

XL-MS has been recognized as an effective source of information about protein structures and interactions. OpenPepXL is a sensitive XL-MS identification software that reports from 7% to 40% more structurally validated cross-links than other tools on data sets with available high-resolution structures for cross-link validation. It is open source and has been built as part of the OpenMS suite of tools. OpenPepXL supports all common operating systems and open data formats.
Keywords: Protein cross-linking, crosslinking, protein structure, structural biology, tandem mass spectrometry, XL-MS
Graphical Abstract

Highlights
OpenPepXL is a new XL-MS identification tool with a high sensitivity.
It is available for all common operating systems and remote computing environments.
OpenPepXL is open source and supports open OpenPepXL is available as part of OpenMS data formats like mzML and mzIdentML.
Abstract
Cross-linking MS (XL-MS) has been recognized as an effective source of information about protein structures and interactions. In contrast to regular peptide identification, XL-MS has to deal with a quadratic search space, where peptides from every protein could potentially be cross-linked to any other protein. To cope with this search space, most tools apply different heuristics for search space reduction. We introduce a new open-source XL-MS database search algorithm, OpenPepXL, which offers increased sensitivity compared with other tools. OpenPepXL searches the full search space of an XL-MS experiment without using heuristics to reduce it. Because of efficient data structures and built-in parallelization OpenPepXL achieves excellent runtimes and can also be deployed on large compute clusters and cloud services while maintaining a slim memory footprint. We compared OpenPepXL to several other commonly used tools for identification of noncleavable labeled and label-free cross-linkers on a diverse set of XL-MS experiments. In our first comparison, we used a data set from a fraction of a cell lysate with a protein database of 128 targets and 128 decoys. At 5% FDR, OpenPepXL finds from 7% to over 50% more unique residue pairs (URPs) than other tools. On data sets with available high-resolution structures for cross-link validation OpenPepXL reports from 7% to over 40% more structurally validated URPs than other tools. Additionally, we used a synthetic peptide data set that allows objective validation of cross-links without relying on structural information and found that OpenPepXL reports at least 12% more validated URPs than other tools. It has been built as part of the OpenMS suite of tools and supports Windows, macOS, and Linux operating systems. OpenPepXL also supports the MzIdentML 1.2 format for XL-MS identification results. It is freely available under a three-clause BSD license at https://openms.org/openpepxl.
Cross-Linking Mass Spectrometry (XL-MS) has proven to be a valuable tool in studying the structures and interactions of proteins (1–5). Although XL-MS is maturing as a very useful method, there is space for improvement at every step of the workflow. Especially the enrichment step of cross-linked peptides derived from cross-linked protein samples has profound effects on the XL-MS analysis as well as the following computational identification and the statistics of the FDR of annotated MS2 spectra. In many XL-MS experiments the samples still contain a vast number of noncross-linked, i.e. linear peptides; consequently cross-linked peptides usually occur with low intensities and are thus less likely to be selected for fragmentation in data-dependent acquisition as well. Therefore, precursor and fragment spectra of relatively few cross-links must be identified among a large set of spectra from unmodified peptides. This is one of the issues that make the statistics for post-processing and filtering XL-MS data more difficult when compared with the identification of linear peptides.
Fragment spectra of cross-linked peptides are also more difficult to annotate as they contain fragments from two peptides. Scoring the whole cross-link fragment spectrum match might result in identifications where one peptide sequence is covered by many fragment ions whereas the second peptide is identified by its precursor mass and very few matching fragment ions only. Reliable identification of one of the peptide sequences does not depend on correct identification of the other sequence. It is possible to have an identification of a cross-linked peptide pair with a high score in a database search where the high score is based on a legitimate good match to one correct peptide, but with a bad match to the second peptide. Reliable identification of a cross-link, that is intended to be useful for modeling a protein structure or complex, requires correct identifications for both peptides and hence the whole identification can only be as good as the identification of the worst of the two peptides (6).
The search for two peptides in each fragment spectrum also has implications for the performance of XL-MS identification software. For a given precursor mass in conventional MS-based protein identification, the length of a possibly matching linear peptide can be roughly estimated by applying an 'averagine' model (7). The number of candidates to be considered for matching peptides in database search primarily depends on the width of the precursor mass tolerance window and the size of the protein database. In XL-MS the mass distributes across two peptides and only the sum of their masses plus the mass of the cross-linker is known. The computational search space contains all possible combinations of cross-linked peptides whose sum of masses lies within the precursor mass window. Searching all combinations of peptides rather than just linearly scanning all peptides requires efficient algorithms to perform a search on acceptable time scales.
The most obvious solution used by some XL-MS search tools is a brute-force enumeration of all peptide-peptide pairs and filtering them by precursor mass (8). Searches can be sped up by using stable-isotope labeled cross-linkers in the cross-linking experiment (9, 10). Such labeling makes cross-linked spectra easily identifiable on the MS1 level and thus reduces the number of corresponding MS2 spectra to be searched by the database search tool. Several conventional linear peptide search tools (11) as well as xQuest (9, 10, 12) and pLink2 (13) use pre-calculated fragment ion indices to retrieve peptides from the protein database based on observed fragment ions. Just like StavroX (8), xQuest fragments and scores pairs of peptides at a time. Therefore, the use of labeled linkers combined with an ion index limits its computational memory consumption and makes it applicable to large protein databases. Another method for reducing the large search space is to use multi-pass scoring. A first scoring step based on a quick heuristic or a partial score can substantially reduce the number of candidates subjected to full scoring, thus reducing the overall runtime. For example, Kojak (14), XiSearch (15), and pLink2 (13) start with a linear peptide search using an open-modification search strategy. Kojak uses a few hundred of the top-scoring peptides and combines them into pairs fitting the precursor mass, whereas XiSearch and pLink2 only keep a certain number of these and search the entire database again for the second peptide. The existing algorithms constrain the search space for their full scoring. That means they do not apply their final, most discriminative score to every candidate cross-link within the precursor tolerance window. This might prematurely dismiss some candidate peptides that would have a high score as a peptide pair and reduce sensitivity in favor of efficiency. It was previously shown that one of the two peptides of a correctly identified cross-link might not be found within the first few hundred or even thousand peptides by pre-scoring linear peptides (16). Our own experiments have also shown that it is not rare to find thousands of peptide pairs with at least 3 matched fragments for each peptide for one fragment spectrum and a middle-sized database of fewer than 500 proteins (data not shown).
Sensitivity is defined as the proportion of real cross-links in a data set identified by a search tool. Unfortunately, it is difficult to calculate the true number of real cross-links in a data set, because the crystal structures are often incomplete, especially for the larger complexes. The theoretical number of possible cross-links for most protein complexes is very high and only a small fraction of them is usually identified. Also, this number is the same for any fixed sample or searched database and does not affect the comparison of tools. Therefore, in this study we use the number of reported cross-links from the target protein database given a fixed FDR threshold as a substitute for the real sensitivity of a search.
In this work we introduce OpenPepXL, an efficient open-source software for identification of cross-linked peptides in fragment mass spectra. It is based on a full exploration of all possible candidate cross-link peptide pairs for each precursor mass in order to achieve high sensitivity, but because of efficient index data structures and search algorithms, it can achieve much improved runtimes. OpenPepXL supports both labeled and label-free, mono- and heterobifunctional noncleavable cross-linkers. It is based on the OpenMS software framework (17) and makes use of multi-core architectures using the OpenMP API. OpenPepXL is part of The OpenMS Proteomics Pipeline (TOPP) that includes tools for labeled and label-free quantification, pre- and post-processing, and visualization of spectra and identification data. It can be installed on all major operating systems (Windows, macOS, and Linux) and is compatible with most computing clusters and cloud services for large-scale data analysis. It can be run as a command-line tool with a preconfigured file containing the settings, or as part of a workflow built using the graphical user interface of the free to use KNIME Analytics Platform (18). OpenPepXL supports several output formats for XL-MS identification data such as the MzIdentML 1.2 format (19), the xQuest XML output format and simple text-based tabular formats. The output can, therefore, be easily integrated into many existing XL-MS data analysis pipelines and is also compatible with the public repository PRIDE (20) which is part of ProteomeXchange (21). We compare OpenPepXL to other commonly used tools for identification of noncleavable cross-linkers (pLink2 (13), XiSearch (15), Kojak (14), StavroX (8), and xQuest (9)) on a diverse set of XL-MS experiments and show that it tends to be more sensitive while still achieving very good runtimes. OpenPepXL is available under a three-clause BSD license at https://www.openms.de/openpepxl/.
MATERIALS AND METHODS
Algorithm Overview
OpenPepXL belongs to the category of algorithms that score an entire candidate molecule of two peptides covalently linked with a cross-linker against an experimental spectrum without doing an open-modification search for linear peptides first. In this sense it has more in common with xQuest (9) and StavroX (8) than with pLink2 (13), Kojak (14), or XiSearch (15). OpenPepXL keeps a list of all linear peptides with modifications and their masses after in silico digestion of the protein database. The candidate peptide pairs are then enumerated for each MS2 spectrum precursor mass (Fig. 1). This way only the necessary pairs are created. By using the indices of the linear peptide table to reference the peptides in a pair, only a minimal amount of additional memory is required for this candidate peptide pair enumeration. Loop-links and mono-links are also considered in this step. Then theoretical spectra containing all linear and cross-linked fragments expected from the peptide pair are generated. By default, b- and y-ion series including neutral losses of NH3 and H2O are considered, but a-, c-, x- and z-ions can also be generated to accommodate different fragmentation methods. A spectrum matching algorithm matches peaks between these theoretical and the experimental spectra. From the number of matched peaks the match-odds score for a candidate peptide pair is calculated (more on the score below).
Fig. 1.
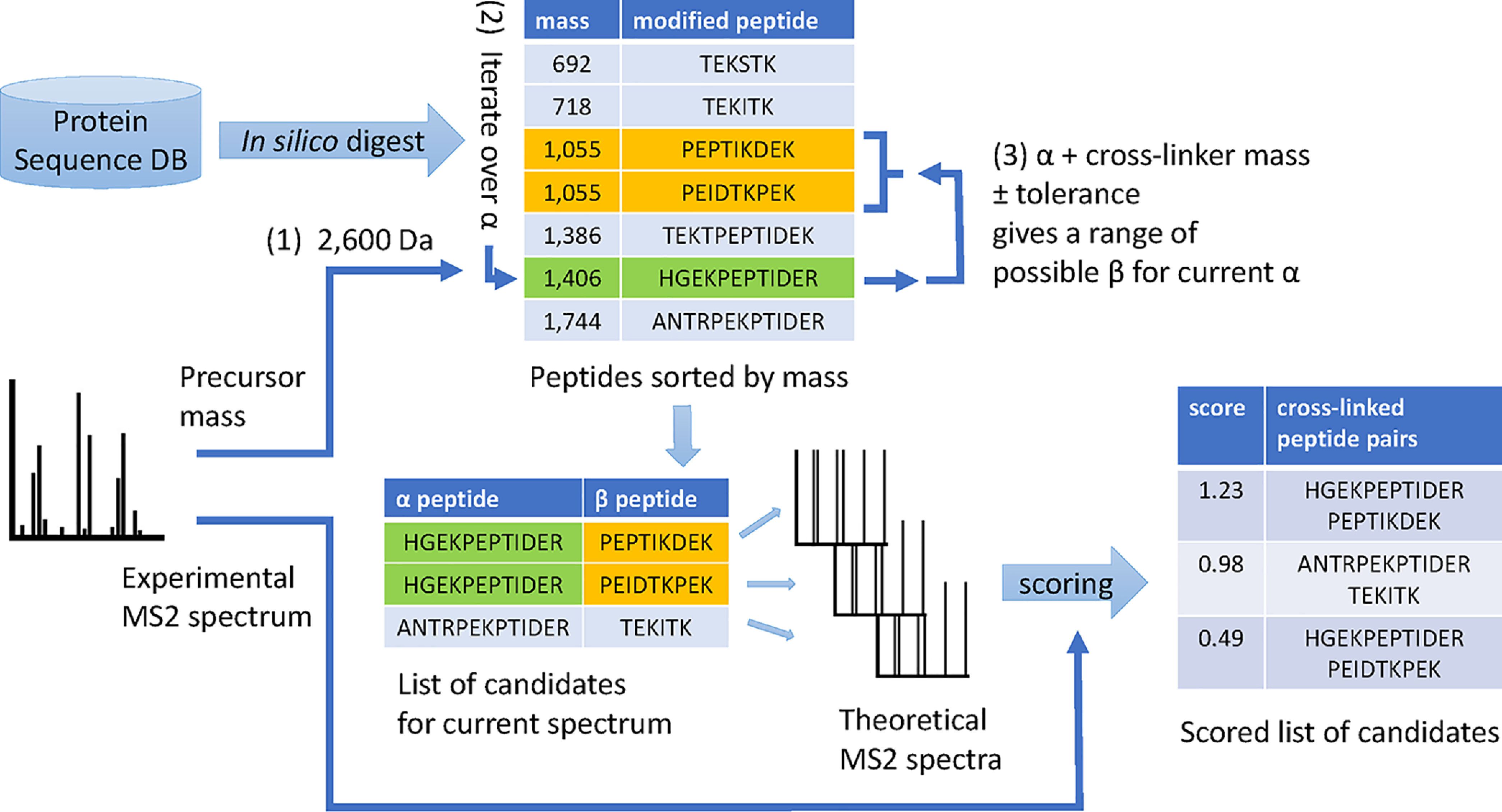
Overview of peptide pair candidate enumeration and identification in OpenPepXL. After in silico digestion a database of modified peptides sorted by mass is kept. For each MS2 spectrum the precursor mass (1) is used to determine the mass range for α peptides (heavier). Iterating through this list (2), for each α peptide, the mass range for β peptides is determined (3) and a list of pairs is enumerated. For each candidate pair, theoretical spectra are generated and scored against one experimental MS2 spectrum (label free experiment) or one linear-ion and one cross-linked ion spectrum (labeled cross-linker experiments).
For experiments using labeled cross-linkers a few additional pre-processing steps are necessary. To pair MS2 spectra of the same peptide pairs linked by light and heavy isotope labeled cross-linkers the MS1 features across mass traces and retention time have to be detected and paired. We use the OpenMS tool for MS1 labeling (FeatureFinderMultiplex) to detect pairs of MS1 features from light and heavy cross-links based on the characteristic mass shift. OpenPepXL then maps MS2 spectrum precursors to their respective features. MS2 spectra mapped to feature pairs are then paired up and processed (Fig. 2) to get peak sets from linear and cross-linked fragments with reduced noise. When matching theoretical spectra against these peak sets, only linear theoretical fragment peaks are matched against the experimental linear peaks and vice versa. This preprocessing step is derived from the xQuest algorithm and focuses the matching and scoring to smaller sets of peaks to reduce the chance of false-positive peak matches. The scores of the linear and cross-linked ion matches are combined to one score before the ranking and filtering of candidates.
Fig. 2.

Preprocessing of experimental spectrum pairs for experiments with labeled linkers. DSS D0/D12 is used as an example. Two experimental spectra from the same peptide pair but a different linker mass are matched without a mass shift and with a mass shift of the label mass difference considering multiple charges. The result is a linear ion spectrum with unknown charges and a cross-linked ion spectrum with known ion charges. This allows for a more constrained and targeted matching to theoretical peaks.
Match-Odds Score
The match-odds score used in OpenPepXL is based on the score of the same name from the xQuest algorithm (9). It is based on the probability of a random match between any peak from the experimental fragment ion spectrum and any peak in the theoretical fragment ion spectrum, given the mass tolerance window tol, mass range r, the number of peaks in the theoretical fragment spectrum s and the number of considered charges for all theoretical peaks c. The probability of one random match to a fragment ion peak is calculated as:
| (1) |
The cumulative distribution function of a binomial distribution with sample size s and probability p is used to determine the probability of getting more than k matched peaks between the experimental and theoretical fragment ion spectra by random chance:
| (2) |
This probability will decrease toward 0 for higher numbers of k where a smaller probability denotes a better match, because it is less likely to have happened by chance. With the -log() function the probability is turned into a score with higher numbers denoting a better match:
| (3) |
We call this the match-odds score m and it is combined with the precursor error pe (difference between theoretical and experimental precursor mass in ppm) in the following formula to get the final OpenPepXL score:
| (4) |
This formula was determined by an agreement between a linear regression and a linear discriminant analysis done to find the best linear combination to separate target from decoy hits on several XL-MS data sets (refer to supplemental Methods for more details).
Mass Spectrometry of CRM Complex
The trimeric complex of human CRM1, SNP1 and Ran carrying a Q69L mutation was cross-linked with bis(sulfosuccinimidyl)suberate (BS3) and injected into an EASY-nLC 1000 HPLC system coupled to a Q Exactive mass spectrometer (Thermo Fisher Scientific) in duplicates under three normalized collision energy (NCE) conditions using a 50-min method. MS1 and MS2 resolution were set to 70,000 and 17,500, respectively. Fifteen most abundant precursors with charge of 3-7 were selected for MS2 fragmentation at NCE 20, 24 or 28% (refer to the supplemental Methods for more details on experimental procedure). For the protein database only the three UniProt sequences O14980, O95149 and P62826 were used. They were manually modified to reflect the modifications made during the protein expression and purification (22). The MS proteomics data including the modified protein sequences have been deposited to the ProteomeXchange Consortium (21) via the PRIDE (20) partner repository with the data set identifier PXD014359.
Public Data Sets
In addition to the CRM complex data set described above, three data sets were downloaded from public repositories or kindly provided to us by other laboratories.
We chose a more complex publicly available data set derived from a BS3-cross-linked crude ribosomal fraction obtained by size exclusion chromatography of HEK293 cell lysate (ProteomeXchange ID PXD006131) (23). The resulting sample was a complex mixture of more than 1700 proteins, which were quantified by label-free quantification of linear peptides. With this data set several protein databases were provided. Starting from one containing the 32 most abundant proteins and doubling in size up to the 512 most abundant proteins. We searched the HCD fragmented subset of this data set consisting of about 170.000 HCD fragmented MS2 spectra against a database of the 128 most abundant proteins and 128 reversed sequence decoys.
Additionally, we analyzed a data set with labeled DSS-d0/d12 and PDH-d0/d10 (pimelic acid dihydrazide) cross-linkers. Commercial Bovine Serum Albumin (BSA; Sigma-Aldrich) was cross-linked with labeled DSS or PDH cross-linker in separate experiments. Both samples were independently analyzed using HCD fragmentation and high-resolution MS/MS detection (Orbitrap Fusion Lumos) or ion trap CID fragmentation with low-resolution MS/MS detection (Orbitrap Elite). This data set was published previously as part of a larger study (24) and kindly provided to us by Alexander Leitner upon request.
Furthermore, we used a cross-linked synthetic peptides data set published by Beveridge et al. (ProteomeXchange ID PXD014337) (25). Instead of using proteins digested by trypsin, tryptic peptides from the S. pyogenes Cas9 protein with one internal lysine each were synthesized in that study. The peptide termini were modified to make sure that DSS could not cross-link to the N termini or C-terminal lysines. The peptides were kept in 12 separate groups without overlapping peptide sequences. Each group was cross-linked with DSS and the cross-linked peptide solutions were mixed before the MS data acquisition of three technical replicates. This means that identified cross-links with the two cross-linked peptides coming from the same group are almost certainly valid identifications, whereas cross-links between peptides from different groups are certain to be false identifications. The protein database we used was the S. pyogenes Cas9 sequence with 10 additional proteins from the supplemental material of the original publication of this data set.
Data Processing
The .RAW files of all data sets were converted into mzML, mzXML, and MGF files using MSConvertGUI from the ProteoWizard toolkit version 3.0.10577. The binary encoding precision was set to 64-bit. Writing an index and TPP compatibility were turned on. No compression was used for mzML files. Reversed sequence decoy protein databases were generated from the target protein databases using the TOPP tool DecoyDatabase. Because it creates its own decoys, only the target database was provided to pLink2. OpenPepXLLF 1.1 (OpenPepXL Label-Free) with the TOPP tool XFDR for False Discovery Rate (FDR) estimation, XiSearch 1.6.731 with xiFDR 1.1.27 for FDR estimation, TPP 5.1.0 with Kojak 1.6.0 and PeptideProphet for FDR estimation, xQuest 2.1.3 with xProphet for FDR estimation as well as pLink 2.3.5 and StavroX 3.6.6.5 with their built-in FDR estimation algorithms were used to identify cross-links in the label-free data sets. The parameters of the different tools were set to equal values where possible and to reasonable or similar values otherwise (supplemental Table S1, Table S2, Table S3). Additional filtering and post-processing were partly done with the TOPP tools IDFilter, IDMerger and TextExporter for OpenPepXL and xQuest output and otherwise with R scripts. An FDR cutoff of 5% on the cross-link spectrum match (CSM) level was applied to every tool and all data sets unless indicated otherwise. Additionally, after this cutoff only unique residue pairs (URPs) supported by at least two of the remaining CSMs were kept. Also, a filter for link distance was applied to intra-peptide links or loop-links. Linked residue pairs were only kept, if they were at least 4 residues apart in the database sequence. This was done to further harmonize the tool results, because this cutoff was different among the tools and linked residue pairs with short sequence distances are not very informative. All tools were compared on the same Windows 10 PC with an Intel(R) Core(TM) i5-6500 CPU and 8 GB of RAM using one CPU core.
The data sets with labeled cross-linkers were only processed with OpenPepXL and xQuest. The TOPP tool FeatureFinderMultiplex was used to detect pairs of MS1 features for OpenPepXL. Otherwise, the same processing steps and filtering rules as for the first two label-free data sets were applied.
The synthetic peptides data set was processed with OpenPepXL with search settings and filter criteria matching those used in the original publication of this data set in Beveridge et al. (25). Search results for the other tools were taken from the publication. This includes the 5 and 1% CSM-FDR results from the search against the S. pyogenes Cas9 sequence with 10 additional proteins. For this data set only the CSM-FDR cutoff was applied and the other filtering steps skipped to make the results directly comparable to those from the original publication.
The MS proteomics data from the CRM data set, including search results from all tools compared in this study have been deposited to the ProteomeXchange Consortium (21) via the PRIDE (20) partner repository with the data set identifier PXD014359. The reanalyzed ribosomal fraction data set was deposited with identifier PXD014520 and the BSA data set with identifier PXD014523. The OpenPepXL results for the synthetic peptide data set were deposited with identifier PXD021417.
Sensitivity and Specificity
In this study we use the number of reported cross-links under a fixed FDR threshold from the target protein database as a substitute for the real sensitivity of a search. Because of the tradeoff between sensitivity and specificity, we compare the sensitivity of all tools at the same FDR setting of 5% at the CSM level. Additionally, only URPs matched to at least two spectra are kept. For OpenPepXL we also recalculate the FDR at unique link level by keeping the decoy hits through the filtering steps and recalculating the FDR for the filtered list of URPs. Where it is possible, we validate the URPs against previously published structural data. For the synthetic peptide data set, it is possible to validate the identified cross-links more objectively than using protein structures.
Structural Validation
TopoLink (26) was used for an analysis of solvent accessible surface distances (SASD) between cross-linked residues. A cutoff of 35 Å was chosen. SASD was measured between Cβ atoms while ignoring all side chains beyond their Cβ atoms.
UCSF Chimera (27) with the Xlink Analyzer (28) plugin was used for a visualization of the identified cross-links on the PDB structures. A Euclidean distance cutoff of 35 Å was chosen for the link coloring. Cross-links consistent with the structures were colored blue, inconsistent cross-links red.
The CRM data set was validated on the x-ray crystallography structure with PDB ID 3GJX (22) and the BSA data set was validated on chain A from the x-ray crystallography structure with PDB ID 4F5S. The ribosomal fraction data set was validated on a larger set of x-ray crystallography and cryo-EM structures.
RESULTS
Benchmark Results
In order to assess the performance of OpenPepXL, we compared it to five currently popular XL-MS search engines (StavroX (8), xQuest (9, 10, 12), pLink2[13], Kojak (14) and XiSearch (15)) on a number of data sets. To the extent possible, the tools were used with settings as similar as possible (see supplemental Tables S1, S2 and S3 for all settings). The data sets used differ in size and complexity: Applying OpenPepXL to the more complex ribosomal fraction sample with thousands of proteins gives insights into sensitivity and performance, but we could only structurally verify about one third of the cross-links. Hence, a second comparison assesses both sensitivity and specificity on the highly purified sample of the CRM complex with a known three-dimensional structure. Lastly OpenPepXL is applied to data generated with labeled cross-linkers and a different cross-linker chemistry to demonstrate its versatility.
To assess the sensitivity of OpenPepXL compared with other tools, we ran a search on the ribosomal fraction data set. About 170,000 MS2 spectra were searched against a protein database of 128 target and 128 decoy proteins on a desktop PC with 8 GB of memory. The 128 target protein database was the largest database that OpenPepXL and XiSearch could handle in a reasonable runtime of less than 3 days. OpenPepXL identified 110 unique residue pairs (URPs), followed by pLink2 (13) with 102 (Fig. 3A). The calculated URP level FDR (URP-FDR) for OpenPepXL after applying the filters was estimated to be 8.8%. A Venn-Diagram showing the overlap of identifications between the tools is shown in supplemental Fig. S5. StavroX (8) could not finish the search because of computer memory requirements. xQuest (9) did not exceed the available memory, but the search was canceled after a week, because the projected remaining runtime under these conditions was unreasonable. Here it has to be noted, that xQuest can be parallelized and can run on a cluster, so in general finishing this search with the limited amount of computer memory is probably within its capabilities. It can also analyze most data sets with labeled cross-linkers within feasible runtimes. The sensitivity of OpenPepXL comes at the cost of a full search of the squared search space and the increased runtime associated with that. To analyze the ribosomal fraction data set using one CPU core pLink2 took 15 min, Kojak about 3 h, OpenPepXL 28 h and XiSearch 36 h (Fig. 3B). OpenPepXL can also be installed on Linux computing clusters and a speedup by a factor of 15 can be achieved by running the tool on 25 cores (supplemental Fig. S1). A structural validation of the ribosomal fraction data set proved to be difficult because most of the identified cross-links linked residue pairs that were not resolved in existing PDB structures. Results from the links that could be validated are shown in supplemental Figs. S3 and S4. Curiously, none of the links found by any of the tools were inconsistent with the structures.
Fig. 3.

Results from the analysis of the ribosomal fraction data set. A, Numbers of identified unique residue pairs (URPs) in the ribosomal fraction data set with a target database of 128 proteins. OpenPepXL identified 110 URPs, pLink2 identified 102 URPs, Kojak 67 URPs and XiSearch 54 URPs. Structural verification of these cross-links is presented in supplemental Figs. S3 and S4. StavroX exceeded the available memory of 8 GB and could not finish the search. xQuest did not exceed the available memory, but the search was canceled because the projected runtime under these conditions was unreasonable. B, Runtimes in hours needed to analyze the ribosomal fraction data set with a database of 128 target and 128 decoy proteins using one CPU core. pLink2 only took 15 min. Kojak took 3 h, OpenPepXL 28 h and XiSearch 36 h.
To show that the cross-links reported by OpenPepXL are useful for modeling protein structures and that the high sensitivity does not result from just reporting more false-positives, a highly purified sample of the trimeric CRM complex with known three-dimensional structure was measured and analyzed by all compared tools. OpenPepXL and pLink2 each reported 78 URPs in total, Kojak reported 61. The cross-links that could be mapped on the structure were 45 URPs for OpenPepXL, 41 URPs for Kojak, followed by pLink2 with 40 URPs (Fig. 4A). The calculated URP- FDR for OpenPepXL after applying the filters was estimated to be 12%. These URPs were validated by calculating the solvent accessible surface distance (SASD) between the linked residues and applying a cutoff of 35 Å. The SASD was calculated on the structure with PDB ID 3GJX. OpenPepXL reported one URP that is inconsistent with the structure, Kojak only reported consistent URPs and pLink2 reported 2 URPs that are inconsistent with the structure (Fig. 4A, Fig. 5). The error rate of OpenPepXL on this data set is approximately equal to that of other tools with comparable sensitivity. OpenPepXL identified 14 URPs, that were not identified by any of the other tools and 6 of them were structurally validated (supplemental Fig. S6). The annotated spectra of the highest-scoring CSMs for each of those 14 URPs are shown in supplemental Figs. S9–S22.
Fig. 4.

Results from the analysis of the CRM complex and BSA data sets. A, Numbers of identified URPs in the CRM data set. Identified URPs that link residues covered by the PDB structure 3GJX were analyzed by TopoLink. The red bars are the proportion of URPs linking residues that are either not solvent accessible, or are farther away than 35 Å according to the SAS distance. The green bars are the proportion of URPs that were not covered by the structure. OpenPepXL identified 78 URPs. 44 URPs were validated and one link is inconsistent with the structure (IWS) with a distance of 37.4 Å between linked residues. Kojak identified 61 URPs of which 41 were validated. pLink2 identified 78 URPs of which 38 were validated and two are IWS, including the same 37.4 Å link as OpenPepXL and an additional IWS link with a 40.4 Å distance. StavroX identified 48 URPs of which 28 were validated and one is IWS. XiSearch found 36 URPs of which 24 were validated and xQuest found 16 of which 14 were validated. B, Numbers of identified URPs in the BSA data set. OpenPepXL and xQuest were compared on ion trap and orbitrap fragment spectra data with two different labeled linkers DSS-d0/d12 and PDH-d0/d10. Identified cross-links that link residues covered by the PDB structure 4F5S were analyzed by TopoLink. The red bars are the proportion of URPs linking residues that are either not solvent accessible, or are farther away than 35 Å according to the SAS distance. OpenPepXL identified a total of 65 URPs in the DSS orbitrap data set, including three IWS links, all of them below a distance of 40 Å. It identified 22 URPs in the PDH orbitrap data set, including one IWS link with a distance of 59.3 Å. xQuest Identified 16 URPs in the PDH orbitrap data set, including 3 IWS links. It identified 21 URPs in the DSS orbitrap data set, including one IWS link. xQuest identified 9 URPs in the PDH ion trap data set and 57 URPs in the DSS ion trap data set, including one IWS link with a distance of 70.2 Å.
Fig. 5.

Cross-links mapped to a PDB structure of the CRM complex. Cross-links identified in the CRM data set with (A) OpenPepXL, (B) Kojak and (C) pLink2, mapped onto the PDB structure 3GJX. Cross-links spanning a Euclidean distance of more than 35 Å are colored red. Those spanning a smaller distance are colored blue.
To assess the sensitivity of OpenPepXL on data with labeled cross-linkers and different linking reaction chemistries, the BSA data sets cross-linked with the labeled cross-linkers DSS-d0/d12 and PDH-d0/d10 were analyzed with OpenPepXL and xQuest. xQuest has been developed especially for stable isotopically labeled cross-linkers. The score of OpenPepXL was calibrated using HCD fragmented MS2 spectra recorded by orbitrap instruments. Also, the spectrum alignment and deisotoping algorithms in OpenPepXL rely on high-resolution fragment spectra. Meanwhile, xQuest is mostly used for CID fragmented MS2 spectra recorded by ion trap instruments. xQuest also does not apply deisotoping but relies mainly on the stable isotope labels for denoising spectra and does not have a feature to correct for misassigned monoisotopic peaks. We obtained a data set, where equal samples were cross-linked with two different labeled cross-linkers and analyzed using both instrument types. For this data set with a very simple target system we chose to search not only for lysine and N-terminal DSS cross-links, but also included serine, threonine and tyrosine as potential linking sites. For the samples cross-linked with PDH we set aspartic acid, glutamic acid and the C terminus as potential cross-linking sites. OpenPepXL identified 65 DSS and 22 PDH URPs in the HCD fragmented orbitrap spectra, whereas xQuest identified 57 DSS and 9 PDH URPs in the CID fragmented ion trap spectra (Fig. 4B). The calculated URP-FDR for OpenPepXL after applying the filters for the DSS orbitrap and PDH orbitrap data sets were estimated to be 1.5% and 7.1% respectively. These URPs were validated using chain A of the structure with PDB ID 4F5S. OpenPepXL reported three DSS URPs and one PDH URP exceeding the SASD cutoff of 35 Å. xQuest identified one DSS URP exceeding the cutoff (Fig. 4B, Fig. 6).
Fig. 6.

Cross-links mapped to a PDB structure of BSA. Cross-links identified in the BSA data set and mapped onto chain A of PDB structure 4F5S. Cross-links spanning a Euclidean distance of more than 35 Å are colored red. Those spanning a smaller distance are colored blue. A, DSS URPs identified by OpenPepXL in the orbitrap data set. B, PDH URPs identified by OpenPepXL in the orbitrap data set. C, DSS URPs identified by xQuest in the ion trap data set. D, PDH URPs identified by xQuest in the ion trap data set.
Structural validation of cross-links using rigid structures cannot account for protein dynamics and the formation of nonspecific cross-links. Additionally, cross-links have a high tendency to form in regions of proteins for which we have no structural data, e.g. in very flexible or unstructured regions. A good example of this can be seen in our structural validation of cross-links for the ribosomal fraction data set (supplemental Fig. S3). Therefore we chose to assess OpenPepXL on a synthetic peptide data set that allows objective validation of reported cross-links independent from available structural information. For this data set all data except for OpenPepXL was taken from Beveridge et al. (25) and therefore xQuest was omitted from the comparison, because it was not considered in that publication. For Kojak results were only available at the unique cross-link level from the 5% CSM-FDR search. From the several available FDR control methods available for Kojak, the results from Percolator with using only unique cross-links was chosen for the comparison. By design of this synthetic data set, it only has two levels of comparison: CSMs and unique links. In this case, unique cross-linked peptide pairs, unique cross-links, and unique residue pairs (URPs) are equivalent. The results are shown in Fig. 7 as well as supplemental Figs. S7 and S8 and supplemental Tables S4–S7. At 5% FDR OpenPepXL reported on average 242 validated URPs with an average calculated URP level FDR of 7.9%. pLink2 reported on average 217 validated URPs with an average calculated URP level FDR of 11.4% (Fig. 7, supplemental Table S5). At 1% FDR OpenPepXL reported on average 168 validated URPs with an average calculated URP level FDR of 1.7%. pLink2 reported on average 207 validated URPs with an average calculated URP level FDR of 6.9% (supplemental Table S7). This data set has a much stronger overlap in reported URPs between the tools compared with the other data sets in this study (supplemental Fig. S7). At 5% FDR OpenPepXL finds 22 URPs that are not found by either pLink2, StavroX or XiSearch. 17 of those were also identified by the Kojak search with a very high average calculated FDR of 22.7%. Looking at the difference between the 5 and 1% FDR searches, the 5% FDR search results show a clear pattern in the validated links between the replicates (Fig. 7). For each tool the second replicate has the most reported links and the third replicate the fewest. The 1% FDR search results look noisier (supplemental Fig. S8, supplemental Tables S6 and S7). The pattern in the differences between the replicates is almost unrecognizable. Although pLink2 reported the highest numbers of cross-links, it also had an unusually high calculated FDR that reached 4.1% at CSM level and 11.6% at URP level for the third replicate.
Fig. 7.
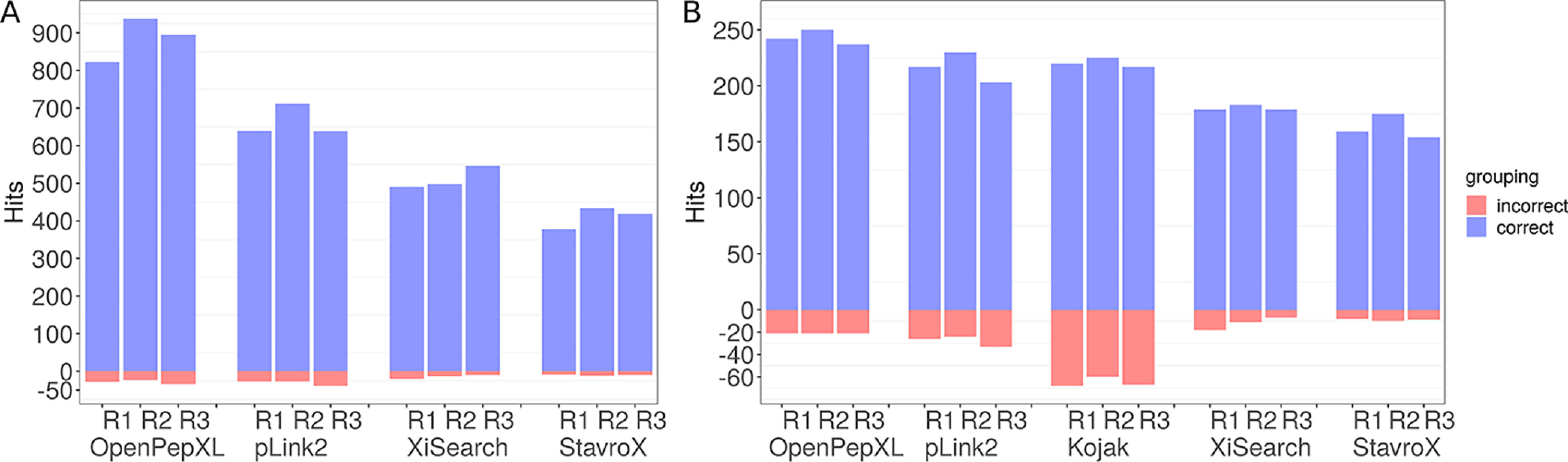
Results from the analysis of the synthetic peptides data set at a 5% FDR cutoff. All three replicates R1, R2 and R3 are shown. The blue bars show the number of valid CSMs/cross-links and the red bars on the negative y axis show the number of false-positive identifications. All data except for OpenPepXL was taken from Beveridge et al. (25). xQuest was omitted because it was not considered in that publication. A, Number of reported CSMs. The exact numbers are in supplemental Table S4. B, Number of identified URPs. The exact numbers are in supplemental Table S5.
We also looked at the numbers of spectra utilized by OpenPepXL and the other tools (supplemental Fig. S8). 5022 MS spectra were recorded for the first of the three replicates. OpenPepXL assigned a result to a total of 4185 spectra. 2029 of those were targets and 2156 were decoys. Validated cross-links above the 5% FDR cutoff were assigned to 822 spectra. OpenPepXL reported 80 validated URPs below the cutoff. pLink2 assigned a result to a total of 1389 spectra. 1006 of those were targets and 384 were decoys. Validated cross-links above the cutoff were assigned to 639 spectra and below the 5% FDR cutoff 4 additional URPs were reported. XiSearch assigned a result to 4363 spectra. 1686 of those were targets and 2677 were decoys. 491 were assigned to validated cross-links above the cutoff and 11 additional URPs were reported below the 5% FDR cutoff. Although XiSearch assigned results to the most MS spectra, it assigned four times as many decoys as targets. This probably makes it very stringent compared with the other tools. Its calculated FDR values on this data set on CSM and URP level are lower than for Kojak and pLink2, but not very different from OpenPepXL (supplemental Tables S4 and S5). pLink2 seems to assign results to very few spectra, even without applying an FDR cutoff. That is partly because it uses several heuristics to filter out spectra and peptides before the actual search and many potential candidates are not kept long enough to reach the FDR estimation step. This approach makes it very fast, but it also means that some of the correct CSMs were probably already filtered out even before the FDR was estimated and the small number of decoys might be the reason for its slightly less stringent FDR control compared with OpenPepXL, XiSearch and StavroX. OpenPepXL had an almost 1:1 distribution of targets and decoys. Among the compared tools it assigned the most targets and validated cross-links to spectra. Many of the correctly assigned CSMs are based mostly on the precursor mass without enough fragment matches for a confident identification. These are then filtered out after FDR estimation and represent the 80 validated URPs below the 5% FDR cutoff. At the same time this also leads to more correct CSMs and URPs being reported above the cutoff. In respect to this comparison, OpenPepXL assigned the most correct identifications to spectra, but there might still be room for improvement in separating correct from incorrect CSMs.
OpenPepXL Features
OpenPepXL can be installed on most current computing environments based on current versions of Windows, macOS and Linux. It is applicable to all labeled and label-free noncleavable cross-linkers. It makes use of labeled linkers to constrain the search space to improve runtimes and denoise MS2 spectra, in a similar way as xQuest does. OpenPepXL is to our knowledge the only tool that is able to effectively combine the match confidence of high resolution orbitrap fragment spectra with the additional benefits from stable isotope labeled cross-link spectra preprocessing.
To move the field of XL-MS toward maturity, it is necessary for as many analysis tools as possible to support standardized file formats that are agreed upon by members of the community. OpenMS supports most of the open file formats specified by the HUPO-PSI like mzML for raw MS data and the MS identification data format MzIdentML. This support was extended to include the XL-MS data extension of the MzIdentML 1.2 specification (19).
The OpenMS Proteomics Pipeline (TOPP) contains many additional tools for MS data processing and analysis, including correction of monoisotopic peak assignment and several quantification methods. OpenPepXL is fully integrated into this pipeline and can be easily combined with many of these tools to build complex processing pipelines.
TOPP includes the graphical visualization tool TOPPView for spectra and peptide identifications. It was extended for XL-MS data and can visualize the MS1 features on an MS1 map, MS1 and MS2 peak spectra including the precursor isolation window of an MS2 spectrum, fragment annotations on matched MS2 peaks and the sequence coverage for both cross-linked peptides (Fig. 8). The spectrum visualization allows zooming and the peak labels are fully editable and movable to aid in manual validation and preparation of images for publication. Manually added or edited annotations can be saved in the OpenMS internal proteomics identification file format idXML.
Fig. 8.
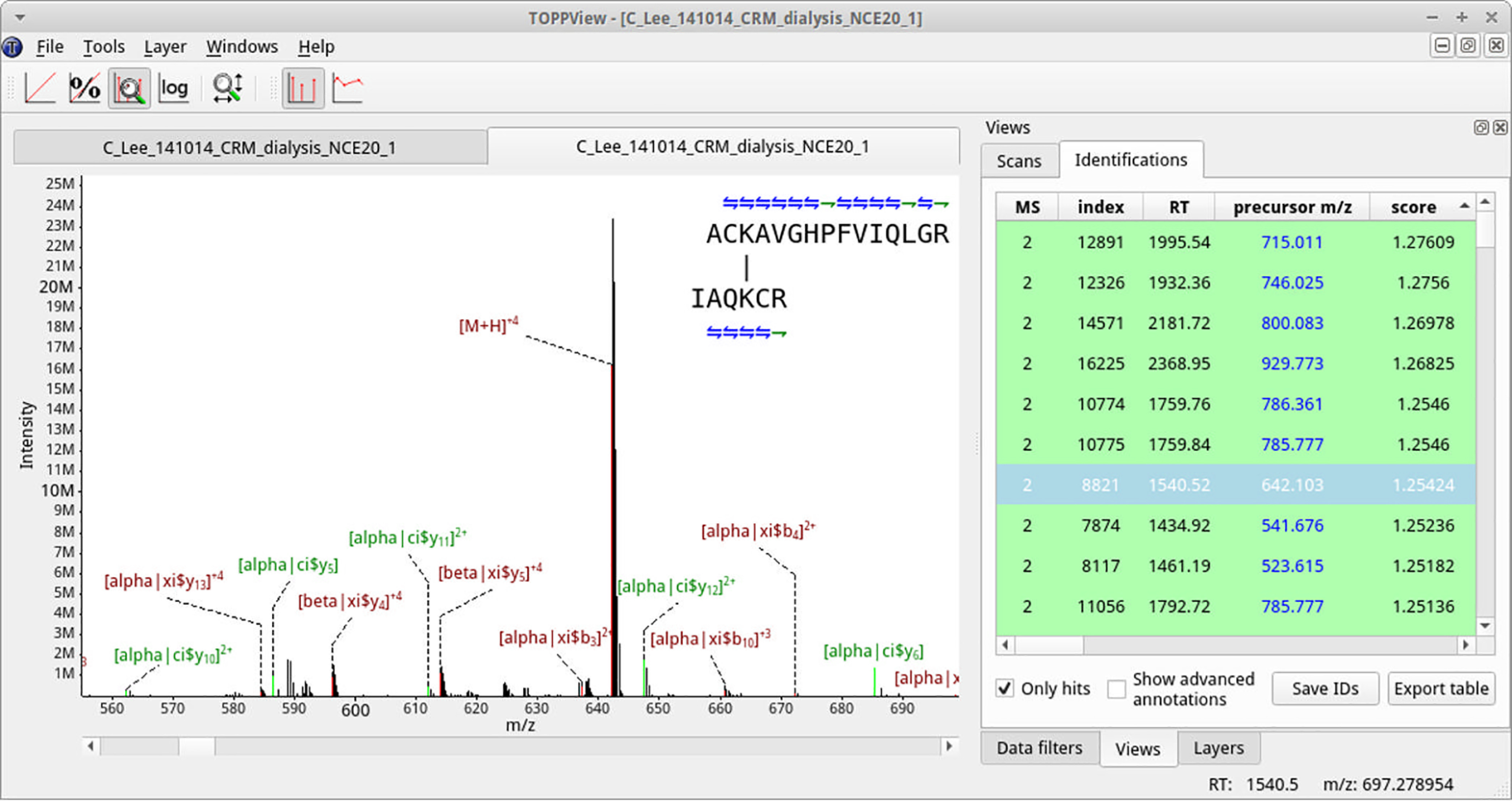
Visualization of Spectra with annotated matched peaks and peptide sequence coverage in TOPPView. On the right side is the table of identifications containing a description of the identified species and several match quality metrics. On the left side is the annotated spectrum with a sequence coverage indicator. A one sided arrow means the fragment starting at the marked residue and containing the rest of the peptide or peptide pair in the direction of the arrow was matched. A double arrow means fragments starting at the marked residue and containing the rest of the peptide or peptide pair in both directions were matched.
DISCUSSION & CONCLUSION
OpenPepXL is a new XL-MS identification algorithm with improved sensitivity at feasible runtime. It is available as open-source software for all major operating systems and compliant with HUPO-PSI standard formats. In our benchmark, OpenPepXL turned out to be a very sensitive XL-MS identification algorithm. It is just as effective on labeled cross-linker data as on label-free data. Its error rate is also similar to other tools with comparable sensitivity. The increased sensitivity is most likely because of the unconstrained search on the complete, quadratic search space. The specificity of OpenPepXL is a consequence of a thorough spectrum matching algorithm that considers relative mass tolerances and ion charge states determined from isotopic patterns or preprocessing of spectra pairs from labeled linkers. The combination of the exploration of the entire search space, very strict criteria for matching peaks between theoretical and experimental spectra and efficient data structures and algorithms makes OpenPepXL a sensitive tool with feasible runtime and memory requirements. OpenPepXL is faster than XiSearch and xQuest, but falls behind Kojak and especially pLink2. Because of efficient data structures and built-in parallelization OpenPepXL achieves very good speedups even on large compute clusters and cloud services while maintaining its slim memory footprint. The increased computational effort for the complete exploration of the quadratic search space can thus be compensated in most cases. This is not the case for several of the other tools, as e.g. pLink2 is only available as a Windows executable and pLink2, StavroX and XiSearch depend on a GUI and are therefore not compatible with many remote computing environments. The implementation of OpenPepXL still has room for improvement and we are looking into ways to make it more efficient without sacrificing its unique sensitivity. Some concepts already common to proteomics data analysis like sequence tags and ion indices are already employed by several of the other XL-MS identification tools and we plan to implement these ideas into OpenPepXL in the future, as long as they are not detrimental to the final output. OpenPepXL is free to use, modify and redistribute for private, academic and commercial applications under the three clause BSD license.
DATA AND SOFTWARE AVAILABILITY
The MS proteomics data from the CRM data set, including search results from all tools compared in this study have been deposited to the ProteomeXchange Consortium (21) via the PRIDE (20) partner repository with the data set identifier PXD014359.
The MS proteomics data from the ribosomal fraction data set (raw data originally from PXD006131 (23)), including search results from all tools compared in this study have been deposited to the ProteomeXchange Consortium (21) via the PRIDE (20) partner repository with the data set identifier PXD014520.
The MS proteomics data from the BSA data set, including search results from OpenPepXL and xQuest have been deposited to the ProteomeXchange Consortium (21) via the PRIDE (20) partner repository with the data set identifier PXD014523.
The MS proteomics data from the synthetic peptide data set (raw data originally from PXD014337 (25)), including search results from OpenPepXL have been deposited to the ProteomeXchange Consortium (21) via the PRIDE (20) partner repository with the data set identifier PXD021417.
Software: OpenPepXL is free to use, modify and redistribute for private, academic and commercial applications under the three clause BSD license. Installers for Windows, macOS and Linux, as well as the source code are linked at https://www.openms.org/openpepxl/.
Supplementary Material
Acknowledgments
We thank Alexander Leitner from the ETH Zürich for providing us with the BSA data set.
Footnotes
This article contains supplemental data.
Funding and additional information—R.F. and H.U. are supported by a grant from the Deutsche Forschungsgemeinschaft (SFB860). T.S. and O.K. were funded by the German Federal Ministry of Education and Research (BMBF) under FKZ 031A535A (German Network for Bioinformatics).
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
Abbreviations—The abbreviations used are:
- FDR
- False Discovery Rate
- XL-MS
- Cross-linking coupled with mass spectrometry
- MS
- Mass Spectrometry
- CSM
- Cross-link Spectrum Match
- URP
- Unique Residue Pair
- MS1
- Precursor spectrum, measurement of full species
- MS2
- tandem MS spectrum, MS/MS spectrum
- DSS
- disuccinimidyl suberatel
- BS3
- bis(sulfosuccinimidyl)suberate
- PDH
- pimelic acid dihydrazide
- IWS
- inconsistent with the protein structure.
REFERENCES
- 1. Liu F., and Heck A. J. (2015) Interrogating the architecture of protein assemblies and protein interaction networks by cross-linking mass spectrometry. Curr. Opin. Struct. Biol. 35, 100–108 [DOI] [PubMed] [Google Scholar]
- 2. Sinz A., Arlt C., Chorev D., and Sharon M. (2015) Chemical cross-linking and native mass spectrometry: A fruitful combination for structural biology. Protein Sci. 24, 1193–1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Leitner A., Faini M., Stengel F., and Aebersold R. (2016) Crosslinking and mass spectrometry: an integrated technology to understand the structure and function of molecular machines. Trends Biochem. Sci. 41, 20–32 [DOI] [PubMed] [Google Scholar]
- 4. O'Reilly F. J., and Rappsilber J. (2018) Cross-linking mass spectrometry: methods and applications in structural, molecular and systems biology. Nat. Struct. Mol. Biol. 25, 1000–1008 [DOI] [PubMed] [Google Scholar]
- 5. Chavez J. D., and Bruce J. E. (2019) Chemical cross-linking with mass spectrometry: a tool for systems structural biology. Curr. Opin. Chem. Biol. 48, 8–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Trnka M. J., Baker P. R., Robinson P. J. J., Burlingame A. L., and Chalkley R. J. (2014) Matching cross-linked peptide spectra: only as good as the worse identification. Mol. Cell. Proteomics 13, 420–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Senko M. W., Beu S. C., and McLaffertycor F. W. (1995) Determination of monoisotopic masses and ion populations for large biomolecules from resolved isotopic distributions. J. Am. Soc. Mass Spectrom. 6, 229–233 [DOI] [PubMed] [Google Scholar]
- 8. Gotze M., Pettelkau J., Schaks S., Bosse K., Ihling C. H., Krauth F., Fritzsche R., Kühn U., and Sinz A. (2012) StavroX—a software for analyzing crosslinked products in protein interaction studies. J. Am. Soc. Mass Spectrom. 23, 76–87 [DOI] [PubMed] [Google Scholar]
- 9. Rinner O., Seebacher J., Walzthoeni T., Mueller L. N., Beck M., Schmidt A., Mueller M., and Aebersold R. (2008) Identification of cross-linked peptides from large sequence databases. Nat. Methods 5, 315–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Leitner A., Walzthoeni T., and Aebersold R. (2014) Lysine-specific chemical cross-linking of protein complexes and identification of cross-linking sites using LC-MS/MS and the xQuest/xProphet software pipeline. Nat. Protoc. 9, 120–137 [DOI] [PubMed] [Google Scholar]
- 11. Kong A. T., Leprevost F. V., Avtonomov D. M., Mellacheruvu D., and Nesvizhskii A. I. (2017) MSFragger: ultrafast and comprehensive peptide identification in mass spectrometry–based proteomics. Nat. Methods 14, 513–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Walzthoeni T., Claassen M., Leitner A., Herzog F., Bohn S., Förster F., Beck M., and Aebersold R. (2012) False discovery rate estimation for cross-linked peptides identified by mass spectrometry. Nat. Methods 9, 901–903 [DOI] [PubMed] [Google Scholar]
- 13. Chen Z.-L., Meng J.-M., Cao Y., Yin J.-L., Fang R.-Q., Fan S.-B., Liu C., Zeng W.-F., Ding Y.-H., Tan D., Wu L., Zhou W.-J., Chi H., Sun R.-X., Dong M.-Q., and He S.-M. (2019) A high-speed search engine pLink 2 with systematic evaluation for proteome-scale identification of cross-linked peptides. Nat. Commun. 10, 3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hoopmann M. R., Zelter A., Johnson R. S., Riffle M., MacCoss M. J., Davis T. N., and Moritz R. L. (2015) Kojak: efficient analysis of chemically cross-linked protein complexes. J. Proteome Res. 14, 2190–2198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fischer L., and Rappsilber J. (2017) Quirks of error estimation in cross-linking/mass spectrometry. Anal. Chem. 89, 3829–3833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dai J., Jiang W., Yu F., and Yu W. (2019) Xolik: finding cross-linked peptides with maximum paired scores in linear time. Bioinformatics 35, 251–257 [DOI] [PubMed] [Google Scholar]
- 17. Rost H. L., Sachsenberg T., Aiche S., Bielow C., Weisser H., Aicheler F., Andreotti S., Ehrlich H.-C., Gutenbrunner P., Kenar E., Liang X., Nahnsen S., Nilse L., Pfeuffer J., Rosenberger G., Rurik M., Schmitt U., Veit J., Walzer M., Wojnar D., Wolski W. E., Schilling O., Choudhary J. S., Malmström L., Aebersold R., Reinert K., and Kohlbacher O. (2016) OpenMS: a flexible open-source software platform for mass spectrometry data analysis. Nat. Methods 13, 741–748 [DOI] [PubMed] [Google Scholar]
- 18. Berthold M. R. (2008) KNIME: The Konstanz Information Miner. Data Analysis, Machine Learning and Applications. Studies in Classification, Data Analysis, and Knowledge Organization. Springer, Berlin, Heidelberg [Google Scholar]
- 19. Vizcaino J. A., Mayer G., Perkins S., Barsnes H., Vaudel M., Perez-Riverol Y., Ternent T., Uszkoreit J., Eisenacher M., Fischer L., Rappsilber J., Netz E., Walzer M., Kohlbacher O., Leitner A., Chalkley R. J., Ghali F., Martínez-Bartolomé S., Deutsch E. W., and Jones A. R. (2017) The mzIdentML data standard version 1.2, supporting advances in proteome informatics. Mol. Cell. Proteomics 16, 1275–1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Riverol Y., Csordas A., Bai J., Bernal-Llinares M., Hewapathirana S., Kundu D. J., Inuganti A., Griss J., Mayer G., Eisenacher M., Pérez E., Uszkoreit J., Pfeuffer J., Sachsenberg T., Yilmaz S., Tiwary S., Cox J., Audain E., Walzer M., Jarnuczak A. F., Ternent T., Brazma A., and Vizcaíno J. A. (2019) The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 47, D442–D450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Deutsch E. W., Csordas A., Sun Z., Jarnuczak A., Perez-Riverol Y., Ternent T., Campbell D. S., Bernal-Llinares M., Okuda S., Kawano S., Moritz R. L., Carver J. J., Wang M., Ishihama Y., Bandeira N., Hermjakob H., and Vizcaíno J. A. (2017) The ProteomeXchange consortium in 2017: supporting the cultural change in proteomics public data deposition. Nucleic Acids Res. 45, D1100–D1106 gkw936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Monecke T., Güttler T., Neumann P., Dickmanns A., Görlich D., and Ficner R. (2009) Crystal structure of the nuclear export receptor CRM1 in complex with Snurportin1 and RanGTP. Science 324, 1087–1091 [DOI] [PubMed] [Google Scholar]
- 23. Kolbowski L., Mendes M. L., and Rappsilber J. (2017) Optimizing the parameters governing the fragmentation of cross-linked peptides in a tribrid mass spectrometer. Anal. Chem. 89, 5311–5318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Iacobucci C., Piotrowski C., Aebersold R., Amaral B. C., Andrews P., Bernfur K., Borchers C., Brodie N. I., Bruce J. E., Cao Y., Chaignepain S., Chavez J. D., Claverol S., Cox J., Davis T., Degliesposti G., Dong M.-Q., Edinger N., Emanuelsson C., Gay M., Götze M., Gomes-Neto F., Gozzo F. C., Gutierrez C., Haupt C., Heck A. J. R., Herzog F., Huang L., Hoopmann M. R., Kalisman N., Klykov O., Kukačka Z., Liu F., MacCoss M. J., Mechtler K., Mesika R., Moritz R. L., Nagaraj N., Nesati V., Neves-Ferreira A. G. C., Ninnis R., Novák P., O'Reilly F. J., Pelzing M., Petrotchenko E., Piersimoni L., Plasencia M., Pukala T., Rand K. D., Rappsilber J., Reichmann D., Sailer C., Sarnowski C. P., Scheltema R. A., Schmidt C., Schriemer D. C., Shi Y., Skehel J. M., Slavin M., Sobott F., Solis-Mezarino V., Stephanowitz H., Stengel F., Stieger C. E., Trabjerg E., Trnka M., Vilaseca M., Viner R., Xiang Y., Yilmaz S., Zelter A., Ziemianowicz D., Leitner A., and Sinz A. (2019) The first community-wide, comparative cross-linking mass spectrometry study. Anal. Chem. 91, 6953–6961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Beveridge R., Stadlmann J., Penninger J. M., and Mechtler K. (2020) A synthetic peptide library for benchmarking crosslinking-mass spectrometry search engines for proteins and protein complexes. Nat. Commun. 11, 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ferrari A. J. R., Clasen M. A., Kurt L., Carvalho P. C., Gozzo F. C., and Martínez L. (2019) TopoLink: evaluation of structural models using chemical crosslinking distance constraints. Bioinformatics 35, 3169–3170 [DOI] [PubMed] [Google Scholar]
- 27. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 28. Kosinski J., von Appen A., Ori A., Karius K., Müller C. W., and Beck M. (2015) Xlink Analyzer: software for analysis and visualization of cross-linking data in the context of three-dimensional structures. J. Struct. Biol. 189, 177–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The MS proteomics data from the CRM data set, including search results from all tools compared in this study have been deposited to the ProteomeXchange Consortium (21) via the PRIDE (20) partner repository with the data set identifier PXD014359.
The MS proteomics data from the ribosomal fraction data set (raw data originally from PXD006131 (23)), including search results from all tools compared in this study have been deposited to the ProteomeXchange Consortium (21) via the PRIDE (20) partner repository with the data set identifier PXD014520.
The MS proteomics data from the BSA data set, including search results from OpenPepXL and xQuest have been deposited to the ProteomeXchange Consortium (21) via the PRIDE (20) partner repository with the data set identifier PXD014523.
The MS proteomics data from the synthetic peptide data set (raw data originally from PXD014337 (25)), including search results from OpenPepXL have been deposited to the ProteomeXchange Consortium (21) via the PRIDE (20) partner repository with the data set identifier PXD021417.
Software: OpenPepXL is free to use, modify and redistribute for private, academic and commercial applications under the three clause BSD license. Installers for Windows, macOS and Linux, as well as the source code are linked at https://www.openms.org/openpepxl/.