

To systematically identify the substrates of RNF146 that are targeted for degradation, quantitative proteome and transcriptome analyses were performed in RNF146 KO and TNKS1/2 DKO cells. We identified 160 potential substrates of RNF146, which include many known substrates of RNF146 and TNKS1/2 as well as 122 potential TNKS-independent substrates of RNF146.
Keywords: RNF146, TNKS, E3 ubiquitin ligase, substrates, label-free quantification, substrate identification, cancer biology, mass spectrometry, protein degradation, ubiquitin, UbiquitinasesS
Graphical Abstract

Highlights
Proteome analyses reveal RNF146 and TNKS1/2 substrates targeted for degradation.
RNF146 KO and TNKS1/2 DKO cells display significantly different proteomes.
RNF146 has both TNKS-dependent and -independent substrates.
Abstract
Specific E3 ligases target tumor suppressors for degradation. Inhibition of such E3 ligases may be an important approach to cancer treatment. RNF146 is a RING domain and PARylation-dependent E3 ligase that functions as an activator of the β-catenin/Wnt and YAP/Hippo pathways by targeting the degradation of several tumor suppressors. Tankyrases 1 and 2 (TNKS1/2) are the only known poly-ADP-ribosyltransferases that require RNF146 to degrade their substrates. However, systematic identification of RNF146 substrates have not yet been performed. To uncover substrates of RNF146 that are targeted for degradation, we generated RNF146 knockout cells and TNKS1/2-double knockout cells and performed proteome profiling with label-free quantification as well as transcriptome analysis. We identified 160 potential substrates of RNF146, which included many known substrates of RNF146 and TNKS1/2 and 122 potential TNKS-independent substrates of RNF146. In addition, we validated OTU domain-containing protein 5 and Protein mono-ADP-ribosyltransferase PARP10 as TNKS1/2-independent substrates of RNF146 and SARDH as a novel substrate of TNKS1/2 and RNF146. Our study is the first proteome-wide analysis of potential RNF146 substrates. Together, these findings not only demonstrate that proteome profiling can be a useful general approach for the systemic identification of substrates of E3 ligases but also reveal new substrates of RNF146, which provides a resource for further functional studies.
Protein ubiquitination is one of the most common post-translational modifications, and it participates in a variety of cellular functions (1). Depending on the type of ubiquitination, ubiquitinated proteins are subjected to degradation or other functions, such as signal transduction (2). Proteolysis, which is mediated by the ubiquitin-proteasome system (UPS), is a major protein degradation system that maintains the homeostasis of the proteome (3). Deregulation of UPS has been shown to contribute to various human diseases, such as cancer and neurodegeneration (4, 5).
Protein ubiquitination is carried out by a cascade of sequential enzymes, E1 ubiquitin-activating enzyme, E2 ubiquitin-conjugating enzyme, and E3 ubiquitin-ligase (6). Proteins can be monoubiquitylated, multiubiquitylated, and polyubiquitylated by this cascade of enzymatic reactions. Among these enzymes, E3 ligase is the key enzyme that determines the specificity of these reactions because it recruits a particular target protein and transfers ubiquitin from the E2-conjugating enzyme to the target protein. About 600 E3 ligases have been identified in the human genome (7). They can be classified into 3 types based on their ubiquitin transfer domain: RING E3 ligases, HECT E3 ligases, and RBR E3 ligases (8). Most E3 ligases are RING E3 ligases. They contain either the zinc-binding domain, called the RING domain, or the U-box domain, which shares a similar structure with RING but does not contain zinc. The responsibility of the RING domain is to bind to E2 enzymes and transfer ubiquitin to substrates (9).
The E3 ligase RNF146, which is responsible for PARylation-dependent ubiquitination, is a special RING E3 ligase that contains 2 functional domains, RING domain and WWE domain (10). Poly-ADP-ribosylated (PARylated) proteins can be recognized by RNF146 with the binding of WWE domain to PARylated substrates. This binding functions as an allosteric signal to activate the RING domain (10). Although RNF146 can undergo auto-ubiquitination in the absence of PARylation, binding to PARylated chains greatly enhances its autoubiquitination (11–13). It is known that PARylation of substrates by Tankyrase 1 and 2 (TNKS1/2) is required for their ubiquitination by RNF146 (11). In fact, RNF146 and TNKS1/2 exist in a complex in which substrate selection and PARylation are determined by TNKS1/2 (10). TNKS1/2 are the only poly-ADP-ribosyltransferases that are known to be responsible for the PARylation and subsequent degradation of RNF146 substrates, and RNF146 is the only known E3 ligase for TNKS1/2. Thus, the known substrates of TNKS1/2, including AXIN1, AXIN2, AMOT proteins, TERF1, Golgin 45 (BLZF1), CASC3, and TNKS1/2, are also the substrates of RNF146. Although RNF146 normally targets substrates for degradation, different types of ubiquitin linkage promoted by RNF146 have also been reported (10, 12). For example, TNKS2 can be ubiquitinated by “Lys-48” and “Lys-63” chains (12). LKB1 is a novel TNKS1/2-dependent substrate of RNF146 that undergoes Lys-63 ubiquitination. This type of modification regulates the activity, but not the degradation, of LKB1, XRCC1, KU70, and LIG3 (13, 14).
RNF146 overexpression may enhance tumor development in lung cancer (52). RNF146 substrates determine the functions of RNF146. For example, RNF146 is an activator of the Wnt signaling pathway, because it degrades AXIN1 and AXIN2, which are key proteins required for β-catenin destruction (12). RNF146 also regulates the ubiquitination of PARP1, XRCC1, and KU70; protects against cell death induced by N-methyl-N-nitro-N-nitrosoguanidine; and promotes cell survival after irradiation (13). RNF146 enhances the oncogenic activity of YAP, which is the key downstream effector of Hippo pathway, by working together with TNKS1/2 to promote the degradation of angiomotin family proteins (AMOT, AMOTL1, and AMOTL2) (15). RNF146 is also known to be involved in normal physiological activities through its ability to target BLZF1 and CASC3 for degradation and therefore control the Golgi structure and pre-mRNA splicing (11).
Given that many reported substrates of RNF146 play critical functions in tumorigenesis, the discovery of additional substrates may be important for further studies of RNF146's functions and its inhibition in cancer treatment. However, to our knowledge, no systematic identification of RNF146 substrates has been performed. Therefore, it is unknown whether the functions of RNF146 and TNKS1/2 are exclusively linked. To systematically identify the substrates of RNF146 that are targeted for degradation, we generated RNF146 knockout (KO) cells and TNKS1/2 double KO (DKO) cells and performed a quantitative analysis of the proteome in these KO cells, with or without treatment with the TNKS1/2 inhibitor XAV939. We also performed transcriptome analysis using those KO cells to distinguish changes in protein or mRNA level. These results revealed 160 potential substrates of RNF146, which include many known and several unknown substrates of RNF146. These data sets will help investigators further define the TNKS1/2-dependent and -independent substrates and functions of RNF146.
EXPERIMENTAL PROCEDURES
Cell Culture and Transfection
293A cells were purchased from ATCC (Manassas, VA) and cultured in Dulbecco's modified Eagle's medium (DMEM) plus 10% fetal bovine serum and penicillin and streptomycin at 37 °C in 5% CO2 (v/v). RNF146 KO, TP53 KO, and TP53TNKS1/2 TKO cell lines were generated using CRISPR/Cas9 gene editing technology. All KO cell lines were validated by Western blot analysis and sequencing.
Polyethyleneimine was used for plasmid transfections. In brief, 10 μg of polyethyleneimine and 2 μg of indicated plasmid were mixed well with 300 μl of Opti-MEM ThermoFisher Scientific (Waltham, MA) and incubated for 15 min at ambient temperature. The mixture was then added to the 6-well plate for 24 h.
Chemicals
XAV939 and olaparib were obtained from Selleck Chemicals. All MS (MS)-grade solvents, including methanol, acetonitrile (ACN), formic acid, and water, were purchased from ThermoFisher Scientific (Waltham, MA). Urea, ammonium bicarbonate, trifluoroacetic acid, ammonium hydroxide (NH4OH), DTT, and iodoacetamide were obtained from Sigma-Aldrich. Trypsin Protease, MS Grade was obtained from ThermoFisher Scientific (Waltham, MA). Lysyl Endopeptidase, MS Grade (Lys-C) was purchased from Fujifilm Wako Chemicals (Richmond, VA). cOmplete Protease Inhibitor mixture was purchased from Roche.
Antibodies and Plasmid
pSpCas9 (BB)-2A-Puro (PX459) was purchased from Addgene. RNF146 constructs, including RNF146-ΔWWE (deleting amino acids 104-159) and RNF146-ΔRING (deleting amino acids 36-78) mutants, were obtained from Dr. Nan Li (16). The expression constructs for these proteins were generated by the GATEWAY cloning system (Invitrogen, Carlsbad, CA). In brief, sequences encoding these proteins were inserted to pDONOR201 vector by BP reaction. After being validated by sequencing, these sequences were recombined into Gateway-compatible destination vectors for the expression of N-terminal-tagged fusion proteins.
Antibodies that recognize the following proteins were used: PARP1 (#9542S), OTUD5 (#20087S), AXIN2 (#2151S), NFATC2 (#4389S), AMOT (#43130S), and MYC (#2272S) (Cell Signaling Tech); TNKS1/2 (H350) (Santa Cruz Biotechnology); DUSP9 (ab1943550 from Abcam); tubulin (T6199), β-actin (A5441), Flag (M2) (F3165), and AMOTL2 (HPA063027) (Sigma-Aldrich); PARP2 (AB_2793328) (Active Motif); and SARDH (A305-855A-M) (Bethyl Laboratories, Inc.).
Real-Time qPCR
After the indicated treatments, cells were washed with cold PBS twice. Total RNAs in each condition were extracted with TRIZOL reagent (Invitrogen). Reverse transcription of RNA to cDNA was performed with a High-Capacity cDNA Reverse Transcription Kit following the manufacturer's instructions (ThermoFisher Scientific, 4368814). Real-time PCR was performed using PowerUp™ SYBR™ Green Master Mix (ThermoFisher Scientific) and the 7500 real-time PCR system (Applied Biosystems) to measure the mRNA level of OTUD5. The 2 − ΔΔCt method was used to quantify gene expression, which was normalized with β-actin. The primer pairs used in this study were β-actin, GCCGACAGGATGCAGAAGGAGATCA/AAGCATTTGCGGTGGACGATGGA; OTUD5: ACTTCTGCAGTGGAACCCAT/GTTTGAATGATGGCAGGCCC.
CRISPR/Cas9-Mediated Gene KO
Special guide RNAs against the targeted gene were introduced into digested PX459 and then transiently transfected into 293A cells in 6-well plates. Twenty-four hours after transfection, cells were selected by puromycin (2 mg/ml) for 2 days and then seeded at 1 cell per well in 96-well plates. Clones were verified by Western blot analysis and sequencing.
Western Blot Analysis
After being washed with cold PBS twice, cells were directly lysed in plates with SDS sample loading buffer (50 mm Tris-Hcl [pH 6.8], 2% SDS, 10% glycerol, 4% 2-mercaptoethanol, and 0.025% Bromphenol blue) and then boiled for 15 min in 99 °C. A Western blot analysis was performed using standard SDS-polyacrylamide gel. Proteins were transferred into PVDF membrane (Millipore) from polyacrylamide gels, membrane would be blotted with antibodies as indicated.
Sample Preparation for MS Data Analysis
293A, RNF146 KO, TP53 KO, and TP53 TNKS1/2 TKO cells were mock treated or treated with XAV939 (10 μm) for 24 h. After being collected from dishes, cells were washed with cold PBS twice and lysed in chilled lysis buffer (8.0 M urea in 0.1 M NH4HCO3, supplemented with 1 × protease inhibitor mixture). The cells were incubated on ice for 30 min and then sonicated at 4 °C (2-min cycles of 5 s on and 10 s off at 30% output power for a tip-probe sonicator). After centrifugation, the supernatant was collected, and the protein concentration was determined by BCA protein assay following the manufacturer's instructions. The supernatant was diluted to equal protein concentrations. Equal proteins from each sample were reduced with 5 mm DTT at 56 °C for 30 min, alkylated with 15 mm iodoacetamide at ambient temperature in the dark for 30 min, and then quenched by 15 mm DTT. Samples were sequentially digested by Lys-C (enzyme/proteins, 1:100) for 4 h and then trypsin (enzyme: proteins, 1:50) overnight at 37 °C. Before trypsin digestion, the urea concentration was diluted into < 2 M with 0.1 M NH4HCO3. The digestion was quenched by trifluoroacetic acid to a final 0.1% concentration.
Cell Proliferation Assay
Four cell lines (293A, RNF146KO, TP53KO, TP53TNKS1/2TKO) were dissociated from dishes by trypsin digestion and seeded into 24-well cell culture plates (1 × 105 cells/well). Cells were counted every 2 days using a Bio-Rad cell counter (Bio-Rad Laboratories, Hercules, CA). Growth curves were drawn based on live cell numbers for 8 days.
RNA-Seq and Data Analysis
Four cell lines (293A, RNF146KO, TP53KO, TP53TNKS1/2TKO) were seeded into 6-well plates and allowed to grow (three biological replicates). RNeasy Mini Kit (Qiagen, 74104) and RNase-Free DNase (Qiagen, 79254) were used to extract total RNA according to the manufacturer's instructions. The library was prepared with the Illumina TruSeq Stranded Total RNA Library Prep kit including rRNA depletion and sequencing at NextSeq 500 (Illumina) to generate 75 bp from paired-ends.
The raw data were processed with FastQC to filter data. Genome mapping was carried out using Star software (v2.7.2b) and the human reference genome (UCSC hg38). The mapped reads were assembled into transcripts and genes with the human genome annotation file (genecode.v28.annotation.gtf) using HTseq software (v0.11.0). DESeq2 (v 1.28.1) was used to analysis the fold change and adjusted p value of transcripts or genes. The significantly changed genes were filtered with adjusted p-value ≤ 0.01, fold change cutoff of 2.
High-pH HPLC Fractionation
The tryptic peptides was desalted with the Sep-Pak SPE column (Waters, Milford, MA) and then dried with a SpeedVac. To decrease the sample complexity, the tryptic peptides were pre-separated by high-pH reverse-phase HPLC with a Waters XBridge C18 column (3.7-μm particles, 4.6 × 250 mm) (17, 18). Eluents were collected every 1 min in an 80-min gradient from 2% to 80% of buffer B (2% H2O, 98% ACN; buffer A: 98% H2O, 2% ACN, NH4OH, pH 10.5) at a flow rate of 0.7 ml/min. Ten fractions were pooled from collected eluents using a previously reported method (19).
NanoHPLC-MS/MS Analysis and Mass Spectrometry Data Analysis
Fractions from the above fractionation were dissolved with solvent A (0.1% formic acid in H2O) and loaded into a homemade reversed-phase trap-column (75 μm ID × 4 cm length, 1.9 μm C18). The peptides were separated with a homemade reversed-phase 25-cm analytical column (75 μm ID, 1.9 μm C18) in a 65-min gradient from 5% to 50% solvent B (0.1% formic acid in 80% ACN) with a constant flow rate of 300 nL/min using the EASY-nLC 1200 system (Thermo Fisher Scientific, Waltham, MA). The temperature of the analytical column was maintained at 50 °C by a column oven. The eluted peptides were ionized and sprayed into a Q Exactive HF-X mass spectrometer (Thermo Fisher Scientific, Waltham, MA) via a nanospray ion source in a positive mode. Data-dependent mode was set in the MS, the precursor scan was set as 375–1500 m/z with a resolution of 70,000 at m/z 200 and 100 ms of maximum injection time, and the automatic gain control target was 1e6. The 40 most intense ions above 1.5e4 were isolated and sequentially fragmentized by higher collision dissociation with normalized collision energy of 28%, with 1 m/z isolation windows and 60 s of dynamic exclusion time. Ion fragments were detected in the Orbitrap at a resolution of 17,500 at m/z 200 with an automatic gain control 1e6 and 100 ms of maximum injection time. Precursor ions with 1 charge or 5 or more charges were excluded for fragmentation.
The acquired MS/MS raw data were processed using MaxQuant software (version 1.6.5.0) and searched against the human proteomes database from uniprot (January 26, 2019, updated, 93,798 sequences) with a reversed decoy database by Andromeda search engine (20). The search was set at a false-discovery rate 0.01 for both the proteins and peptides, with a minimum length of 7 amino acids. Matching between runs was performed for each batch of data with default parameters, and label-free quantification (LFQ) was performed with a minimum ratio count of 2 (21). Cysteine carbamidomethylation was set as a fixed modification, and N-terminal acetylation and methionine oxidation were set as variable modifications. Peptides without modification were used for quantification. The precursor error tolerance was ±7 ppm with a fragment ion ± 20 ppm. Trypsin/P was chosen as the cleavage enzyme, and 2 maximum missing cleavages were allowed for each peptide in the database search. The proteingroup.txt file was used to next analysis.
Statistical Analysis
Proteins annotated with “reverse,” “potential contamination,” and “only identified by site” were removed. Proteins with an LFQ value in each condition were kept for next analysis. All statistical analyses were performed using Perseus software (version 1.5.8.0) (22). Data reproducibility was determined by calculating the Pearson correlation of the LFQ intensities of any 2 LC–MS/MS runs. The PCA component analysis was based on the LFQ intensities of all data. The differential proteins were calculated by a t test with a p-value ≤ 0.01, fold change cutoff of 2 and unique peptides ≥2.
Experimental Design and Statistical Rationale
For the MS analysis, the sample size is four for each data set, and a total of 8 samples in this study. For each sample, three biological replicates from the same single clone were collected at different time. Control samples are parental cell lines in each data set. Data statistical analyses were performed using Perseus software (version 1.5.8.0) (22). For RNA-seq data, there were four samples. For each sample, three biological replicates were collected and analyzed.
RESULTS
Quantitative Proteomic Profiling Reveals Potential Substrates of RNF146 and TNK1/2 Targeted for Degradation
To systematically identify proteins that were targeted for degradation by RNF146, we first used CRISPR/Cas9 genome-editing technology and successfully generated HEK293A-derived RNF146KO cells, which were confirmed by Western blot (Fig. 1A and supplemental Fig. S1). RNF146 specifically binds to PARylated proteins and mediates their ubiquitination and subsequent degradation (23). TNKS1/2 are the only known poly-ADP-ribosyltransferases that are responsible for substrates to be PARylated and then recognized by RNF146 for ubiquitination and degradation (11). However, the expression of TNKS and RNF146 and their correlation appear to be different in normal tissue versus in tumor tissues based on the TCGA and CPTAC databases (supplemental Fig. S2A and S2B). Moreover, RNF146 KO led to reduced cell proliferation, whereas TNKS1/2 DKO did not show any significant grow defect in p53 KO background (supplemental Fig. S2C). To specifically identify TNKS1/2-dependent and -independent substrates of RNF146, we also generated TNKS1/2DKO in HEK293A_TP53KO cells (Fig. 1A and supplemental Fig. S1). XAV939 specifically inhibits TNKS activity and is therefore known to increase the protein level of several substrates of TNKS (15, 23). Therefore, we treated each cell line with/without XAV939. The dose and time point for XAV939 treatment (10 μm, 24 h) were based on previous reports (24, 25). An MS-based label-free quantitative proteomics analysis was performed to profile the proteome change in each condition (Fig. 1B).
Fig. 1.
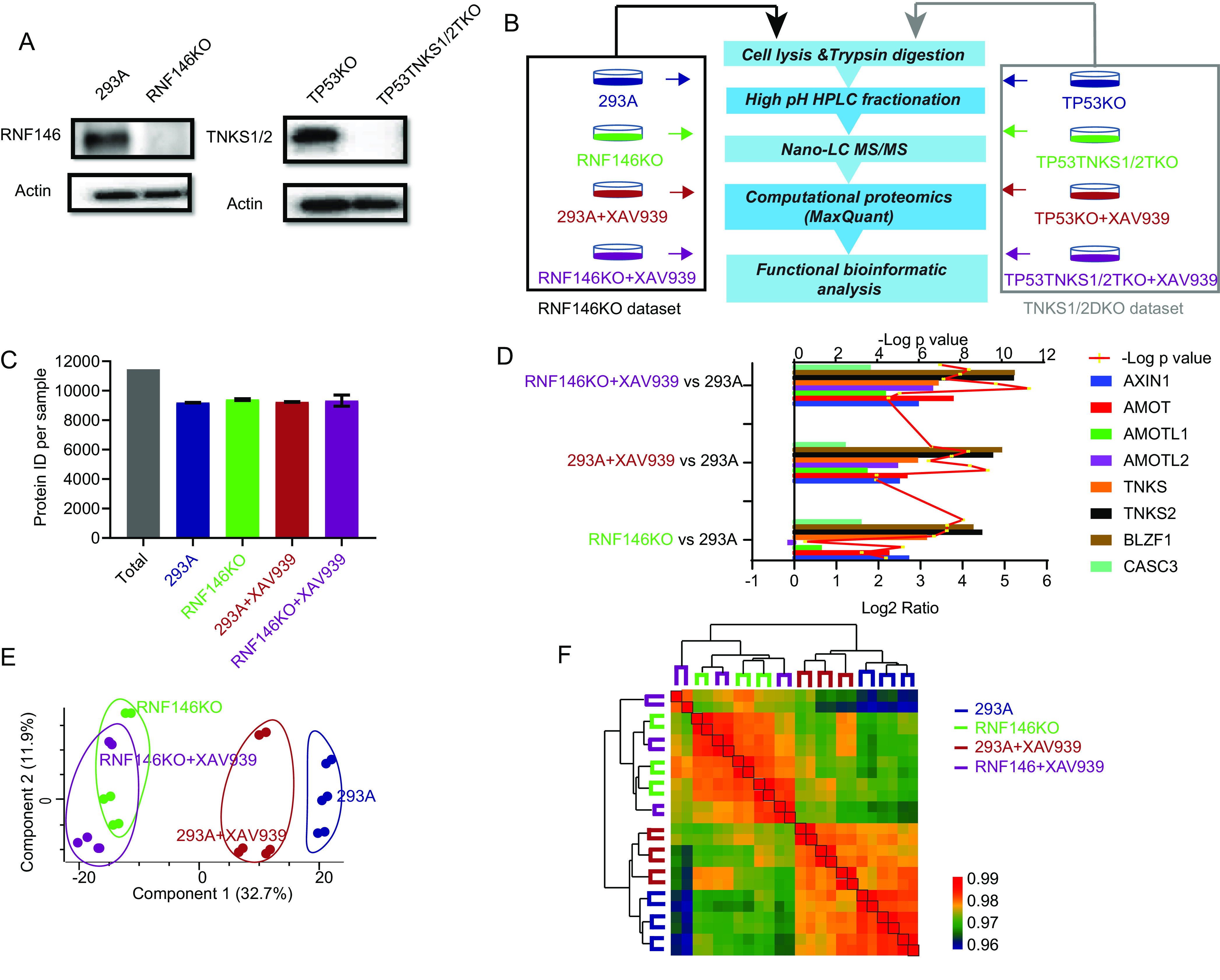
Label-free quantitative proteomics analysis of RNF146KO cells. A, Immunoblot analysis of whole cell lysates prepared from RNF146 KO and TNKS1/2 DKO cells created by CRISPR-Cas9 technology, conducted using the indicated antibodies. B, Proteomic workflow for label-free quantification. Each cell line was mock treated or treated with TNKSi (XAV939, 10 μm, 24 h). Equal amounts of proteins (peptides) from each condition were pre-fractionated by basic HPLC and then combined into 10 fractions. Each condition was performed using 3 biological replicates. The RNF146 KO data set and TNKS1/2 DKO data sets were separated by black and gray rectangles, respectively. C, The number of total quantified proteins of each sample in RNF146 data set. D, The known substrates of TNKS1/2 and RNF146 were used as positive controls, as shown by the Log2 ratio in the RNF146 KO data set. The y axis was shown for each condition compared with those in 293A cells. The line was shown for t test p value. E, Samples in the RNF146 KO data set were separated into 2 parts (WT and KO) using 2-dimensional principle component analyses. F, Heatmap shows the Pearson correlation among biological replicates and unsupervised clustering of samples in RNF146KO data set.
We generated 2 data sets, an RNF146KO data set and a TNKS1/2DKO data set. To ensure data reproducibility, proteins were extracted in 3 biological replicates in each cell line and condition. Equal amounts of proteins from each condition were digested and fractionated into 10 final fractions using high-pH RP-HPLC. After the MS analysis, the raw data were searched by MaxQuant (version 1.6.5.0) against the Uniprot Human database (January 26, 2019, updated) with a protein and peptide false discovery rate of 1%. To profile the proteome, we used “Match between Runs” in each data batch.
We totally identified 11,460 protein groups and average ∼9500 protein in each condition in RNF146KO data set (Fig. 1C). To determine the quality of these data sets, we evaluated some known substrates of TNKS and RNF146, such as AXIN1, BLZF1, AMOT, AMOTL1, and AMOTL2 (based Uniprot function annotation and (15)). As expected, the changing of these proteins (except AMOTL2 in the RNF146 KO cell line) was consistent with that in previous reports (Fig. 1D). Principal component analysis revealed that the samples could be distinguished into, RNF146KO and RNF146KO+XAV939, 293A+XAV939, and 293A groups (Fig. 1E). Although XAV939 treatment somewhat separated 293A and 293A+XAV939 samples, we could not separate RNF146KO samples with or without XAV939 treatment. Further, unsupervised cluster based on Pearson correlation of these samples revealed similar patterns (Fig. 1F). Each sample displayed high reproducibility with Pearson's r > 0.96; the replicates in each sample had a higher correlation. Together, these results demonstrate the high quality of RNF146KO data set.
In TNKS1/2DKO data set, a total of 11,241 protein groups were identified with ∼9300 protein groups in each sample (Fig. 2A). The changes of the known substrates in TNKS1/2DKO data set was consistent with that in previous reports (Fig. 2B). Similarly, the principal component analysis clearly separated TP53KO and TP53TNKS1/2TKO cell lines but could not separate samples with or without XAV939 treatment (Fig. 2C). The reproducibility of these samples was high (Pearson's r > 0.96) and unsupervised cluster showed similar patterns with principal component analysis (Fig. 2D). Therefore, the quality of our TNKS1/2DKO data set is also high and worth further analysis.
Fig. 2.
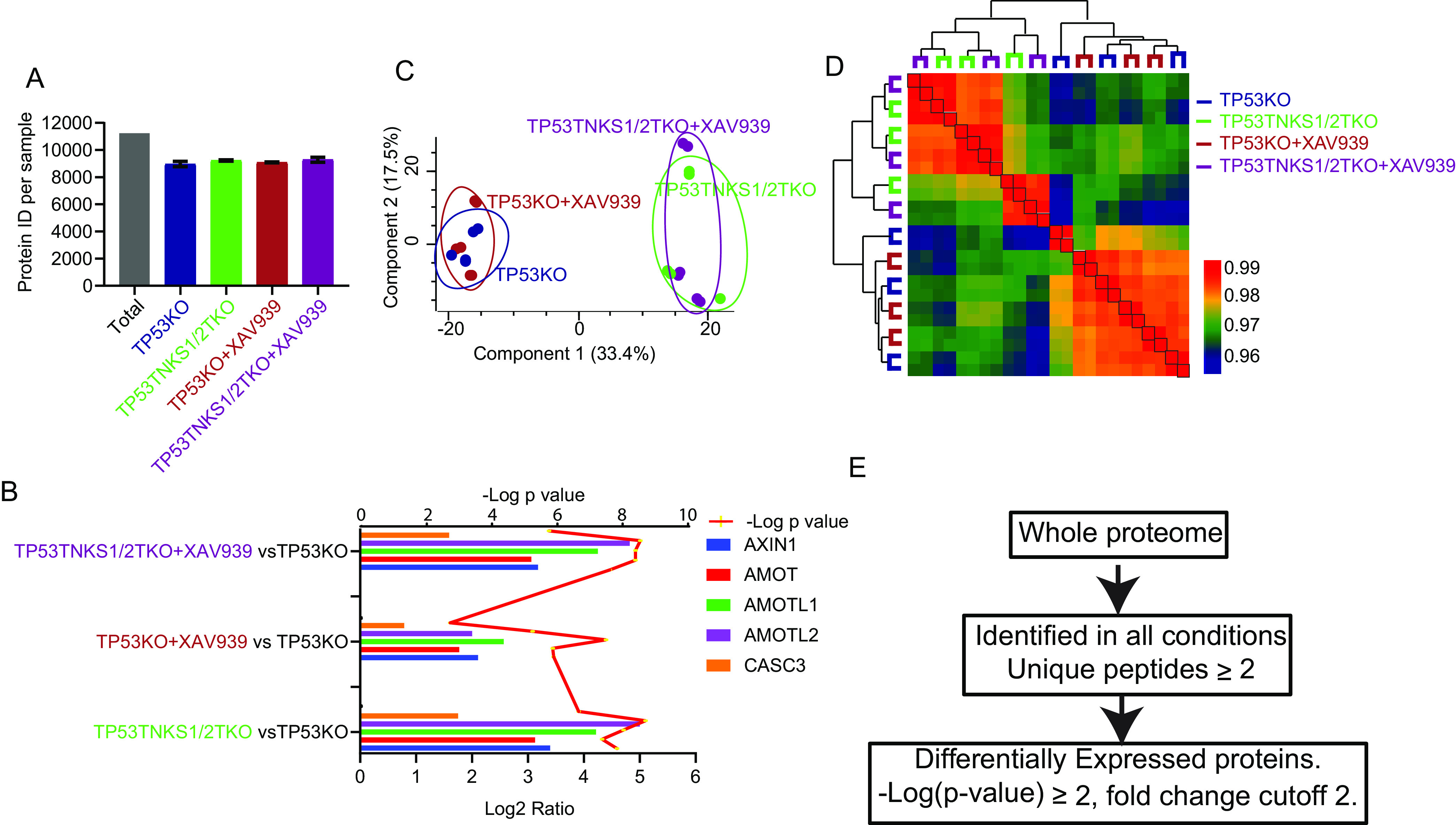
Label-free quantitative proteomics analysis of TNKS1/2DKO cells. A, The number of total quantified proteins of each sample in TNKS1/2DKO data set. B, The known substrates of TNKS1/2 and RNF146 were used as positive controls, as shown by the Log2 ratio in the RNF146 KO data set. The y axis was shown for each condition compared with those in TP53KO cells. The line was shown for t test p value. C, Samples in the TNKS1/2 DKO data set were separated into 2 parts (WT and KO) by 2-dimensional principle component analyses. D, Heatmap shows the Pearson correlation among biological replicates and unsupervised clustering of samples in TNKS1/2DKO data set. E, Outline of analysis in which proteins were significantly and differentially expressed in each data set.
To identify substrates of RNF146 and/or TNKS1/2 that are targeted for degradation, we processed the data using the following criteria to define differential proteins (Fig. 2E). To achieve high-quality quantification, we selected proteins with an LFQ value in all conditions. After filtering, there were 7238 (RNF146KO data set) and 7130 (TNKS1/2DKO data set) quantifiable proteins in the 2 data sets (supplemental Tables S1 and S2). Among those proteins, significant changes between two conditions were determined using t test, filtered by p-value ≤ 0.01, a fold change cutoff of 2 and unique peptides ≥2. These criteria led to 210 (RNF146KO data set) and 188 (TNKS1/2DKO data set) differentially expressed proteins when we compared XAV939 treatment and/or KO cells with control cells in these 2 data sets. In the RNF146KO data set, 161 and 43 proteins were significantly upregulated and downregulated, respectively, in RNF146KO versus 293A; 35 and 3 in 293A+XAV939 versus 293A (supplemental Table S3). In the TNSK1/2DKO data set, 123 and 65 proteins were significantly upregulated and downregulated, respectively, in TP53TNKS1/2TKO versus TP53KO; 3 upregulated proteins in TP53KO+XAV939 versus TP53KO (supplemental Table S4).
Proteomic Analysis Reveals Targets Regulated by XAV939
XAV939 is a specific TNKS inhibitor commonly used to study the functions of TNKS1/2 (23). In this study, we generated 2 data sets with 2 control cell lines (293A and TP53KO). In our data, several known substrates of TNKS and RNF146 were upregulated by XAV939 in both data sets (Figs. 1D and 2C), although XAV939 treatment only slightly affected the proteomes of 293A and TP53KO cells (supplemental Fig. S3A). Nevertheless, 35 and 3 proteins were significantly upregulated in 293A+XAV939 and TP53KO+XAV939 cells respectively, compared with those in controls (supplemental Fig. S3A). The known substrates of TNKS1/2 appeared as the top upregulated proteins in both data sets. Together, these data suggest that XAV939 treatment was effective and showed high specificity in this study.
Angiomotin family proteins (AMOT, AMOTL1 and AMOTL2) are known substrates of TNKS1/2 that can be upregulated by XAV939 (15). However, in our data sets, the AMOTL2 level was not increased in RNF146KO cells; in fact, it even decreased slightly. This may be because of a negative feedback loop of Hippo/YAP pathway, because although all three AMOT family proteins are negative regulators of YAP activity, AMOTL2 appears to be transcriptionally upregulated by YAP activation (26). Nevertheless, XAV939 treatment significantly increased the level of AMOTL2 in RNF146 KO cells (Fig. 1D). Although, knockout of RNF146 significantly increased AMOT and AMOTL1, treatment with XAV939 appeared to further enhance these increases in RNF146KO cells (Fig. 1D and supplemental Fig. S3B). In addition, when the targets of XAV939, i.e. TNKS1/2, were absent, XAV939 treatment did not affect the proteome (supplemental Fig. S3B). Together, these data may indicate that besides RNF146, there may be an additional E3 ligase that acts with TNKS1/2. However, the role of this putative ligase may be limited and only appear to be involved in the regulation of AMOT family proteins.
To further characterize the upregulated proteins following XAV939 treatment, we combined all the upregulated proteins in supplemental Fig. S3A and S3B into a list (i.e. Upregulated proteins in 293A+XAV939 versus 293A, TP53KO+XAV939 versus TP53KO, RNF146KO+XAV939 versus RNF146KO, TP533TNKS1/2TKO+XAV939 versus TP53TNK1/2TKO). This list contains 36 proteins (supplemental Table S5) and most of them are the upregulated proteins in 293A+XAV939 versus 293A group. We first performed pathway and process enrichment analysis by Metascape (27). As expected, the top enrichment pathway was WNT pathway (supplemental Fig. S3C). The known substrates of TNKS1/2 contain a consensus tankyrase binding site (RXXG[P/A/C]XG); an earlier study suggested that proteins containing this binding site are potential substrates of TNKS1/2 (28). Therefore, we compared the upregulated proteins identified in our study with those predicted substrates. We also compared our data with our previously published list of high-confidence interaction proteins (HCIPs) of TNKS1/2 (29), because potential substrates of TNKS1/2 should also interact with TNKS1/2. Indeed, we identified 9 proteins upregulated by XAV939 treatment that also present in HCIPs of TNKS and are predicted TNKS1/2 substrates (supplemental Fig. S3D), 7 of which are known substrates of TNKS, indicating the high quality of our data. The other 2 proteins, FAM117B and GLCCI1, are paralogs of each other. They were both slightly upregulated in TP53 KO cells treated with XAV939 (fold changes = 1.63 and 1.79, -Log p-values = 4.52 and 4.79, respectively). These results suggest that these 2 proteins are likely substrates of TNKS1/2 and are regulated by XAV939. In addition, we found another 7 upregulated proteins as predicted substrates of TNKS, because they contain the consensus tankyrase binding site (28). We anticipate that these are also potential TNKS substrates regulated by XAV939.
Proteomic Analyses Reveal Targets Regulated by TNKS1/2
As for the TNKS1/2 data set, in theory, TNKS1/2DKO cells and XAV939-treated cells should have similar proteomes. However, this was not the case in our study (Fig. 2C). There were more upregulated proteins in TP53TNKS1/2TKO cells than those in TP53KO+XAV939 cells when they were compared with TP53KO cells (Fig. 3A and supplemental Fig. S3A). Most upregulated proteins in TP53TNKS1/2TKO cells were still upregulated when the proteome of TP53TNKS1/2TKO cells was compared with that of TP53KO+XAV939 cells, including several known substrates (supplemental Fig. S4A). XAV939 treatment in TP53TNKS1/2TKO cells did not affect most of the upregulated proteins of TP53TNKS1/2TKO compared with TP53KO cells (supplemental Fig. S4B).These data indicate that inhibition of TNKS1/2 by XAV939 did not mimic TNKS1/2 DKO. More substrates are regulated by TNKS1/2 based on TNKS1/2DKO cells, but these substrates did not seem to be affected by XAV939 treatment, presumably because XAV939 did not completely block TNKS1/2 activity and the residual activities of TNKS1/2 in the presence of XAV939 are sufficient for modifying and resulting in the degradation of some TNKS substrates. Indeed, XAV939 treatment in TP53TNKS1/2TKO cells did not lead to any significant change in the proteome of these cells (supplemental Fig. S3B), suggesting that XAV939 is a specific TNKS inhibitor.
Fig. 3.

Protein changed by double knockout of TNKS1/2. A, Volcano plot shows the significant change in differentially expressed proteins in TP53TNKS1/2TKO versus TP53KO cells. Each dot represents a protein. The brown and olive dot indicate significantly upregulated and downregulated proteins, respectively. The known substrates of TNKS1/2 are labeled with gene names. y axis is the –Log p-value, and x axis is the Log2 fold change. Tests were performed using a t test filtered by p-value ≤ 0.01, a fold change cutoff of 2 and unique peptide ≥ 2. B, The top 5 upregulated proteins in TP53TNKS1/2TKO versus TP53KO cells shown with Log2 fold change and –Log p value. C, Immunoblot assay of whole cell extracts for SARDH and NFATC2 in TP53KO and TP53TNKS1/2TKO cells plus XAV939 treatment (10 μm, 24 h). TNKS1/2, AMOT, and AXIN2 were used as positive controls, and β-actin was used as the loading control. D, Reintroduction of TNKS could rescue the overexpression of SARDH and NFATC2.
To systematically reveal the substrates of TNKS1/2, we analyzed the significant differently expressed proteins in TP53TNKS1/2TKO versus TP53KO (Fig. 3A). We then focused on the 123 upregulated proteins in TP53TNKS1/2TKO versus TP53KO as shown in Fig. 3A. One third of those upregulated proteins contain the consensus tankyrase binding site (supplemental Fig. S4C). Moreover, four of the top 5 upregulated proteins contain the consensus TNKS binding site (Fig. 3B and supplemental Fig. S4C). Two are known TNKS substrates, AMOTL1 and AXIN1; the other three are periplakin (PPL), nuclear factor of activated T-cells, cytoplasmic 2 (NFATC2) and sarcosine dehydrogenase, mitochondrial (SARDH). PPL is a component of cornified envelopes (30), whereas NFATC2 acts as a transcription factor and promotes invasive migration (31, 32). PPL has 2 high-confidence TNKS binding sites, whereas NFATC2 has 1. Although these 2 proteins are not present in the list of HCIPs of TNKS1/2, the change in NFATC2 was confirmed by Western blot analysis (Fig. 3C and supplemental Fig. S4D). SARDH is a mitochondrial protein that catalyzes the oxidative demethylation of sarcosine (33). Although this protein did not contain the consensus TNKS binding site, the change in SARDH protein level was confirmed by immunoblotting (Fig. 3C). However, the change in the SARDH and NFATC2 protein levels was not affected by XAV939 treatment, again suggesting that not all TNKS1/2 substrates are affected by XAV939. Alternatively, TNKS1/2 may have some catalytic independent functions that remain to be elucidated. We will consider these possibilities in the Discussion.
To uncover the potential substrates of TNSK1/2, we also compared transcript changes with proteome changes (supplemental Fig. S4E and supplemental Table S6). There was low correlation (∼0.2) between these changes, indicating that post-transcriptional control mechanisms play major roles in the regulation of TNKS1/2 proteome. The low correlation between changes in RNA and protein levels was also reported in an early study (34). One hundred sixteen out of the 123 upregulated proteins including the known TNKS substrates did not show significant change in their transcript level (supplemental Table S6). These results suggest that the change of TNKS1/2 proteome is mainly controlled by post-transcriptional regulation, although a few changed proteins including PPL also showed change in their transcript level.
To further validate SARDH and NFATC2 as substrates of TNKS1/2, we examined SARDH and NFATC2 levels in 293A_TP53TNKS1/2TKO cells that had been transfected with control vector or vectors encoding TNKS. As shown in Fig. 3D, SARDH and NFATC2 levels significantly decreased with reconstitution of TNKS. These results indicate that SARDH and NFATC2 are likely to be regulated by TNKS1/2.
RNF146 Has TNKS-Independent Substrates
RNF146 is a PARylation-dependent E3 ligase (10). Binding with the PARylated chain is an allosteric signal that activates RNF146. TNKS1/2 and RNF146 work together to ribosylate, ubiquitinate, and eventually degrade their substrates. For now, RNF146 is the only E3 ligase that is known to be responsible for the ubiquitination and degradation of TNKS1/2 substrates. TNKS1/2 is also the only known ADP-ribosyltransferase that works with RNF146. However, RNF146KO cells did not show significantly upregulated AMOTL1 and AMOTL2, which are known TNKS1/2 substrates (Fig. 1D and 4A). In addition, AMOT, TNKS2, and BLZF1 were modestly upregulated in RNF146KO+XAV939 cells compared with in RNF146KO cells (Fig. 1D). These data imply that TNKS may have substrates that are not exclusively regulated by RNF146. Similarly, it is also possible that RNF146 may target additional substrates beyond those that are regulated by TNKS1/2.
Fig. 4.
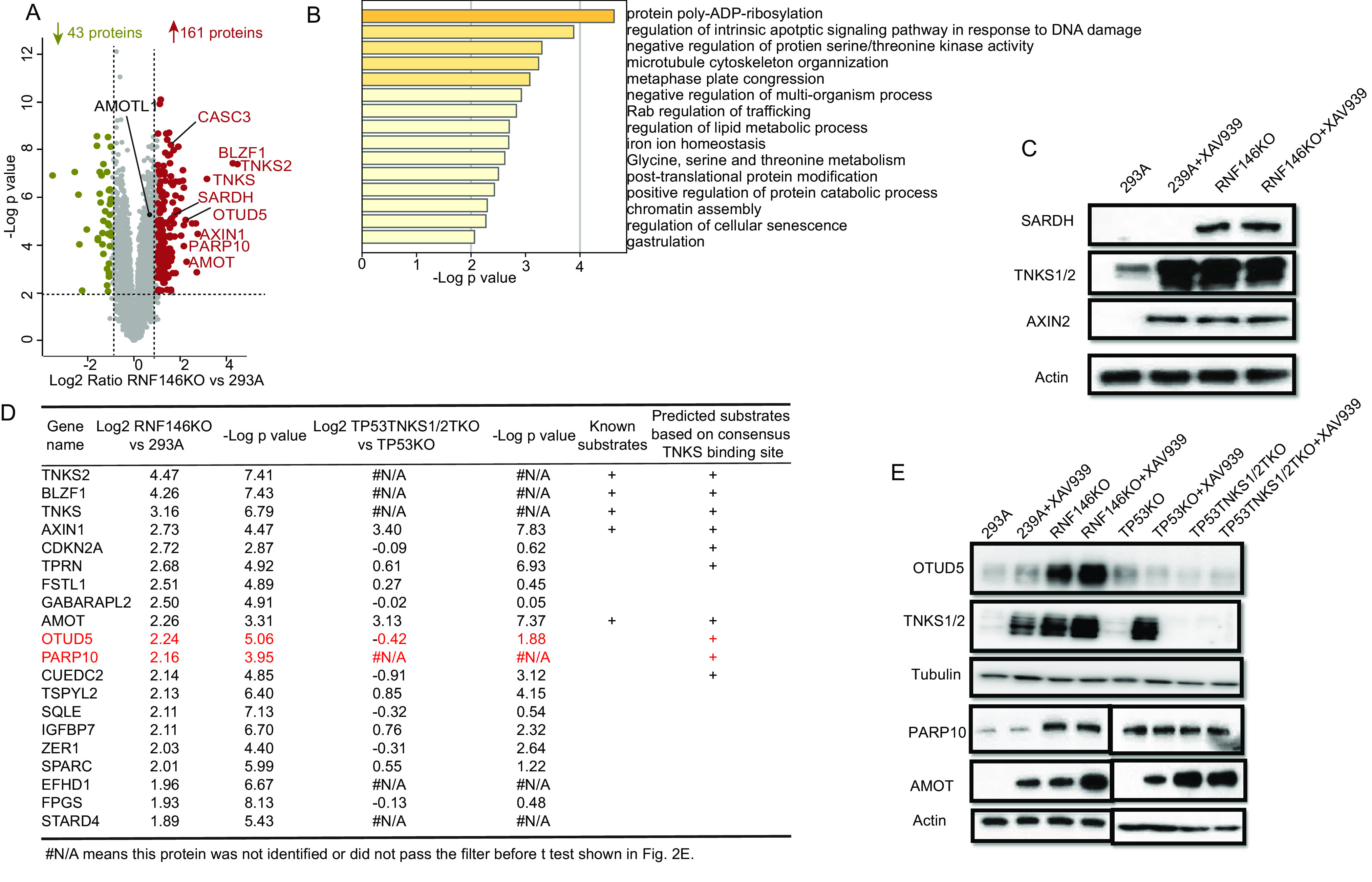
Proteins targeted for degradation by RNF146. A, Volcano plot shows the significant change in differentially expressed proteins in RNF146KO versus 293A cells. Each dot represents a protein. The brown and olive dots indicate significantly upregulated and downregulated proteins, respectively. The known substrates of RNF146 are labeled with gene names. y axis is the –Log p-value, and x axis is the Log2 fold change. Tests were performed using a t test filtered by p-value ≤ 0.01, a fold change cutoff of 2 and unique peptide ≥ 2. B, Biological process enrichment analysis by Metascape for upregulated proteins. C, Immunoblot assay of whole cell extracts for SARDH prepared using 293A, 293A+XAV939, RNF146KO, and RNF146KO+XAV939 cells. TNKS1/2 and AXIN2 were used as positive controls; and β-actin was used as the loading control. D, The top20 upregulated protein in RNF146KO versus 293A. E, OTUD5 was upregulated in 293A_RNF146 KO and 293A_RNF146 KO+XAV939 cells, but not in other cells. TNKS1/2 were used as positive controls, and Tubulin was used as the loading control. PARP10 was significantly increased in RNF146KO cells, but not in TNKS1/2 DKO cells or XAV939-treated cells. AMOT was included as a positive control, and β-actin was used as the loading control.
To identify the potential substrates of RNF146, we first analyzed significant differently expressed proteins in RNF146KO versus 293A and obtained a list of 161 upregulated proteins and 43 downregulated proteins (Fig. 4A). We used the Metascape to analyze process enrichment for upregulated proteins and showed that protein poly-ADP-ribosylation is at the top of these processes (Fig. 4B). We then compared these upregulation proteins with upregulated proteins in cells with double knockout of TNKS1/2 (supplemental Fig. S5A). However, the overlap between knockout of RNF146 and double knockout of TNKS1/2 was low, with only 11 proteins upregulated in both conditions (supplemental Fig. S5A); this finding suggests that RNF146 and TNKS1/2 have different substrates, which we explored further. Four of the 11 overlapped proteins contain the consensus tankyrase binding site, including 3 well-known TNKS substrates (AXIN1, AMOT and CASC3). There were also 2 significantly upregulated proteins, monoglyceride lipase (MGLL), which is a cytosol protein that controls the fatty acid level and promotes cancer cell migration (35) and SARDH (supplemental Fig. S5A). Interestingly, neither contains a tankyrase binding site. SARDH catalyzes the oxidative demethylation of sarcosine and is highly increased during prostate cancer progression (36). We showed in Fig. 3 that SARDH is regulated by TNKS1/2. Here, we further validated the increase of SARDH in RNF146 KO cells by Western blot analysis (Fig. 4C). These findings suggest that RNF146 and TNKS1/2 partly control cancer progression by regulating cancer metabolism.
To further validate the 161 upregulated proteins, we analyzed the correlation of transcript level changes with the proteome level changes (supplemental Fig. S5C and supplemental Table S7). Similarly, the correlation was low (∼0.2), indicating RNF146 regulates the proteome level but not transcription. Except KIF1A, 160 out of 161 proteins, did not show significant transcript level change (supplemental Table S7).
To identify the TNKS1/2-independent substrates of RNF146, we first compared the top 20 upregulated proteins RNF146KO cells and TP53TNKS1/2TKO cells. As expected, known substrates of TNKS showed up in both the top 20 proteins in RNF146KO cells and in TP53TNKS1/2TKO (Fig. 4D). However, five of the top 20 upregulated proteins in TP53TNKS1/2TKO cells showed decrease trend in RNF146KO cells, including the proto-oncogene PCK1 and JUN (Table I). These results demonstrated the differential regulation of protein abundance by RNF146 and TNKS1/2 and the importance to reveal TNKS-independent substrates of RNF146.
Table I. The top 20 upregulated proteins in TP53TNKS1/2 TKO cells.
| Gene Name | Log2 TP53TNKS1/2TKO versus TP53KO | -Log p value | Log2 RNF146KO versus 293A | -Log p value | Known Substrates | Predicted Substrates Based on consensus TNKS binding site |
|---|---|---|---|---|---|---|
| AMOTL1 | 4.22 | 8.03 | 0.65 | 5.28 | + | + |
| SARDH | 3.82 | 7.10 | 1.75* | 5.29 | ||
| PPL | 3.51 | 7.74 | N/A# | N/A# | + | |
| AXIN1 | 3.40 | 7.83 | 2.73* | 4.47 | + | + |
| NFATC2 | 3.17 | 5.07 | N/A# | N/A# | + | |
| AMOT | 3.13 | 7.37 | 2.26* | 3.31 | + | + |
| S100A16 | 2.37 | 4.74 | −0.87 | 2.40 | ||
| EPHA2 | 2.29 | 5.55 | −0.45 | 2.75 | ||
| TGFB1I1 | 2.15 | 5.47 | 0.74 | 3.22 | ||
| NAB2 | 2.15 | 7.01 | 0.44 | 3.07 | ||
| PCK1 | 2.08 | 4.26 | −3.54* | 6.91 | + | |
| HMGA2 | 2.07 | 4.50 | −0.61 | 2.80 | + | |
| FAT1 | 2.04 | 3.98 | N/A# | N/A# | + | |
| COL6A2 | 2.02 | 6.29 | N/A# | N/A# | ||
| JUN | 1.99 | 8.35 | −1.02* | 4.48 | ||
| MGLL | 1.99 | 6.76 | 1.79* | 2.84 | ||
| CORO2B | 1.94 | 4.38 | 0.51 | 1.13 | ||
| TIMP1 | 1.93 | 6.47 | 1.36* | 6.69 | ||
| ALDH1A2 | 1.93 | 7.22 | N/A# | N/A# | ||
| TAGLN | 1.93 | 2.58 | 1.17* | 4.68 |
#N/A means this protein was not identified or did not pass the filter before t test shown in the Fig. 2E.
*means this protein was significantly different expression.
To further define the TNKS-independent substrates, proteins upregulated in TNKS1/2 DKO cells were excluded from the list of 161 proteins upregulated in RNF146 KO cells. This provided us a list of 122 potential substrates of RNF146 that are not regulated by TNKS1/2 KO (supplemental Fig. S5A and supplemental Table S7). We chose 2 proteins, protein mono-ADP-ribosyltransferase PARP10 (PARP10) and OTU domain-containing protein 5 (OTUD5), for further validation, because they are among the top 20 upregulated proteins in RNF146 KO cells (Fig. 4D, labeled with red color). Both OTUD5 and PARP10 level increased significantly in RNF146KO cells but not in TNKS1/2KO cells (Fig. 4E). We reason that there are additional substrates of RNF146 that are independent of TNKS1/2.
Validation of RNF146 Substrates
As mentioned above, we validated the upregulation of 3 potential new substrates of TNKS and RNF146 (i.e. SARDH, PARP10, and OTUD5) by Western blot analysis. Because OTUD5 appears to be regulated by RNF146 but not by TNKS, we further determined whether OTUD5 is a potential substrate of RNF146. OTUD5 is a deubiquitinating enzyme with a preference for Lys63 linkages (37). OTUD5 negatively regulates type I interferon and interleukin-10 responses by deubiquitylating TRAF3, which diminishes IL-17A production (38–40). There is also evidence that OTUD5 can deubiquitinate and stabilize TP53 (41). We further examined how the OTUD5 level was controlled by RNF146. RNA isolation from various cell lines, followed by an RT-qPCR analysis, revealed that the OTUD5 mRNA level did not change in RNF146 or other KO cell lines with or without TNKSi treatment (Fig. 5A). We next determined the stability of OTUD5 by treating cells with translation inhibitor cycloheximide. Cycloheximide treatment led to a rapid decline in OTUD5 level in WT but not RNF146KO cells (Fig. 5B), which was like that of TNKS1/2, a known substrate of RNF146. These data suggest that like TNKS1/2, OTUD5 is likely regulated by RNF146 via ubiquitination and degradation.
Fig. 5.
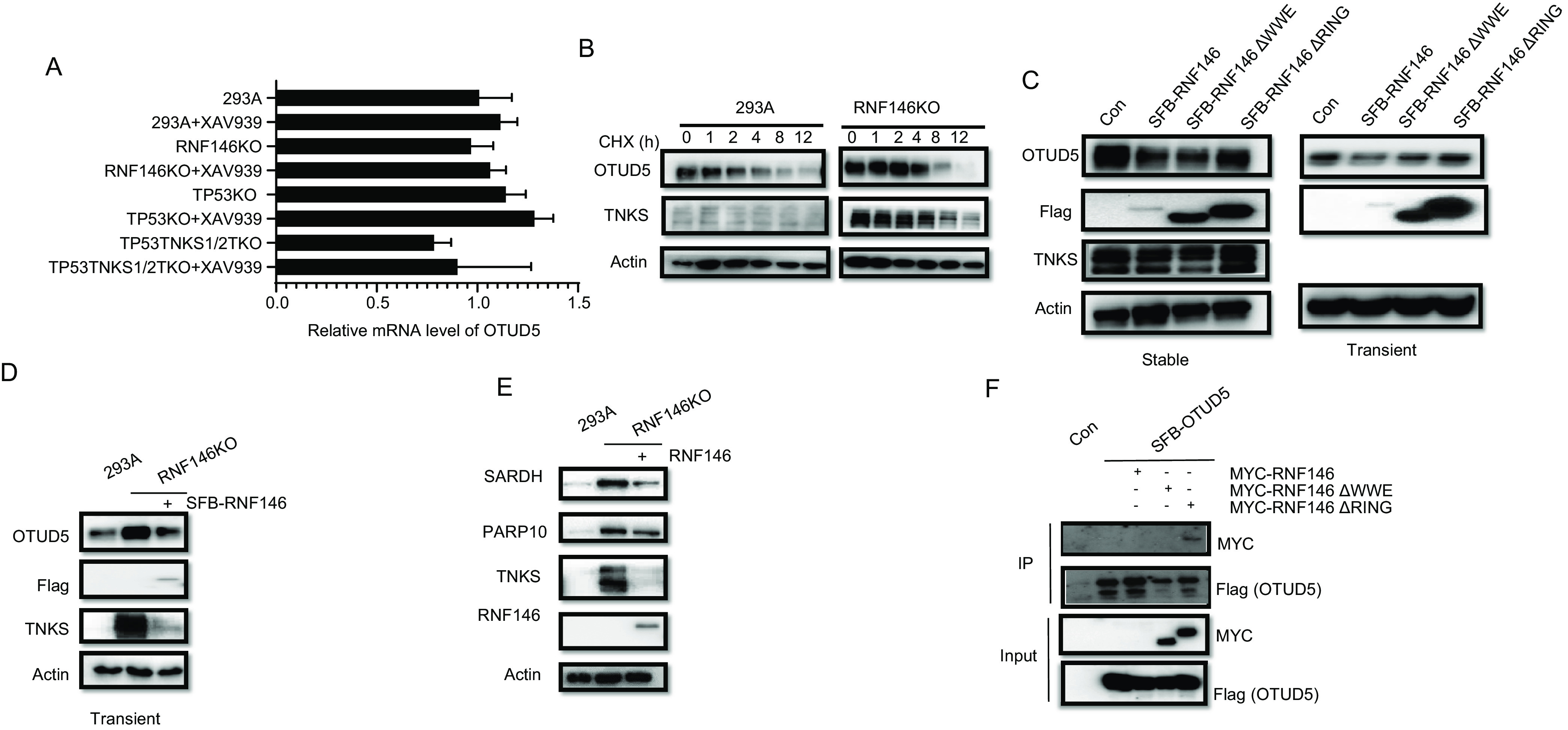
OTUD5 is a potential TNKS-independent target of RNF146. A, mRNA levels of OTUD5 were assessed by real-time qPCR after the indicated treatments and are presented as means ± S.E. of 3 biological replicates. B, Immunoblot assay of whole cell extracts for OTUD5 and TNKS expression was conducted in 293A and RNF146 KO cells treated with 50 µg/ml cycloheximide for the indicated times. C, Immunoblot assay of 293A cells with overexpression of N-SFB-RNF146, N-SFB-RNF146 ΔWWE, or N-SFB-RNF146 ΔRING with the indicated antibody (left: stable overexpression; right: transient overexpression). D, Reintroduction of N-SFB-RNF146 into RNF146 KO cells was validated by immunoblotting with the indicated antibodies. E, Reintroduction of RNF146 could rescue the overexpression of SARDH and PARP10 in RNF146 KO cells. F, Co-immunoprecipitation of OTUD5 with RNF146. RNF146 and RNF146 mutants were fused with N-terminal MYC tags and transiently expressed in 293T cells, whereas OTUD5 with N-terminal S protein, Flag, streptavidin-binding peptide (SFB) tag. We harvested cells after 24 h, pulled down the overexpressed proteins with streptavidin beads, and analyzed them via Western blot analysis.
To further validate OTUD5 as a substrate of RNF146, we examined OTUD5 levels in 293A and RNF146KO cells that had been transfected with control vector or vectors encoding WT RNF146. As the WWE domain of RNF146 is required for the recognition of ribosylated proteins and the RING domain is critical for its E3 ligase activity (11), we generated RNF146 △WWE and RNF146 △RING deletion mutants. As shown in Fig. 5C, the OTUD5 protein level was decreased in 293A cells expressing WT RNF146. Similar results were obtained in RNF146KO cells (Fig. 5D). We also determined the levels of PARP10 and SARDH in RNF146 KO cells that had been reconstituted with RNF146. Both proteins were upregulated in KO cells, but downregulated when we reintroduced RNF146 (Fig. 5E). Together with the data presented in Fig. 4E and 4F, our data indicate that SARDH is a common substrate of TNKS1/2 and RNF146, whereas PARP10 is a TNKS1/2-independent substrate of RNF146.
Next, we examined the interaction between OTUD5 and RNF146 by performing co-immunoprecipitation experiments. RNF146 can degrade itself; therefore, the expression level of WT RNF146 is usually extremely lower than that of mutants (39). As shown in Fig. 5F, OTUD5 co-immunoprecipitated with RNF146 △RING mutant, but not with RNF146 △WWE mutant, when these mutants were expressed at similar levels, suggesting that OTUD5 may be ribosylated before it could bind to and be ubiquitinated by RNF146. Besides TNKS, PARP1 also modify substrates by poly(ADP)-ribosylation, and there is evidence that RNF146 can interact with PARP1 (13, 42). Thus, we determined whether the OTUD5 level would change in the presence of PARP1 inhibitor olaparib. As shown in supplemental Fig. S5B, treatment with olaparib did not change the protein level of OTUD5, indicating that OTUD5 is not regulated by PARP1.
DISCUSSION
In this study, we performed an in-depth label-free quantitative proteomics analysis to identify RNF146 substrates. In total, more than ten thousand proteins were identified in 2 data sets. After data analysis using stringent statistical criteria, 161 proteins were upregulated in the RNF146KO cells, which include known substrates of TNKS1/2. Compared with RNA-seq data and protein upregulated in TNKS1/2 DKO, we compiled a list of 160 proteins that are potential substrates of RNF146, and 122 potential TNKS-independent substrates of RNF146 (supplemental Table S7). We validated one of the novel substrates, SARDH, which is shared by TNKS1/2 and RNF146. However, a significant fraction of proteins that were upregulated in RNF146 KO cells were not upregulated in TNKS1/2 DKO cells compared with their control cells (supplemental Fig. S5A), indicating that RNF146 and TNKS1/2 may have distinct substrates. Indeed, we confirmed that OTUD5 and PARP10 are potential TNKS1/2-independent substrates of RNF146. These data together suggest that RNF146 has both TNKS-dependent and -independent roles in the cell (Fig. 6). Our data provide a rich resource for further functional studies of RNF146.
Fig. 6.

Proposed model for different RNF146 and TNKS substrates. Proteins are poly-ADP-ribosylated by TNKS1/2, or other poly-ADP-ribose polymerase, or mono-ADP-ribosylated by PARP10 then poly-ADP-ribosylated. Those poly-ADP-ribosylated protein are recognized and ubiquitylated by RNF146. RNF146 can make two type ubiquitination, K48 and K63-linked ubiquitination. Proteins with K48-linked ubiquitination are subjected to degradation, like PARP10, OTUD5 and SARDH in this study.
E3 ligases such as RNF146 are known to play critical functions in many cellular processes. Identifying substrates of these E3 ligases is critical for understanding their roles in human physiology and pathology. There are 3 general methods for identifying substrates of E3 ligases with MS analysis: affinity-purification MS (AP-MS), di-gly remnant affinity purification proteomics, and differential expression proteomics (43); unfortunately, each method has limitations. AP-MS is carried out on the basis of ligase-substrate interactions. However, most E3 ligases only interact with their substrates weakly or transiently (43). In addition, many E3 ligases undergo auto-ubiquitination and degradation. For example, RNF146 degrades itself; therefore, it is difficult to obtain cell lines with significant levels of WT RNF146 (Fig. 5). The low abundance of E3 ligases makes it more difficult to identify ligase-substrate interactions via AP-MS. Di-gly affinity purification followed by MS is a powerful technique that can be used for the global examination of ubiquitylation sites regulated by E3 ligases. However, it is costly because of the use of antibodies and the substantial amount of proteins that are ubiquitinated. In addition, although more than 20,000 ubiquitylation sites can be identified in any given experiment, their coverage is still low (7). We often rediscover abundant ubiquitination events but do not identify those that are regulated under a particular condition or by a specific E3 ligase. Moreover, the di-gly peptide enrichment procedure may be biased because it relies on antibody that could preferentially recognize some but not other di-gly peptides. Differential expression proteomics may be a better method, at least for E3 ligases such as RNF146 that target proteins for degradation. With the recent development of high-resolution MS, proteome-wide in-depth mapping of substrates of a given E3 ligase is now possible, especially by integrating high-pH HPLC fractionation with a nano-HPLC–MS/MS analysis (44, 45).
RNF146 is activated when it binds to PARylated proteins (10), indicating that it specifically ubiquitinates and degrades proteins that are PARylated. TNKS1/2 are the only known poly-ADP-ribosyltransferases that are responsible for PARylated substrates targeted by RNF146 for ubiquitination and degradation. Therefore, we compared proteomes in TNKS1/2DKO cells with those in RNF146KO cells. Several earlier studies, including one published by our group, attempted to identify substrates of TNKS1/2 using TNKS1/2DKO cells or by treating cells with XAV939 (11, 23, 29, 46). These studies revealed many known TNKS substrates and some novel ones, including Notch1/2/3, NKD1/2, and HectD1, which broadened our understanding of TNKS1/2's functions. We generated RNF146 knockout cell line in 239A and TNKS1/2 double knock out cell line in TP53 knockout 293A. In this study, we determined global changes in protein level in RNF146KO versus 293A, TP53TNKS1/2TDKO versus TP53KO cells treated with/without XAV939. Our study not only revealed the change in many known substrates but also some potential substrates of TNKS. However, Notch1/2/3, NKD1/2, and HectD1 were not significantly changed in our data sets. There are several potential explanations for this discrepancy. First, we used a different cell line (i.e. 293A) in this study. Second, the low abundance of these proteins and their modest changes in previous studies may have eliminated these proteins in our data sets because we used stringent criteria for our data analysis. Third, although the specific TNKS1/2 inhibitor XAV939 worked well in our experiments, cells treated with XAV939 and TNKS1/2DKO cells showed significant differences (supplemental Fig. S4A). It is unlikely that other poly [ADP-ribose] polymerases may compensate the function of TNKS1/2 as we did not identify any PARP family member upregulated by XAV939 treatment. In addition, we did not observe any indication for such compensation, because there is small difference between TNKS knockout cells treated with or without XAV939 (supplemental Fig. S3B). Although we cannot rule out this possibility that XAV939 may inhibit other PARPs. However, even if it were true, such inhibitory effect may not be reflected on protein abundance. The likely explanation is that XAV939 inhibits but does not abolish TNKS1/2 activity. Indeed, several known substrates of TNKS were further upregulated in TNKS1/2 DKO cells compared with those in cells treated with XAV939 (supplemental Fig. S4A). Another nonexclusive explanation is that TNKS enzymes may have noncatalytic function, which we have not formally tested by performing side-by-side proteome analysis using TNKS1/2 knockout cells reconstituted with WT or catalytic inactive mutants of TNKS or TNKS2.
Besides known substrates of TNKS1/2, three additional proteins, SARDH, PPL, and NFATC2, were at the top of the list of upregulated proteins that were identified in TNKS1/2 DKO cells (Fig. 3B). We confirmed the changed levels of SARDH and NFATC2 by Western blot analysis (Fig. 3C). Interestingly, SARDH does not contain the consensus TNK binding site. Thus, it is possible that some TNKS substrates may lack the known TNKS binding motif or have degenerate ones that are not easy to detect. TNKS1/2 may function as oncogenes to promote tumor growth on the basis of the functions of their well-known substrates. However, these newly identified substrates of TNKS may complicate this hypothesis. For example, NFATC2 not only promotes breast cancer cell invasiveness and migration but also enhances tumor-initiating phenotypes in lung adenocarcinoma (47–50). PPL, an adhesion molecule, enhances attachment of squamous cancer cells (51). PPL is also a potential biomarker for poor prognosis in esophageal squamous cell carcinoma (52). SARDH metabolizes sarcosine. High levels of sarcosine enhance the progression of prostate cancer, and low expression of SARDH was reported in prostate cancer (36, 53). Overexpression of SARDH can inhibit tumor growth in prostate cancer xenografts (53). These data suggest that the functions of TNKS1/2 are more complex than we anticipated.
The focus of this study is on RNF146. We have only limited knowledge about RNF146 and its substrates. RNF146 shares some substrates with TNKS1/2, such as angiomotin and axin family proteins. However, we found that the overlap of upregulated proteins in RNF146KO cells with those in TNKS1/2DKO cells was low (supplemental Fig. S5A). Of the top 20 upregulated proteins in RNF146 KO cells (Fig. 4D), tops are well-known substrates of TNKS1/2 and RNF146, whereas the others do not appear to be regulated by TNKS1/2, including p14ARF, FSTL1, OTUD5, and PARP10, suggesting that RNF146 may have functions other than those controlled by TNKS (Table I). We experimentally validated the change in OTUD5 and PARP10 protein levels and showed that the upregulation of OTUD5 and PARP10 could be reversed by the reintroduction of RNF146 in RNF146 KO cells (Fig. 4E and Fig. 5). OTUD5 functions as a negative regulator of type I interferon (35–37) and is involved in stabilizing TP53 (38). PARP10 may have 2 roles in cancer, because it potentially suppresses tumor metastasis by inhibiting Aurora A activity and enhances tumorigenesis by alleviating replication stress (54, 55). These results showed the possibility of different substrates of RNF146 and TNKS1/2. Further comparison of data sets in RNF146KO and TNKS1/2DKO cells revealed some additional common substrates of RNF146 and TNKS1/2. For example, SARDH and MGLL are among the top upregulated proteins in both RNF146 KO and TNKS1/2 DKO cells. We validated the result by showing that the increase in SARDH protein level was reversed by the reintroduction of RNF146 in RNF146KO cells (Fig. 4E and 5E). Although MGLL promotes cancer pathogenesis and aggression, SARDH may function as a tumor suppressor gene (35, 56). These data indicate that detailed functional analyses are needed before any conclusions can be drawn based on their potential enzyme-substrate relationship.
As mentioned above, many upregulated proteins in RNF146KO cells are not regulated by TNKS1/2. It is likely other poly [ADP-ribose] polymerases may work with RNF146 to degrade proteins (Fig. 6). Besides TNKS1/2, PARP10 is the only PARP family protein upregulated by RNF146 knockout. It is reasonable to speculate that PARP10 may function with RNF146, although PARP10 is only known to mediate mono-ADP-ribosylation but not poly-ADP-ribosylation (57, 58). More evidences are needed to test the functional relationship between PARP10 and RNF146. How RNF146 functions independent of TNKS remains to be determined. However, the identification of this list of potential RNF146 substrates that are independent of TNKS should facilitate future mechanistic studies of RNF146 and its substrates in various cellular processes.
Comparative analysis of proteome and transcriptome showed low correlation, indicating that TNKS1/2 and/or RNF146 knockout-induced proteome changes mainly occur at protein level, but not indirectly via changes in mRNA level. For example, only 7 out of the 123 upregulated proteins in TNKS1/2 DKO cells showed significant upregulation at their mRNA level, and only one of the 161 upregulated proteins in RNF146KO cells showed significant change in its mRNA level. The possible explanation is that TNKS1/2 and RNF146 mainly target proteins for modification and degradation, but do not play any major role in transcriptional regulation.
Although our present study uncovered systematically identified substrates of TNKS1/2 and RNF146, there are several limitations in our data sets. We only used one cell line 293A in our study. We chose 293A, because this cell line is widely used and was derived from normal embryonic kidney epithelial cells, which may express most if not all of the proteins. The diploid state of these cells also ensure that our experiments would not be affected by any gene expression alternations because of genomic mutations and/or aneuploidy. However, we may have missed some cell- or tissue-specific substrates. Moreover, we were initially concerned whether we were able to create viable TNKS1/2 DKO cell lines, given the known functions of TNKS in WNT, Hippo and other signaling pathways. Thus, we created TNKS1/2 DKO in P53 KO background. Therefore, data derived from TNKS1/2 DKO were all performed and analyzed in the same P53 KO background, whereas cells with or without RNF146 KO in the presence or absence of XAV939 treatment were performed in parental 293A cells and analyzed separately. Unfortunately, this experimental design introduced additional variation in our data sets. In addition, the technology we used has its own limitations. Despite the rapid advance in MS technology, current MS-based proteomics still cannot achieve complete coverage of the whole proteome. Therefore, proteins at low abundance are likely lost in our quantitative proteome analysis, which may account for some of the difference in our data sets versus the ones reported by previous studies (16, 46). Especially, some known TNKS substrates including TNKS2 and BLZF1 are expressed at very low levels, which prevent them to be detected in some our samples, similar to an early study (59). However, these proteins are present in data sets obtained from TNKS1/2 DKO cells or following XAV939 treatment. Unfortunately, because they are not present in control data sets, these proteins are not included in your analysis. For example, BLZF1 was in the top change protein in RNF146KO data set; whereas not in TNKS1/2DKO data set. There were missing value for BLZF1 in TP53KO cells of TNKS1/2DKO data set. In addition, the proteome approach that we used here cannot be used for substrates that are not targeted for degradation. Moreover, the potential substrates identified in our study may or may not be direct targets of RNF146.
Our study is the first proteome-wide analysis of potential RNF146 substrates, which should facilitate future mechanistic studies of the TNKS-dependent and -independent functions of RNF146. Our results also demonstrate that proteome profiling can be extended to other E3 ligases and deubiquitinating enzymes (DUB) and allow the unbiased identification of E3 ligase and/or DUB substrates that are regulated by protein degradation.
DATA AVAILABILITY
The MS/MS raw data and processed data reported in this study have been deposited to MassIVE data sets with the dataset identifier MSV000084692. The transcription data were deposited into GEO database with accession No. GSE156101.
Supplementary Material
Acknowledgments
We thank all the members of the Chen laboratory for their help and constructive discussions. We also thank Ann Sutton from the Dept. of Scientific Publications at The University of Texas MD Anderson Cancer Center for editing the manuscript.
Footnotes
This article contains supplemental Figures and Tables.
Funding and Additional Information—This work was supported by internal MD Anderson research support and the Pamela and Wayne Garrison Distinguished Chair in Cancer Research to J.C. J.C. also received support from CPRIT (RP160667) and National Institutes of Health grants (CA210929, CA216911, and CA216437) and MD Anderson's National Institutes of Health Cancer Center Support Grant (CA016672).
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
Abbreviations—The abbreviations used are:
- TNKS1/2
- tankyrases 1 and 2
- RNF146
- E3 ligase RNF146
- MS
- mass spectrometry
- AXIN1
- axin inhibition protein 1
- BLZF1
- golgin 45
- AMOT
- angiomotin
- AMOTL1/2
- angiomotin-like protein 1 and 2
- PPL
- periplakin
- NFATC2
- nuclear factor of activated T cells 2
- OTUD5
- OTU domain-containing protein 5
- PARP1
- poly(ADP-Ribose) polymerase family member 1
- PARP10
- Poly(ADP-Ribose) polymerase family member 10
- LFQ
- label-free quantification
- KO
- knockout
- DKO
- double knockout.
REFERENCES
- 1. Ciechanover A. (1998) The ubiquitin-proteasome pathway: on protein death and cell life. EMBO J. 17, 7151–7160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Komander D., and Rape M. (2012) The ubiquitin code. Annu. Rev. Biochem. 81, 203–229 [DOI] [PubMed] [Google Scholar]
- 3. Hershko A., and Ciechanover A. (1998) The ubiquitin system. Annu. Rev. Biochem. 67, 425–479 [DOI] [PubMed] [Google Scholar]
- 4. Jung C.-R., Hwang K.-S., Yoo J., Cho W.-K., Kim J.-M., Kim W. H., and Im D.-S. (2006) E2-EPF UCP targets pVHL for degradation and associates with tumor growth and metastasis. Nat. Med. 12, 809–816 [DOI] [PubMed] [Google Scholar]
- 5. Tan Z., Sun X., Hou F.-S., Oh H.-W., Hilgenberg L. G. W., Hol E. M., van Leeuwen F. W., Smith M. A., O'Dowd D. K., and Schreiber S. S. (2007) Mutant ubiquitin found in Alzheimer's disease causes neuritic beading of mitochondria in association with neuronal degeneration. Cell Death Differ. 14, 1721–1732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nakayama K. I., and Nakayama K. (2006) Ubiquitin ligases: cell-cycle control and cancer. Nat. Rev. Cancer 6, 369–381 [DOI] [PubMed] [Google Scholar]
- 7. Iconomou M., and Saunders D. N. (2016) Systematic approaches to identify E3 ligase substrates. Biochem. J. 473, 4083–4101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Morreale F. E., and Walden H. (2016) Types of ubiquitin ligases. Cell 165, 248–248.e1 [DOI] [PubMed] [Google Scholar]
- 9. Berndsen C. E., and Wolberger C. (2014) New insights into ubiquitin E3 ligase mechanism. Nat. Struct. Mol. Biol. 21, 301–307 [DOI] [PubMed] [Google Scholar]
- 10. DaRosa P. A., Wang Z., Jiang X., Pruneda J. N., Cong F., Klevit R. E., and Xu W. (2015) Allosteric activation of the RNF146 ubiquitin ligase by a poly(ADP-ribosyl)ation signal. Nature 517, 223–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang Y., Liu S., Mickanin C., Feng Y., Charlat O., Michaud G. A., Schirle M., Shi X., Hild M., Bauer A., Myer V. E., Finan P. M., Porter J. A., Huang S.-M. A., and Cong F. (2011) RNF146 is a poly(ADP-ribose)-directed E3 ligase that regulates axin degradation and Wnt signaling. Nat. Cell Biol. 13, 623–629 [DOI] [PubMed] [Google Scholar]
- 12. Callow M. G., Tran H., Phu L., Lau T., Lee J., Sandoval W. N., Liu P. S., Bheddah S., Tao J., Lill J. R., Hongo J.-A., Davis D., Kirkpatrick D. S., Polakis P., and Costa M. (2011) Ubiquitin ligase RNF146 regulates tankyrase and Axin to promote Wnt signaling. PLoS ONE. 6, e22595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kang H. C., Lee Y.-I., Shin J.-H., Andrabi S. A., Chi Z., Gagné J.-P., Lee Y., Ko H. S., Lee B. D., Poirier G. G., Dawson V. L., and Dawson T. M. (2011) Iduna is a poly(ADP-ribose) (PAR)-dependent E3 ubiquitin ligase that regulates DNA damage. Proc. Natl. Acad. Sci. U S A 108, 14103–14108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li N., Wang Y., Neri S., Zhen Y., Fong L. W. R., Qiao Y., Li X., Chen Z., Stephan C., Deng W., Ye R., Jiang W., Zhang S., Yu Y., Hung M.-C., Chen J., and Lin S. H. (2019) Tankyrase disrupts metabolic homeostasis and promotes tumorigenesis by inhibiting LKB1-AMPK signaling. Nat. Commun. 10, 4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang W., Li N., Li X., Tran M. K., Han X., and Chen J. (2015) Tankyrase Inhibitors Target YAP by Stabilizing Angiomotin Family Proteins. Cell Rep. 13, 524–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li N., Zhang Y., Han X., Liang K., Wang J., Feng L., Wang W., Songyang Z., Lin C., Yang L., Yu Y., and Chen J. (2015) Poly-ADP ribosylation of PTEN by tankyrases promotes PTEN degradation and tumor growth. Genes Dev. 29, 157–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kim S. C., Sprung R., Chen Y., Xu Y., Ball H., Pei J., Cheng T., Kho Y., Xiao H., Xiao L., Grishin N. V., White M., Yang X.-J., and Zhao Y. (2006) Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol. Cell. 23, 607–618 [DOI] [PubMed] [Google Scholar]
- 18. Qian L., Nie L., Chen M., Liu P., Zhu J., Zhai L., Tao S.-C., Cheng Z., Zhao Y., and Tan M. (2016) Global profiling of protein lysine malonylation in Escherichia coli reveals its role in energy metabolism. J. Proteome Res. 15, 2060–2071 [DOI] [PubMed] [Google Scholar]
- 19. Song C., Ye M., Han G., Jiang X., Wang F., Yu Z., Chen R., and Zou H. (2010) Reversed-phase-reversed-phase liquid chromatography approach with high orthogonality for multidimensional separation of phosphopeptides. Anal. Chem. 82, 53–56 [DOI] [PubMed] [Google Scholar]
- 20. Tyanova S., Temu T., and Cox J. (2016) The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 11, 2301–2319 [DOI] [PubMed] [Google Scholar]
- 21. Cox J., Hein M. Y., Luber C. A., Paron I., Nagaraj N., and Mann M. (2014) Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteomics 13, 2513–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tyanova S., Temu T., Sinitcyn P., Carlson A., Hein M. Y., Geiger T., Mann M., and Cox J. (2016) The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740 [DOI] [PubMed] [Google Scholar]
- 23. Huang S.-M. A., Mishina Y. M., Liu S., Cheung A., Stegmeier F., Michaud G. A., Charlat O., Wiellette E., Zhang Y., Wiessner S., Hild M., Shi X., Wilson C. J., Mickanin C., Myer V., Fazal A., Tomlinson R., Serluca F., Shao W., Cheng H., Shultz M., Rau C., Schirle M., Schlegl J., Ghidelli S., Fawell S., Lu C., Curtis D., Kirschner M. W., Lengauer C., Finan P. M., Tallarico J. A., Bouwmeester T., Porter J. A., Bauer A., and Cong F. (2009) Tankyrase inhibition stabilizes axin and antagonizes Wnt signaling. Nature 461, 614–620 [DOI] [PubMed] [Google Scholar]
- 24. Lanier M., Schade D., Willems E., Tsuda M., Spiering S., Kalisiak J., Mercola M., and Cashman J. R. (2012) Wnt inhibition correlates with human embryonic stem cell cardiomyogenesis: a structure-activity relationship study based on inhibitors for the Wnt response. J. Med. Chem. 55, 697–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shultz M. D., Kirby C. A., Stams T., Chin D. N., Blank J., Charlat O., Cheng H., Cheung A., Cong F., Feng Y., Fortin P. D., Hood T., Tyagi V., Xu M., Zhang B., and Shao W. (2012) [1,2,4]triazol-3-ylsulfanylmethyl)-3-phenyl-[1,2,4]oxadiazoles: antagonists of the Wnt pathway that inhibit tankyrases 1 and 2 via novel adenosine pocket binding. J. Med. Chem. 55, 1127–1136 [DOI] [PubMed] [Google Scholar]
- 26. Wang W., Xiao Z.-D., Li X., Aziz K. E., Gan B., Johnson R. L., and Chen J. (2015) AMPK modulates Hippo pathway activity to regulate energy homeostasis. Nat. Cell Biol. 17, 490–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhou Y., Zhou B., Pache L., Chang M., Khodabakhshi A. H., Tanaseichuk O., Benner C., and Chanda S. K. (2019) Metascape provides a biologist-oriented resource for the analysis of systems-level data sets. Nat. Commun. 10, 1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Guettler S., LaRose J., Petsalaki E., Gish G., Scotter A., Pawson T., Rottapel R., and Sicheri F. (2011) Structural basis and sequence rules for substrate recognition by Tankyrase explain the basis for cherubism disease. Cell 147, 1340–1354 [DOI] [PubMed] [Google Scholar]
- 29. Li X., Han H., Zhou M.-T., Yang B., Ta A. P., Li N., Chen J., and Wang W. (2017) Proteomic analysis of the human tankyrase protein interaction network reveals its role in pexophagy. Cell Rep. 20, 737–749 [DOI] [PubMed] [Google Scholar]
- 30. Ruhrberg C., Hajibagheri M. A., Parry D. A., and Watt F. M. (1997) Periplakin, a novel component of cornified envelopes and desmosomes that belongs to the plakin family and forms complexes with envoplakin. J. Cell Biol. 139, 1835–1849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yiu G. K., Kaunisto A., Chin Y. R., and Toker A. (2011) NFAT promotes carcinoma invasive migration through glypican-6. Biochem. J. 440, 157–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bettelli E., Dastrange M., and Oukka M. (2005) Foxp3 interacts with nuclear factor of activated T cells and NF-kappa B to repress cytokine gene expression and effector functions of T helper cells. Proc. Natl. Acad. Sci. U S A 102, 5138–5143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Eschenbrenner M., and Jorns M. S. (1999) Cloning and mapping of the cDNA for human sarcosine dehydrogenase, a flavoenzyme defective in patients with sarcosinemia. Genomics 59, 300–308 [DOI] [PubMed] [Google Scholar]
- 34. Sacco F., Silvestri A., Posca D., Pirrò S., Gherardini P. F., Castagnoli L., Mann M., and Cesareni G. (2016) Deep proteomics of breast cancer cells reveals that metformin rewires signaling networks away from a pro-growth state. Cell Syst. 2, 159–171 [DOI] [PubMed] [Google Scholar]
- 35. Nomura D. K., Long J. Z., Niessen S., Hoover H. S., Ng S.-W., and Cravatt B. F. (2010) Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell 140, 49–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sreekumar A., Poisson L. M., Rajendiran T. M., Khan A. P., Cao Q., Yu J., Laxman B., Mehra R., Lonigro R. J., Li Y., Nyati M. K., Ahsan A., Kalyana-Sundaram S., Han B., Cao X., Byun J., Omenn G. S., Ghosh D., Pennathur S., Alexander D. C., Berger A., Shuster J. R., Wei J. T., Varambally S., Beecher C., and Chinnaiyan A. M. (2009) Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature 457, 910–914 [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 37. Virdee S., Ye Y., Nguyen D. P., Komander D., and Chin J. W. (2010) Engineered diubiquitin synthesis reveals Lys29-isopeptide specificity of an OTU deubiquitinase. Nat. Chem. Biol. 6, 750–757 [DOI] [PubMed] [Google Scholar]
- 38. Kayagaki N., Phung Q., Chan S., Chaudhari R., Quan C., O'Rourke K. M., Eby M., Pietras E., Cheng G., Bazan J. F., Zhang Z., Arnott D., and Dixit V. M. (2007) DUBA: a deubiquitinase that regulates type I interferon production. Science 318, 1628–1632 [DOI] [PubMed] [Google Scholar]
- 39. Gonzalez-Navajas J. M., et al. (2010) Interleukin 1 receptor signaling regulates DUBA expression and facilitates Toll-like receptor 9-driven antiinflammatory cytokine production. J. Exp. Med. 207, 2799–2807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rutz S., Kayagaki N., Phung Q. T., Eidenschenk C., Noubade R., Wang X., Lesch J., Lu R., Newton K., Huang O. W., Cochran A. G., Vasser M., Fauber B. P., DeVoss J., Webster J., Diehl L., Modrusan Z., Kirkpatrick D. S., Lill J. R., Ouyang W., and Dixit V. M. (2015) Deubiquitinase DUBA is a post-translational brake on interleukin-17 production in T cells. Nature 518, 417–421 [DOI] [PubMed] [Google Scholar]
- 41. Luo J., Lu Z., Lu X., Chen L., Cao J., Zhang S., Ling Y., and Zhou X. (2013) OTUD5 regulates p53 stability by deubiquitinating p53. PLoS ONE. 8, e77682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ahel I., Ahel D., Matsusaka T., Clark A. J., Pines J., Boulton S. J., and West S. C. (2008) Poly(ADP-ribose)-binding zinc finger motifs in DNA repair/checkpoint proteins. Nature 451, 81–85 [DOI] [PubMed] [Google Scholar]
- 43. Rayner S. L., Morsch M., Molloy M. P., Shi B., Chung R., and Lee A. (2019) Using proteomics to identify ubiquitin ligase-substrate pairs: how novel methods may unveil therapeutic targets for neurodegenerative diseases. Cell. Mol. Life Sci. 76, 2499–2510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jiang Y., Sun A., Zhao Y., Ying W., Sun H., Yang X., Xing B., Sun W., Ren L., Hu B., Li C., Zhang L., Qin G., Zhang M., Chen N., Zhang M., Huang Y., Zhou J., Zhao Y., Liu M., Zhu X., Qiu Y., Sun Y., Huang C., Yan M., Wang M., Liu W., Tian F., Xu H., Zhou J., Wu Z., Shi T., Zhu W., Qin J., Xie L., Fan J., Qian X., and He F. (2019) Proteomics identifies new therapeutic targets of early-stage hepatocellular carcinoma. Nature 567, 257–261 [DOI] [PubMed] [Google Scholar]
- 45. Ni X., Tan Z., Ding C., Zhang C., Song L., Yang S., Liu M., Jia R., Zhao C., Song L., Liu W., Zhou Q., Gong T., Li X., Tai Y., Zhu W., Shi T., Wang Y., Xu J., Zhen B., and Qin J. (2019) A region-resolved mucosa proteome of the human stomach. Nat. Commun. 10, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bhardwaj A., Yang Y., Ueberheide B., and Smith S. (2017) Whole proteome analysis of human tankyrase knockout cells reveals targets of tankyrase-mediated degradation. Nat. Commun. 8, 2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Xiao Z.-J., Liu J., Wang S.-Q., Zhu Y., Gao X.-Y., Tin V. P.-C., Qin J., Wang J.-W., and Wong M. P. (2017) NFATc2 enhances tumor-initiating phenotypes through the NFATc2/SOX2/ALDH axis in lung adenocarcinoma. Elife 6, e26733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kim G.-C., Kwon H.-K., Lee C.-G., Verma R., Rudra D., Kim T., Kang K., Nam J. H., Kim Y., and Im S.-H. (2018) Upregulation of Ets1 expression by NFATc2 and NFKB1/RELA promotes breast cancer cell invasiveness. Oncogenesis 7, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jauliac S., López-Rodriguez C., Shaw L. M., Brown L. F., Rao A., and Toker A. (2002) The role of NFAT transcription factors in integrin-mediated carcinoma invasion. Nat. Cell Biol. 4, 540–544 [DOI] [PubMed] [Google Scholar]
- 50. Yoeli-Lerner M., Yiu G. K., Rabinovitz I., Erhardt P., Jauliac S., and Toker A. (2005) Akt blocks breast cancer cell motility and invasion through the transcription factor NFAT. Mol. Cell 20, 539–550 [DOI] [PubMed] [Google Scholar]
- 51. Tonoike Y., Matsushita K., Tomonaga T., Katada K., Tanaka N., Shimada H., Nakatani Y., Okamoto Y., and Nomura F. (2011) Adhesion molecule periplakin is involved in cellular movement and attachment in pharyngeal squamous cancer cells. BMC Cell Biol. 12, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yamada K., Hagiwara T., Inazuka F., Sezaki T., Igari T., Yokoi C., Nohara K., Yamashita S., Dohi T., and Kawamura Y. I. (2018) Expression of the desmosome-related molecule periplakin is associated with advanced stage and poor prognosis of esophageal squamous cell carcinoma. Transl. Cancer Res. 7, 79–87 [Google Scholar]
- 53. Khan A. P., Rajendiran T. M., Ateeq B., Asangani I. A., Athanikar J. N., Yocum A. K., Mehra R., Siddiqui J., Palapattu G., Wei J. T., Michailidis G., Sreekumar A., and Chinnaiyan A. M. (2013) The role of sarcosine metabolism in prostate cancer progression. Neoplasia 15, 491–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhao Y., Hu X., Wei L., Song D., Wang J., You L., Saiyin H., Li Z., Yu W., Yu L., Ding J., and Wu J. (2018) PARP10 suppresses tumor metastasis through regulation of Aurora A activity. Oncogene 37, 2921–2935 [DOI] [PubMed] [Google Scholar]
- 55. Schleicher E. M., Galvan A. M., Imamura-Kawasawa Y., Moldovan G.-L., and Nicolae C. M. (2018) PARP10 promotes cellular proliferation and tumorigenesis by alleviating replication stress. Nucleic Acids Res. 46, 8908–8916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Xiang W., Shi R., Kang X., Zhang X., Chen P., Zhang L., Hou A., Wang R., Zhao Y., Zhao K., Liu Y., Ma Y., Luo H., Shang S., Zhang J., He F., Yu S., Gan L., Shi C., Li Y., Yang W., Liang H., and Miao H. (2018) Monoacylglycerol lipase regulates cannabinoid receptor 2-dependent macrophage activation and cancer progression. Nat. Commun. 9, 2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kleine H., Poreba E., Lesniewicz K., Hassa P. O., Hottiger M. O., Litchfield D. W., Shilton B. H., and Lüscher B. (2008) Substrate-assisted catalysis by PARP10 limits its activity to mono-ADP-ribosylation. Mol. Cell. 32, 57–69 [DOI] [PubMed] [Google Scholar]
- 58. Rosenthal F., Feijs K. L. H., Frugier E., Bonalli M., Forst A. H., Imhof R., Winkler H. C., Fischer D., Caflisch A., Hassa P. O., Lüscher B., and Hottiger M. O. (2013) Macrodomain-containing proteins are new mono-ADP-ribosylhydrolases. Nat. Struct. Mol. Biol. 20, 502–507 [DOI] [PubMed] [Google Scholar]
- 59. Geiger T., Wehner A., Schaab C., Cox J., and Mann M. (2012) Comparative proteomic analysis of eleven common cell lines reveals ubiquitous but varying expression of most proteins. Mol. Cell. Proteomics 11, M111.014050–014050 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The MS/MS raw data and processed data reported in this study have been deposited to MassIVE data sets with the dataset identifier MSV000084692. The transcription data were deposited into GEO database with accession No. GSE156101.