 94–1036, 0.35 μg), LRR deletion mutant (ΔLRR; amino acids 1–741, 0.6 mg), pyrin domain (amino acids 1–93, 0.25 μg), NOD domain (amino acids 220–536, 1.2 μg), LRR domain (amino acids 742–991, 0.75 μg) by Lipofectamine LTX (Invitrogen). The total amount of transfected DNA was normalized with the empty vector pCDNA3.
Immunoprecipitation
Cells were lysed in ice-cold lysis buffer (50 mM Tris, pH 7.4, 2 mM EDTA, 150 mM NaCl, 0.5% Nonidet P-40, 1 × EDTA-free Roche protease inhibitor cocktail). Cell lysates were clarified by centrifugation (15,000 g) at 4°C for 10 min. Pre-cleared cell lysates were directly incubated with streptavidin beads at 4°C overnight or with anti-HA (1:200) antibody or control IgG at 4°C overnight, the proteins bound by antibody were pulled down by protein G beads and subjected to immunoblotting analysis.
RACK1 knockdown in mouse macrophages
A pool of RACK1 siRNA (Darmacon, ON-TARGETplus RACK1 siRNA pool) was used to silence the expression of RACK1 in iBMDM. iBMDM (2 × 106 cells) were transfected with 30 nM siRNA using Amaxa cell line Nucleofector kit V according to the manufacturer’s instructions. A pool of control siRNA (Darmacon, ON-TARGETplus Non-targeting pool) was used as a negative control. Cells were then plated in 6-well plates (5 × 105 cells per well) and incubated for 48 h after transfection, followed by stimulation with LPS or LPS plus NLRP3 activators. For RACK1 knockdown and rescue experiments, a single RACK1 siRNA (GUAGAUGAAUUGAAGCAAG) was used to silence the expression of endogenous RACK1. After knockdown, RACK1 was then rescued by infection with lentivirus containing vector pHIV-EGFP for siRNA resistant HA-tagged wild-type RACK1 or mutant RACK1 (RACK1 R36D/K38E) defective in ribosomal binding for 24 h. For the shRNA mediated knockdown, lentiviral pLKO.1 plasmids targeting mouse Rack1 (shRACK1 #1, TRCN0000287170; shRACK1#2, TRCN0000294547) were purchased from Sigma. pLKO scramble (Addgene 1864) was used as a negative control. Lentiviruses were produced in HEK293T cells by using package vectors pCMV-dR8.2 dvpr and pCMV-VSV-G, and concentrated by Lenti-X concentrator (Clontech). Gene knockdown in primary macrophages was performed as previously reported and puromycin-selected macrophages were used for experiments (He et al., 2016b).
Inflammasome activation, cytokine measurement and cytotoxicity detection
Immortalized macrophages or primary macrophages were plated in 12-well plates (5 × 105 cells per well) with 1% FBS IMDM (GIBCO) medium. Primary macrophages were plated with differential medium. Culture medium was replaced with 0.5 mL serum-free IMDM per well after overnight culture. Cells were then primed with 200 ng Ml−1 ultrapure LPS for 4 h, followed by stimulation with PBS (mock), 5 mM ATP (30 min), 5 μM nigericin (1 h), 0.5 μM gramicidin (1 h), poly(dA:dT) (2 μg mL−1, 4 h), Salmonella (MOI = 10, 1 h), LLOMe (2 μM, 4 h), silica (500 μg mL−1, 4 h). After stimulation, culture supernatants and cell lysates were collected separately or combined together for immunoblotting analysis. For cytokine measurement and cytotoxicity detection, 96-well plates (5 × 104 cells per well) were used and culture supernatants were analyzed with ELISA kits (R&D system) or cytotoxicity detection kit (TakaRa). For the non-canonical inflammasome activation, macrophages were plated in 12-well plates with 1% FBS IMDM (GIBCO) medium. Culture medium was replaced with 0.5 mL serum-free IMDM per well after overnight culture. Cells were primed with Pam3CSK (300 ng mL−1) for 6 h and then transfected with DOTAP (7.5 μL per well) alone or DOTAP plus LPS-SM (0.5 μg per well) for 4 h. Culture supernatants and cell lysates were collected for immunoblotting.
ASC speck staining and ASC oligomer cross-linking
Macrophages were plated on an 8-well permanox chamber slide (Thermo Scientific, 177445) overnight. Cells were primed with 200 ng mL−1LPS for 4 h, then stimulated with ATP (5 μM, 30 min), nigericin (5 μM, 1 h), or infected with Salmonella (MOI = 10, 1 h). After stimulation, cells were fixed with 4% paraformaldehyde, followed by permeabilization with 0.1% Triton X-100 and blocked with PBS buffer containing 3% BSA. Cells were incubated with anti-ASC antibody at 4°C overnight and then incubated with Alexa Fluor-488-conjugated secondary antibody at room temperature for 1 h. Nuclei were stained by DAPI. Images were captured by an Olympus Fluo-View 500 confocal microscope system.
For ASC oligomer cross-linking, macrophages in 6-well plates were stimulated as previously indicated. Cells were lysed with PBS buffer containing 0.5% Triton X-100, and the cell lysates were centrifuged at 6,000 g for 15 min at 4°C. The supernatants and pellet fractions were termed as the Triton X-100-soluble and Triton X-100-insoluble fractions, respectively. The Triton X-100-insoluble fractions were washed with PBS twice and cross-linked for 30 min at room temperature with 2 mM bis[sulfosuccinimidy] suberate (BS3). The cross-linked pellets were spun down at 6,000 g for 15 min and dissolved in SDS sample buffer for immunoblotting analysis.
Blue native PAGE and 2D PAGE
Blue native gel electrophoresis was performed as previously described (Swamy et al., 2006). Macrophages were plated in 6-well plates (1 × 106 per well) overnight and stimulated as indicated on the second day. After stimulation, cells were washed once with cold PBS and then lysed in ice-cold native lysis buffer (20 mM Bis-tris, 500 mM ε-aminocaproic acid, 20 mM NaCl, 10% (w/v) glycerol, 0.5% digitonin, 0.5 mM Na3VO4, 1 mM PMSF, 0.5 mM NaF, 1 × EDTA-free Roche protease inhibitor cocktail, pH 7.0) for 15 min on ice. Cell lysates were clarified by centrifugation at 20,000 g for 30 min at 4°C; Proteins were separated in 4%–12% blue native PAGE and then analyzed by western blot. For the 2D PAGE, the natively resolved gel slice was loaded into the well of 4%–12% SDS–PAGE gel after soaked in 1 × Laemmli buffer as previously described (Swamy et al., 2006).
RACK1 knockdown in HEK293T cells and bioluminescence resonance energy transfer (BRET) assay
HEK293T cells stably expressing YFP-NLRP3-Luc (NLRP3 tagged with N-terminal YFP and C-terminal Luciferase) (Tapia-Abellán et al., 2019), were transfected using Lipofectamine2000 (Life Technologies) with indicated concentrations of RACK1 siRNA (Sigma, 5´-CGAUUUGUGGGCCAUACCA-3´) or 200 nM of control siRNA (Sigma, SIC001). After 2 days, total RNA was obtained from transfected cells using the RNeasy Mini kit (QIAGEN) according to manufacturer instructions and was quantified using a NanoDrop 2000 (Thermo Fisher Scientific). Reverse transcription was performed with an iScriptTM cDNA Synthesis kit (BioRad) according to the manufacturer’s recommendations. Quantitative PCR was performed with SYBR Premix ExTaq (Takara) and specific primers (Sigma, KiCqStart Primers). The relative RACK1 gene expression levels were shown after normalization to the endogenous GAPDH expression.
For BRET assay, cells were transfected with 200 nM of RACK1 or control siRNA for 2 days and then seeded on a poly-L-lysine-coated white 96-well plate the day before the assay. The signal from BRET was read after 5 min of coelenterazine-h (5 μM, Life Technologies) addition. Luminescence was detected at 37°C in a Synergy Mx plate reader (Biotek) using two filters for emission at 485 ± 20 nm and 530 ± 20 nm. The BRET ratio was calculated as the difference between the 528 nm and 485 nm emission ratio of R-Luc and YFP-NLRP3 fusion protein and the 530 nm and 485 nm emission ratio of the R-Luc protein alone. Results are expressed in milliBRET (mBRET) units normalized to a basal signal as we have previously described (Martín-Sánchez et al., 2016).
QUANTIFICATION AND STATISTICAL ANALYSIS
No statistical methods were used to predetermine sample size or to include or exclude samples. Data are expressed as mean ± SD. Statistical analysis was performed using unpaired two-tailed Student’s t test or Mann-Whitney test by GraphPad Prism. A p value less than 0.05 was considered statistically significant.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-HA | GenScript | Cat# A01244–100: RRID: AB_1289306 |
| Mouse monoclonal anti-Flag | GenScript | Cat# A00187–200; RRID: AB_1720813 |
| Mouse monoclonal anti-NLRP3(Cryo-2) | Adipogen | Cat# AG-20B-0014-C100; RRID: AB_2490202 |
| Rabbit monoclonal anti-NEK7 | Abcam | Cat# ab133514; RRID: AB_2877625 |
| Rabbit monoclonal anti-GSDMD | Abcam | Cat# ab209845; RRID: AB_2783550 |
| Rabbit monoclonal anti-RACK1 | Cell Signaling | Cat# 5432; RRID: AB_10705522 |
| Rabbit monoclonal anti-S6 Ribosomal Protein (RPS6) | Cell Signaling | Cat# 2217S; RRID: AB_331355 |
| Mouse monoclonal IgG2b isotype control | Cell signaling | Cat# 53484S; RRID: AB_2799435 |
| Goat polyclonal anti-IL-1β | R&D system | Cat# AF-401-NA; RRID: AB_416684 |
| Rat polyclonal anti-ASC | Dr. Gabriel Nunez | N/A |
| Rabbit polyclonal anti-Caspase-1 | Dr. Gabriel Nunez | N/A |
| THE™ beta Actin Antibody [HRP] | GenScript | Cat# A00730–200; RRID: AB_914100 |
| Bacterial and Virus Strains | ||
| Salmonella strain SL1344 | Dr. Denise Monack | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| High-capacity streptavidin agarose resin | ThermoFisher | Cat# 20359 |
| High-capacity streptavidin agarose resin | GenScript | Cat# L00353 |
| S-protein agarose beads | EMD Millipore | Cat# 69704 |
| Protein G beads | GenScript | Cat# L00209 |
| LPS (Escherichia coli 055:B5, Ultrapure) | InvivoGen | Cat# tlrl-pb5lps |
| LPS-SM (Salmonella Minnesota R595,Ultrapure) | InvivoGen | Cat# tlrl-smlps |
| poly(dA:dT)/lyovec | InvivoGen | Cat# tlrl-patc |
| ATP | Sigma | Cat# A2383 |
| Gramicidin | Sigma | Cat# G5002 |
| L-leucyl-L-leucine methyl ester (LLOMe) | Sigma | Cat# L7393 |
| Nigericin | EMD Millipore | Cat# 481990 |
| bis[sulfosuccinimidy] suberate (BS3) | ThermoFisher | Cat# 21580 |
| Silica | US Silica | Cat# Min-U-Sil 5 |
| C-1 | Tocris | Cat# 0543 |
| GF 109203X | Tocris | Cat# 0741 |
| Go 6983 | Tocris | Cat# 2285 |
| Lipofectamine LTX | ThermoFisher | Cat# 15338100 |
| Lipofectamine 2000 | ThermoFisher | Cat# 11668019 |
| DOTAP | Biontex | Cat# T010–2.0 |
| Lenti-X concentrator | Clontech | Cat# 631232 |
| SYBR Premix ExTaq | Takara | Cat# RR820B |
| jetPEI solution | Polyplus Transfection | Cat# 101–10N |
| Critical Commercial Assays | ||
| Mouse TNF-α ELISA Kit | R&D Systems | Cat# DY410–05 |
| Mouse IL-1β ELISA Kit | R&D System | Cat# DY401–05 |
| LDH Cytotoxicity Detection Kit | TakaRa Bio | Cat# MK401 |
| QuikChange II XL Site-Directed Mutagenesis Kit | Agilent | Cat# 20051 |
| Cell line Nucleofector kit V | Lonza | Cat# 90279050 |
| ON-TARGETplus RACK1 siRNA pool | Darmacon | Cat# L-062125-00-0005 |
| ON-TARGETplus Non-targeting pool | Darmacon | Cat# D-001810-10-20 |
| RNeasy Mini kit | QIAGEN | Cat# 74104 |
| iScriptTM cDNA Synthesis kit | BioRad | Cat# 1708890 |
| KiCqStart Primers | Sigma, | Cat# KSPQ12012 |
| Experimental Models: Cell Lines | ||
| Immortalized bone marrow-derived macrophages (iBMDM) | (He et al., 2016b) | N/A |
| Nlrp3−/− iBMDM | (He et al., 2016b) | N/A |
| NEK7 KO iBMDM | (He et al., 2016b) | N/A |
| NLRP3 R258W iBMDM | Dr. Gabriel Nunez | N/A |
| HEK293T | ATCC | Cat# CRL-3216 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | Jackson Laboratories | Cat# 000664 |
| Oligonucleotides | ||
| Mouse RACK1 siRNA for in vivo: 5’ GUAGAUGAAUUGAAGCAAG 3’ | Biosynthesis | N/A |
| Control siRNA for in vivo: 5’ CUUGCUUCAAUUCAUCUAC 3’ | Biosynthesis | N/A |
| Human RACK1 siRNA: 5’ CGAUUUGUGGGCCAUACCA 3’ | Sigma | N/A |
| Control siRNA | Sigma | Cat# SIC001 |
| Recombinant DNA | ||
| RACK1 cDNA plasmid (pEGFP-N1-RACK1) | Addgene | Cat# 41088 |
| pHIV-EGFP | Addgene | Cat# 21373 |
| pHIV-NLRP3-SFP | (He et al., 2016b) | N/A |
| pHIV-NEK7-SFP | This paper | N/A |
| pHIV-ASC-SFP | This paper | N/A |
| pHIV-RACK1-HA | This paper | N/A |
| pHIV-RACK1R36D/K38E-HA | This paper | N/A |
| pCDNA3-NLRP3-Flag | (He et al., 2014) | N/A |
| pCDNA3-NLRP3-Δpyrin -Flag | (He et al., 2014) | N/A |
| pCDNA3-NLRP3-ΔLRR -Flag | (He et al., 2014) | N/A |
| pCDNA3-NLRP3-LRR-Flag | (He et al., 2014) | N/A |
| pCDNA3-NLRP3-NOD-Flag | (He et al., 2014) | N/A |
| pCDNA3-NLRP3-pyrin-Flag | (He et al., 2014) | N/A |
| pLKO.1-shRACK1 #1 | Sigma | TRCN0000287170 |
| pLKO.1-shRACK1 #2 | Sigma | TRCN0000294547 |
| pCMV-dR8.2 dvpr | Addgene | Cat# 8455 |
| pCMV-VSV-G | Addgene | Cat# 8454 |
| pLKO scramble | Addgene | Cat# 1864 |
| pcDNA3.1- YFP-NLRP3-Luc | (Tapia-Abellán et al., 2019) | N/A |
| Software and Algorithms | ||
| ImageJ | https://imagej.nih.gov | N/A |
| GraphPad Prism 7.0 | GraphPad Software | N/A |
| Other | ||
| 8-well permanox chamber slide | Thermo Scientific | Cat# 177445 |
| PVDF membrane | EMD Millipore | Cat# IPVH00010 |
Highlights.
RACK1 interacts with NLRP3 and NEK7
RACK1 depletion specifically impairs NLRP3 inflammasome activation
RACK1 promotes NLRP3 active conformation and inflammasome assembly
ACKNOWLEDGMENTS
We thank Devon Jeltema, Nathan Kelley, Yanhai Xie, and Aliza Fatema for their technical assistance. This work was funded in part by National Institutes of Health grants (R01AI148544 to Y.H, R01AI06331 to G.N.), Wayne State startup funds (Y.H), a Ministerio de Economía, Industria y Competitividad grant (SAF2017-88276-Rto P.P.), a Fundación Séneca grant (20859/PI/18 to P.P), and a European Research Council grant (ERC-2013-CoG 614578 to P.P.). pEGFP-N1-RACK1 (Addgene plasmid 41088) was a gift from Anna Huttenlocher. pHIV-EGFP (Addgene plasmid 21373) was a gift from Bryan Welm and Zena Werb. pCMV-dR8.2 dvpr (Addgene plasmid 8455), and pCMVVSV-G (Addgene plasmid 8454) were gifts from Bob Weinberg. We also thank Nathan Kelley, Amina Wofford, and Raghavendar Thipparthi for their editing help.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108405.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Adams DR, Ron D, and Kiely PA (2011). RACK1, A multifaceted scaffolding protein: structure and function. Cell Commun. Signal 9, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky IE, Palm NW, Sadanand S, Ryndak MB, Sutterwala FS, Flavell RA, Bliska JB, and Medzhitov R (2010). A Yersinia effector protein promotes virulence by preventing inflammasome recognition of the type III secretion system. Cell Host Microbe 7, 376–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broz P, and Dixit VM (2016). Inflammasomes: mechanism of assembly, regulation and signalling. Nat. Rev. Immunol 16, 407–420. [DOI] [PubMed] [Google Scholar]
- Brydges SD, Mueller JL, McGeough MD, Pena CA, Misaghi A, Gandhi C, Putnam CD, Boyle DL, Firestein GS, Horner AA, et al. (2009). Inflammasome-mediated disease animal models reveal roles for innate but not adaptive immunity. Immunity 30, 875–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Z, Li JF, Niu Y, Zhang XC, Woody OZ, Xiong Y, Djonović S, Millet Y, Bush J, McConkey BJ, et al. (2015). Pathogen-secreted proteases activate a novel plant immune pathway. Nature 521, 213–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corsini E, Battaini F, Lucchi L, Marinovich M, Racchi M, Govoni S, and Galli CL (1999). A defective protein kinase C anchoring system underlying age-associated impairment in TNF-αlpha production in rat macrophages. J. Immunol 163, 3468–3473. [PubMed] [Google Scholar]
- Coyle SM, Gilbert WV, and Doudna JA (2009). Direct link between RACK1 function and localization at the ribosome in vivo. Mol. Cell. Biol 29, 1626–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng M, Guo H, Tam JW, Johnson BM, Brickey WJ, New JS, Lenox A, Shi H, Golenbock DT, Koller BH, et al. (2019). Platelet-activating factor (PAF) mediates NLRP3-NEK7 inflammasome induction independently of PAFR. J. Exp. Med 216, 2838–2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan JA, Bergstralh DT, Wang Y, Willingham SB, Ye Z, Zimmermann AG, and Ting JPY (2007). Cryopyrin/NALP3 binds ATP/dATP, is an ATPase, and requires ATP binding to mediate inflammatory signaling. Proc. Natl. Acad. Sci. USA 104, 8041–8046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes-Alnemri T, Wu J, Yu JW, Datta P, Miller B, Jankowski W, Rosenberg S, Zhang J, and Alnemri ES (2007). The pyroptosome: a supra-molecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 14, 1590–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry AM, O’Regan L, Sabir SR, and Bayliss R (2012). Cell cycle regulation by the NEK family of protein kinases. J. Cell Sci 125, 4423–4433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groß CJ, Mishra R, Schneider KS, Médard G, Wettmarshausen J, Dittlein DC, Shi H, Gorka O, Koenig PA, Fromm S, et al. (2016). K+ efflux-independent NLRP3 inflammasome activation by small molecules targeting mitochondria. Immunity 45, 761–773. [DOI] [PubMed] [Google Scholar]
- Guo H, Callaway JB, and Ting JP (2015). Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat. Med 21, 677–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Franchi L, and Núñez G (2013). TLR agonists stimulate Nlrp3-dependent IL-1β production independently of the purinergic P2X7 receptor in dendritic cells and in vivo. J. Immunol 190, 334–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Varadarajan S, Muñoz-Planillo R, Burberry A, Nakamura Y, and Núñez G (2014). 3,4-methylenedioxy-b-nitrostyrene inhibits NLRP3 inflammasome activation by blocking assembly of the inflammasome. J. Biol. Chem 289, 1142–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Hara H, and Núñez G (2016a). Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem. Sci 41, 1012–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Zeng MY, Yang D, Motro B, and Núñez G (2016b). NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 530, 354–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman HM, Mueller JL, Broide DH, Wanderer AA, and Kolodner RD (2001). Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat. Genet 29, 301–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley N, Jeltema D, Duan Y, and He Y (2019). The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int. J. Mol. Sci 20, 3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HD, Kong E, Kim Y, Chang JS, and Kim J (2017). RACK1 depletion in the ribosome induces selective translation for non-canonical autophagy. Cell Death Dis. 8, e2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroha K, Akamatsu M, Dimitrova L, Ito T, Kato Y, Shirahige K, and Inada T (2010). Receptor for activated C kinase 1 stimulates nascent polypep-tide-dependent translation arrest. EMBO Rep. 11, 956–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang T, Lee JPW, Elgass K, Pinar AA, Tate MD, Aitken EH, Fan H, Creed SJ, Deen NS, Traore DAK, et al. (2018). Macrophage migration inhibitory factor is required for NLRP3 inflammasome activation. Nat. Commun 9, 2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Thome S, Ma X, Amrute-Nayak M, Finigan A, Kitt L, Masters L, James JR, Shi Y, Meng G, and Mallat Z (2017). MARK4 regulates NLRP3 positioning and inflammasome activation through a microtubule-dependent mechanism. Nat. Commun 8, 15986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamidipudi V, and Cartwright CA (2009). A novel pro-apoptotic function of RACK1: suppression of Src activity in the intrinsic and Akt pathways. Onco-gene 28, 4421–4433. [DOI] [PubMed] [Google Scholar]
- Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, and Dixit VM (2006). Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440, 228–232. [DOI] [PubMed] [Google Scholar]
- Martín-Sánchez F, Compan V, and Pelegrín P (2016). Measuring NLR oligomerization, III: detection of NLRP3 complex by bioluminescence resonance energy transfer. Methods Mol. Biol 1417, 159–168. [DOI] [PubMed] [Google Scholar]
- Martinon F, Burns K, and Tschopp J (2002). The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 10, 417–426. [DOI] [PubMed] [Google Scholar]
- Mayor A, Martinon F, De Smedt T, Pétrilli V, and Tschopp J (2007). A crucial function of SGT1 and HSP90 in inflammasome activity links mammalian and plant innate immune responses. Nat. Immunol 8, 497–503. [DOI] [PubMed] [Google Scholar]
- McCahill A, Warwicker J, Bolger GB, Houslay MD, and Yarwood SJ (2002). The RACK1 scaffold protein: a dynamic cog in cell response mechanisms. Mol. Pharmacol 62, 1261–1273. [DOI] [PubMed] [Google Scholar]
- Meng G, Zhang F, Fuss I, Kitani A, and Strober W (2009). A mutation in the Nlrp3 gene causing inflammasome hyperactivation potentiates Th17 cell-dominant immune responses. Immunity 30, 860–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, and Núñez G (2013). K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38, 1142–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima A, Chen L, Thao NP, Fujiwara M, Wong HL, Kuwano M, Umemura K, Shirasu K, Kawasaki T, and Shimamoto K (2008). RACK1 functions in rice innate immunity by interacting with the Rac1 immune complex. Plant Cell 20, 2265–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabl J, Leibundgut M, Ataide SF, Haag A, and Ban N (2011). Crystal structure of the eukaryotic 40S ribosomal subunit in complex with initiation factor 1. Science 331, 730–736. [DOI] [PubMed] [Google Scholar]
- Ron D, Chen CH, Caldwell J, Jamieson L, Orr E, and Mochly-Rosen D (1994). Cloning of an intracellular receptor for protein kinase C: a homolog of the beta subunit of G proteins. Proc. Natl. Acad. Sci. USA 91, 839–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan Y, Sun L, Hao Y, Wang L, Xu J, Zhang W, Xie J, Guo L, Zhou L, Yun X, et al. (2012). Ribosomal RACK1 promotes chemoresistance and growth in human hepatocellular carcinoma. J. Clin. Invest 122, 2554–2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samir P, Kesavardhana S, Patmore DM, Gingras S, Malireddi RKS, Karki R, Guy CS, Briard B, Place DE, Bhattacharya A, et al. (2019). DDX3X acts as a live-or-die checkpoint in stressed cells by regulating NLRP3 inflammasome. Nature 573, 590–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid-Burgk JL, Chauhan D, Schmidt T, Ebert TS, Reinhardt J, Endl E, and Hornung V (2016). A genome-wide CRISPR (clustered regularly inter-spaced short palindromic repeats) screen identifies NEK7 as an essential component of NLRP3 inflammasome activation. J. Biol. Chem 291, 103–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder K, and Tschopp J (2010). The inflammasomes. Cell 140, 821–832. [DOI] [PubMed] [Google Scholar]
- Sengupta J, Nilsson J, Gursky R, Spahn CM, Nissen P, and Frank J (2004). Identification of the versatile scaffold protein RACK1 on the eukaryotic ribosome by cryo-EM. Nat. Struct. Mol. Biol 11, 957–962. [DOI] [PubMed] [Google Scholar]
- Sharif H, Wang L, Wang WL, Magupalli VG, Andreeva L, Qiao Q, Hauenstein AV, Wu Z, Núñez G, Mao Y, and Wu H (2019). Structural mechanism for NEK7-licensed activation of NLRP3 inflammasome. Nature 570, 338–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma D, and Kanneganti TD (2016). The cell biology of inflammasomes: mechanisms of inflammasome activation and regulation. J. Cell Biol 213, 617–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, and Shao F (2015). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665. [DOI] [PubMed] [Google Scholar]
- Shi H, Wang Y, Li X, Zhan X, Tang M, Fina M, Su L, Pratt D, Bu CH, Hildebrand S, et al. (2016). NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat. Immunol 17, 250–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stebbins EG, and Mochly-Rosen D (2001). Binding specificity for RACK1 resides in the V5 region of beta II protein kinase C. J. Biol. Chem 276, 29644–29650. [DOI] [PubMed] [Google Scholar]
- Subramanian N, Natarajan K, Clatworthy MR, Wang Z, and Germain RN (2013). The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell 153, 348–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutterwala FS, Ogura Y, Szczepanik M, Lara-Tejero M, Lichtenberger GS, Grant EP, Bertin J, Coyle AJ, Galán JE, Askenase PW, and Flavell RA (2006). Critical role for NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity 24, 317–327. [DOI] [PubMed] [Google Scholar]
- Swamy M, Siegers GM, Minguet S, Wollscheid B, and Schamel WW (2006). Blue native polyacrylamide gel electrophoresis (BN-PAGE) for the identification and analysis of multiprotein complexes. Sci. STKE 2006, pl4. [DOI] [PubMed] [Google Scholar]
- Swanson KV, Deng M, and Ting JP (2019). The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat. Rev. Immunol 19, 477–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapia-Abellán A, Angosto-Bazarra D, Martínez-Banaclocha H, de Torre-Minguela C, Cerón-Carrasco JP, Pérez-Sánchez H, Arostegui JI, and Pelegrin P (2019). MCC950 closes the active conformation of NLRP3 to an inactive state. Nat. Chem. Biol 15, 560–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorslund SE, Edgren T, Pettersson J, Nordfelth R, Sellin ME, Ivanova E, Francis MS, Isaksson EL, Wolf-Watz H, and Fällman M (2011). The RACK1 signaling scaffold protein selectively interacts with Yersinia pseudotuberculosis virulence function. PLoS ONE 6, e16784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volta V, Beugnet A, Gallo S, Magri L, Brina D, Pesce E, Calamita P, Sanvito F, and Biffo S (2013). RACK1 depletion in a mouse model causes lethality, pigmentation deficits and reduction in protein synthesis efficiency. Cell. Mol. Life Sci 70, 1439–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyleta ML, Wong J, and Magun BE (2012). Suppression of ribosomal function triggers innate immune signaling through activation of the NLRP3 inflammasome. PLoS ONE 7, e36044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Meszaros G, He WT, Xu Y, de Fatima Magliarelli H, Mailly L, Mihlan M, Liu Y, Puig Gámez M, Goginashvili A, et al. (2017). Protein kinase D at the Golgi controls NLRP3 inflammasome activation. J. Exp. Med 214, 2671–2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Tardivel A, Thorens B, Choi I, and Tschopp J (2010). Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol 11, 136–140. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code.