

Abstract
Grafted mesenchymal stem cells (MSCs) yield neuroprotection in preclinical stroke models by secreting extracellular vesicles (EVs). The neuroprotective cargo of EVs, however, has not yet been identified. To investigate such cargo and its underlying mechanism, primary neurons were exposed to oxygen‐glucose‐deprivation (OGD) and cocultured with adipose‐derived MSCs (ADMSCs) or ADMSC‐secreted EVs. Under such conditions, both ADMSCs and ADMSC‐secreted EVs significantly reduced neuronal death. Screening for signalling cascades being involved in the interaction between ADMSCs and neurons revealed a decreased autophagic flux as well as a declined p53‐BNIP3 activity in neurons receiving either treatment paradigm. However, the aforementioned effects were reversed when ADMSCs were pretreated with the inhibitor of exosomal secretion GW4869 or when Hrs was knocked down. In light of miR‐25‐3p being the most highly expressed miRNA in ADMSC‐EVs interacting with the p53 pathway, further in vitro work focused on this pathway. Indeed, a miR‐25‐3p oligonucleotide mimic reduced cell death, whereas the anti‐oligonucleotide increased autophagic flux and cell death by modulating p53‐BNIP3 signalling in primary neurons exposed to OGD. Likewise, native ADMSC‐EVs but not EVs obtained from ADMSCs pretreated with the anti‐miR‐25‐3p oligonucleotide (ADMSC‐EVsanti‐miR‐25‐3p) confirmed the aforementioned in vitro observations in C57BL/6 mice exposed to cerebral ischemia. The infarct size was reduced, and neurological recovery was increased in mice treated with native ADMSC‐EVs when compared to ADMSC‐EVsanti‐miR‐25‐3p. ADMSCs induce neuroprotection by improved autophagic flux through secreted EVs containing miR‐25‐3p. Hence, our work uncovers a novel key factor in naturally secreted ADMSC‐EVs for the regulation of autophagy and induction of neuroprotection in a preclinical stroke model.
Keywords: cerebral ischemia; extracellular vesicles, adipose‐derived MSCs; neurological recovery; autophagy
1. INTRODUCTION
Mesenchymal stem cells (MSCs) have been thoroughly analysed in preclinical stroke models, revealing both neuroprotection and increased neurological recovery after transplantation in vivo (Chen et al., 2001; Gervois et al., 2016; Lucia Maria Ferri et al., 2016). These grafted stem cells are neither integrated into residing neural networks nor do they take over functional characteristics of lost neurons (Fernández‐Susavila, Bugallo‐Casal, Castillo, & Campos, 2019). Instead, transplanted cells work through indirect ways (Baraniak & Mcdevitt, 2010; Liang, Ding, Zhang, Tse, & Lian, 2014). Recently, bilayer‐structured vesicles ranging in size from 30 nm to 1000 nm and containing a defined set of cargo such as non‐coding RNAs, DNAs, and proteins were defined as extracellular vesicles (EVs) (Kim et al., 2013; Mathivanan, Fahner, Reid, & Simpson, 2012; Théry et al., 2018). These EVs derived from MSCs or other stem cell sources appear to be non‐inferior with regard to their therapeutic potential against cerebral ischemia when compared to their host cells (Cunningham, Redondo‐Castro, & Allan, 2018; Doeppner et al., 2015; Xin et al., 2013). The neuroprotective cargo of MSCs‐derived EVs, however, has not been sufficiently identified.
Recent studies have shown that EVs regulate protein expression of recipient cells and modify cell characteristics through microRNA transfer (Phinney et al., 2015; Tkach & Théry, 2016). MicroRNAs in turn bind to untranslated regions and in some cases also to coding regions of mRNAs, thus post‐transcriptionally regulating gene expression (Lim et al., 2005; Vemuganti, 2010). Interestingly, autophagy – a self‐degradative process – is one target of miRNA regulation (Frankel & Lund, 2012). Recent work emphasized the role of autophagy to be a key regulator of ischemic stroke (Wang et al., 2018), unveiling a new range of potential therapeutic targets for neuroprotection.
Autophagy includes a process of forming an isolation membrane that spherically expands in order to engulf materials designated for degradation, which gives rise to a double‐membrane vesicle called the autophagosome. The latter will deliver these engulfed materials to the lysosome for later degradation (Yoshimori, 2007). Under conditions of cerebral ischemia, autophagy acts as a double‐edged sword for neuronal survival (Chen, Sun, Liu, & Sun, 2014). A modest induction of autophagy upon induction of hypoxia or cerebral ischemia has been associated with enhanced cell viability and decreased infarct size (Carloni et al., 2010; Dai et al., 2017; Wang et al., 2012). Nevertheless, prolonged oxygen and glucose deprivation or stroke event propels excessive autophagy, turning temporarily activated and protective autophagy to chronic activation, which in turn contributes to significant cell death (He et al., 2012; Mo, Fang, He, & Zhang, 2012; Shi et al., 2012; Xin et al., 2011). The role of autophagy in stem cell‐induced and even more so in EV‐induced neuroprotection against stroke as well as the underlying mechanisms still remain elusive. Hence, the present work analyzes whether or not the application of ADMSC‐EVs affects the autophagic response under conditions of both in vitro hypoxia and in vivo cerebral ischemia with regard to cell survival and underlying mechanisms.
2. MATERIALS AND METHODS
2.1. Experimental in vivo paradigm and induction of cerebral ischemia
All studies were performed with governmental approval according to the NIH guidelines for the care and use of laboratory animals, following both the ARRIVE and the STAIR guidelines. Male C57BL/6 mice aged 10–12 weeks (Janvier labs, Le Genest‐Saint‐Isle, France) were kept under circadian rhythm and had free access to food and water. At all stages of the study, the researchers were blinded from the experimental conditions. The mice were randomly assigned to the various treatment groups. The result of the power calculation was 0.8732706 for Western blot analysis, assuming an effect size of 0.75. The result of the power calculation was 0.9105700 for both behavioural test analysis and immunofluorescence analysis, assuming an effect size of 0.70. Precise numbers of animals used are given for each condition in the figure legends and in the Supplementary Table S1 including survival rates of mice.
The induction of cerebral ischemia was performed via middle cerebral artery occlusion (MCAO), as described previously (Doeppner et al., 2015). In brief, silicon‐coated monofilament (Doccol Corp., Sharon, MA, USA) was inserted into the right CCA and then gently moved forward towards the offspring of the right middle cerebral artery (MCA). During the experiment, the laser Doppler flow (LDF) was recorded with a flexible probe (Perimed AB, Järfälla, Sweden) covering the core of the right MCA territory. One hour after monofilament insertion, reperfusion was initiated by monofilament removal, and the LDF recordings were continued for an additional 15 min before the wounds were carefully sutured.
The first set of mice underwent MCAO followed by injection of PBS (control) or ADMSC‐EVs. EVs were released by 2 × 106 ADMSCs diluted in 200 μl of PBS corresponding to 10 μg of EVs. Injection was done by cannulating the right femoral vein, and the injection rate was 200 μl per 10 min. Mice received such a treatment either at the beginning of the reperfusion or 12 h after reperfusion, followed by a survival of 24 h (eight mice per group). The brains were removed and used for infarct volume analysis as described later.
The second set of mice underwent sham surgery or MCAO followed by PBS (control) or ADMSC‐EV treatment immediately at the beginning of the reperfusion or 12 h after reperfusion. Animals in the sham group underwent all surgical procedures for MCAO induction except for occlusion of the MCA. All these groups were allowed to survive for 24 h post‐stroke (six mice per group). These brains were used for Western blot analysis.
The third set of mice (six mice per group) was subjected to 1 h of ischemia (except for the sham group) followed by 6, 24, 48 or 72 h of reperfusion. All these groups were also used for Western blot analysis.
The fourth set of mice was exposed to MCAO followed by intraperitoneal injections of PBS (control) or 3‐Methyladenine (3‐MA, 15 mg/kg; Merck Group, Darmstadt, Germany) immediately at the beginning of the reperfusion or 12 h after reperfusion. The mice were allowed to survive for one day (six mice per group) or fourteen days (ten mice per group). These mice were used for Western blot analysis, behavioural analyses and histochemical studies of brain injury.
The fifth set of mice was exposed to MCAO followed by administration of PBS (control), ADMSC‐EVs, ADMSC‐EVNC and ADMSC‐EVanti‐miR‐25 (anti‐miR‐25‐3p oligonucleotides or negative control oligonucleotides were transfected into ADMSCs before harvesting EVs) via cannulation of the right femoral vein in the anesthetized mice 12 h after reperfusion. The mice were allowed to survive for one day (six mice per group) or for fourteen days (ten mice per group). These mice were used for Western blot analysis, behavioural analyses and histochemical studies of brain injury.
2.2. Adipose‐derived mesenchymal stem cells (ADMSCs) isolation and characterization
ADMSCs were isolated, as described previously (Ahmadian Kia et al., 2011). In brief, adipose tissue was harvested from wildtype C57BL/6 mice and washed with PBS. Thereafter, the tissue was mechanically chopped before digestion with 0.1 % collagenase I (Gibco, Darmstadt, Germany) for 1 h at 37°C with intermittent shaking. The digested tissue was washed with high glucose Dulbecco's modified Eagle's medium (DMEM; Merck Group, Darmstadt, Germany) containing 10 % fetal bovine serum (FBS; Merck Group, Darmstadt, Germany) and then centrifuged at 1,000 rpm for 5 min to remove mature adipocytes. The cell pellet was resuspended in DMEM supplemented with 10 % FBS and 1% penicillin/streptomycin (PS; Gibco, Darmstadt, Germany) in a 37°C incubator with 5 % CO2. The adherent cells were passaged with 0.25 % trypsin containing 0.02 % EDTA (Sigma‐Aldrich, St. Louis, MO, USA) for 3–4 passages.
Cytometric evaluation of ADMSC surface profiles was carried out at passage 2. After two days of culture, the cells were washed with PBS and fixed in 4 % paraformaldehyde for 30 min. ADMSCs were identified with fluorescein isothiocyanate (FITC)‐conjugated antibodies against CD29, CD34, CD45, CD90 (BD Biosciences, San Jose, CA, USA) and with phycoerythrin (PE)‐conjugated antibodies against CD105 (Ebioscience, San Diego, CA, USA). The percentage of adhering cells that stained for CD29, D34, CD45, CD90, and CD105, respectively, was quantified by FlowJO software (version 8.0). The results are shown in Figure S1.
2.3. Preparation of primary cortical neurons
For the preparation of cortical neurons, pregnant C57BL/6 mice were killed by CO2 inhalation at embryonic day 17. Embryos were dissected, and tissue pieces were trypsinized and dissociated using a fire‐polished Pasteur pipette. Cells were seeded on poly‐L‐ornithine/laminin (Sigma‐Aldrich, St. Louis, MO, USA)‐coated 6 or 24 well plates at a density of 100,000/cm2 containing neuroblast medium (Gibco, Darmstadt, Germany) with additional transferrin (Sigma‐Aldrich, St. Louis, MO, USA), penicillin/streptomycin (PS; Gibco, Darmstadt, Germany), L‐glutamine (Seromed, Dollnstein, Germany), and B27 supplement (Gibco, Darmstadt, Germany). Cells were used for subsequent experiments after 4 days of cell culture.
2.4. Standard and oxygen‐glucose‐deprivation (OGD) model
For the standard (normoxia) condition, cells were maintained under an ambient atmosphere in an incubator at 37°C with 5 % CO2. For the induction of OGD, primary neurons were incubated at 37°C in a hypoxic chamber (Toepffer Laborsysteme GmbH, Göppingen, Germany) (0.5 % O2, 5 % CO2) in Sterofundin medium (Braun, Melsungen, Germany) containing 1 mM mannitol (Serag‐Wiessner, Naila, Germany) for different durations of time. Afterwards, the cells were re‐incubated under standard cell culture conditions for 24 h (reoxygenation).
2.5. ADMSC–primary neuron coculture and EV treatment
ADMSCs were plated onto 6‐well (2 × 104 cells/insert) transwell (3 μm pore size; Costar, Maryland, USA). After 24 h, the primary neurons were added to the plates, and the cells were cultured under the indicated (normoxia or hypoxia) conditions. To illustrate whether the effects of ADMSC‐EVs were dose‐dependent, three different ADMSC‐EV concentrations were tested: ADMSC‐EVs low (EVs equivalent to 2 × 103 ADMSCs, 0.01 μg), AMDSC‐EVs medium (EVs equivalent to 2 × 104 ADMSCs, 0.1 μg), ADMSC‐EVs high (EVs equivalent to 2 × 105 ADMSCs, 1 μg). The EVs were added to primary neurons at the beginning of the OGD and at the beginning of the reoxygenation.
2.6. ADMSC‐EV assays – enrichment, characterization, purification, uptake and inhibition
Details can be found in Supplementary Materials and Methods S1.
2.7. Autophagic flux
For autophagic flux assessments in primary neurons, cells were cultured on coverslips for 3 days and transfected with RFP‐GFP‐LC3B using the Autophagy Tandem Sensor RFP‐GFP‐LC3B Kit (Thermo Fisher Scientific, Waltham, Massachusetts, USA). After 24 h, the cells were cultured under the indicated experimental conditions (i.e., normoxia or OGD condition, with or without ADMSC‐EVs) and fixed with 4 % paraformaldehyde (PFA). Images were obtained with a fluorescence microscope, and the number of autophagosomes and autolysosomes were quantified in each cell (20‐25 cells per experimental group) in 3 independent experiments. For assessments of autophagic flux by LC‐3 II, Bafilomycin A1 (Sigma‐Aldrich, St. Louis, MO, USA) was used 3 h before harvesting. Western blots were performed, thereafter.
2.8. Knockdown and overexpression
MiR‐25b‐3p mimicking oligonucleotides, anti‐miR‐25‐3p oligonucleotides, siRNAs against Hrs and their control oligonucleotides (RiboBio, Guangzhou, China) were transfected into primary neurons or ADMSCs by using Turbofect Transfection reagents (Thermo Fisher Scientific, Waltham, Massachusetts, USA) according to the manufacturer's instructions. After 24 h of transfection, the cells were used for subsequent experiments.
2.9. Real‐time qRT‐PCR
Total RNA was isolated using Trizol reagent (Invitrogen, Waltham, California, USA) according to the manufacturer's instructions. The KAPA SYBR® FAST One‐Step Kit for LightCycler®480 (Merck Group, Darmstadt, Germany) was used to perform qRT‐PCR as the manufacturer's instructions request, and the PCR primers were purchased from Eurofins Genomics (Ebersberg, Germany). MicroRNA expression was normalized to U6 and let‐7a expression and calculated via the standard 2−ΔΔCt method.
2.10. RNase A and TritonX‐100‐treatment
To degrade RNA, 500 μl of ADMSC‐EVs (derived from 10 × 106 ADMSCs) were separated into four groups and each group was incubated in the presence of 20 μg/ml RNase A (Invitrogen, Waltham, California, USA) and/or with 1% TritonX‐100 (Sigma‐Aldrich, St. Louis, MO, USA) for 30 min at 37°C under gentle agitation. RNase digestion was stopped by addition 1.25 μl/ml RNasin (Sigma‐Aldrich, St. Louis, MO, USA) and RNA‐isolation was performed as previous described. All treated EVs were used immediately or were stored at ‐80 °C.
2.11. Cell survival
Cell viability was measured by a colorimetric assay by using the MTT (thiazolyl blue tetrazolium, Sigma‐Aldrich, St. Louis, MO, USA) viability assay according to the protocol (Venkataramani et al., 2018). Cell viability data are presented as relative changes in percent compared to untreated controls. Furthermore, cell death rate was also determined via fluorescence microscopy by using a LIVE/DEAD Viability kit (Lonza, Basel, Switzerland) as directed by the manufacturer's instructions; living cells were identified with calcein AM (4 μM, green fluorescence), and dead cells were identified with ethidium homodimer‐1 (2 μM, red fluorescence). Three independent experiments were conducted and ≥200 cells were evaluated for each condition.
2.12. Assessment of infarct volumes
One day after MCAO, the infarct volume was assessed by 2,3,5‐triphenyl tetrazolium chloride (TTC) staining in 2‐mm thick coronal brain slices. The infarct proportion was calculated by the formula: corrected percentage of infarct volume = (contralateral hemispheric volume – ipsilateral non‐infarcted volume) / contralateral hemispheric volume x 100.
2.13. Analysis of post‐stroke motor coordination deficits
Mice were trained on days one and two before the induction of stroke to ensure proper test behaviour. The actual tests for analysis of motor coordination were performed at four, seven, ten and fourteen days using the tight rope test and the corner turn test, as previously described (Doeppner, Kaltwasser, Bähr, & Hermann, 2014). The details can be found in Supplementary Table S2.
2.14. Protein extracts and Western Blot analyses
Details can be found in Supplementary Materials and Methods S2.
2.15. Immunofluorescence staining
Brain injury as indicated by neuronal density was evaluated in 16 μm cryostat sections stained with a rabbit monoclonal anti‐NeuN antibody (1:300; Merck Group, Darmstadt, Germany), which was detected by a donkey anti‐rabbit Alexa Fluor 488 antibody (Invitrogen, Waltham, California, USA). Quantitative analyses for immunohistochemical stainings were performed defining five regions of interests (ROIs) at anterior‐posterior +0.14 mm, medial‐lateral ±1.15 to +2.25 mm, and dorsal‐ventral ‐2.25 to +3.25 mm. The number of NeuN+ cells was investigated by manual cell counting. The analysis was performed with a fluorescence microscope (Zeiss, Jena, Germany). Five sections per mouse were analyzed, and the mean neuronal densities were determined for all ROIs.
2.16. Statistical analysis
All statistical analyses were performed using GraphPad Prism Software (GraphPad Software Inc., San Diego, CA, USA). For comparison of two groups, the two‐tailed independent Student's t‐test was used. For comparison of three or more groups, a one‐way analysis of variance (ANOVA) followed by the Tukey's post‐hoc‐test was performed. The power calculations were performed using G*Power software. Unless otherwise stated, data are presented as mean values with SD. A P value of <0.05 was considered statistically significant.
3. RESULTS
3.1. Purification, Isolation, and Characterization of ADMSC‐EVs
We first enriched ADMSC‐EVs from conditioned medium of ADMSCs using two well established methods (UC and PEG). Complete cell culture medium has been processed in the same PEG‐based manner as conditioned cell culture medium (CCM). The subsequent characterization of such enriched ADMSC‐EVs included transmission electron microscopy (TEM), nanosight tracking analysis (NTA), and Western blotting. In line with previous reports (Zheng et al., 2020), Western blotting analysis on the expression patterns of so called EV surface markers revealed CD9, CD63, Alix, and TSG101 but not albumin, Histone, and TOMM20 abundance in these ADMSC‐EVs (Figure 1a). The data showed no difference between the UC and the PEG method. The results from the NTA revealed the majority of ADMSC‐EVs to be about 100 nm in size in both groups (Figure 1b). TEM analysis showed no significant morphological differences between UC‐EVs and PEG‐EVs, as both preparations contained smaller and larger vesicles (Figure 1c).
FIGURE 1.
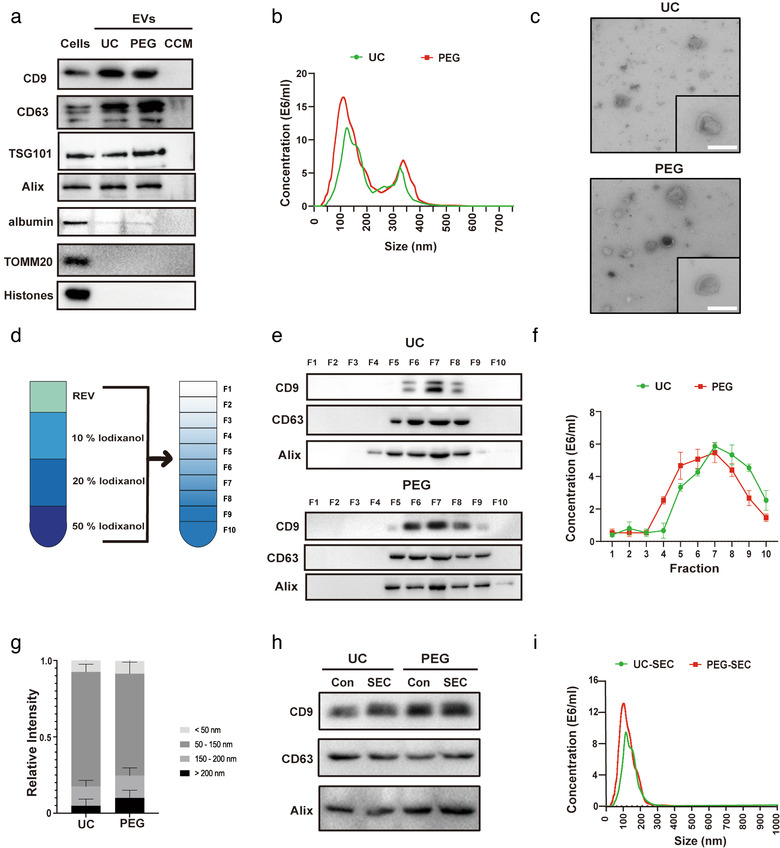
Characterization and purification of ADMSC‐EVs. Adipose‐derived mesenchymal stem cells (ADMSCs) were cultured under standard cell culture conditions, and conditioned cell medium (CCM) was obtained after passage three. CCM was used for the enrichment of extracellular vesicles (EVs) using either the differential centrifugation (i.e. ultracentrifugation, UC) or the polyethylene glycol (PEG) method. (a) Western blot analysis of EVs against exosomal markers of CD9, CD63, TSG101 and Alix, with albumin, TOMM20 and Histones being used as negative markers. Western blots were performed on total cell lysates (cells), EV lysates (UC and PEG) and the CCM. (b) Nanoparticle tracking analysis (NTA) from enriched EVs (UC and PEG) depicting size distribution patterns. (c) Representative transmission electron microscopy (TEM) analysis from EVs enriched by either UC or the PEG method. Scale bar, 100 nm. (d) Resuspended EVs isolated by UC or PEG were allowed to float into an overlayed iodixanol gradient to purify and isolate the small extracellular vesicle (sEV) population. (e) The iodixanol cushion gradient fractions for UC and PEG were analysed by Western blotting (fraction 1–10) using exosome markers. Equivalent volumes of each fraction were loaded per lane. Representative images were shown for CD9, CD63 and Alix which were enriched in fraction seven. (f) NTA was used to assess EV concentrations for each fraction (fraction 1–10). The two‐tailed independent Student's t‐test was used. (g) Representative size distribution of EVs isolated by UC or PEG from their corresponding fraction seven gradients. (h) Western blotting against EV markers (CD9, CD63, and Alix) were performed on ADMSC‐EVs isolated by UC or PEG after (or without, Con) size exclusion chromatography (SEC) purification. (i) The size analysis of SEC purified EVs (UC and PEG) was again done by means of NTA
Because PEG protocols may be incapable of discriminating EVs from non‐EV nanoparticles and protein (Patel et al., 2019), our next goal was to set up another separation assay to determine which are the biologically relevant nanoparticles of the aforementioned samples, i.e., UC and PEG samples. Density gradients like iodixanol are a classic means to separate membrane‐enclosed vesicles according to their floatation speed and equilibrium density (Colombo, Raposo, & Théry, 2014). We first used a self‐forming iodixanol (OptiPrep) gradient to subfractionate EVs isolated by UC or PEG (Figure 1d). After subfractionation, ten fractions were recovered and analysed for the presence of protein markers like CD9, CD63, and Alix in both UC and PEG fractions. As shown in Figure 1e, the samples recovered floated mostly in fraction seven in both UC and PEG groups. Consistent with the Western blot results, NTA revealed a vast majority of vesicles to be in fraction seven, regardless of the enrichment method chosen (Figure 1f). These vesicles found in fraction seven typically had a diameter of 30–150 nm (Figure 1g). Thus, floatation into iodixanol gradients allows separation of subtypes of EVs displaying different buoyant densities and sizes, with small EVs (sEVs, 50 to 200 nm diameter) strongly enriched in the seventh fraction regardless of the isolation method.
In subsequent experiments, we collected layer seven as the purified sEVs (iodixanol) for further experiments. Size exclusion chromatography (SEC) is considered to be one of the best methods for isolating and purifying EVs from different matrices (Stranska et al., 2018). Then, EVs isolated by UC or PEG were applied to the SEC column and collected after filtration. UC‐SEC and PEG‐SEC were characterized based on the size distribution and the presence of the EV‐enriched proteins. Western blot analysis revealed that both UC‐SEC and PEG‐SEC were enriched for EV markers (e.g., CD9, CD63, and Alix), showing no difference between filtered and untreated ones (Figure 1h). Figure 1i shows a representative size distribution profile of UC‐SEC and PEG‐SEC based on NTA, which reflects a similar size distribution pattern of UC‐SEC and PEG‐SEC with the highest peak at approximately 100 nm.
3.2. ADMSC‐EVs protect neurons from oxygen‐glucose‐deprivation (OGD) injury
EVs enriched from bone marrow‐derived MSCs induce neuroprotection and enhance neurological recovery in preclinical stroke models, as previously described by us and others (Bang & Kim, 2019; Doeppner et al., 2015; Xin et al., 2013). The present study, however, analysed the underlying mechanisms of ADMSC‐EV‐induced neuroprotection using both in vitro and in vivo stroke models. Primary neurons exposed to OGD followed by 24 h of reoxygenation displayed a significant extent of cell injury, depending on the duration of the OGD (Figure 2a). Although recent evidence suggests a role for autophagy to be involved in cell injury due to hypoxia or ischemia, published data appears to be contradictory with regard to autophagy being beneficial or detrimental (Chen et al., 2014; Wei, Wang, & Miao, 2012). We, therefore, analysed autophagy levels using the expression of the autophagy‐associated protein LC3‐II in our OGD model. LC3‐II levels significantly correlated with the extent of cell injury, i.e., LC3‐II was significantly more abundant in neurons exposed to 10 h of OGD than in neurons exposed to 4 h or 8 h of OGD (Figure 2b‐c). Further, in vitro experiments were performed using the 10 h OGD experimental paradigm only. Treatment of primary neurons exposed to OGD with ADMSCs or with EVs isolated from UC or PEG yielded a significant reduction of cell injury when compared to controls (Figure 2d). No differences between the various methods used were observed. Interestingly, ADMSC‐EV treatment was not inferior to ADMSC treatment with regard to survival rates of neurons exposed to OGD (Figure 2d).
FIGURE 2.
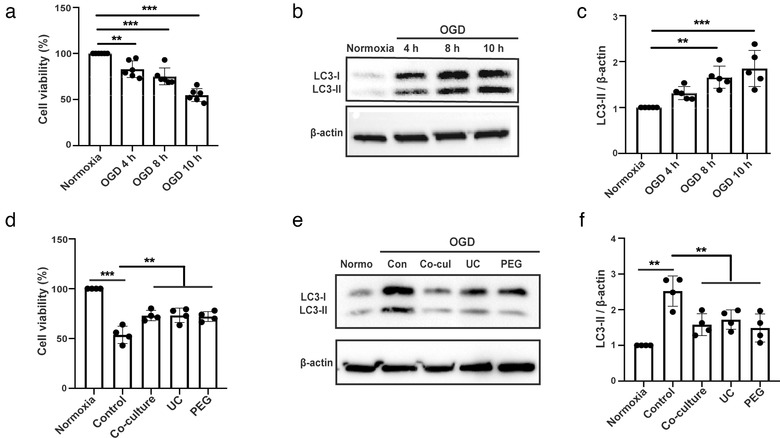
ADMSC‐EVs protect neurons from oxygen‐glucose deprivation (OGD) injury through autophagy regulation. Neuroprotective effects of ADMSC‐EVs in cultivated mouse primary cortical neurons were detected by cell viability assays. Primary neurons were incubated for 4 days as mentioned in the materials and methods part. (a) Cell viability was analysed in primary neurons exposed to 4, 8 and 10 h of OGD followed by 24 h of reoxygenation using the MTT assay (n = 3). Cells incubated under standard cell culture conditions (‘Normoxia’) were defined as 100 % cell survival. (b‐c) Both qualitative and quantitative analysis of autophagy levels as indicated by the abundance of the autophagy associated protein LC3‐II under the aforementioned time points (n = 3). Cells incubated under standard cell culture conditions (‘’Normoxia ’) were used as negative control. (d) The neuroprotective effect of ADMSC‐EVs in cultured primary neurons was evaluated by the MTT assay. After 4 days of cell culture, cells were exposed to 10 h of OGD followed by 24 h of reoxygenation, as mentioned by “OGD” for the later assays. Cell viability was analysed in neurons incubated with either PBS (Control), ADMSCs (Co‐culture) or ADMSC‐EVs (EVs) isolated by UC or PEG after induction of OGD followed by reoxygenation. PBS, ADMSCs and EVs were given at the beginning of both hypoxia and reoxygenation. Cells incubated under standard cell culture conditions (Normoxia) were defined as 100 % cell survival (n = 3). (e‐f) LC3 levels were evaluated by Western blotting in OGD exposed primary neurons treated with ADMSCs (Co‐culture) or ADMSC‐EVs isolated by UC or PEG. ADMSCs and EVs were given at the beginning of both hypoxia and reoxygenation. Neurons treated with PBS under OGD conditions served as positive control (Control). Cells incubated under standard cell culture conditions (Normoxia) were used as negative control (n = 3). A representative Western blot is shown in (e), whereas the quantitative analysis for LC3‐II is shown in (f). One‐way ANOVA followed by the Tukey's post‐hoc‐test was used. Data are shown as mean ± SD. Data are statistically different from each other with *P < 0.05, **P < 0.01, and ***P < 0.001
3.3. ADMSC‐derived EVs inhibit autophagic flux and protect primary neurons from OGD‐injury through regulation of autophagy
In order to study whether or not ADMSC‐derived EVs enhance the resistance of primary neurons against OGD by regulating autophagic activity, autophagy levels were assessed using the expression of autophagy‐associated protein LC3‐II. When primary neurons were exposed to OGD and co‐cultured with ADMSCs or treated with ADMSC‐derived EVs enriched from UC or PEG, the stroke‐induced increased protein abundance of LC3‐II was reversed (Figure 2e‐f). We also compared the effects of the concentrates from iodixanol separated layers one, three, and seven on alleviating cell damage after iodixanol density gradient ultracentrifugation (DGUC). Not surprisingly, the seventh layer of concentrates, but not the first or the third layer which occupied a typical sEV diameter, has a significant effect on enhancing cell viability. This effect is the same as the unstratified one (Figure S2A). Consistent with the results from DGUC, SEC filtered EVs have the same effects as the unfiltered EVs with regard to reducing cell damage after OGD, regardless of the enrichment method chosen (Suppl. Fig. S2B). More importantly, all of the manipulated EVs (iodixanol and SEC) can down‐regulate neuronal autophagy levels after OGD, and the effects were not inferior to the unpurified one (Figure S2C‐D). Combined with the aforementioned results, we showed that sEVs are the key compounds mediating reduced levels of autophagy and neuroprotection. Importantly, we confirmed that sEVs are the key compounds in both EV‐isolation methods (UC and PEG), and that these two methods display no significant difference between each other. We therefore used the PEG method to enrich ADMSC‐EVs for the remainder of the study.
To confirm the physiological properties of ADMSC‐EVs in the aforementioned OGD model, the ADMSC‐EV uptake in primary neurons was studied using DiI staining. Indeed, primary neurons displayed an intracellular uptake of EVs after OGD (Figure S2E). We then asked the question whether or not EV‐induced effects are dose‐dependent. Hence, three different doses (2 × 103, 2 × 104, and 2 × 105 cell equivalents) of ADMSC‐EVs were used in the OGD model. ADMSC‐EVs significantly repressed the LC3‐II expression (Figure S2F‐G) and rescued cell injury (Figure S2H) in a dose‐dependent manner. Although we do not have clear evidence, it appears to be feasible that high dosages (2 × 105) might lead to excessively down‐regulated autophagy levels, resulting in the loss of further protection from EVs to neurons after OGD. On the contrary, low dosages (2 × 103) of EVs result in an insufficient concentration of either cargo proteins or non‐coding RNAs, thus failing to induce neuroprotection. However, the medium dosages (2 × 104) of ADMSC‐EVs showed beneficial effects regarding cell survival. Further experiments were therefore performed choosing the medium dosage.
In addition, we also measured the cell survival after co‐culturing neurons with ADMSC‐EVs that were pretreated with either RNase A, the detergent Triton X‐100 (to disrupt the lipid bilayer of vesicles), both of them, or with the untreated EVs after OGD as shown in Figure S2I. When EVs were pretreated with RNase A, but not with Triton X‐100, and co‐cultured with primary neurons, there was no significant difference between RNase A treated and the untreated group in terms of cell viability after OGD. Pretreating EVs with Triton X‐100 alone resulted in some decrease in neuroprotection. Conversely, pretreating EVs with Triton X‐100 and RNase A led to near‐total elimination of the protective properties of EVs. These results support the hypothesis that EVs are likely to be the therapeutically efficacious substance in our OGD model.
Although LC3‐II, which is specifically recruited to the autophagosome membrane (Mizushima & Yoshimori, 2007), is a good marker for analyzing autophagy, the static nature of such measurements is hard to interpret. As a matter of fact, an increase in LC3‐II levels can either be a consequence of enhanced autophagosome formation or due to a lack of autophagosome fusion with lysosomes (du Toit, Hofmeyr, Gniadek, & Loos, 2018). To distinguish between these two states of autophagy processing, we evaluated the effect of autophagy by treating primary neurons with bafilomycin A1 (BafA1), which is a potent V‐ATPase inhibitor that impedes autophagosome‐lysosome fusion. If the breakdown of autophagosomes is blocked, the absolute LC3‐II accumulation (BafA1 treated minus the untreated one) indicates how many new autophagosomes are being produced over a particular time. The magnitude of BafA1‐induced LC3‐II accumulation under OGD conditions was significantly lower in primary neurons cultured with either ADMSCs or EVs when compared to controls (Figure 3a‐b). Furthermore, we employed a tandem fluorescence RFP‐GFP‐LC3B reporter system to monitor the autophagic flux in primary neurons. Incubation of primary neurons exposed to OGD with ADMSC‐EVs yielded a reduced number of both autophagosomes and autolysosomes (Figure 3c‐d).
FIGURE 3.
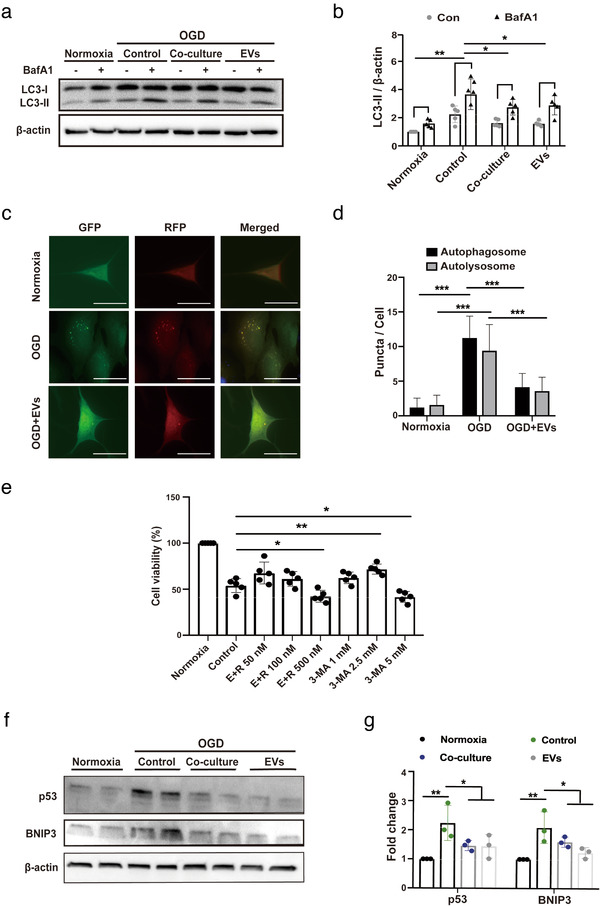
ADMSC‐EVs inhibit autophagic flux and increase cell viability through p53 and BNIP3 signalling. (A‐B) Assessment of the autophagic flux was done using bafilomycin A1 (BafA1) in the aforementioned groups (Normoxia, Control, Co‐culture and EVs). BafA1 was added 3 h before harvesting the cells. LC3 levels were evaluated again by Western blotting in the presence of DMSO or BafA1 (n = 3). Quantitative analysis of LC3‐II blotting is shown in (b). (c‐d) Autophagosomes (yellow) and autolysosomes (red) were detected in OGD‐exposed primary neurons that express mRFP‐GFP‐LC3. The neurons were treated with PBS or ADMSC‐EVs. PBS and EVs were given at the beginning of hypoxia and reoxygenation. Cells incubated under standard cell culture conditions (Normoxia) were used as negative control. Scale bar, 10 μm. The number of autophagosomes and autolysosomes in each cell (20‐30 cells per group) was quantified in (d) (n = 3). (e) The impact of autophagy on neuronal survival after OGD was evaluated using different concentrations of the autophagy inhibitor 3‐MA in comparison to neurons treated with the solvent DMSO using the MTT assay. ADMSC‐EVs together with different concentrations of the autophagy stimulator rapamycin (E+R; n = 3) were also used on primary neurons exposed to OGD. All the drugs were given twice, at the beginning of hypoxia and reoxygenation. Cells incubated under standard cell culture conditions (Normoxia) were defined as 100 % cell survival. (f‐g) Both p53 and BNIP3 were evaluated by Western blotting in OGD exposed primary neurons treated with PBS, ADMSCs (Co‐culture) or ADMSC‐EVs (EVs). Neurons treated with PBS under OGD conditions served as positive control (Control). Cells incubated under standard cell culture conditions (Normoxia) were used as negative control (n = 3). EVs were given at the beginning of hypoxia and reoxygenation. The quantitative analysis of p53 and BNIP3 Western blotting is shown in (g). One‐way ANOVA followed by the Tukey's post‐hoc‐test, data are given as mean ± SD. Data are statistically different from each other with *P < 0.05, **P < 0.01, and ***P < 0.001
To further reveal the relationship between ADMSC‐EVs and autophagy, the autophagy activator rapamycin and the inhibitor 3‐MA were used in primary neurons exposed to OGD (Figure 3e). The combined treatment of neurons with different concentrations of rapamycin and ADMSC‐EVs resulted in a reversal of the former EV‐induced neuroprotection, indicating that ADMSC‐EVs apparently act in an opposite way than rapamycin. On the contrary, inhibition of autophagy using 3‐MA significantly increased neuron viability in a concentration dependent manner (Figure 3e). The protective impact of 3‐MA, however, was lost when 3‐MA was applied in a concentration of 5 mM, suggesting that over‐suppression of autophagy may be detrimental for primary neurons. Rather, a moderate decrease in autophagy confers significant protection against OGD‐induced cell death in primary neurons.
3.4. ADMSC‐derived EVs regulate autophagy by signalling pathways involving p53 and B‐cell lymphoma 2–interacting protein 3 (BNIP3)
To understand the mechanisms by which ADMSC‐derived EVs regulate autophagy, we investigated autophagy‐related signalling cascades. Autophagy is known to be regulated by at least four signalling pathways, among which are p53 and BNIP3. Interestingly, protein levels of p53 and BNIP3 were significantly increased in OGD‐exposed primary neurons when compared to standard cell culture conditions (Figure 3f‐g). Incubation of neurons with either ADMSCs or with ADMSC‐EVs significantly decreased the OGD‐induced upregulation of both p53 and BNIP3 (Figure 3f‐g), indicating that ADMSC‐EVs possibly modulate autophagy through p53 and BNIP3 signalling.
3.5. Inhibition of exosome biogenesis reverses ADMSC‐EV‐induced effects in neurons exposed to OGD
As mentioned before, exosomes form an important subgroup of EVs. Two mechanisms of exosome formation have been described by ceramide synthesis (Trajkovic et al., 2008) and by the endosomal sorting complex required for transport (ESCRT) machinery (Hanson & Cashikar, 2012), respectively. To determine the role of exosomes in the regulation of autophagy and neuroprotection in primary neurons exposed to OGD, we inhibited ceramide synthesis with the neutral sphingomyelinase‐targeting inhibitor GW4869 and inhibited ESCRT‐mediated exosome biogenesis by knocking down Hrs, a member of the ESCRT‐0 complex (Tamai et al., 2010). As expected, both Hrs‐KD and GW4869 pre‐treatment led to a large decrease in exosome secretion shown by NTA analysis (Figure 4a), showing no difference between the two methods. Moreover, the effects of ADMSC‐EVs (EVs) on autophagy regulation (Figure 4b‐c) and neuroprotection (Figure 4d) were significantly blocked when ADMSCs were pretreated with the inhibitor of exosomal secretion GW4869 or Hrs‐KD. Then, we applied GW4869 in the coculture model, the effects of ADMSC‐EVs (EVs) on neuroprotection (Figure 4d) and autophagy regulation (Figure 4e‐f) were significantly blocked when ADMSCs were pretreated with the inhibitor of exosomal secretion GW4869.
FIGURE 4.
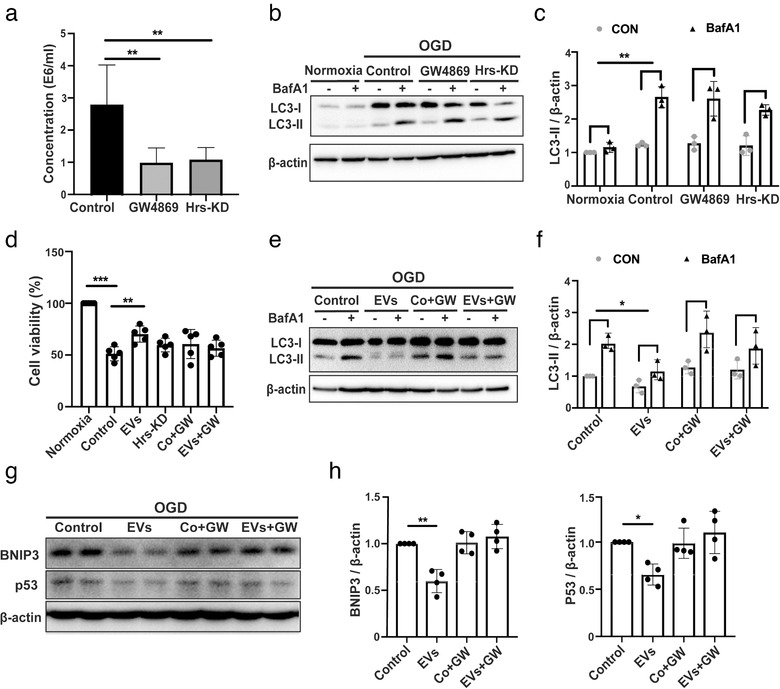
The regulation of the autophagic flux by ADMSC‐EVs depends on exosomes. (a) ADMSC‐derived EVs obtained from ADMSCs pre‐treated with DMSO, GW4869 or Hrs‐siRNA (knockdown, Hrs‐KD) were isolated by the PEG method. The quantification of EV numbers is shown in (A) (n = 5). (b‐c) LC3 levels were detected by Western blotting followed by densitometric analysis in primary neurons exposed to oxygen‐glucose‐deprivation (OGD). OGD‐exposed neurons were either incubated with EVs isolated from ADMSCs pre‐treated with the exosome secretion inhibitor GW4869 (GW4869) or with ADMSC‐derived EVs obtained from ADMSCs that were pre‐transfected with Hrs‐siRNA (Hrs‐KD). All experimental conditions were performed with or without the autophagic flux inhibitor BafA1 (n = 3). (d) Cell viability was examined in OGD‐exposed primary neurons that were treated with PBS, ADMSC‐EVs (EVs), ADMSC‐derived EVs obtained from ADMSCs that were pre‐transfected with Hrs‐siRNA (Hrs‐KD), ADMSCs treated with the exosome secretion inhibitor GW4869 (Co+GW) or ADMSC‐EVs isolated from conditioned medium containing GW4869 (EVs+GW; n = 3). Cells incubated under standard cell culture conditions (Normoxia) were defined as 100 % cell survival. (e‐f) OGD‐exposed neurons were either incubated with ADMSC‐derived EVs (EVs), ADMSCs pre‐treated with the exosome secretion inhibitor GW4869 (Co+GW), or with ADMSC‐derived EVs obtained from ADMSCs that were pretreated with GW4869 (EVs+GW). EVs, GW4869, EVs+GW were given at the beginning of both hypoxia and reoxygenation. All experimental conditions were performed with or without the autophagic flux inhibitor BafA1 (n = 3). BafA1 was added 3 h before harvesting the cells. (g‐h) P53 and BNIP3 were quantified by Western blotting in OGD‐exposed primary neurons co‐incubated with PBS, ADMSC‐EVs (EVs), ADMSCs with GW4869 (Co+GW) or with EVs isolated from ADMSCs pre‐treated with GW4869 (EVs+GW; n = 3). Quantitative analysis of p53 and BNIP3 is shown (h). One‐way ANOVA followed by the Tukey's post‐hoc‐test was used, data are shown as mean ± SD. Data are statistically different from each other with *P < 0.05, **P < 0.01, and ***P < 0.001
Importantly, the incubation with ADMSC‐EVs down‐regulated the abundance of p53 and BNIP3 in neurons exposed to OGD, whereas incubation of neurons with EVs obtained from such pretreated ADMSCs failed to inhibit p53 and BNIP3 protein levels (Figure 4g‐h). Thus, exosomes forming a subgroup of ADMSC‐EVs appear to be critical in mediating the anti‐autophagic activity of ADMSCs in primary neurons exposed to OGD.
3.6. The miR‐25‐3p is abundant in ADMSC‐EVs reducing the autophagic flux in hypoxic primary neurons
To further identify the compounds of EVs that are responsible for ADMSC‐EV‐induced regulation of autophagy, we first explored miRNAs that directly target the p53/BNIP3 autophagy signalling pathway. Out of more than 10 miRNAs known to target p53 (Liu, Zhang, Zhao, & Feng, 2017), we selected 6 candidates miRNA (miR‐25‐3p, miR‐98, miR‐125a‐5p, miR125b‐5p, miR‐214‐3p, and miR‐30) that are more closely related to neurons for further screening. In order to eliminate the bias due to the selection of internal controls, we have selected two internal controls, U6 and let‐7a, for relative quantification of miRNAs (Li et al., 2015). The results from qRT‐PCR analyses indicated that miR‐25‐3p was one of the most abundant miRNAs in ADMSC‐EVs (Figure 5a). In order to show in which physicochemical state miR‐25 can be found in our conditions, we treated EVs with either RNase A, the detergent Triton X‐100 (to disrupt the lipid bilayer of vesicles), both of them, or with the solvent alone (Control). We then measured the levels of miR‐25 by RT‐qPCR. When EVs were treated with RNase A, but not with Triton X‐100, the PCR signal for the miR‐25 remained within the margin of error compared to the signal for solvent treated one. This suggests that RNAs captured by the column were located inside vesicles and were therefore protected by the lipid bilayer from RNase A digestion (Figure 5b). Treating EVs with Triton X‐100 alone resulted in some degradation of RNA, supposedly because disruption of the vesicle membrane exposes the RNA to residual RNase present in the environment. Conversely, treating with Triton X‐100 and RNase A led to near‐total digestion of the RNAs (Figure 5b), confirming that their initial resistance to digestion was due to sequestration within vesicles, which proved miR‐25 to be inside of ADMSC‐EVs. Nevertheless, the intracellular concentration of miR‐25‐3p in primary neurons significantly decreased when these cells where exposed to OGD (Figure 5c). On the contrary, treatment of primary neurons with ADMSC‐EVs significantly increased the concentration of miR‐25‐3p (Figure 5c).
FIGURE 5.
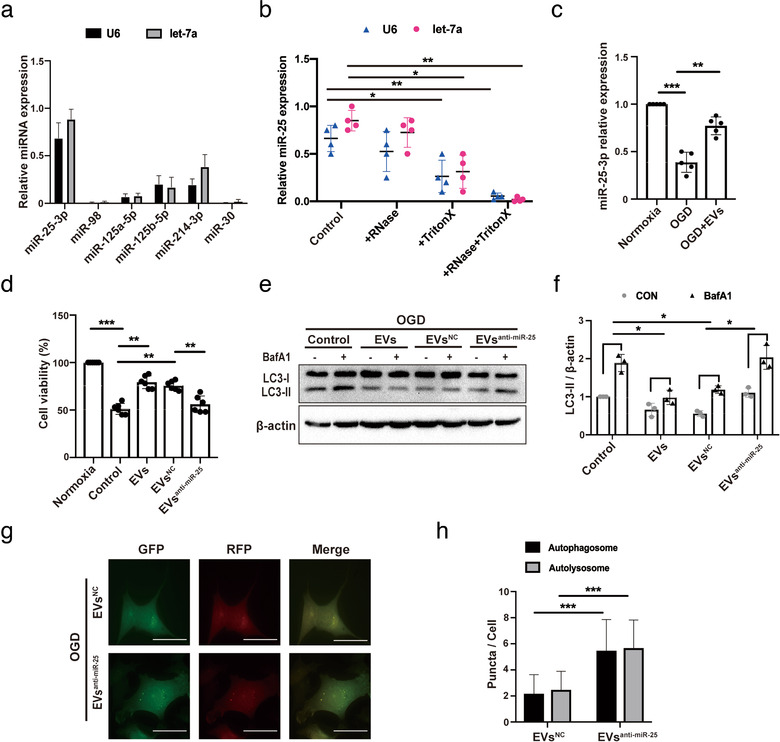
ADMSCs regulate autophagy and induce neuroprotection by miR‐25‐3p. (a) Real time Quantitative Polymerase chain reaction (qRT‐PCR) quantification of miRNA concentrations in EVs obtained from ADMSCs as stated in the materials and methods section. (b) Levels of miR‐25‐3p were detected in ADMSC‐EVs that were pretreated either RNase A (+RNase), the detergent Triton X‐100 (+Triton X), both of them (+RNase+Triton X), or with the solvent alone (Control) (n = 3). (c) Levels of cellular miR‐25‐3p were measured in primary neurons treated with PBS or ADMSC‐EVs in the OGD model by qRT‐PCR. PBS and EVs were given at the beginning of hypoxia and reoxygenation. Data refer to neurons cultured under standard cell culture conditions (Normoxia). (d) Cell viability was examined in primary neurons exposed to OGD that were treated with PBS, EVs obtained from normal ADMSCs (EVs), EVs isolated from ADMSCs that were pretreated with anti‐miR‐25‐3p (EVsanti‐miR‐25) or with EVs isolated from ADMSCs that were pretreated with scramble (EVsNC; n = 3). All of them were given at the beginning of hypoxia and reoxygenation. Cells incubated under standard cell culture conditions (Normoxia) were defined as 100 % cell survival. (e‐f) Western blot identification for LC3 in primary neurons after incubation with different EVs, i.e., EVs isolated from normal ADMSCs (EVs), EVs obtained from ADMSCs that were pretreated with anti‐miR‐25‐3p (EVsanti‐miR‐25) or with EVs isolated from ADMSCs that were pretreated with scramble (EVsNC). OGD experiments were performed with or without BafA1 under each condition (n = 3). Quantitative analysis of LC3‐II is shown in (f). (g‐h) Autophagosomes and autolysosomes were detected in EVsanti‐miR‐25‐treated primary neurons or in EVsNC‐treated neurons that expressed mRFP‐GFP‐LC3 under OGD conditions. The number of autophagosomes and autolysosomes in each cell (20‐30 cells per group) was quantified (n = 3). EVsanti‐miR‐25 and EVsNC were given twice as mentioned before. Scale bar, 10 μm. The two‐tailed independent Student's t‐test was used in H. One‐way ANOVA followed by the Tukey's post‐hoc‐test was used except for H, data are shown as mean ± SD. Data are statistically different from each other with *P < 0.05, **P < 0.01, and ***P < 0.001
To confirm whether or not miR‐25‐3p has a functional role in the aforementioned EV‐induced regulation of autophagy under OGD conditions of neurons, EVs were obtained from ADMSCs that were pretreated with an anti‐miR‐25‐3p oligonucleotide (ADMSC‐EVanti‐miR‐25) or with a scrambled construct as control (ADMSC‐EVNC). The cell viability after OGD was significantly higher in primary neurons cultured with ADMSC‐EVsNC (EVsNC) when compared to PBS‐treated controls (Figure 5d). However, these effects were reversed when the cells were treated with ADMSC‐EVsanti‐miR‐25 (EVsanti‐miR‐25; Figure 5d). The magnitude of BafA1‐induced LC3‐II accumulation under OGD conditions was also significantly lower in primary neurons cultured with ADMSC‐EVsNC when compared to PBS‐treated cells (Figure 5e‐f), whereas these results were again reversed when the neurons were incubated with ADMSC‐EVanti‐miR‐25 (Figure 4e‐f). Likewise, using a luciferase system, a significant increase in the number of autophagosomes and autolysosomes was counted in neurons incubated with ADMSC‐EVsanti‐miR‐25 (EVsanti‐miR‐25) compared to ADMSC‐EVsNC (EVsNC) (Figure 5g‐h). Thereafter, the effects of miR‐25‐3p on the regulation of autophagy were further analysed, using a miR‐25‐3p mimic and its inhibitor directly in primary neurons that were exposed to OGD. Hypoxic neurons transfected with the miR‐25‐3p mimic recapitulated the inhibitive effects on both autophagy regulation (Figure S3A‐B) and autophagic flux (Figure S3C‐D) when compared with the scramble group. Through the live/dead assay, we found that the transfection itself did not increase cell death compared with the untransfected group, suggesting that the autophagy regulating effect was due to miR‐25a itself but not due to the protein loss after cell death (Figure S3E‐F). Conversely, transfection of neurons with the miR‐25 inhibitor yielded opposite results (Figure S3G‐H). Being consistent with the autophagic flux, the levels of p53 and BNIP3 were decreased in response to the treatment with the miR‐25‐3p mimic (Figure S3I‐J), while abundance of these proteins was enhanced when the neurons were treated with the miR‐25‐3p inhibitor (Figure S3 H‐L). Finally, incubation of neurons exposed to hypoxia with the miR‐25‐3p mimic resulted in increased cell viability (Figure S3M), whereas inhibition of miR‐25‐3p decreased neuron viability (Figure S3N).
3.7. ADMSC‐EVs induce sustained neuroprotection and promote neurological recovery after stroke in mice
In light of the aforementioned in vitro data on primary neurons, we investigated whether or not ADMSC‐EV administration improves post‐stroke neurological recovery by modulating autophagic activity. We first checked the EV biodistribution patterns under ischemic conditions. Consistent with previous results (Zagrean, Hermann, Opris, Zagrean, & Popa‐Wagner, 2018), ADMSC‐EVs indeed do reach the central nervous system, at least under ischemic stroke conditions (Figure 6a). Following a previously published protocol (Doeppner et al., 2015), EVs released by 2 × 106 ADMSCs diluted in 200 μl of PBS were systemically administered immediately at the beginning of the reperfusion or at 12 h after reperfusion. Mice that received ADMSC‐EVs immediately during the beginning of the reperfusion exhibited significantly smaller infarct volumes when compared to PBS controls (Figure 6b‐c). Likewise, treatment of stroke mice with EVs at 12 h also yielded significantly reduced infarct volumes when compared to controls, with no significant difference between the two EV groups (Figure 6b‐c). Along with such a reduction of acute brain injury, the behavioural test analyses revealed better test scores of mice treated with EVs at either time point (Figure 6d‐e). Of note, this better test performance in both the corner turn and the tight rope test was long‐lasting and stable until the end of the observation period of 14 days. We subsequently analysed neuronal survival in the ischemic striatum at 14 days after the stroke. In line with the reduction of neurological impairment, increased neuronal densities were found in mice treated with ADMSC‐EVs of both injection time points (Figure 6f‐g). Conclusively, ADMSC‐derived EVs reduce post‐stroke brain injury on both the histological and the functional level in experimental stroke models.
FIGURE 6.
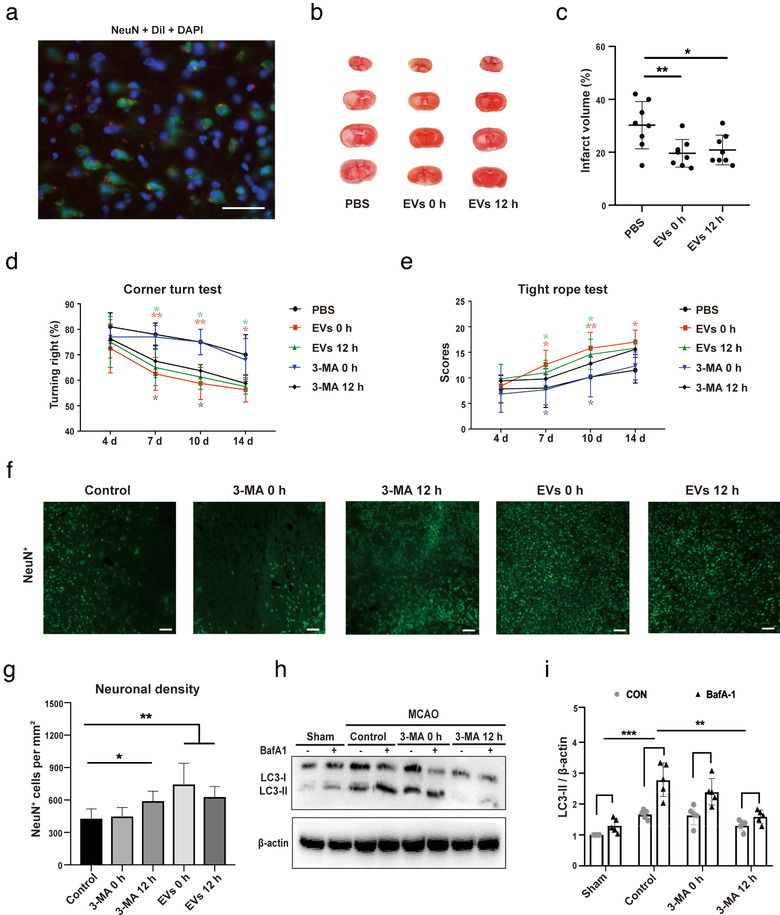
EV‐induced regulation of autophagy reduces post‐stroke brain injury and improves neurological recovery. (a) Representative immunofluorescence images displaying the biodistribution of ADMSC‐EVs within the ischemic hemisphere. DiI (red spots), NeuN (green) and DAPI (blue). Scale bars, 25 μm. (b‐c) Neuroprotective effects of ADMSC‐EVs in mice exposed to 1 h of middle cerebral artery occlusion (MCAO) followed by 24 h of reperfusion were evaluated by TTC staining. ADMSC‐EVs were injected at the beginning of the reperfusion (EVs 0 h) or at 12 h after reperfusion (EVs 12 h). Mice treated with PBS served as control (n = 8 per group). Quantitative analysis of the infarct size is shown in (c). (d‐e) Mice (n = 10 per condition) were exposed to 1 h of MCAO with subsequent reperfusion for 14 days during which the corner turn test (d) and the tight rope test (e) were performed. Mice received systemic delivery of ADMSC‐EVs or of 3‐MA (15 mg/kg) immediately at the beginning of the reperfusion (EVs 0 h) or 12 h after reperfusion (EVs 12 h). Control mice received PBS only. Motor coordination tests were done at 4, 7, 10 and 14 days after cerebral ischemia. Both EVs 0 h and EVs 12 h groups showed significant improvement in the tight rope test compared to the PBS control group. On day fourteen, only EVs 0 h significantly improved tight rope performance. In the corner turn test, the EVs 0 h and 12 h group showed improvement on day seven, ten and fourteen compared to the PBS control group, and the 3‐MA 12 h group showed improvement on day seven and day ten compared to the PBS control group. (f‐g) The neuronal density was measured in mice treated with PBS (Control), ADMSC‐EVs or of 3‐MA. EVs and 3‐MA were systemically injected at the beginning of the reperfusion or 12 h after reperfusion. NeuN staining within the ischemic lesion site was done on day 14 (n = 10 per group). Scale bars, 50 μm. (g‐h) The autophagic flux was evaluated with BafA1 in MCAO mice that received 3‐MA injection immediately at the beginning of (3‐MA 0 h) or 12 h after reperfusion (3‐MA 12 h). PBS was given to control animals (n = 6 per group). Quantitative analysis of LC3‐II is shown (h). One‐way ANOVA followed by the Tukey's post‐hoc‐test was used, data are shown as mean ± SD. Data are statistically different from each other with *P < 0.05, **P < 0.01, and ***P < 0.001
3.8. ADMSC‐EV administration after MCAO reduces autophagic flux through p53‐BNIP3 signalling
To further confirm whether autophagy inhibition contributes to neuroprotection and functional improvement, we used 3‐MA to inhibit autophagy after MCAO, and then assessed neuronal survival as well as neurological recovery. Treatment of mice with the inhibitor of autophagy, 3‐MA, at 12 h post‐stroke confirmed the aforementioned results of the behavioural tests, thus further supporting the hypothesis that inhibition of autophagy contributes to neurological recovery upon stroke induction (Figure 6d‐e). Whereas delivery of 3‐MA at 12 h post‐stroke yielded better neurological recovery in these mice when compared to PBS controls, delivery of 3‐MA immediately at the beginning of the reperfusion only partially enhanced neurological recovery (Figure 6d‐e). Analysis of neuronal survival at 14 days post‐stroke showed increased neuronal densities in mice treated with 3‐MA at 12 h, whereas treatment immediately at the beginning of the reperfusion did not result in neuroprotection (Figure 6f‐g). Again, treatment of stroke mice with 3‐MA at 12 h significantly reduced the autophagic flux 24 h post‐surgery as indicated when BafA1 was simultaneously used in these animals. The LC3‐II levels were significantly lower in these mice as compared to the PBS group (Figure 6h‐i). In contrast to this, treatment with 3‐MA immediately during the beginning of the reperfusion had no impact on the autophagic flux. These results suggest that modulating post‐stroke over‐activated autophagic activity induces neuroprotection in mice after induction of MCAO.
To further investigate whether or not neuroprotection caused by ADMSC‐EVs is associated with a regulation of autophagy, the temporal resolution patterns of autophagy levels in non‐treated stroke mice were assessed. Western blotting against LC3‐II revealed a peak of protein abundance at 24 h post‐stroke when compared to sham animals (Figure S4A‐B). Treatment of mice with ADMSC‐EVs immediately at the beginning of the reperfusion (EVs 0 h; Figure S4C‐D) or at 12 h (EVs 12 h; Figure S4E‐F) significantly reduced the stroke‐induced activation of autophagy as measured at 24 h post‐stroke. These observations suggest that ADMSC‐EVs significantly reduce the stroke‐induced autophagic flux in mice.
We next detected the expression of p53 and BNIP3 in vivo. Consistent with our in vitro data, both p53 and BNIP3 protein levels were increased in the infarct region of MCAO mice. Application of ADMSC‐EVs at the aforementioned two injection time points (0 h and 12 h) significantly reduced abundance of both p53 and BNIP3 in stroke mice (Figure S4G‐H).
3.9. Post‐stroke autophagic regulation by ADMSC‐EVs depends on miR‐25‐3p
In order to confirm that the decline in autophagic flux associated with ADMSC‐EV administration is mediated by the EV‐delivery of miR‐25‐3p, mice were injected with either PBS, ADMSC‐EVs, ADMSC‐EVs that had been pretreated with anti‐miR25‐3p (EVsanti‐miR25), or with ADMSC‐EVs that had been pretreated with the control oligonucleotide (EVsNC). The injection was done 12 h after MCAO surgery. After 24 h of reperfusion, the BafA1‐induced LC3‐II accumulation in the ischemic striatum was significantly lower in both the ADMSC‐EV and the ADMSC‐EVsNC groups compared to the PBS group (Figure 7a‐b). However, treatment with ADMSC‐EVsanti‐miR25 failed to show similar effects on the autophagic flux (Figure 7a‐b).
FIGURE 7.
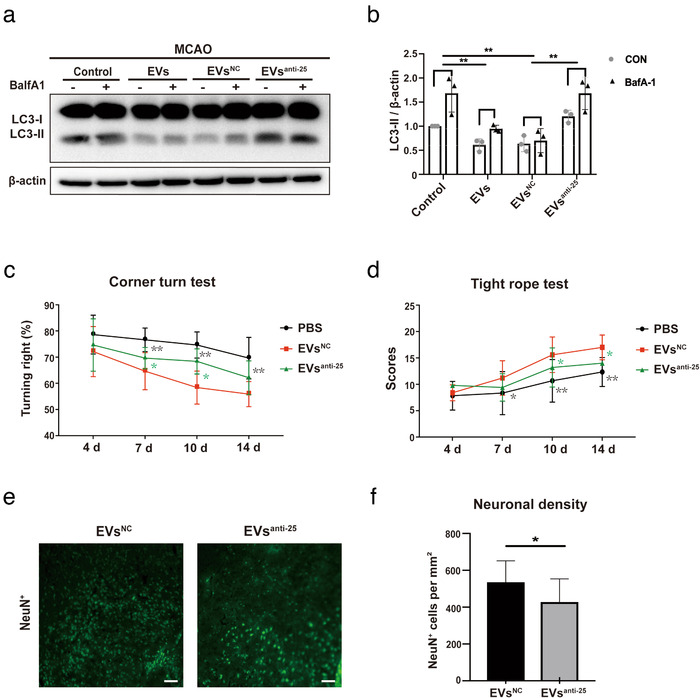
Loss of miR‐25‐3p in ADMSC‐EVs diminishes post‐stroke EV‐induced regulation of autophagy and neuroprotection. (a‐b) The autophagic flux was evaluated with BafA1 in MCAO (middle cerebral artery occlusion) mice that either received PBS or ADMSC‐EVs 12 h after the induction of ischemic stroke, including ADMSC‐EVs, ADMSC‐EVsNC, and ADMSC‐EVsanti‐miR‐25 (n = 6 per group). BafA1 was injected 3 h before sacrifice. Quantitative analysis of LC3‐II is shown in (b). (c‐d) Motor coordination was evaluated by the tight rope test and by the corner turn test at four, seven, ten and fourteen days after cerebral ischemia to verify the effects of ADMSC‐EVsNC and ADMSC‐EVsanti‐miR‐25 in MCAO animals compared to PBS group. In the corner turn test, the EVsNC group showed improvement on day seven, ten and fourteen compared with PBS control group. The EVsNC group showed improvement on day seven and day ten compared to the EVsanti‐miR‐25 group. EVsNC group showed significant improvement in the tight rope test compared to the PBS control group. EVsNC groups also showed significant better tight rope performance compared to EVsanti‐miR‐25 on day ten and fourteen. (e‐f) The neuronal density in ADMSC‐EVsNC and ADMSC‐EVsanti‐miR‐25 was measured on day fourteen as indicated by NeuN staining within the ischemic lesion site (n = 10 per group). The quantitative analysis of neuronal density is shown in (f). Scale bars, 50 μm. The two‐tailed independent Student's t‐test was used in. One‐way ANOVA followed by the Tukey's post‐hoc‐test was used except for F, data are shown as mean ± SD. Data are statistically different from each other with *P < 0.05, **P < 0.01, and ***P < 0.001
Consistently, neurological recovery as assessed by the corner turn and the tight rope test was enhanced in mice treated with ADMSC‐EVsNC when compared to both controls and mice treated with ADMSC‐EVsanti‐miR25 (Figure 7c‐d). Analysis of the neuronal density 14 days after MCAO revealed that mice treated with ADMSC‐EVsNC displayed reduced brain injury compared to animals treated with ADMSC‐EVsanti‐miR25 Figure 7e‐f). Conclusively, ADMSC‐derived EVs thus reduce brain injury on both the histological and the functional level. The anti‐autophagic activity associated with ADMSC‐EV administration after MCAO is at least partially mediated by the EV transfer of miR‐25‐3p.
4. DISCUSSION
EVs from various tissue sources induce both neuroprotection and neuroregeneration in preclinical stroke models. The underlying mechanisms of such EV‐induced beneficial effects under stroke conditions, however, remain elusive. Using both an in vitro and an in vivo stroke model, the present work identified a new mode of action by which EVs derived from ADMSCs mediate post‐stroke neuroprotection and neurological recovery. The findings presented herein suggest that ADMSC‐EVs are able to inhibit ischemia‐induced autophagy. A key mechanism of ADMSCs‐induced neuroprotection involves the EV‐mediated transfer of miR‐25‐3p from ADMSCs to the neuron, where it interferes with the p53/BNIP3 signalling pathway (Figure 8).
FIGURE 8.
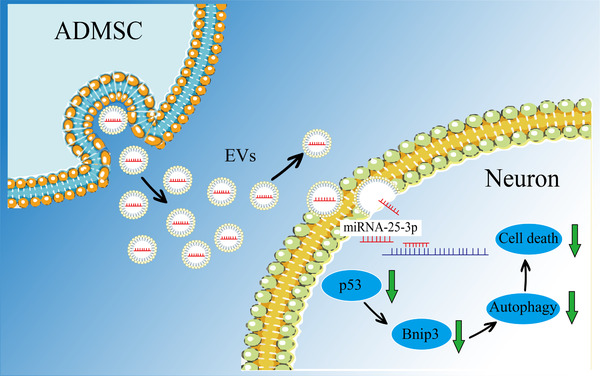
A schematic diagram showing the role of miR‐25‐3p derived from ADMSC‐EVs in a preclinical stroke model. ADMSCs release EVs enriched with miR‐25‐3p, which are uptaken by neurons. In neurons, miR‐25‐3p induces degradation of the mRNA of p53, resulting in the downregulation of the p53 protein level and subsequent reduction of BNIP3. The inhibition of BNIP3, in turn, further reduces the levels of autophagy which exerts the neuroprotective effect
Intercellular transfer of RNA by small EVs has opened a new avenue of research centred on these tiny cellular particles in diverse physiological systems. However, these small size particles have imposed numerous technical hurdles for their isolation and study. Related to EV isolation, the most popular method remains differential ultracentrifugation (UC). UC, however, is limited in scale and cumbersome (Lötvall et al., 2014). Other concentration methods have therefore to be adopted for concentrating these vesicles. The use of polyethylene glycol (PEG) has recently gained popularity due to its rapid and simple‐to‐use nature. Previous work from our own group systematically analysed the PEG precipitation approach in direct comparison with standard EV enrichment procedures on HEK293T cells (Ludwig et al., 2018). We revealed the PEG approach to be not inferior to standard EV enrichment procedures using HEK293T cells. In the present study, we also compared two different isolation methods, UC and PEG enrichment in the enrichment of ADMSC‐EVs. The purification rates obtained by these methods were high for ADMSC‐EVs, as indicated in the Western blot results. Especially, EV negative markers such as Albumin, TOMM20, and Histones were close to the detection threshold or not detectable at all in our UC and PEG method group. Likewise, the distribution and morphological patterns were similar between EVs enriched with either UC or PEG.
The PEG method has been reported to retain certain amount of protein contaminants (Patel et al., 2019), and these co‐precipitated protein complexes may have related functions and affect the experimental outcome. To obtain what fraction is the active fraction mediating neuroprotection, it was necessary to combine several separation procedures and to follow a nonbiased approach (Freitas et al., 2019; Kowal et al., 2016). We showed that density gradient ultracentrifugation (DGUC) or size exclusion chromatography (SEC) can separate sEVs from other co‐precipitates, which was indicated by strong signals of EV markers and typical particle diameters of 30–150 nm after purification. Moreover, the further purified sEV showed a similar performance on alleviating cell damage and reducing autophagy levels after ischemic stroke compared to unpurified EVs, suggesting that sEV are the therapeutically efficacious factors in our stroke models. In particular, both DGUC and SEC have a positive effect on the determination of functional fractions for large‐scale isolation of EVs from conditioned media by applying precipitation using PEG coupled with a DGUC or a SEC approach. Therefore, PEG offers an option to remedy the limitations of ultracentrifugation for the initial concentration step.
The pathophysiology of cerebral ischemia involves a plethora of different signalling cascades (Fricker, Tolkovsky, Borutaite, Coleman, & Brown, 2018). Among these different mechanisms, more recent data suggests a role of autophagy being an interesting target for stroke treatment (Descloux, Ginet, Clarke, Puyal, & Truttmann, 2015; Puyal, Ginet, Grishchuk, Truttmann, & Clarke, 2012; Wang et al., 2018). However, the precise role of autophagy under such ischemic stroke conditions is still under debate, i.e., autophagy might have both beneficial and detrimental effects. Some studies found pharmacological induction of autophagy by rapamycin to reduce apoptotic and necrotic cell death during hypoxia, whereas others reported inhibition of autophagy by 3‐MA to enhance cell death (Carloni et al., 2010; Wang et al., 2012) . Contrary to this, many groups reported that an increase in autophagy contributes to cell death. Using a model of excitotoxicity, the application of the glutamate receptor activator resulted in the death of cortical neurons, whereas delivery of the autophagy inhibitor 3‐MA or the genetic knockdown of Atg7 and beclin‐1 yielded neuroprotection (Ginet et al., 2014). Similar findings were supported by other models of cerebral ischemia (Koike et al., 2008; Xing et al., 2012).
In our study, autophagy was strongly activated upon induction of either cerebral ischemia or oxygen‐glucose‐deprivation (OGD). Activation of autophagy under such conditions represented a dynamic evolutionary process with a peak at 24 h after MCAO induction. Interestingly, down‐regulation of autophagy using 3‐MA at the onset of reperfusion did not induce any significant effect on brain injury, whereas 3‐MA delivery at 12 h after reperfusion significantly improved both the neurological outcome and the neuronal density. In vitro studies confirmed the beneficial effect of inhibition of autophagy using 3‐MA in a dose dependent manner. The lack of 3‐MA when given immediately after the stroke in our experimental setting is most likely a consequence of the rather low half‐life time of 3‐MA with about 2–3 h (Sweet, Carda, & Small, 1981). As such, 3‐MA is already metabolized before the peak of post‐stroke autophagic activity is reached at 24 h. In light of both previous studies and the present work, it stands to reason that basal levels of autophagy are essential for proper neuronal function, with moderate increases of autophagic activity during mild conditions of ischemia, hypoxia, or nutrient deprivation still allowing neurons to maintain a sufficiently high energy supply (Kim et al., 2018). More severe noxious stimuli such as acute ischemic stroke, however, ensue an increase in autophagic flux that may culminate in autophagic cell death (Ginet et al., 2014; Liu & Levine, 2015). Consequently, a well‐balanced inhibition of autophagy under stroke conditions seems to be in order (Shi et al., 2012).
Whereas pharmacological drugs such as 3‐MA might only transiently affect post‐stroke autophagy due to metabolization and short half‐life time, stem cells like ADMSCs including their secretion products such as EVs are known to induce long‐term effects under various circumstances (Lai, Yeo, Tan, & Lim, 2013; Tian et al., 2018). Indeed, the application of ADMSCs resulted in a pronounced downregulation of the autophagy marker LC3‐II under both in vitro and in vivo conditions, contributing to both neuroprotection in vitro and in vivo as well as to neurological recovery in vivo. Interestingly, the inhibition of exosome secretion from ADMSCs resulted in a therapeutic loss of both such pretreated ADMSCs and their enriched EVs. The latter suggests a role for exosomes as a subgroup of the EV family that might be responsible for the biological effects observed in the study.
Inhibiting autophagy itself, however, does not exclusively predict the therapeutic impact of such a kind of therapy. Rather, the success of such a therapeutic intervention depends on the precise autophagic signaling pathway that is supposed to be inhibited. Multiple reports indicate that cerebral ischemia activates HIF‐1α which induces p53 or BNIP3 expression (Althaus et al., 2006; Li, Sun, Ni, Chen, & Guo, 2013). The induced p53 stabilization as a consequence of up‐regulated HIF‐1α promotes further expression of BNIP3 (Wang et al., 2013; Xin et al., 2011). The latter contains a single Bcl‐2 homology 3 (BH3) domain and belongs to the Bcl‐2 family proteins that serve as an important target gene of HIF‐1α (Chinnadurai, Vijayalingam, & Gibson, 2008). BNIP3 can compete with beclin‐1 for binding to Bcl‐2. Released beclin‐1, in turn, thus triggers autophagy and cell death (Glick, Barth, & Macleod, 2010; He & Klionsky, 2009). Herein, both p53 and BNIP3 levels were increased upon induction of hypoxia or cerebral ischemia, and ADMSC‐EVs reversed the aforementioned increase of these proteins. The present study thus shows that autophagy inhibition plays a critical role in ADMSC‐EV‐based therapy of ischemic stroke, although other autophagy‐related signalling pathways being regulated by ADMSC‐EVs cannot be completely excluded.
The role of EVs in intracellular signalling, as well as their therapeutic potential for the treatment of ischemic stroke (Cunningham et al., 2018; Webb et al., 2018), has only recently become a prominent topic of research. Previous work from our own group and from other scientists have shown that EVs have both proangiogenic and anti‐inflammatory properties (Dabrowska, Andrzejewska, Lukomska, & Janowski, 2019; Doeppner et al., 2015; Keshtkar, Azarpira, & Ghahremani, 2018; Xin et al., 2013). As indicated before, EVs do not only carry diverse sets of proteins (Doeppner et al., 2017; Doeppner et al., 2012; Doeppner et al., 2009; Doeppner, Kaltwasser, Fengyan, Hermann, & Bähr, 2013) but also contain non‐coding RNA and DNA, among which microRNAs are of particular interest (Bertoli, Cava, & Castiglioni, 2015). The latter guides the binding of the RNA‐induced silencing complex to regions of partial complementarity located mainly within 3′untranslated regions (UTRs) of target messenger RNA (mRNA) molecules, resulting in mRNA degradation and/or translational inhibition (Bartel, 2009). We have already demonstrated that miR‐124, which is among the most abundant microRNAs in the adult mammalian brain (Cao, Pfaff, & Gage, 2007; Mishima, Mizuguchi, Kawahigashi, Takizawa, & Takizawa, 2007) affects a plethora of signaling molecules such as inhibition of deubiquitination of Usp14, significantly contributing to the reduction of post‐stroke brain injury in rodents (Doeppner et al., 2013). Moreover, it is well known that miRNAs impact the core autophagy pathway, including p53 (Frankel & Lund, 2012). Since EVs are able to transfer miRNA cargo to target cells (Abels & Breakefield, 2016), it is fair enough to hypothesize that ADMSC‐EVs yield protection of neurons and brain tissue using this way of action. We herein demonstrated that ADMSC‐EVs improve neurological recovery by impeding autophagy and the key anti‐autophagic and cytoprotective compound of ADMSC‐EV cargo may be miR‐25‐3p. The latter is one of several miRNAs found in EVs that target p53. Previous research on miR‐25‐3p has primarily focused on its biological function under conditions of cancer (Zeng et al., 2018; Zhang et al., 2019). To the best of our knowledge, our results from both in vitro and in vivo data suggested for the first time that miR‐25‐3p likely contributes to the pathogenesis of stroke. To test the hypothesis that miR‐25‐3p was responsible for ADMSC‐EV‐mediated neuroprotection, ADMSC‐EVs that had been pretreated with anti‐miR‐25‐3p were administered via femoral vein injection. As expected, the miR‐25‐3p knockdown abolished the therapeutic effects of ADMSC‐EVs, as evidenced by the markedly decreased neuronal density and the lack of motor coordination recovery.
In conclusion, the findings presented herein provide novel insides into the mechanisms by which EVs induce therapeutic actions under experimental stroke settings. The data provides evidence that native ADMSC‐EVs yield neuroprotection and enhanced neurological recovery by inhibiting ischemia‐induced autophagy. Inhibition of autophagy, in turn, is mediated by EVs transferring miR‐25‐3p from ADMSCs to their target cells. The latter results in interference with the p53/BNIP3 signaling pathway. These novel observations on EVs derived from stem cells might lead to the development of new therapeutic targets and strategies for the treatment of ischemic stroke.
AUTHOR CONTRIBUTION STATEMENT
Research and experiments were performed by Kuang, Zheng, Zhang, Ai, Venkataramani and Doeppner. Design and concept of the study were from Kuang and Doeppner. Kuang, Doeppner, Hermann, Kilic, Majid, and Bähr wrote the manuscript. Financial support was provided by Bähr and Doeppner.
CONFLICT OF INTEREST
The authors declare to have no conflict of interest of any kind.
ETHICAL APPROVAL
This article does not contain any studies with human participants performed by any of the authors. All animal experiments were performed with governmental approval according to the NIH guidelines for the care and use of laboratory animals. Both the STAIR criteria and the ARRIVE guidelines have been followed.
Supporting information
Supporting Information S1.
Supporting Information S2.
ACKNOWLEDGEMENTS
We thank Irina Graf and Regine Kruse (both Goettingen, Germany) for excellent technical assistance. This study was funded by TÜBITAK (to TRD).
Open access funding enabled and organized by Projekt DEAL.
Kuang Y, Zheng S, Zhang L, et al. Adipose‐derived mesenchymal stem cells reduce autophagy in stroke mice by extracellular vesicle transfer of miR‐25. J Extracell Vesicles. 2020;10:e12024 10.1002/jev2.12024
Correction added on 25 November 2020, after first online publication: Projekt Deal funding statement has been added.
REFERENCES
- Abels, E. R. , & Breakefield, X. O. (2016). Introduction to extracellular vesicles: Biogenesis, RNA cargo selection, content, release, and uptake. Cellular and Molecular Neurobiology, 36(3), 301–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmadian Kia, N. , Bahrami, A. R. , Ebrahimi, M. , Matin, M. M. , Neshati, Z. , Almohaddesin, M. R. , … Bidkhori, H. R. (2011). Comparative analysis of chemokine receptor's expression in mesenchymal stem cells derived from human bone marrow and adipose tissue. Journal of Molecular Neuroscience, 44(3), 178–185. [DOI] [PubMed] [Google Scholar]
- Althaus, J. , Bernaudin, M. , Petit, E. , Toutain, J. , Touzani, O. , & Rami, A. (2006). Expression of the gene encoding the pro‐apoptotic BNIP3 protein and stimulation of hypoxia‐inducible factor‐1alpha (HIF‐1alpha) protein following focal cerebral ischemia in rats. Neurochemistry International, 48(8), 687–695. [DOI] [PubMed] [Google Scholar]
- Bang, O. Y. , & Kim, E. H. (2019). Mesenchymal stem cell‐derived extracellular vesicle therapy for stroke: Challenges and progress. Frontiers in Neurology, 10, 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baraniak, P. R. , & Mcdevitt, T. C. (2010). Stem cell paracrine actions and tissue regeneration. Regenerative Medicine, 5(1), 121–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel, D. P. (2009). MicroRNAs: Target recognition and regulatory functions. Cell, 136(2), 215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertoli, G. , Cava, C. , & Castiglioni, I. (2015). MicroRNAs: New biomarkers for diagnosis, prognosis, therapy prediction and therapeutic tools for breast cancer. Theranostics, 5(10), 1122–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao, X. , Pfaff, S. L. , & Gage, F. H. (2007). A functional study of miR‐124 in the developing neural tube. Genes & Development, 21(5), 531–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carloni, S. , Girelli, S. , Scopa, C. , Buonocore, G. , Longini, M. , & Balduini, W. (2010). Activation of autophagy and Akt/CREB signaling play an equivalent role in the neuroprotective effect of rapamycin in neonatal hypoxia‐ischemia. Autophagy, 6(3), 366–377. [DOI] [PubMed] [Google Scholar]
- Chen, J. , Li, Y. , Wang, L. , Zhang, Z. , Lu, D. , Lu, M. , & Chopp, M. (2001). Therapeutic benefit of intravenous administration of bone marrow stromal cells after cerebral ischemia in rats. Stroke; A Journal of Cerebral Circulation, 32(4), 1005–1011. [DOI] [PubMed] [Google Scholar]
- Chen, W. , Sun, Y. , Liu, K. , & Sun, X. (2014). Autophagy: A double‐edged sword for neuronal survival after cerebral ischemia. Neural Regeneration Research, 9(12), 1210–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnadurai, G. , Vijayalingam, S. , & Gibson, S. B. (2008). BNIP3 subfamily BH3‐only proteins: Mitochondrial stress sensors in normal and pathological functions. Oncogene, 27(Suppl 1), S114–S127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo, M. , Raposo, G. , & Théry, C. (2014). Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annual Review of Cell and Developmental Biology, 30, 255–289. [DOI] [PubMed] [Google Scholar]
- Cunningham, C. J. , Redondo‐Castro, E. , & Allan, S. M. (2018). The therapeutic potential of the mesenchymal stem cell secretome in ischaemic stroke. Journal of Cerebral Blood Flow and Metabolism, 38(8), 1276–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabrowska, S. , Andrzejewska, A. , Lukomska, B. , & Janowski, M. (2019). Neuroinflammation as a target for treatment of stroke using mesenchymal stem cells and extracellular vesicles. Journal of Neuroinflammation, 16(1), 178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai, S.‐H. , Chen, T. , Li, X. , Yue, K.‐Y. , Luo, P. , Yang, L.‐K. , … Jiang, X. F. (2017). Sirt3 confers protection against neuronal ischemia by inducing autophagy: Involvement of the AMPK‐mTOR pathway. Free Radical Biology and Medicine, 108, 345–353. [DOI] [PubMed] [Google Scholar]
- Descloux, C. , Ginet, V. , Clarke, P. G. H. , Puyal, J. , & Truttmann, A. C. (2015). Neuronal death after perinatal cerebral hypoxia‐ischemia: Focus on autophagy‐mediated cell death. International Journal of Developmental Neuroscience, 45, 75–85. [DOI] [PubMed] [Google Scholar]
- Doeppner, T. R. , Doehring, M. , Bretschneider, E. , Zechariah, A. , Kaltwasser, B. , Müller, B. , … Michel, U. (2013). MicroRNA‐124 protects against focal cerebral ischemia via mechanisms involving Usp14‐dependent REST degradation. Acta Neuropathologica, 126(2), 251–265. [DOI] [PubMed] [Google Scholar]
- Doeppner, T. R. , Doehring, M. , Kaltwasser, B. , Majid, A. , Lin, F. , Bähr, M. , … Hermann, D. M. (2017). Ischemic post‐conditioning induces post‐stroke neuroprotection via Hsp70‐mediated proteasome inhibition and facilitates neural progenitor cell transplantation. Molecular Neurobiology, 54(8), 6061–6073. [DOI] [PubMed] [Google Scholar]
- Doeppner, T. R. , Ewert, T. A. S. , Tönges, L. , Herz, J. , Zechariah, A. , Elali, A. , … Bähr, M. (2012). Transduction of neural precursor cells with TAT‐heat shock protein 70 chaperone: Therapeutic potential against ischemic stroke after intrastriatal and systemic transplantation. Stem Cells, 30(6), 1297–1310. [DOI] [PubMed] [Google Scholar]
- Doeppner, T. R. , Herz, J. , Görgens, A. , Schlechter, J. , Ludwig, A.‐K. , Radtke, S. , … Hermann, D. M. (2015). Extracellular vesicles improve post‐stroke neuroregeneration and prevent postischemic immunosuppression. Stem Cells Translational Medicine, 4(10), 1131–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doeppner, T. R. , Kaltwasser, B. , Bähr, M. , & Hermann, D. M. (2014). Effects of neural progenitor cells on post‐stroke neurological impairment‐a detailed and comprehensive analysis of behavioral tests. Frontiers in Cellular Neuroscience, 8, 338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doeppner, T. R. , Kaltwasser, B. , Fengyan, J. , Hermann, D. M. , & Bähr, M. (2013). TAT‐Hsp70 induces neuroprotection against stroke via anti‐inflammatory actions providing appropriate cellular microenvironment for transplantation of neural precursor cells. Journal of Cerebral Blood Flow and Metabolism, 33(11), 1778–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doeppner, T. R. , Nagel, F. , Dietz, G. P.h , Weise, J. , Tönges, L. , Schwarting, S. , & Bähr, M. (2009). TAT‐Hsp70‐mediated neuroprotection and increased survival of neuronal precursor cells after focal cerebral ischemia in mice. Journal of Cerebral Blood Flow and Metabolism, 29(6), 1187–1196. [DOI] [PubMed] [Google Scholar]
- du Toit, A. , Hofmeyr, J. S. , Gniadek, T. J. , & Loos, B. (2018). Measuring autophagosome flux. Autophagy, 14(6), 1060–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández‐Susavila, H. , Bugallo‐Casal, A. , Castillo, J. , & Campos, F. (2019). Adult stem cells and induced pluripotent stem cells for stroke treatment. Frontiers in Neurology, 10, 908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankel, L. B. , & Lund, A. H. (2012). MicroRNA regulation of autophagy. Carcinogenesis, 33(11), 2018–2025. [DOI] [PubMed] [Google Scholar]
- Freitas, D. , Balmaña, M. , Poças, J. , Campos, D. , Osório, H. , Konstantinidi, A. , … Reis, C. A. (2019). Different isolation approaches lead to diverse glycosylated extracellular vesicle populations. Journal of Extracellular Vesicles, 8(1), 1621131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricker, M. , Tolkovsky, A. M. , Borutaite, V. , Coleman, M. , & Brown, G. C. (2018). Neuronal cell death. Physiological Reviews, 98(2), 813–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gervois, P. , Wolfs, E. , Ratajczak, J. , Dillen, Y. , Vangansewinkel, T. , Hilkens, P. , … Struys, T. (2016). Stem cell‐based therapies for ischemic stroke: Preclinical results and the potential of imaging‐assisted evaluation of donor cell fate and mechanisms of brain regeneration. Medicinal Research Reviews, 36(6), 1080–1126. [DOI] [PubMed] [Google Scholar]
- Ginet, V. , Spiehlmann, A. , Rummel, C. , Rudinskiy, N. , Grishchuk, Y. , Luthi‐Carter, R. , … Puyal, J. (2014). Involvement of autophagy in hypoxic‐excitotoxic neuronal death. Autophagy, 10(5), 846–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glick, D. , Barth, S. , & Macleod, K. F. (2010). Autophagy: Cellular and molecular mechanisms. Journal of Pathology, 221(1), 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson, P. I. , & Cashikar, A. (2012). Multivesicular body morphogenesis. Annual Review of Cell and Developmental Biology, 28, 337–362. [DOI] [PubMed] [Google Scholar]
- He, C. , & Klionsky, D. J. (2009). Regulation mechanisms and signaling pathways of autophagy. Annual Review of Genetics, 43, 67–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, S. , Wang, C. , Dong, H. , Xia, F. , Zhou, H. , Jiang, X. , … Xu, H. (2012). Immune‐related GTPase M (IRGM1) regulates neuronal autophagy in a mouse model of stroke. Autophagy, 8(11), 1621–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshtkar, S. , Azarpira, N. , & Ghahremani, M. H. (2018). Mesenchymal stem cell‐derived extracellular vesicles: Novel frontiers in regenerative medicine. Current Stem Cell Research & Therapy, 9(1), 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, D.‐K. , Kang, B. , Kim, O. Y. , Choi, D.‐S. , Lee, J. , Kim, S. R. , … Gho, Y. S. (2013). EVpedia: An integrated database of high‐throughput data for systemic analyses of extracellular vesicles. Journal of Extracellular Vesicles, 20384 2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, K.‐A. , Shin, D. , Kim, J.‐H. , Shin, Y.‐J. , Rajanikant, G. K. , Majid, A. , … Bae, O.‐N. (2018). Role of autophagy in endothelial damage and blood‐brain barrier disruption in ischemic stroke. Stroke; A Journal of Cerebral Circulation, 49(6), 1571–1579. [DOI] [PubMed] [Google Scholar]
- Koike, M. , Shibata, M. , Tadakoshi, M. , Gotoh, K. , Komatsu, M. , Waguri, S. , … Uchiyama, Y. (2008). Inhibition of autophagy prevents hippocampal pyramidal neuron death after hypoxic‐ischemic injury. American Journal of Pathology, 172(2), 454–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowal, J. , Arras, G. , Colombo, M. , Jouve, M. , Morath, J. P. , Primdal‐Bengtson, B. , … Théry, C. (2016). Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proceedings of the National Academy of Sciences of the United States of America, 113(8), E968–E977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai, R. C. , Yeo, R. W. Y. , Tan, K. H. , & Lim, S. K. (2013). Exosomes for drug delivery ‐ a novel application for the mesenchymal stem cell. Biotechnology Advances, 31(5), 543–551. [DOI] [PubMed] [Google Scholar]
- Li, A. , Sun, X. , Ni, Y. , Chen, X. , & Guo, A. (2013). HIF‐1alpha involves in neuronal apoptosis after traumatic brain injury in adult rats. Journal of Molecular Neuroscience, 51(3), 1052–1062. [DOI] [PubMed] [Google Scholar]
- Li, Y. , Zhang, L. , Liu, F. , Xiang, G. , Jiang, D. , & Pu, X. (2015). Identification of endogenous controls for analyzing serum exosomal miRNA in patients with hepatitis B or hepatocellular carcinoma. Disease Markers, 2015, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang, X. , Ding, Y. , Zhang, Y. , Tse, H.‐F. , & Lian, Q. (2014). Paracrine mechanisms of mesenchymal stem cell‐based therapy: Current status and perspectives. Cell Transplantation, 23(9), 1045–1059. [DOI] [PubMed] [Google Scholar]
- Lim, L. P. , Lau, N. C. , Garrett‐Engele, P. , Grimson, A. , Schelter, J. M. , Castle, J. , … Johnson, J. M. (2005). Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature, 433(7027), 769–773. [DOI] [PubMed] [Google Scholar]
- Liu, J. , Zhang, C. , Zhao, Y. , & Feng, Z. (2017). MicroRNA Control of p53. Journal of Cellular Biochemistry, 118(1), 7–14. [DOI] [PubMed] [Google Scholar]
- Liu, Y. , & Levine, B. (2015). Autosis and autophagic cell death: The dark side of autophagy. Cell Death and Differentiation, 22(3), 367–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lötvall, J. , Hill, A. F. , Hochberg, F. , Buzás, E. I. , Di Vizio, D. , Gardiner, C. , … Théry, C. (2014). Minimal experimental requirements for definition of extracellular vesicles and their functions: A position statement from the International Society for Extracellular Vesicles. Journal of Extracellular Vesicles, 3, 26913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucia Maria Ferri, A. , Bersano, A. , Lisini, D. , Boncoraglio, G. , Frigerio, S. , & Parati, E. (2016). Mesenchymal Stem Cells for Ischemic Stroke: Progress and Possibilities. Current Medicinal Chemistry, 23(16), 1598–1608. [DOI] [PubMed] [Google Scholar]
- Ludwig, A.‐K. , De Miroschedji, K. , Doeppner, T. R. , Börger, V. , Ruesing, J. , Rebmann, V. , … Giebel, B. (2018). Precipitation with polyethylene glycol followed by washing and pelleting by ultracentrifugation enriches extracellular vesicles from tissue culture supernatants in small and large scales. Journal of Extracellular Vesicles, 7(1), 1528109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathivanan, S. , Fahner, C. J. , Reid, G. E. , & Simpson, R. J. (2012). ExoCarta 2012: Database of exosomal proteins, RNA and lipids. Nucleic Acids Research, 40(Database issue), D1241–D1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishima, T. , Mizuguchi, Y. , Kawahigashi, Y. , Takizawa, T. , & Takizawa, T. (2007). RT‐PCR‐based analysis of microRNA (miR‐1 and ‐124) expression in mouse CNS. Brain Research, 1131(1), 37–43. [DOI] [PubMed] [Google Scholar]
- Mizushima, N. , & Yoshimori, T. (2007). How to interpret LC3 immunoblotting. Autophagy, 3(6), 542–545. [DOI] [PubMed] [Google Scholar]
- Mo, Z.‐T. , Fang, Y‐Q. , He, Y.‐P. , & Zhang, S. (2012). beta‐Asarone protects PC12 cells against OGD/R‐induced injury via attenuating Beclin‐1‐dependent autophagy. Acta Pharmacologica Sinica, 33(6), 737–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel, G. K. , Khan, M. A. , Zubair, H. , Srivastava, S. K. , Khushman, M. ’. D. , Singh, S. , & Singh, A. P. (2019). Comparative analysis of exosome isolation methods using culture supernatant for optimum yield, purity and downstream applications. Scientific Reports, 9(1), 5335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phinney, D. G. , Di Giuseppe, M. , Njah, J. , Sala, E. , Shiva, S. , St Croix, C. M. , … Ortiz, L. A. (2015). Mesenchymal stem cells use extracellular vesicles to outsource mitophagy and shuttle microRNAs. Nature Communications, 6, 8472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puyal, J. , Ginet, V. , Grishchuk, Y. , Truttmann, A. C. , & Clarke, P. G. H. (2012). Neuronal autophagy as a mediator of life and death: Contrasting roles in chronic neurodegenerative and acute neural disorders. The Neuroscientist, 18(3), 224–236. [DOI] [PubMed] [Google Scholar]
- Shi, R. , Weng, J. , Zhao, L. , Li, X.‐M. , Gao, T.‐M. , & Kong, J. (2012). Excessive autophagy contributes to neuron death in cerebral ischemia. CNS Neuroscience & Therapeutics, 18(3), 250–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranska, R. , Gysbrechts, L. , Wouters, J. , Vermeersch, P. , Bloch, K. , Dierickx, D. , … Snoeck, R. (2018). Comparison of membrane affinity‐based method with size‐exclusion chromatography for isolation of exosome‐like vesicles from human plasma. Journal of Translational Medicine, 16(1). 10.1186/s12967-017-1374-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweet, J. M. , Carda, B. , & Small, G. D. (1981). Repair of 3‐methyladenine and 7‐methylguanine in nuclear DNA of Chlamydomonas: Requirement for protein synthesis. Mutation Research, 84(1), 73–82. [DOI] [PubMed] [Google Scholar]
- Tamai, K. , Tanaka, N. , Nakano, T. , Kakazu, E. , Kondo, Y. , Inoue, J. , … Sugamura, K. (2010). Exosome secretion of dendritic cells is regulated by Hrs, an ESCRT‐0 protein. Biochemical and Biophysical Research Communications, 399(3), 384–390. [DOI] [PubMed] [Google Scholar]
- Théry, C. , Witwer, K. W. , Aikawa, E. , Alcaraz, M. J. , Anderson, J. D. , Andriantsitohaina, R. , … Zuba‐Surma, E. K. (2018). Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. Journal of Extracellular Vesicles, 7(1), 1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian, T. , Zhang, H.‐X. , He, C.‐P. , Fan, S. , Zhu, Y.‐L. , Qi, C. , … Gao, J. (2018). Surface functionalized exosomes as targeted drug delivery vehicles for cerebral ischemia therapy. Biomaterials, 150, 137–149. [DOI] [PubMed] [Google Scholar]
- Tkach, M. , & Théry, C. (2016). Communication by Extracellular Vesicles: Where We Are and Where We Need to Go. Cell, 164(6), 1226–1232. [DOI] [PubMed] [Google Scholar]
- Trajkovic, K. , Hsu, C. , Chiantia, S. , Rajendran, L. , Wenzel, D. , Wieland, F. , … Simons, M. (2008). Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science, 319(5867), 1244–1247. [DOI] [PubMed] [Google Scholar]
- Vemuganti, R. (2010). The MicroRNAs and Stroke: No Need to be Coded to be Counted. Translational Stroke Research, 1(3), 158–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkataramani, V. , Doeppner, T. R. , Willkommen, D. , Cahill, C. M. , Xin, Y. , Ye, G. , … Rogers, J. T. (2018). Manganese causes neurotoxic iron accumulation via translational repression of amyloid precursor protein and H‐Ferritin. Journal of Neurochemistry, 147(6), 831–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, E. Y. , Gang, H. , Aviv, Y. , Dhingra, R. , Margulets, V. , & Kirshenbaum, L. A. (2013). p53 mediates autophagy and cell death by a mechanism contingent on Bnip3. Hypertension, 62(1), 70–77. [DOI] [PubMed] [Google Scholar]
- Wang, P. , Guan, Y.‐F. , Du, H. , Zhai, Q.‐W. , Su, D.‐F. , & Miao, C.‐Y. (2012). Induction of autophagy contributes to the neuroprotection of nicotinamide phosphoribosyltransferase in cerebral ischemia. Autophagy, 8(1), 77–87. [DOI] [PubMed] [Google Scholar]
- Wang, P. , Shao, B. Z. , Deng, Z. , Chen, S. , Yue, Z. , & Miao, C. Y. (2018). Autophagy in ischemic stroke. Progress in Neurobiology, 163–164, 98–117. [DOI] [PubMed] [Google Scholar]
- Webb, R. L. , Kaiser, E. E. , Jurgielewicz, B. J. , Spellicy, S. , Scoville, S. L. , Thompson, T. A. , … Stice, S. L. (2018). Human neural stem cell extracellular vesicles improve recovery in a porcine model of ischemic stroke. Stroke; A Journal of Cerebral Circulation, 49(5), 1248–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei, K. , Wang, P. , & Miao, C.‐Y. (2012). A double‐edged sword with therapeutic potential: An updated role of autophagy in ischemic cerebral injury. CNS Neuroscience & Therapeutics, 18(11), 879–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin, H. , Li, Y. , Cui, Y. , Yang, J. J. , Zhang, Z. G. , & Chopp, M. (2013). Systemic administration of exosomes released from mesenchymal stromal cells promote functional recovery and neurovascular plasticity after stroke in rats. Journal of Cerebral Blood Flow and Metabolism, 33(11), 1711–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin, X‐Y. , Pan, J. , Wang, X.‐Q. , Ma, J.‐F. , Ding, J.‐Q. , Yang, G.‐Y. , & Chen, S.‐D. (2011). 2‐methoxyestradiol attenuates autophagy activation after global ischemia. Canadian Journal of Neurological Sciences, 38(4), 631–638. [DOI] [PubMed] [Google Scholar]
- Xing, S. , Zhang, Y. , Li, J. , Zhang, J. , Li, Y. , Dang, C. , … Zeng, J. (2012). Beclin 1 knockdown inhibits autophagic activation and prevents the secondary neurodegenerative damage in the ipsilateral thalamus following focal cerebral infarction. Autophagy, 8(1), 63–76. [DOI] [PubMed] [Google Scholar]
- Yoshimori, T. (2007). Autophagy: Paying Charon's toll. Cell, 128(5), 833–836. [DOI] [PubMed] [Google Scholar]
- Zagrean, A.‐M. , Hermann, D. M. , Opris, I. , Zagrean, L. , & Popa‐Wagner, A. (2018). Multicellular crosstalk between exosomes and the neurovascular unit after cerebral ischemia. Therapeutic implications. Frontiers in Neuroscience, 12, 811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, Z. , Li, Y. , Pan, Y. , Lan, X. , Song, F. , Sun, J. , … Liang, L. (2018). Cancer‐derived exosomal miR‐25‐3p promotes pre‐metastatic niche formation by inducing vascular permeability and angiogenesis. Nature Communications, 9(1), 5395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, J. , Bai, R. , Li, M. , Ye, H. , Wu, C. , Wang, C. , … Lin, D. (2019). Excessive miR‐25‐3p maturation via N(6)‐methyladenosine stimulated by cigarette smoke promotes pancreatic cancer progression. Nature Communications, 10(1), 1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, X. , Zhang, L. , Kuang, Y. , Venkataramani, V. , Jin, F. , Hein, K. , … Doeppner, T. R. (2020). Extracellular vesicles derived from neural progenitor cells—A preclinical evaluation for stroke treatment in mice. Translational Stroke Research. 10.1007/s12975-020-00814-z [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information S1.
Supporting Information S2.