

Advanced data integration methods have been here used for a multi-level joint analysis of transcriptomics and proteomics changes on the mouse brain at the hyper-acute phase of stroke. By doing so, an integrated network of inter-correlated and strongly deregulated genes and proteins after ischemia has been identified. Among the conforming elements, CTNND2 and GADD45G have proved to be promising blood biomarkers to be used for the diagnosis and prognosis of ischemic stroke disease, respectively.
Keywords: Stroke biology, biomarkers, animal models, systems biology, bioinformatics, biomarker: diagnostic, biomarker: prognostic
Graphical Abstract

Highlights
Multi-omics analyses of transcriptomics and proteomics data in the mouse ischemic brain.
CTNND2 blood content might be potentially useful for an acute ischemic stroke diagnosis.
GADD45G circulating levels could have a role in acute ischemic stroke prognosis.
Abstract
Stroke remains a leading cause of death and disability worldwide. Despite continuous advances, the identification of key molecular signatures in the hyper-acute phase of ischemic stroke is still a primary interest for translational research on stroke diagnosis, prognosis, and treatment. Data integration from high-throughput -omics techniques has become crucial to unraveling key interactions among different molecular elements in complex biological contexts, such as ischemic stroke. Thus, we used advanced data integration methods for a multi-level joint analysis of transcriptomics and proteomics data sets obtained from mouse brains at 2 h after cerebral ischemia. By modeling net-like correlation structures, we identified an integrated network of genes and proteins that are differentially expressed at a very early stage after stroke. We validated 10 of these deregulated elements in acute stroke, and changes in their expression pattern over time after cerebral ischemia were described. Of these, CLDN20, GADD45G, RGS2, BAG5, and CTNND2 were next evaluated as blood biomarkers of cerebral ischemia in mice and human blood samples, which were obtained from stroke patients and patients presenting stroke-mimicking conditions. Our findings indicate that CTNND2 levels in blood might potentially be useful for distinguishing ischemic strokes from stroke-mimicking conditions in the hyper-acute phase of the disease. Furthermore, circulating GADD45G content within the first 6 h after stroke could also play a key role in predicting poor outcomes in stroke patients. For the first time, we have used an integrative biostatistical approach to elucidate key molecules in the initial stages of stroke pathophysiology and highlight new notable molecules that might be further considered as blood biomarkers of ischemic stroke.
Stroke remains a major leading cause of death and disability worldwide (1) and is a major public health problem that contributes to rising costs of healthcare in many developed countries (2). Effective therapies based on reperfusion mechanisms are currently used (3, 4), but only a small percentage of ischemic stroke patients currently benefit from them. The reason is the narrow therapeutic time window and absolute contraindications for those who present risk of bleeding (5, 6). Thus, new therapeutic strategies are needed to protect the ischemic brain.
More prompt and accurate diagnoses of ischemic stroke might increase the number of patients that benefit from therapies. At present, stroke diagnosis is based on neurological exploration and costly imaging techniques (7), which are still scarcely accessible in pre-hospital settings, primary healthcare centers, and developing countries. Blood biomarkers are expected to become potential substitutes for these neuroimaging approaches (8–10) because they might accelerate the distinction of stroke from other neurological disorders that present similar clinical symptoms. Furthermore, blood biomarkers might also accelerate stroke treatment, help with patient monitoring for adverse effects, and help to anticipate poor patient prognoses at later stages of the disease (11, 12). With this goal, better characterization of the stroke pathophysiology is required to facilitate the identification of molecular indicators of ischemic stroke and provide novel insights into potential therapeutic targets. Such efforts could help to restrain or even reverse the progression of ischemic damage.
In this context, -omics techniques have emerged as sophisticated large-scale analytical tools that enable the accurate identification of biological changes in the brain at different molecular levels (13). Joint analyses of multiple data sets from different –omics techniques are becoming crucial to unravel the relationships among different molecular components and globally interpret all findings in a complex biological context (14). Thus, the combination of multi-omics techniques through integrative analyses is expected to enhance the comprehension of the molecular dynamics underlying ischemic stroke and might provide a framework where the complexity of the interactive molecular networks prevails over the individual alterations of each component separately.
The aim of this study was to identify the main transcriptomics and proteomics changes that occur in the brains of mice during the hyper-acute phase of cerebral ischemia. The goal is to determine potential biomarkers of ischemic stroke. We aimed to combine data from both –omics techniques using an integrative analysis to reveal potential networks of inter-connected genes and proteins with substantial involvement in the very first stages of the stroke pathology. This integrative approach has helped us to identify a set of molecules that are positively replicated and could be further explored in the brains and blood of mice at different time points after cerebral ischemia. Finally, we analyzed the performance of several highlighted candidates as biomarkers for the diagnosis or prognosis of ischemic stroke in a clinical setting.
EXPERIMENTAL PROCEDURES
Experimental Design and Statistical Rationale
The study being presented here is divided into 3 main sections: a first discovery phase and a second replication phase, both conducted in brain mouse samples, and a third qualification phase conducted on mice and human brain and blood samples. Sample size for the initial discovery phase was selected based on technical considerations for the proteomics and transcriptomics approaches. Once candidates were selected for replication, the minimum sample size to replicate the results from the discovery phase was estimated for each candidate separately (power of 80%, α = 0.05) (Ene 3.0, GlaxoSmithKline, UK). To reach a successful replication of at least half of the initially selected candidates, a sample size of 6 animals per group was chosen. The number of mouse and human samples used for the qualification phase was subjected to sample availability. Details about statistical analyses and demographic and clinical data are given below.
Animals
All animal procedures were conducted in compliance with the Spanish legislation and in accordance with the Directives of the European Union and were approved by the Ethics Committee of the Vall d'Hebron Research Institute. C57BL/6J male mice were used for the experiments (8/12-week-old; Janvier Labs, France). Animals were kept in a climate-controlled environment on a 12-h light/12-h dark cycle. Food and water were available ad libitum. Analgesia (Buprenorfine, 0.05 mg/kg, s.c, Divasa Farma-Vic S.A, Spain) was administered to all animals to minimize pain and discomfort. Anesthesia (isoflurane, 4% for induction, 2% for maintenance in air, Abbot Laboratories, Spain) was given via facemask during all surgical procedures described below. All experiments were conducted in a randomized manner and in adherence to the ARRIVE guidelines (15).
A total of 37 animals were used to complete the whole study. In the discovery phase, 15 animals were needed. Among them, 3 were excluded after applying the following criteria: incomplete occlusion or reperfusion after removal of the filament (n = 2) and death during the experimental protocol (n = 1). In the replication phase, 18 animals were used. From those, 6 were excluded because of incomplete occlusion or reperfusion after removal of the filament (n = 2), death during the experimental protocol (n = 3) or poor brain perfusion during euthanasia (n = 1).
Transient Cerebral Ischemia Model (tMCAO)
Transient infarction in the middle cerebral artery territory was induced for 90 min by introducing an intraluminal filament (Doccol 602256PK10Re), as described elsewhere (16). Only animals that exhibited a reduction of CBF of 80% after filament introduction and a recovery of 75% after filament removal were included in the study. After reperfusion, animals could recover for 30 min or 4.5 h (corresponding with 2 h or 6 h from the beginning of the MCA occlusion), according to their experimental group. Sham surgery was performed by the same surgical procedures without insertion of the nylon-coated filament.
Brain and Blood Collection and Extraction of Protein and RNA
Animals were deeply anesthetized at their respective times and blood samples were collected through cardiac puncture, contained in EDTA collection tubes and centrifuged at 1500 × g at 4 °C for 15 min. The top layer containing the platelet-poor plasma fraction was stored at −80 °C until further use. Then, animals were transcardially perfused with cold saline to remove blood from brain vessels. Immediately after perfusion, mouse brains were quickly removed and sectioned into 1 mm slices in cold conditions. The slice corresponding to the bregma anatomical point (as the core of the infarct tissue (17)) was carefully dissected to isolate the right (ipsilateral, IP) and left (contralateral, CL) hemispheres separately (supplemental Fig. S1). Each hemisphere was flash-frozen in liquid nitrogen and stored at −80 °C. Flash-frozen tissues were pulverized into powder in liquid nitrogen, and total fractions of protein and RNA were then isolated using the MirVana™ Paris™ (Thermo Fisher Scientific Inc., MA). RNA and protein fractions were kept at −80 °C until further use.
Discovery Phase Study
The discovery phase was performed on RNA and protein extracts from brain samples from 8 ischemic animals euthanatized 2 h after the MCAO onset. Four sham-control animals were also included in this phase to discard the selection of gene and protein candidates differentially altered because of other phenomena rather than the ischemic insult.
Transcriptomics Study
Total RNA concentrations from mouse brain samples were measured with a Nanodrop 1000 Spectrophotometer (ThermoFisher) and RNA integrity was assessed using the Agilent 2100 BioAnalyzer (Agilent Technologies, USA). The Genechip© Mouse Clariom S 24 × arrays plate (Affymetrix, ThermoFisher) was used to analyze gene expression patterns on a whole-genome scale on a single array. Starting material was 100 ng of total RNA of each sample. Briefly, sense ssDNA was generated from total RNA with the GeneChip WT Plus Reagent Kit (Affymetrix) according to the manufacturer's instructions. Then, sense ssDNA was fragmented, labeled and hybridized to the arrays with the GeneChip WT Terminal Labeling and Hybridization Kit (Affymetrix). Arrays plate was scanned and processed with Affymetrix GeneChip Command Console to obtain expression array intensity .cel files.
Statistical and bioinformatics analyses of microarray data were performed using custom scripts in R language version 3.4.2 (R Core Team, 2017, Vienna, Austria) with common Bioconductor packages. In brief, after following a standard quality control (18), Robust Multi-array Average algorithm (19) was used for pre-processing microarray data to perform background adjustment, normalization and summarization of probe set expression values. Then, genes whose standard deviation (S.D.) was below the 50 percentile of all S.D., without a known Entrez Gene database identifier and without a valid annotation to the Gene Ontology database were filtered out from the whole data set and the final amount of 9324 genes was considered for the statistical analysis. Selection of differentially expressed elements was based on a linear model analysis with empirical Bayes modification for the variance estimates (20). False Discovery Rate (FDR)-based corrections for multiple testing were also calculated (Adj. p-value) (21). Fold-change (FC) was calculated by dividing the IP to the CL expression value for each animal. Data on the brain transcriptomic profile after stroke was analyzed by Ingenuity Pathway Analysis (IPA, Qiagen, USA) (supplemental Methods).
Proteomics Study
Protein extracts from mouse brain samples were quantified using the BCA Protein Assay (ThermoFisher). Twenty µg of each sample were resolved in 12% SDS-polyacrylamide gels under reducing conditions and stained with colloidal Coomassie G250 (Bio-Rad, CA). Each lane was divided in five segments that were individually processed to perform in-gel digestion of the proteins. Briefly, stained gel fragments were cut into small pieces, washed with 50 mm ammonium bicarbonate/50% ethanol and dehydrated with ethanol. Reduction was performed by incubating samples with 10 mm DTT (DTT) for 1h at 56 °C, followed by alkylation with 55 mm iodoacetamide for 30 min at dark. After washing and dehydration with acetonitrile, gel pieces were covered with 2.7 ng/μl trypsin (Promega, USA) in 25 mm ammonium bicarbonate (300 µL) and digestion was run overnight at 37 °C. Peptide extraction was carried out by incubation at 37 °C with acetonitrile (150 µL) and further incubation with 0.2% trifluoroacetic acid (400 µL). The eluted peptides were dried in a Savant™ SpeedVac™ High Capacity Concentrator (ThermoFisher) and stored at −20 °C until further use.
Tryptic digests from excised bands were analyzed using a LC–MS (LC–MS) approach in a linear trap quadrupole (LTQ) Orbitrap Velos mass spectrometer (ThermoFisher). Peptide mixtures were fractionated by on-line nanoflow liquid chromatography using an EASY-nLC system (Proxeon Biosystems, ThermoFisher) with a two-linear-column system: digests were loaded at 4 µL/min onto a trapping guard column (EASY-column, 20 × 0.1 mm, packed with Reprosil C18, 5 μm particle size) and then eluted from the analytical column (EASY-column, 100 × 0.75 mm, packed with Reprosil C18, 3 μm particle size). Separation was achieved by using a mobile phase of 0.1% formic acid in water (Buffer A) and acetonitrile with 0.1% formic acid (Buffer B) and applying a linear gradient from 0 to 35% of buffer B for 120 min at a flow rate of 300 nL/min. Ions were generated applying a voltage of 1.9 kV to a stainless steel nano-bore emitter.
The mass spectrometer was operated in a data-dependent mode. A scan cycle was initiated with a full-scan MS (MS) spectrum (from m/z 300 to 1600) acquired in the Orbitrap with a resolution of 30,000. The 20 most abundant ions were selected for collision-induced dissociation fragmentation in the LTQ when their intensity exceeded a minimum threshold of 1000 counts, excluding singly charged ions. Automatic gain control (AGC) target values were set to 1 × 106 ions for survey MS and 5000 ions for MS/MS experiments. The maximum ion accumulation time was 500 and 200 ms in the MS and MS/MS modes, respectively. The normalized collision energy was set to 35%, and one microscan was acquired per spectrum. Ions subjected to MS/MS with a relative mass window of 10 ppm were excluded from further sequencing for 20 s. For all precursor masses a window of 20 ppm and isolation width of 2 Da was defined. Orbitrap measurements were performed enabling the lock mass option (m/z 445.120024) for survey scans to improve mass accuracy.
Progenesis® QI for proteomics software v3.0 (Nonlinear dynamics, UK) was used for MS data analysis using default settings. The results from each of the five gel fractions were independently analyzed and all MS runs were automatically aligned to a selected reference sample. Alignments were then manually supervised and automatically normalized to all features. A first normalization (within-fraction) of MS signals, based on the median of ratiometric distribution of the abundance measurements, was performed automatically by the Progenesis® QI for proteomics software. Only features within the 400 to 1600 m/z range, from 5 to 115 min of retention time, and with positive charges between 2 to 4 were considered for identification and quantification. Peaklists were generated using Proteome Discoverer 2.1 (Thermo Fisher) and analyzed using the Mascot search engine (v5.1, Matrix Science, UK). Protein identification was carried out using the SwissProt-MusMusculus database (2017_10: 16.942 entries), setting precursor mass tolerance to 10 ppm and fragment mass tolerance to 0.8 Da. Oxidized methionine was considered as variable amino acid modification and carbamidomethylation of cysteines as fixed modification. Trypsin was selected as the enzyme allowing up to two missed cleavage. Significant threshold for protein identification was set to p < 0.05 for the probability-based Mascot score. Peptide and protein identifications were further filtered to < 1%FDR as measured by a concatenated target-decoy database search strategy. Finally, the five fractions were combined into one single Progenesis experiment and a second (between-fraction) normalization was automatically performed by the software to compensate for different running conditions. Label-free protein abundance quantification was based on the sum of the peak areas within the isotope boundaries of peptide ion peaks. Proteins identified by identical peptide sets were grouped to satisfy the principles of parsimony. Only those proteins quantified and identified with at least 2 unique and nonconflicting peptides (it is, features assigned unambiguously to peptides belonging to the protein, as assessed by Progenesis software) were considered for the statistical analysis (44.8%, 2485 proteins). Protein and peptide identifications are provided in supplemental Table S1.
Data were further processed using custom scripts in R language version 3.4.2 (R Core Team, 2017). Protein abundance values were log-10 transformed and column-wise standardized (22). Selection of differentially expressed proteins was based on a linear model analysis implemented in the Bioconductor limma package (23). False Discovery Rate (FDR)-based corrections for multiple testing were also calculated (Adj. p-value) (21). Fold-change (FC) was calculated by dividing the IP to the CL expression value for each animal. Data on the brain proteomic profile after stroke was analyzed by Ingenuity Pathway Analysis (IPA, Qiagen, Germany) (supplemental Methods).
Integrative Analyses
For the integrative analyses only samples from ischemic animals were considered. All features (genes and proteins) with a nonadjusted p-value <0.05 were included in the analyses. Individual pre-processing of ischemia-related differentially expressed gene and protein data sets through Principal Component Analysis (PCA) resulted in the identification of two samples (corresponding to the IP and CL hemispheres from one single animal) behaving as outliers, which were beforehand excluded for the integrative analyses (supplemental Fig. S2). Thus, the integrative analyses of significant differentially expressed genes and proteins were only performed with samples from 7 MCAO animals. Besides, the protein TXN2 was momentarily precluded from these integrative analyses, because its relatively high fold change expression in the ischemic hemisphere masked all other candidates with a similar abundance profile when data were integrated.
Standardized and normalized gene and proteins data sets were used for the integrative analysis. Three different integrative tools were employed, all of them implementing omics-focused versions of already known dimension reduction techniques. First, Multiple Co-Inertia Analysis (MCIA (24)) included in made4 R package (v1.52.0 (25)) was performed to maximize the covariance between gene and protein data sets for each group of samples. To that end, data were matched according to the measurements (samples) and weight-equalized. Then, data were transformed into comparable lower dimensional spaces and a generalization of the Co-Intertia Analysis (CIA) was applied to provide a simultaneous ordination of measurements (samples) and features (genes and proteins) within the same hyperspace. Second, mixOmics R package (v6.3.1, http://mixomics.org) was used to perform a regularized Canonical Correlation Analysis (rCCA (26)) between gene and protein data sets. In brief, data were properly transposed and scaled and the tuning of the rCCA parameters were iterated until achieving a cross-validation score of 0.8 (lambda 1 = 0.1318 and lambda 2 = 0.001). Resulting relevance network was plotted for a correlation cutoff of R ≥ 0.75. Third, the mogsa R package (v1.12.2 (27)) was employed to annotate and weight genes and proteins data sets against gene sets from the Gene Ontology (GO). This analysis was based on the application of the Multiple Factor Analysis (28) and was fed with the gene sets corresponding to GO annotations in Mus musculus for Hallmark (v5.2 (29)) and Broad Institue's C2 Cannonical Pathways molecular signature databases (http://software.broadinstitute.org/gsea/login.jsp, v5.2). Default parameters were used, applying weighting of the individual data sets to prevent data sets with more features or different scales dominating the MFA results. Only those gene sets significantly enriched in our joint data set (genes and proteins) that depict any of our 18 selected candidates from the correlation-based integrative network are shown. The mogsa R package also generated gene set scores (GSS) by computing all gene and protein contributions to each gene set found to be enriched (27). GSS were further decomposed with respect to each data set (genes and proteins).
Replication Phase Study
The replication phase was conducted on brain samples from 6 ischemic animals obtained 2 h after ischemia. Replicated candidates were further evaluated on other brain samples from 6 ischemic animals collected 6 h after ischemic onset. RNA and protein fractions were obtained from the mouse brains following the same procedure as in the discovery phase.
Digital Multiplexed Gene Expression Assay
The Nanostring's nCounter® XT Assay (NanoString Technologies®, USA) was performed at the Genomics Core Facility, Center Esther Koplowitz, Barcelona (Spain). The assay was conducted according to manufacturer's instructions. Briefly, Nanostring's nCounter Elements™ XT probes were created (Integrated DNA Technologies, S.L, Spain) for candidate genes from the discovery phase, along with 3 housekeeping genes (beta-2-microglobulin (B2m), glyceraldehyde-3-phosphate dehydrogenase (Gapdh) and peptidyl-prolyl cis-trans isomerase A (Ppia)) (supplemental Table S2). One hundred ng of the total RNA from mouse brain samples was hybridized with the complete Master Mix containing the pool of probes. Then, samples were loaded onto the NanoString PrepStation, placed into the nCounter cartridge and transferred to the nCounter digital analyzer for image capture and data acquisition of fluorescent reporters. Measurements were taken at high sensitivity with 280 fields of view. Data analysis and normalization with standard procedures was performed with the NanoString-supplied software (v3.0). Briefly, following background subtraction, raw intensity values (RNA counts) were normalized to the geometric mean of the positive control spike-in RNAs, followed by a second normalization to the geometric mean of the housekeeping genes. Values were finally expressed as fold-change of the IP compared with the CL hemispheres for each animal. Differences in gene levels between IP and CL hemispheres were assessed by paired t test (normally distributed variables) or Wilcoxon signed-rank test (nonnormally distributed variables). Comparisons between fold-change expressions at 2 h and at 6 h were evaluated through the Student t test (normally distributed variables) or the Mann-Whitney test (nonnormally distributed variables). All p-values were then corrected by the false discovery rate (Adj. p-values).
Targeted Mass Spectrometry: Parallel Reaction Monitoring (PRM)
This proteomic experiment was categorized as a Tier 2 targeted MS measurement, as previously published elsewhere (30). Protein extracts from mouse brain samples were subjected to buffer exchange to 8M Urea in 50 mm ammonium bicarbonate using 3KDa cutoff Amicon Ultra ultrafiltration devices (Merck-Millipore, USA). Afterward, total protein content was quantified using the RCDC kit (Bio-Rad), and 4 µg of each protein extract were processed. Samples were reduced with 10 mm DTT for 1h, and then alkylated with 20 mm iodoacetamide for 30 min at dark. Carbamidomethylation reaction was quenched by addition of 35 mm N-acetyl-l-cysteine for 15 min at dark. Samples were diluted with 50 mm ammonium bicarbonate to a final concentration of 1M Urea before being digested with trypsin in a ratio of 1:20 (w/w) overnight at 37 °C. The reaction was stopped with 0.5% formic acid, and the tryptic digest was kept at −20 °C until further analysis.
Proteotypic peptides from the selected protein candidates, along with 3 housekeeping proteins (B2M, GAPDH and PPIA), were selected from the LC–MS experimental results on the discovery phase or from MS data repositories (Peptide Atlas, SRM Atlas) (supplemental Table S2). Isotopically labeled versions of target peptide sequences were purchased from ThermoFisher. Analysis of the heavy labeled peptides spiked into a pool of the samples yielded concentration response curves with high correlation coefficients (typically >0.9) and low coefficients of variation (≤20%) over a 10-fold concentration range.
Prior to LC–MS analysis, known amounts of each labeled peptide (ranging from 1-18000 fmol per µg of digest) were mixed with sample digests, from which 500 ng were analyzed using LTQ Orbitrap Velos MS. In brief, peptide mixtures were fractionated in the EASY-nLC 1000 system: digests were loaded at 4 µL/min onto a trapping guard column (Acclaim PepMap 100 nanoviper, 20 × 0.75 mm, packed with C18, 3 μm particle size; ThermoFisher) and then eluted from the analytical column (250 × 0.75 mm, packed with Reprosil Pur C18-AQ, 3 μm particle size; Dr. Maisch GmbH). Separation was finally achieved by the same procedure used for the discovery phase LC–MS analysis (4.2.2.).
The LTQ Orbitrap Velos MS was operated in parallel reaction monitoring (PRM) mode. PRM was used to acquire full MS/MS spectra of the target peptides, from which a precursor ion mass list was generated based on their sequence into Skyline (v4.1.0. MacCross Lab Software, WA). AGC target value was set for 5000 ions in MS/MS. The maximum ion accumulation time was 50 ms. Normalized collision energy was set to 38% and one microscan was acquired per spectrum. For all precursor masses an isolation width of 2 Da was defined.
Raw data were imported to Skyline software to analyze the results in a blind manner. Chromatographic ion extractions of the 3 to 5 transitions that gave the highest intensities were used to quantify each peptide (available in the Panorama Public repository). For peptides giving significant amounts of two different charge state ions, signals were acquired for both. Dot product values of the selected transitions compared with the standard peptide were used as a criterion to remove signals with potential interferences. Ratios between the unlabeled endogenous peptide and the labeled internal standard (L/H) were then used to calculate endogenous peptide levels within each sample. In brief, amounts of each endogenous peptide were extrapolated from their respective concentration-response curves and normalized to the geometric abundance average of the housekeeping proteins. For those proteins with more than one representative peptide, the geometric mean of all peptide values was also computed for each sample. All samples were run twice and the mean protein value for each sample was finally obtained. Final values were expressed as fold-change of the IP compared with the CL hemisphere. Differences in protein levels between IP and CL hemispheres were assessed by paired t test (normally distributed variables) or Wilcoxon signed-rank test (nonnormally distributed variables). Comparisons between fold-change expressions at 2 h and at 6 h were evaluated through the Student t test (normally distributed variables) or the Mann-Whitney test (nonnormally distributed variables). All p-values were then corrected by the false discovery rate (Adj. p-values).
Western Blot
BAG5 and SRGAP2 were evaluated by means of Western Blot. Detailed methodology is described in supplemental Methods.
Qualification Phase Study
The qualification phase was conducted on brain and blood samples from the same ischemic and sham-control animals obtained 2 h (n = 8) and 6 h (n = 6) after ischemia and from sham-control animals (n = 4). Outstanding candidates were further evaluated as stroke blood biomarkers on human blood samples from ischemic stroke patients and patients suffering from stroke-mimicking conditions.
Human Blood Samples
All human studies were approved by the Ethics Committee of Vall d'Hebron Hospital (PR[AG]157/2011) and written informed consent was obtained from all subjects or relatives in accordance with the Declaration of Helsinki (1964) and its later amendments or comparable ethical standards. All patients were admitted to the emergency department of the Vall d'Hebron Hospital (Barcelona, Spain) from August 2012 to June 2015 within the first 6 h after neurological symptoms onset. On admission, patients underwent a standardized protocol of brain imaging to differentially diagnose ischemic stroke and stroke-mimicking conditions (mimics). Trained neurologists assessed stroke severity using the National Institutes of Health Stroke Scale (NIHSS) and obtained demographic and clinical data from all patients. Eighty-nine ischemic stroke patients and 40 other patients suffering from stroke-mimicking conditions (mimics) were included in this study. Fifteen (16.8%) out of the 89 stroke patients received the standard thrombolytic treatment (intravenous 0.9 mg/Kg recombinant tissue-plasminogen activator), and 5 (5.6%) underwent mechanical thrombectomy to remove the arterial clot. Only 3 ischemic stroke patients (3.4%) receive both thrombolytic therapies. The clinical follow-up of each patient was carried out at hospital discharge. Functional outcome was evaluated according to the modified Rankin Scale (mRS); patients with a mRS score below 2 were classified as “good outcome” and patients with a mRS from 2 to 6 as “poor outcome.”
Blood samples from all patients were drawn on admission (<6 h from symptoms onset) and before administration of any treatment. Samples were collected in EDTA tubes and centrifuged at 1500 × g at 4 °C for 15 min. The top layer containing the platelet-poor plasma fraction was then stored at −80 °C until further use.
Mouse and Human ELISAs
Selected candidates were evaluated by means of commercial ELISA immunoassays, following manufacturer's instructions (supplemental Methods).
Statistical Analyses
SPSS statistical package (v22.0; IBM Corporation, Armonk, NY, USA) and R software were used for statistical analyses and GraphPad Prism (v6.0; GraphPad Software, La Jolla, CA, USA) for creating graphs. Normality was assessed by Shapiro-Wilk and/or Kolmogorov–Smirnov tests. In mice, brain protein levels between the IP and CL hemispheres, and IP/CL ratios between different time points were compared using Student t test and/or Mann-Whitney test. Blood protein levels after MCAO were compared with those after sham-control surgery through One-way ANOVA or Kruskall Wallis, followed by Tukey's multiple comparison test.
For analyzing human data, Chi-squared test was used to assess intergroup differences for categorical variables, expressed as frequencies. Correlations between continuous variables were calculated using Spearman's test (no parametric data). Receiver operating characteristics (ROC) curves were used to obtain the cutoff points of GADD45G and CTNND2 circulating levels with optimal accuracy (both sensitivity and specificity) for each specific end point. To identify whether the biomarkers potentially and significantly improve the capacity of the clinical markers now used to predict stroke diagnosis or stroke prognosis, forward stepwise multivariate logistic regression analyses were performed. All clinical variables associated with each end point at p < 0.1 were included as covariables. Odds ratio and 95% confidence interval (CI) were adjusted by age, sex and NIHSS score at baseline. Using the selected cutoff point, baseline levels of each biomarker were also added on top of their respective clinical model (only clinical) to assess its independent association and to build new predictive models (clinical + biomarker). The likelihood-ratio test was used to assess the goodness of fit of the two predictive models (lmtest package) and the integrated discrimination improvement (IDI) was used to determine the added value of each biomarker to the clinical model alone (Hmisc R package). In all cases, a two-sided p-value <0.05 was considered significant at a 95% confidence level.
RESULTS
Differentially Expressed Genes and Proteins Early after Cerebral Ischemia
A chart of the study workflow is shown in Fig. 1. We elucidated the specific gene and protein changes that occur early in the brain after cerebral ischemia by evaluating mouse-brain samples obtained at 2 h after a surgery to induce ischemia or a sham-control surgery. The changes were identified using transcriptomics and proteomics approaches. After cerebral ischemia, 76 genes and 192 proteins were found to be differentially regulated between IP and CL brain regions (nonadjusted p-values < 0.05; supplemental Table S3). In contrast, only 2 genes and 60 proteins were altered between IP and CL hemispheres after the sham-control surgery (nonadjusted p-value < 0.05). Alteration of the transcriptome and proteome after cerebral ischemia is also evidenced by the differential contribution of genes and proteins to key deregulated pathways after stroke, as shown in supplemental Fig. S3.
Fig. 1.
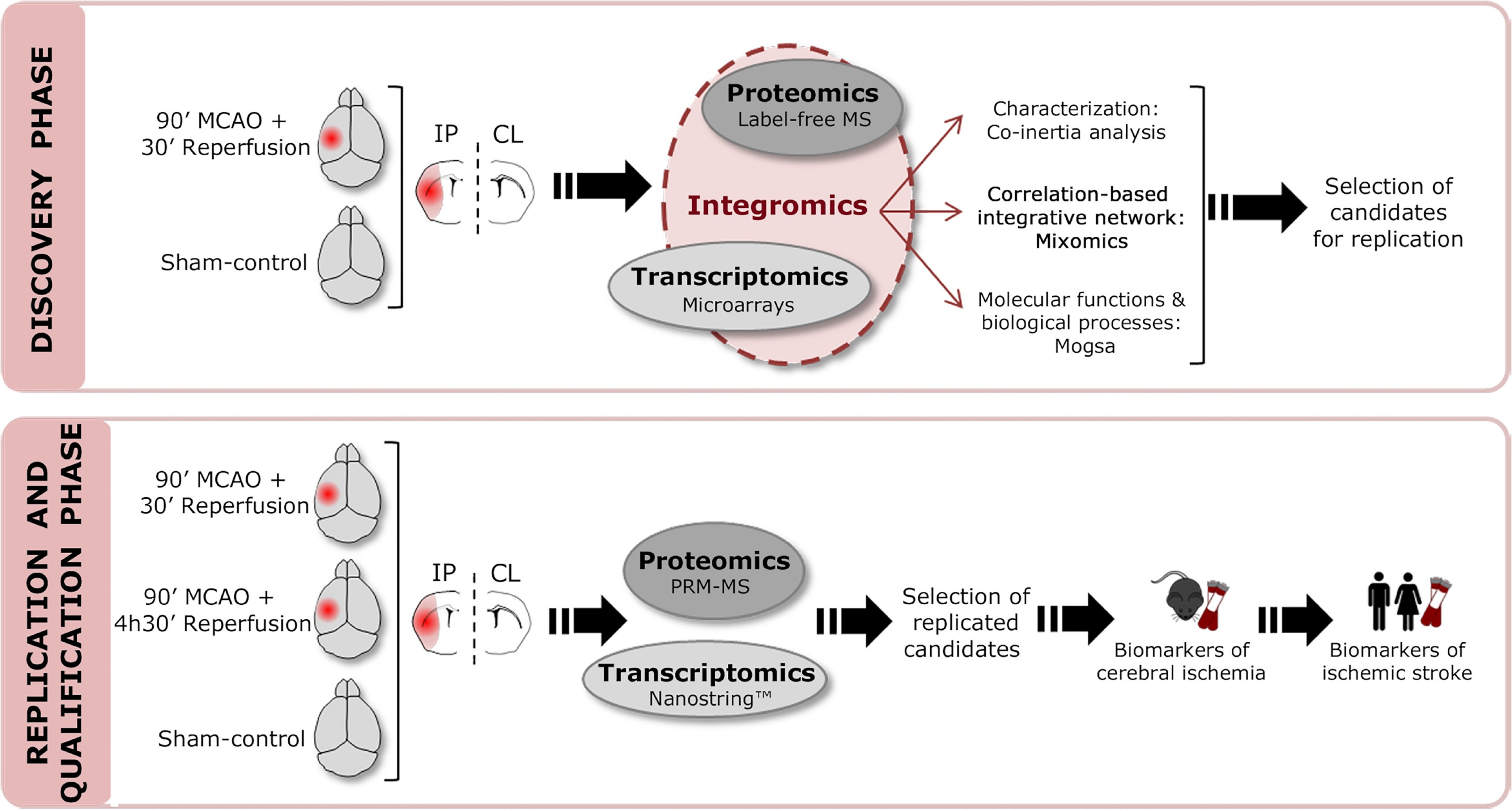
Workflow chart. Schematic representation of the different phases of our study.
Integrative Analysis of Differentially Abundant Proteins and Genes after Cerebral Ischemia
To explore the mechanisms underlying cerebral ischemia more deeply, we conducted a comprehensive integration analysis of both -omics data sets from MCAO animals. Overall, 76 genes and 191 proteins that were deregulated after ischemia (after excluding TXN2; see methods section) were used to combine data in these integrative approaches (Fig. 2). Using MCIA, we examined how the experimental samples behaved when projecting gene and protein components in the same bi-dimensional space (Fig. 2A). We clearly differentiated two separated co-structures corresponding to each brain hemisphere, which confirmed that integrated data from both data sets had the ability to plainly discriminate phenotypical changes because of cerebral ischemia. However, both data sets' structures were substantially different in the bi-dimensional projection and only showed around a 43% overall similarity (RV coefficient = 0.429).
Fig. 2.

Integrative analysis of transcriptomic and proteomic data from the mouse ischemic brain. A, Multiple co-inertia analysis (MCIA), distribution of samples based on their transcriptomic (dot) and proteomic (arrow) information for CL (dark gray) and IP (light gray) samples. Circle and arrows from each sample are joined by a line, the length of which is proportional to the divergence between those samples in the two data sets. B, Relevance network of top correlations between genes (circles) and proteins (rectangles). Dashed lines indicate positive correlations; continuous lines indicate negative correlations. Selected candidates for replication are marked in dark grey. C, Altered biological processes and signaling pathways in which the selected candidates (in bold) are involved. Genes are indicated in italics; proteins are shown in regular type. D, Decomposed gene set score (GSS) of the most outstanding intracellular signaling pathways weighted for genes (light grey) and proteins (dark grey). Means and 95% of confidence intervals are depicted in bar graphs. The higher decomposed GSS, the larger contribution of the data set to the specific process or pathway.
We modeled network-like correlation structures (rCCA) between our protein and gene data sets, through which we identified an integrated network where highly correlated elements are represented (Fig. 2b). Positive and negative correlations were found among 24 genes and 8 proteins. Interestingly, two clusters were visually highlighted around CAMK2A and SRGAP2, with 19 and 10 directly connected genes, respectively. This suggests an important role of these candidates in the pathophysiology of stroke.
From this correlation-based integrated network, 14 representative candidates were selected to replicate their performance in the hyper-acute phase of cerebral ischemia (as shown in gray in Fig. 2B). Furthermore, 3 other candidates were also included because of their leading position in the analysis of the gene individual data set (sorted by p-value: Gadd45g and Rgs2) and in the protein data set (sorted by logFC: TXN2). Overall, 17 candidates (12 genes and 5 proteins) were chosen to be further replicated in a new cohort of ischemic animals (Table I). Gene and protein data sets from sham-control animals were used to discard the selection of any relevant component disturbed because of other reasons rather than cerebral ischemia itself. In this regard, all selected candidates were assumed to be specifically deregulated because of cerebral ischemia because none of them were altered after the sham-control surgery.
Table I. Gene expression and protein abundance of the selected candidates in all study phases. *BAG5 and SRGAP2 replication was conducted using independent western blot experiments, so p-values are not adjusted for multiple comparisons (supplemental Fig. S4). Significant p-values are indicated in bold, #indicates p < 0.1.
| Discovery phase-2 h after MCAO- |
Replication phase-2 h after MCAO- |
Exploration over time-6 h after MCAO- |
2 h versus 6 h |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Fold change (IP/CL) | Raw p-value | Adj. p-value | Fold change (IP/CL) | Raw p-value | Adj. p-value | Fold change (IP/CL) | Raw p-value | Adj. p-value | Raw p-value | Adj. p-value | |
| Gene expression | |||||||||||
| 4933427D14Rik | 0.341 | 0.045 | 0.999 | 0.865 | 0.011 | 0.017 | 0.942 | 0.360 | 0.390 | 0.338 | 0.440 |
| Atf3 | 27.44 | <0.001 | 0.167 | 7.603 | 0.002 | 0.006 | 14.230 | <0.001 | 0.002 | 0.198 | 0.286 |
| Ccl3 | 161.32 | <0.001 | 0.027 | 9.012 | <0.001 | 0.001 | 10.130 | 0.027 | 0.050 | 0.849 | 0.849 |
| Cldn20 | 0.216 | 0.007 | 0.999 | 0.494 | 0.004 | 0.007 | 0.845 | 0.094# | 0.135 | 0.012 | 0.099# |
| Cstad | 0.352 | 0.034 | 0.999 | 0.498 | 0.004 | 0.007 | 0.456 | 0.001 | 0.002 | 0.580 | 0.685 |
| Fosb | 45.74 | <0.001 | 0.793 | 7.407 | <0.001 | 0.001 | 6.349 | 0.002 | 0.006 | 0.683 | 0.740 |
| Gadd45g | 20.06 | 0.005 | 0.999 | 4.010 | <0.001 | 0.001 | 2.129 | <0.001 | 0.001 | 0.015 | 0.099# |
| Gata2 | 0.319 | 0.017 | 0.999 | 0.775 | 0.118 | 0.171 | 1.053 | 0.893 | 0.893 | 0.134 | 0.235 |
| Il1r1 | 0.299 | 0.029 | 0.999 | 0.882 | 0.233 | 0.275 | 1.145 | 0.350 | 0.390 | 0.125 | 0.235 |
| Intu | 0.384 | 0.039 | 0.999 | 0.937 | 0.422 | 0.457 | 1.158 | 0.249 | 0.324 | 0.145 | 0.235 |
| Rgs2 | 43.38 | 0.008 | 0.999 | 3.922 | 0.002 | 0.005 | 2.721 | <0.001 | 0.001 | 0.074# | 0.235 |
| Zfand4 | 0.308 | 0.027 | 0.999 | 0.997 | 0.196 | 0.254 | 0.620 | 0.007 | 0.014 | 0.125 | 0.235 |
| Protein abundance | |||||||||||
| Bag5* | 0.761 | <0.001 | 0.042 | 0.776 | 0.048 | – | 0.558 | <0.001 | – | 0.096# | – |
| Ctnnd2 | 0.921 | 0.001 | 0.220 | 0.778 | 0.028 | 0.084# | 0.960 | 0.457 | 0.686 | 0.054# | 0.162 |
| Camk2a | 0.826 | 0.001 | 0.219 | 0.772 | 0.120 | 0.180 | 0.837 | 0.124 | 0.372 | 0.708 | 0.708 |
| Srgap2* | 1.089 | 0.007 | 0.474 | 1.088 | 0.597 | – | 0.784 | 0.123 | – | 0.159 | – |
| Txn2 | 2.851 | 0.011 | 0.489 | 1.013 | 0.826 | 0.826 | 1.015 | 0.989 | 0.989 | 0.629 | 0.708 |
Further information was also obtained about the main biological processes and molecular functions in which these selected candidates were involved (mogsa). In this regard, the results indicated that some of our selected candidates have a role in the post-stroke impaired neuronal system (Fig. 2C). Others are involved in altered transmission across synapses and the overactivation of the hypoxic pathways, which are all well-known altered functions in acute cerebral ischemia. Moreover, changes in the immune system were already noticeable at this early time point as well, reinforcing its involvement in the acute pathophysiology of stroke. Mediators and contributors of hypoxic signaling pathways were also over-represented in our set of candidates, together with several intracellular signaling pathways, including those involving mitogen-activated protein kinase (MAPK), activator protein-1 (AP-1) transcription factor, G protein–coupled receptors (GPCRs), p53, and tumor necrosis factor alpha (TNFα). Interestingly, deeper exploration of these altered pathways indicated that genes were contributing substantially more than proteins to the alteration of all the specific signaling cascades at the time point examined (Fig. 2D).
Replication and Exploration of the Selected Candidates over Time after Cerebral Ischemia
To confirm the robustness of the microarray and LC–MS results, the selected gene and protein candidates were evaluated in a new cohort of MCAO animals using the Nanostring® nCounter assay and PRM-MS (supplemental Table S2). Eight of the 12 selected genes (66.7%) were correctly replicated in this new cohort of animals: 2 h after MCAO, Ccl3, Atf3, Fosb, Gadd45g, and Rgs2 significantly increased in the IP compared with the CL hemisphere, whereas 4933427D14Rik, Cldn20, and Cstad decreased in the IP versus the CL brain region (Table I). Regarding proteins, 2 out of the 5 selected proteins (40%) were also successfully replicated: both BAG5 and CTNND2 protein levels showed a decrease in the IP hemisphere compared with their CL.
Gene and protein candidates were also explored in the brain at 6 h after MCAO. The brain expression of Ccl3, Atf3, and Fosb remained at high levels at 6 h after the ischemic event, and no differences in the IP/CL ratios were detected between the 2 studied time points (Table I). Conversely, the genes Gadd45g and Rgs2 showed a decrease by half in their fold-change expression at 6 h after MCAO compared with that at 2 h, but the expression remained high in the IP compared with the CL hemisphere.
Regarding the early downregulated genes, 4933427D14Rik no longer maintained the changes between IP and CL hemispheres at 6 h after ischemia. At this later time point, the expression of Cldn20 also returned to normal levels, whereas that of Cstad decreased over time. In terms of protein candidates, BAG5 levels decreased further in the IP than the CL hemisphere at 6 h after MCAO, whereas CTNND2 protein levels reversed their initial downregulation at this later time point (Table I).
CLDN20, GADD45G, and RGS2 Protein Levels in the Brain after Cerebral Ischemia
From all the positively replicated candidates, further attention was focused on Cldn20, Gadd45g, Rgs2, CTNND2, and BAG molecules because all showed an interesting and mostly unknown deregulated expression profile in the brain after ischemia. The gene candidates Cldn20, Gadd45g, and Rgs2 were first evaluated at the protein level. Similar to its gene expression profile, brain levels of CLDN20 protein significantly decreased in the IP hemisphere at 2 h after ischemia (p = 0.013 versus CL) and tended to return to normal levels at a later time (p = 0.057 versus CL) (Fig. 3A). In contrast, the GADD45G protein content did not differ between the IP and CL hemispheres at 2 h after the ischemic insult but showed a significant increase in the IP hemisphere later (p = 0.030 versus CL). No tangible differences were observed in RGS2 protein levels between IP and CL hemispheres at 2 h and 6 h after ischemia.
Fig. 3.

Qualification study of the selected candidates. A, Protein levels of CLDN20, GADD45G and RGS2 in brain after cerebral ischemia. The IP/CL ratio (mean ± S.D.) is shown for each candidate at each examined time point (n = 8 for 2 h post MCAO; n = 6 for 6 h post MCAO). B, Blood protein levels in mice after sham-control surgery (n = 4) and 2 h (n = 8) and 6 h (n = 6) after cerebral ischemia. Graph bars represent mean ± S.D. C, Blood protein levels of CTNND2 in ischemic stroke (n = 71) and mimic patients (n = 36) (median and interquartile range are indicated). ROC curve comparison between the clinical predictive model (grey line) and clinical predictive model with CTNND2 > 1.672 ng/ml (black dashed line) is also shown and the AUC (95% CI) is indicated for each ROC curve. D, Blood protein levels of GADD45G in ischemic stroke patients that showed poor in-hospital functional outcome (n = 48) and patients who did not (n = 27) (median and interquartile range are indicated). ROC curve comparison between the clinical predictive model (grey line) and clinical predictive model with GADD45G > 200.141 pg/ml (black dashed line) is also shown, and the AUC (95% CI) is indicated for each ROC curve. In all cases, *p < 0.05 and #p < 0.1.
Exploration of Selected Candidates as Blood Biomarkers of Cerebral Ischemia in Mice
To study their role as stroke biomarkers, the levels of CLDN20, GADD45G, RGS2, BAG5, and CTNND2 protein were also evaluated in blood samples from mice subjected to cerebral ischemia (at 2 h and 6 h) and sham-control surgery. Unfortunately, CLDN20 levels were not detectable in any of our mouse blood samples. Blood levels of GADD45G and RGS2 were higher 2 h after cerebral ischemia than in sham-control animals (p = 0.043 and p = 0.017, respectively) (Fig. 3B). Later, circulating GADD45G levels remained increased (sham versus 6 h post-MCAO: p = 0.023), whereas RGS2 blood content slightly decreased and no longer differed from RGS2 levels in the sham group. In contrast, the CTNND2 protein content decreased substantially in circulation in mice that underwent stroke surgery compared with the sham-controls, although statistical significance was only reached at 6 h after ischemia (sham versus 6 h post-MCAO: p = 0.032). BAG5 did not differ among the experimental groups.
Exploration of Selected Candidates as Blood Biomarkers of Ischemic Stroke
To further confirm the potential of the candidate biomarkers for ischemic stroke in humans, we evaluated their circulating levels in human blood samples from patients suffering from ischemic stroke (n = 89) and stroke-mimicking conditions (mimics, n = 40). The demographic and clinical characteristics of these patients are shown in supplemental Table S4. In short, ischemic stroke patients had higher NIHSS scores at baseline and a higher prevalence of atrial fibrillation than the mimic group (p = 0.004 and p = 0.003, respectively). No differences were observed for RGS2 between patient groups.
CTNND2 as a Biomarker for Ischemic Stroke Diagnosis
CTNND2 blood levels were assessed in 71 ischemic stroke patients and 36 mimics with very similar demographic and clinical characteristics to the complete cohort of patients (data not shown). At baseline, blood levels of CTNND2 appeared to be higher in ischemic stroke group than the mimic group at just above the margin of statistical significance (p = 0.054) (Fig. 3C). The ischemic stroke group had more severe strokes (according to the NIHSS score) and exhibited a higher prevalence of atrial fibrillation than the mimic group (supplemental Table S4), but differences in the circulating levels of CTNND2 could not be explained by any of these clinical variations (data not shown).
Using ROC curve analysis, we selected a cutoff point for CTNND2 (1.672 ng/ml) with higher sensitivity and specificity to differentiate the ischemic stroke group from the mimic group (62% sensitivity and 74% specificity) (p < 0.001) (Fig. 3C). A multivariate logistic regression analysis was performed to evaluate the capacity of these associated clinical variables to diagnose stroke. The analysis included the NIHSS score at admission, the presence of atrial fibrillation, and adjustments for age and sex. The model confirmed that atrial fibrillation remained as the only independent predictor of ischemic stroke.
To test whether the biomarker improved the performance of the clinical variables, we added CTNND2 > 1.672 ng/ml as a covariable to this predictive clinical model. We found that the biomarker also persisted as an independent factor to discriminate between patients showing stroke-mimicking symptoms and the ischemic stroke group (Table IIA). Notably, the addition of the CTNND2 also improved the goodness-of-fit of the clinical model (p = 0.004) and significantly improved the discriminatory ability to diagnose nonstroke events by 8.84%.
Table II. Predictive comparative models for ischemic stroke diagnosis (A) and post-stroke poor outcome at hospital discharge (B). For logistic regression models, ORadj (95% CI) and p-values are given. Biomarkers were added to clinical logistic regression using the indicated cutoff point. Clinical model is always used as a reference model (Ref.). IDI index is given for events, nonevents, and for the sum of both (with 95% CI). Statistical significant results are highlighted in bold.
| A CTNND2 | Ischemic Stroke Diagnosis |
|
|---|---|---|
| Only Clinical | Clinical + CTNND2 | |
| Logistic regression (OR adj) | ||
| Admission NIHSS score | 0.93 (0.86–1.02), p = 0.118 | 0.95 (0.86–1.04), p = 0.257 |
| Age | 0.98 (0.96–1.01), p = 0.286 | 0.98 (0.95–1.02), p = 0.338 |
| Sex | 1.23 (0.55–2.71), p = 0.613 | 0.95 (0.40–2.49), p = 0.991 |
| Atrial fibrillation | 0.31 (0.11–0.93), p = 0.036 | 0.29 (0.08–1.00), p = 0.050 |
| CTNND2 > 1.672 ng/ml | 0.26 (0.10–0.67), p = 0.005 | |
| IDI statistics | ||
| IDI | Ref. | 9.12% (3.50–14.74), p = 0.001 |
| IDI events | – | 0.29% |
| IDI nonevents | – | 8.84% |
| B GADD45G | Poor outcome at discharge |
|
| Only clinical | Clinical + GADD45G | |
| Logistic regression (OR adj) | ||
| Admission NIHSS score | 1.23 (1.08–1.40), p = 0.001 | 1.51 (1.19–1.90), p = 0.001 |
| Age | 1.06 (1.01–1.11), p = 0.018 | 1.09 (1.01–1.17), p = 0.019 |
| Sex | 1.04 (0.37–2.92), p = 0.940 | 0.70 (0.17–2.90), p = 0.620 |
| GADD45G > 200.141 pg/ml | 13.27 (2.59-67.85), p = 0.002 | |
| IDI statistics | ||
| IDI | Ref. | 15.46% (7.40–23.54), p < 0.001 |
| IDI events | – | 8.39% |
| IDI nonevents | – | 7.08% |
GADD45G as a Biomarker for Ischemic Stroke Prognosis
GADD45G blood levels were assessed in 75 ischemic stroke patients and 38 patients from the mimic group. No differences were observed between these two populations. However, and most interestingly, GADD45G blood levels at baseline were significantly increased in patients who showed poor in-hospital functional outcomes (n = 46, 64%) than those who showed good outcomes (n = 29, p = 0.038) (Fig. 3D) Compared with patients with good functional outcomes, those with poor outcomes were older, had more severe strokes (according to the NIHSS score) and exhibited a nonstatistically significant trend of a lower prevalence of smoking and a higher incidence of atrial fibrillation (supplemental Table S5). However, differences in the circulating levels of GADD45G could not be explained by these demographic and clinical variations (data not shown).
Through ROC curve analysis, we determined a cutoff point for GADD45G (200.141 pg/ml) that was associated with poor functional outcome at hospital discharge, which had 43.8% sensitivity and 81.5% specificity (p = 0.028) (Fig. 3D). A multivariate logistic regression analysis adjusted for age, sex, and NIHSS score at admission confirmed that age and baseline NIHSS score were the only independent clinical predictors of poor functional outcome of the patients at discharge (Table IIB).
The addition of GADD45G > 200.141 pg/ml as a covariable to this predictive clinical model showed that GADD45G strongly remained as an independent predictor of functional outcome at the studied time point. The addition of this biomarker to the predictive clinical model improved the goodness-of-fit of the clinical model alone according to the likelihood ratio test (p < 0.001). Furthermore, GADD45G blood levels substantially increased the discriminatory ability of the clinical model by 15.46%, thus improving the differentiation of patients who would present poor outcome by 8.39%.
DISCUSSION
With the translational aim of identifying potential stroke biomarkers, this study has applied an integrative analysis of massive transcriptomics and proteomics data compiled from mouse brains very acutely after cerebral ischemia. As a result, an integrated network incorporating both inter-connected genes and proteins with a strong involvement in the hyper-acute stages of stroke pathophysiology has been presented. CTNND2 and GADD45G were successfully identified as promising blood biomarkers for acute ischemic stroke diagnosis and prognosis, respectively.
Numerous studies have aimed at unraveling the main molecular changes that occur in the brain following cerebral ischemia. Most of these contributions have been based on the characterization of only protein or gene alterations separately. These efforts have provided knowledge about the pathophysiology of stroke to the point of achieving a reliable (but still incomplete) understanding of the main mechanisms of cerebral ischemia at different molecular levels. Beyond the exploration of individual contributions of genes, proteins, and metabolites to stroke pathology, global comprehension of all their complex interactions, associations, and connections is expected to further improve the identification of biologically relevant disease-associated mediators of stroke. Thus, we used recently developed multivariate projection-based approaches to transcriptomics and proteomics data sets to explore the relationships between genes and proteins.
MCIA pinpointed the separation between the infarcted and healthy regions of the brain, which were clearly marked for both genes and proteins. The overall similarity of both data sets, however, was substantially low, suggesting that genes and proteins might provide different but complementary information to the global integrated system at the specific time points examined. Nevertheless, other biological reasons might also be behind the low correlation coefficient observed, such as the regulation of the translation efficiency, half-life differences between proteins and mRNAs, and proteome turnover, which are processes known to be highly affected by pathological perturbations (31).
We use the mixOmics biostatistical tool to comprehend the underlying correlations between the differentially altered genes and proteins, which emphasized complex unnoticed associations among different molecular elements of stroke pathology. We verified that most of the resulting inter-connected candidates were involved in well-known biological processes that are altered early after cerebral ischemia, including the upregulation of hypoxic pathways, altered synaptic transmission, and unbalanced excitotoxicity mechanisms, as well as the prompt disturbance of the immunological system (32). Indeed, some of these candidates have already been described in the literature as important mediators of the stroke pathology, including Atf3, Ccl3, and CAMK2A, thus supporting the robustness of our findings and providing high reliability to this novel integrative approach (33–37).
It should be stressed that our experiments revealed that genes had a higher contribution to the observed stroke-induced molecular alterations than proteins. One feasible explanation for this observation might be the early time point after ischemia at which we looked for changes. However, many different (an often undervalued) regulatory processes that occur after mRNA expression that strictly control protein abundance might also influence these differential contributions between the gene and protein data sets (38).
Among the 5 candidates, we deeply focused on CLD20. RGS2 and BAG5 did not provide valuable information about the diagnosis or prognosis of ischemic stroke when measured as blood biomarkers. However, all three did show a strong, acute, deregulated protein profile in the brain during the hyper-acute phase after cerebral ischemia, which was replicated throughout the study. Thus, future research could be done to modulate their levels at the therapeutic level.
GADD45G and CTNND2 did appear to be promising blood biomarkers of ischemic stroke prognosis and diagnosis, respectively. GADD45G is a member of the GADD45 family of proteins, which are involved in acute cellular stress response mechanisms, including cell cycle arrest, DNA repair, and apoptosis (39, 40). We have found a pronounced increase in Gadd45g gene expression in the brain early after ischemia. This prompt increase in the mRNA levels was translated into higher GADD45G protein content, as reported previously (41–43).
The exact implication of this GADD45G increase observed in the brain after stroke is still quite unclear. Beyond the recognized role of GADD45 family members during brain development and plasticity (44, 45), GADD45G is also closely related to heart failure because its over-expression results in increased p-38 MAPK-dependent apoptosis, fibrosis, and left ventricular dysfunction, whereas Gadd45g deletion confers resistance to ischemic heart injury (46). In fact, in cancer, there has been a great variety of proposed mechanisms by which GADD45G could induce cell cycle arrest and apoptosis, which all depend on the cell type and the pathological conditions (47). Therefore, despite this pro-apoptotic role still being unexplored in the context of stroke, all evidence of the negative modulation of GADD45G to limit cell death could also be taken into consideration as a therapeutic approach in the development of stroke.
Beyond this plausible therapeutic role, GADD45G has been explored as a blood biomarker. Circulating GADD45G levels did not improve the discrimination of ischemic stroke from stroke-mimicking conditions, but it did show good performance in the estimation of stroke prognosis in the hyperacute phase of the disease, when quick decision-making is essential for thorough management and the prevention of secondary complications. Higher GADD45G blood levels were observed in patients that showed early poor outcomes after stroke.
To the best of our knowledge, no relationship has previously been established between circulating GADD45G and stroke prognosis. In fact, GADD45G has only been associated with the progression of renal disease when measured in urine (48) and has been proposed as a surrogate biomarker of cancer treatment when examined in tumor tissue (49). However, the implications of high circulating GADD45G levels are still unexplored. It is only known that GADD45G is closely related to the exacerbation of inflammation because Gadd45g expression in circulating cells is rapidly triggered by pro-inflammatory cytokines (50). Once expressed, GADD45G also further promotes the upregulation of cytokines such as TNFα and IL-6 (51). Notwithstanding, the usefulness of GADD45G blood levels as a prognosis factor is promising, and further studies to support these new findings would be of great value.
CTNND2 (or neural plakophilin-related Armadillo-repeat protein (NPRAP)) is a neuronal-specific adhesion junction-associated protein that is specifically enriched in the postsynaptic and dendritic compartments (52), where it plays a pivotal role as a signaling sensor and integrator (53). CTNND2 also interacts with and stabilizes brain cadherins by linking them to the actin cytoskeleton and to a wide variety of post-synaptic scaffold molecules, including the ionotropic N-methyl-d-aspartic acid receptor 2A (NR2A) and the metabotropic glutamate receptor 1K (mGluR1K) (54). As an acute response to cerebral ischemia, we have detected a sudden reduction in CTNND2 gene expression and protein abundance in ischemic brains. Our results are supported by those of Jones et al., who found that the glutamate-induced downregulation of CTNND2 in cortical neurons also leads to its dissociation from NR2A and mGluR1K very soon after the insult, when apoptosis is not even noticed. The functional consequence of this reduction in CTNND2 levels after ischemia has not been investigated yet.
CTNND2 is closely related to the Wnt/β-catenin signaling pathway, which regulates angiogenesis, neurogenesis, and cell survival. However, it is still not clear whether it promotes or prevents physiological β-catenin turnover, which would hamper or facilitate the β-catenin-mediated transcription of relevant genes involved in angiogenesis and neurogenesis, respectively (55–57). It might be of interest to investigate the modulation of CTNND2 levels early after stroke to unravel these inconsistencies.
In sharp contrast to the brain, we also found that CTNND2 levels in the blood did show a reliable ability to discriminate the ischemic stroke group from mimic patients. Because CTNND2 expression is highly specific to the brain, one could speculate that this increase in CTNND2 blood content might be a direct consequence of what is being released from the ischemic cerebral tissue. However, different results have been observed in blood between the CTNND2 responses to ischemia in mice and humans, animal studies to unravel the exact CTNND2 provenance in circulation would be quite difficult.
CTNND2 has been previously described as a potential diagnostic biomarker in urine for prostate cancer (58). Beyond cancer, no previous work has outlined a role for CTNND2 as a blood biomarker of disease. Thus, although promising, its usefulness for stroke diagnosis needs to be considered further because it substantially adds value to the discriminatory ability of the predictive diagnosis model built with only clinical variables. Concretely, CTNND2 substantially improved the discrimination of the nonevents, which might also have a vital role in ruling out suspected strokes as early as possible and managing patients according to a nonstroke cerebral pathology.
We have taken advantage of a novel and complementary biostatistical strategy to join and integrate data from transcriptomics and proteomics and identify relevant candidates as acute biomarkers for the diagnosis and prognosis of ischemic stroke. We believe that emerging integrative approaches like the one presented will continue to grow in scale and popularity in coming years. They would complement or even go beyond the traditional biostatistical tools used to analyze single -omics data sets, which could have missed notable and relevant data from each –omics analysis separately.
Along these lines, the integration of other -omics techniques might yield even greater insight into the pathogenesis of stroke. For instance, metabolomics could provide additional information on the functional consequences of these altered gene or protein patterns after stroke. Therefore, profiling of the impaired metabolome in the hyper-acute phase after stroke might strengthen the depiction of the complex stroke phenotype and provide new potential blood biomarkers for this devastating disease. Moreover, further efforts might also be directed toward the exploration of all other pinpointed stroke-related elements that we have encountered as potential therapeutic targets for stroke pathology, as well as their role as stroke biomarkers.
This study also had some limitations that should be taken into consideration. First, the integrative analysis identified network-like correlation structures of inter-connected genes and proteins, from which notable candidates were selected. Although these correlations could be understood as hypothetical functional linkages, they do not strictly imply a direct biological connection among components. Indeed, they have been used as a novel and complementary theory-based statistical strategy to highlight relevant yet undescribed associations among constituents of the stroke pathology, but deep details about the underlying biological interpretation of all these connections still need to be elucidated in the future. Second, time extrapolations between mice and humans are still not well established and shifts over time might have to be considered in a future. Furthermore, because of technical incompatibilities, we were not able to assess infarct volumes in the ischemic animals at the examined time points, which would have also been an interesting parameter to analyze and incorporate into the integrative analysis together with other relevant demographic factors, including age and sex.
In conclusion, this breakthrough integrative approach enabled us to identify, positively replicate, and further explore 18 stroke-associated inter-connected genes and proteins in the ischemic brains of mice over time. Moreover, we took advantage of this approach to explore the promising potential of GADD45G and CTNND2 as blood biomarkers of stroke prognosis and diagnosis, respectively.
DATA AVAILABILITY
The transcriptomics data generated in this work has been deposited in the Gene Expression Omnibus (GEO) database: GSE137595. The LC–MS proteomics data has been deposited to the ProteomeXchange Consortium via the PRIDE repository with the data set identifier PXD016538, and the PRM proteomics data has been submitted to the Panorama Public repository software (ProteomeXchange ID: PXD017280).
Supplementary Material
Acknowledgments
We show our gratitude to the patients included in the study and their relatives.
Footnotes
This article contains supplemental data.
Funding and Additional Information—Neurovascular Research Laboratory acknowledges funding for this project by PI15/00354 and PI18/00804 grants from Fondo de Investigaciones Sanitarias and takes part in the Spanish stroke research network INVICTUS+ (RD16/0019/0021) of the Instituto de Salud Carlos III (co-financed by the European Regional Development Fund, FEDER). A.R is supported by the Miguel Servet contract (CPII15/00003), L. R. is supported by a predoctoral fellowship grant (IFI17/00012) and A. B. is supported by a Juan Rodes research grant (JR16/00008), all three from Instituto de Salud Carlos III. The Proteomics Laboratory at VHIO (member of Proteored) is supported by Grant IPT17/0019-ISCIII-SGEFI/ERDF, from Instituto de Salud Carlos III.
Conflict of interest—The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Abbreviations—The abbreviations used are:
- AGC
- Automatic gain control
- AP-1
- Activator protein-1
- Atf3
- Activating transcription factor 3
- AUC
- Area under the curve
- B2M
- Beta-2-microglobulin
- CAMK2A
- Calcium/calmodulin-dependent protein kinase type II subunit alpha
- Ccl3
- Chemokine ligand 3
- CI
- Confidence interval
- CL
- Contralateral brain hemisphere
- CLDN20
- Claudin-20
- Cstad
- CSA-conditional, T cell activation-dependent geneCTNND2
- DNA
- Deoxyribonucleic acid
- DTT
- Dithiothreitol
- FC
- Fold-change
- GADD45G
- Growth arrest and DNA-damage-inducible protein GADD45 gamma
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- GO
- Gene Ontology
- GPCR
- G protein-coupled receptor
- GSS
- Gene set scores
- IDI
- Integrated discrimination improvement
- IL-6
- Interleukin 6
- IP
- Ipsilateral brain hemisphere
- LC-MS
- Liquid chromatography-mass spectrometry
- LTQ
- inear trap quadrupole
- MAPK
- Mitogen-activated protein kinase
- MCA
- Middle cerebral artery
- MCAO
- Middle cerebral artery occlusion
- MCIA
- Multiple Co-Inertia Analysis
- mGluR1K
- metabotropic glutamate receptor 1K
- mRNA
- messenger ribonucleic acid
- mRS
- Modified Rankin Scale
- MS
- Mass spectrometry
- NHISS
- National Institutes of Health Stroke Scale
- NR2A
- N-methyl-D-aspartic acid receptor 2A
- ORadj
- Adjusted odd ratio
- PCA
- Principal component analysis
- PPIA
- Peptidyl-prolyl cis-trans isomerase A
- PRM
- Parallel reaction monitoring
- rCCA
- regularized Canonical Correlation Analysis
- RGS2
- Regulator of G-protein signaling 2
- RNA
- Ribonucleic acid
- ROC
- Receiver operating characteristics
- SD
- Standard deviation
- SRGAP2
- SLIT-ROBO Rho GTPase-activating protein 2
- ssDNA
- Single-stranded deoxyribonucleic acid
- TNF-α
- Tumor necrosis factor alpha
- TXN2
- Thioredoxin 2.
REFERENCES
- 1. Benjamin E. J., Blaha M. J., Chiuve S. E., Cushman M., Das S. R., Deo R., de Ferranti S. D., Floyd J., Fornage M., Gillespie C., Isasi C. R., Jiménez M. C., Jordan L. C., Judd S. E., Lackland D., Lichtman J. H., Lisabeth L., Liu S., Longenecker C. T., Mackey R. H., Matsushita K., Mozaffarian D., Mussolino M. E., Nasir K., Neumar R. W., Palaniappan L., Pandey D. K., Thiagarajan R. R., Reeves M. J., Ritchey M., Rodriguez C. J., Roth G. A., Rosamond W. D., Sasson C., Towfighi A., Tsao C. W., Turner M. B., Virani S. S., Voeks J. H., Willey J. Z., Wilkins J. T., Wu J. H., Alger H. M., Wong S. S., and Muntner P, American Heart Association Statistics Committee and Stroke Statistics Subcommittee, (2017) Heart disease and stroke statistics-2017 update: a report from the American Heart Association. Circulation 135, e146–e603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wang G., Zhang Z., Ayala C., et al. (2016) Costs of hospitalization for stroke patients aged 18-64 years in the United States. J. Stroke Cerebrovasc. Dis. 3, 973–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fiehler J., and Gerloff C. (2015) Mechanical thrombectomy in stroke. Dtsch. Arztebl. Int. 112, 830–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lansberg M. G., Bluhmki E., and Thijs V. N. (2009) Efficacy and safety of tissue plasminogen activator 3 to 4.5 hours after acute ischemic stroke: a metaanalysis. Stroke 40, 2438–2441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Urra X., Abilleira S., Dorado L., Ribó M., Cardona P., Millán M., Chamorro A., Molina C., Cobo E., Dávalos A., Jovin T. G., and Gallofré M, Catalan Stroke Code and Reperfusion Consortium, (2015) Mechanical thrombectomy in and outside the REVASCAT trial: insights from a concurrent population-based stroke registry. Stroke 46, 3437–3442 [DOI] [PubMed] [Google Scholar]
- 6. Yaghi S., Willey J. Z., Cucchiara B., Goldstein J. N., Gonzales N. R., Khatri P., Kim L. J., Mayer S. A., Sheth K. N., and Schwamm L. H, American Heart Association Stroke Council; Council on Cardiovascular and Stroke Nursing; Council on Clinical Cardiology; and Council on Quality of Care and Outcomes Research, (2017) Treatment and outcome of hemorrhagic transformation after intravenous alteplase in acute ischemic stroke: a scientific statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 48, e343–e361 [DOI] [PubMed] [Google Scholar]
- 7. Yew K. S., and Cheng E. M. (2015) Diagnosis of acute stroke. Am. Fam. Physician. 91, 528–536 [PubMed] [Google Scholar]
- 8. Rothstein L., and Jickling G. C. (2013) Ischemic stroke biomarkers in blood. Biomark. Med. 7, 37–47 [DOI] [PubMed] [Google Scholar]
- 9. Jickling G. C., and Sharp F. R. (2015) Biomarker panels in ischemic stroke. Stroke 46, 915–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Glickman S. W., Phillips S., Anstrom K. J., Laskowitz D. T., and Cairns C. B. (2011) Discriminative capacity of biomarkers for acute stroke in the emergency department. J. Emerg. Med. 41, 333–339 [DOI] [PubMed] [Google Scholar]
- 11. Bustamante A., Sobrino T., Giralt D., García-Berrocoso T., Llombart V., Ugarriza I., Espadaler M., Rodríguez N., Sudlow C., Castellanos M., Smith C. J., Rodríguez-Yánez M., Waje-Andreassen U., Tanne D., Oto J., Barber M., Worthmann H., Wartenberg K. E., Becker K. J., Chakraborty B., Oh S.-H., Whiteley W. N., Castillo J., and Montaner J. (2014) Prognostic value of blood interleukin-6 in the prediction of functional outcome after stroke: A systematic review and meta-analysis. J. Neuroimmunol. 274, 215–224 [DOI] [PubMed] [Google Scholar]
- 12. Simats A., García-Berrocoso T., Penalba A., Giralt D., Llovera G., Jiang Y., Ramiro L., Bustamante A., Martinez-Saez E., Canals F., Wang X., Liesz A., Rosell A., and Montaner J. (2018) CCL23: A new CC chemokine involved in human brain damage. J. Intern. Med. 283, 461–463 [DOI] [PubMed] [Google Scholar]
- 13. Horgan R. P., and Kenny L. C. (2011) SAC review 'Omic' technologies : genomics, transcriptomics, proteomics and metabolomics. Obstetrician Gynaecologist 13, 189–195 [Google Scholar]
- 14. Sun Y. V., and Hu Y.-J. (2016) Integrative analysis of multi-omics data for discovery and functional studies of complex human diseases HHS public access. Adv Genet 93, 147–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kilkenny C., Browne W. J., Cuthill I. C., Emerson M., and Altman D. G. (2010) Improving bioscience research reporting: The arrive guidelines for reporting animal research. PLos Biol. 8, e1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Clark W., Lessov N., Dixon M., and Eckenstein F. (1997) Monofilament intraluminal middle cerebral artery occlusion in the mouse. Neurological Research 19, 641–648 [DOI] [PubMed] [Google Scholar]
- 17. Lee S., Lee M., Hong Y., Won J., Lee Y., Kang S.-G., Chang K.-T., and Hong Y. (2014) Middle cerebral artery occlusion methods in rat versus mouse models of transient focal cerebral ischemic stroke. Neural Regen. Res. 9, 757–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kauffmann A., Gentleman R., and Huber W. (2009) arrayQualityMetrics - A bioconductor package for quality assessment of microarray data. Bioinformatics 25, 415–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Irizarry R. A., Hobbs B., Collin F., Beazer-Barclay Y. D., Antonellis K. J., Scherf U., and Speed T. P. (2003) Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4, 249–264 [DOI] [PubMed] [Google Scholar]
- 20. Smyth G. K. (2004) Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Statistical Appl. Gen. Mol. Biol. 3, 1–25 [DOI] [PubMed] [Google Scholar]
- 21. Benjamini Y., and Hochberg Y. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. Roy. Statistical Soc.: 57, 289–300 [Google Scholar]
- 22. van den Berg R. A., Hoefsloot H. C. J., Westerhuis J. A., Smilde A. K., and van der Werf M. J. (2006) Centering, scaling, and transformations: Improving the biological information content of metabolomics data. BMC Genomics. 7, 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ritchie M. E., Phipson B., Wu D., Hu Y., Law C. W., Shi W., and Smyth G. K. (2015) limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic acids Res. 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Meng C., Kuster B., Culhane A. C., and Gholami A. M. (2014) A multivariate approach to the integration of multi-omics datasets. BMC Bioinformatics. 15, 162 Epub ahead of print [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Culhane A. C., Thioulouse J., Perrière G., and Higgins D. G. (2005) MADE4: an R package for multivariate analysis of gene expression data. Bioinformatics 21, 2789–2790 [DOI] [PubMed] [Google Scholar]
- 26. Lê Cao K.-A., González I., and Déjean S. (2009) integrOmics: an R package to unravel relationships between two omics datasets. Bioinformatics 25, 2855–285610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Meng C., Kuster B., Peters B., Culhane A. C., Moghaddas Gholami A., Gholami A. M., and Moghaddas Gholami A. (2016) moGSA: integrative single sample gene-set analysis of multiple omics data. BioRxiv 046904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Escofier B., and Pagès J. (1994) Multiple factor analysis (AFMULT package). Computational Statistics Data Analysis 18, 121–140 [Google Scholar]
- 29. Liberzon A., Birger C., Thorvaldsdóttir H., Ghandi M., Mesirov J. P., and Tamayo P. (2015) The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Carr S. A., Abbatiello S. E., Ackermann B. L., Borchers C., Domon B., Deutsch E. W., Grant R. P., Hoofnagle A. N., Hüttenhain R., Koomen J. M., Liebler D. C., Liu T., MacLean B., Mani D. R., Mansfield E., Neubert H., Paulovich A. G., Reiter L., Vitek O., Aebersold R., Anderson L., Bethem R., Blonder J., Boja E., Botelho J., Boyne M., Bradshaw R. A., Burlingame A. L., Chan D., Keshishian H., Kuhn E., Kinsinger C., Lee J. S. H., Lee S.-W., Moritz R., Oses-Prieto J., Rifai N., Ritchie J., Rodriguez H., Srinivas P. R., Townsend R. R., Van Eyk J., Whiteley G., Wiita A., and Weintraub S. (2014) Targeted peptide measurements in biology and medicine: Best practices for mass spectrometry-based assay development using a fit-for-purpose approach. Mol. Cell. Proteomics 13, 907–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kumar D., Bansal G., Narang A., Basak T., Abbas T., and Dash D. (2016) Integrating transcriptome and proteome profiling: Strategies and applications. Proteomics 16, 2533–2544 [DOI] [PubMed] [Google Scholar]
- 32. Xing C., Arai K., Lo E. H., and Hommel M. (2012) Pathophysiologic cascades in ischemic stroke. Int. J. Stroke 7, 378–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Waxham M. N., Grotta J. C., Silva A. J., Strong R., and Aronowski J. (1996) Ischemia-induced neuronal damage: a role for calcium/calmodulin-dependent protein kinase II. J. Cereb. Blood Flow Metab. 16, 1–6 [DOI] [PubMed] [Google Scholar]
- 34. Vest R. S., O'Leary H., Coultrap S. J., Kindy M. S., and Bayer K. U. (2010) Effective Post-insult Neuroprotection by a Novel Ca 2+/Calmodulin-dependent Protein Kinase II (CaMKII) Inhibitor. Journal of Biological Chemistry 285, 20675–20682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Takarada T., Kou M., Hida M., Fukumori R., Nakamura S., Kutsukake T., Kuramoto N., Hinoi E., and Yoneda Y. (2016) Protective upregulation of activating transcription factor-3 against glutamate neurotoxicity in neuronal cells under ischemia. J. Neurosci. Res. 94, 378–388 [DOI] [PubMed] [Google Scholar]
- 36. Zhang S.-J., Buchthal B., Lau D., Hayer S., Dick O., Schwaninger M., Veltkamp R., Zou M., Weiss U., and Bading H. (2011) A signaling cascade of nuclear calcium-CREB-ATF3 activated by synaptic NMDA receptors defines a gene repression module that protects against extrasynaptic NMDA receptor-induced neuronal cell death and ischemic brain damage. J. Neurosci. 31, 4978–4990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Takami S., Minami M., Nagata I., Namura S., and Satoh M. (2001) Chemokine receptor antagonist peptide, viral MIP-II, protects the brain against focal cerebral ischemia in mice. J. Cereb. Blood Flow Metab. 21, 1430–1435 [DOI] [PubMed] [Google Scholar]
- 38. Vogel C., and Marcotte E. M. (2012) Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 13, 227–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cretu A., Sha X., Tront J., Hoffman B., and Liebermann D. A. (2009) Stress sensor Gadd45 genes as therapeutic targets in cancer. Cancer Ther. 7, 268–276 [PMC free article] [PubMed] [Google Scholar]
- 40. Salvador J. M., Brown-Clay J. D., and Fornace A. J. (2013) Gadd45 in stress signaling, cell cycle control, and apoptosis. Adv. Exp. Med. Biol. 793, 1–19 [DOI] [PubMed] [Google Scholar]
- 41. Chen J., Uchimura K., Stetler R. A., Zhu R. L., Nakayama M., Jin K., Graham S. H., and Simon R. P. (1998) Transient Global Ischemia Triggers Expression of the DNA Damage-Inducible Gene GADD45 in the Rat Brain. J. Cereb. Blood Flow Metab. 18, 646–657 [DOI] [PubMed] [Google Scholar]
- 42. Nagata T., Takahashi Y., Sugahara M., Murata A., Nishida Y., Ishikawa K., and Asai S. (2004) Profiling of genes associated with transcriptional responses in mouse hippocampus after transient forebrain ischemia using high-density oligonucleotide DNA array. Molecular Brain Research 121, 1–11 [DOI] [PubMed] [Google Scholar]
- 43. Ramsay LAnn., Quillé M. -L., Orset C., Grange P., Rousselet E., Férec C., Le Gac G., Génin E., and Timsit S. (2019) Blood transcriptomic biomarker as a surrogate of ischemic brain gene expression. Ann. Clin. Transl. Neurol. 6, 1681–1695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sarkisian M. R., and Siebzehnrubl D. (2012) Abnormal levels of Gadd45alpha in developing neocortex impair neurite outgrowth. PLoS ONE. 7, e44207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Leach P. T., Poplawski S. G., Kenney J. W., Hoffman B., Liebermann D. A., Abel T., and Gould T. J. (2012) Gadd45b knockout mice exhibit selective deficits in hippocampus-dependent long-term memory. Learn. Mem. 19, 319–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lucas A., Mialet-Perez J., Daviaud D., Parini A., Marber M. S., and Sicard P. (2015) Gadd45γ regulates cardiomyocyte death and post-myocardial infarction left ventricular remodelling. Cardiovasc. Res. 108, 254–267 [DOI] [PubMed] [Google Scholar]
- 47. Flores O., and Burnstein K. L. (2010) GADD45gamma: a new vitamin D-regulated gene that is antiproliferative in prostate cancer cells. Endocrinology 151, 4654–4664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yu S., Cho J., Park I., Kim S. J., Kim H., and Shin G.-T. (2009) Urinary GADD45gamma expression is associated with progression of lgA nephropathy. Am. J. Nephrol. 30, 135–139 [DOI] [PubMed] [Google Scholar]
- 49. Ou D.-L., Shyue S.-K., Lin L.-I., Feng Z.-R., Liou J.-Y., Fan H.-H., Lee B.-S., Hsu C., and Cheng A.-L. (2015) Growth arrest dna damage-inducible gene 45 gamma expression as a prognostic and predictive biomarker in hepatocellular carcinoma. Oncotarget 6, 27953–27965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Thalheimer F. B., Wingert S., De Giacomo P., Haetscher N., Rehage M., Brill B., Theis F. J., Hennighausen L., Schroeder T., and Rieger M. A. (2014) Cytokine-regulated GADD45G induces differentiation and lineage selection in hematopoietic stem cells. Stem Cell Reports. 3, 34–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shin G. T., Lee H. J., and Kim H. (2012) GADD45γ regulates TNF-α and IL-6 synthesis in THP-1 cells. Inflamm. Res. 61, 1195–1202 [DOI] [PubMed] [Google Scholar]
- 52. Matter C., Pribadi M., Liu X., and Trachtenberg J. T. (2009) δ-Catenin Is Required for the Maintenance of Neural Structure and Function in Mature Cortex In Vivo. Neuron 64, 320–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Abu-Elneel K., Ochiishi T., Medina M., Remedi M., Gastaldi L., Caceres A., and Kosik K. S. (2008) A δ-catenin signaling pathway leading to dendritic protrusions. J. Biol. Chem. 283, 32781–32791 [DOI] [PubMed] [Google Scholar]
- 54. Jones S. B., Lanford G. W., Chen Y.-H., Morabito M., Moribito M., Kim K., and Lu Q. (2002) Glutamate-induced δ-catenin redistribution and dissociation from postsynaptic receptor complexes. Neuroscience 115, 1009–1021 [DOI] [PubMed] [Google Scholar]
- 55. Huang F., Chen J., Lan R., Wang Z., Chen R., Lin J., and Fu L. (2019) Hypoxia induced δ-Catenin to enhance mice hepatocellular carcinoma progression via Wnt signaling. Exp. Cell Res. 374, 94–103 [DOI] [PubMed] [Google Scholar]
- 56. Nopparat J., Zhang J., Lu J.-P., Chen Y.-H., Zheng D., Neufer P. D., Fan J. M., Hong H., Boykin C., and Lu Q. (2015) δ-Catenin, a Wnt/β-catenin modulator, reveals inducible mutagenesis promoting cancer cell survival adaptation and metabolic reprogramming. Oncogene 34, 1542–1552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bareiss S., Kim K., and Lu Q. (2010) δ-catenin/NPRAP: A new member of the glycogen synthase kinase-3β signaling complex that promotes β-catenin turnover in neurons. J. Neurosci. Res. 88, 2350–2363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lu Q., Zhang J., Allison R., Gay H., Yang W.-X., Bhowmick N. A., Frelix G., Shappell S., and Chen Y.-H. (2009) Identification of extracellular δ-Catenin accumulation for prostate cancer detection. Prostate 69, 411–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The transcriptomics data generated in this work has been deposited in the Gene Expression Omnibus (GEO) database: GSE137595. The LC–MS proteomics data has been deposited to the ProteomeXchange Consortium via the PRIDE repository with the data set identifier PXD016538, and the PRM proteomics data has been submitted to the Panorama Public repository software (ProteomeXchange ID: PXD017280).