

The global proteome and phosphoproteome of mouse kidney upon septic infection were profiled using high resolution mass spectrometry and label-free quantitation. Novel and less-well studied marker proteins indicative of the onset of acute kidney injury (AKI) and progression to chronic kidney disease (CKD) were revealed and further validated by immunoassays. Signaling pathways associated with oxidative stress and pyroptosis were demonstrated to regulate kidney pathology. The data would serve as a great resource of biomarker discovery of sepsis and kidney disease.
Keywords: Quantification, phosphoproteome, kidney disease, infectious disease, systems biology, biomarker: diagnostic, mass spectrometry, omics, acute kidney infection, chronic kidney disease, label-free quantitation
Graphical Abstract

Highlights
Cecal Ligation Puncture (CLP) mouse model to study sepsis-induced kidney disease.
Quantitative global proteome and phosphoproteome profiling of mouse kidneys.
Highly significant candidate markers for onset and progression of AKI to CKD.
Mechanistic insights into sepsis-associated kidney injuries.
Abstract
Sepsis-induced acute kidney injury (S-AKI) is the most common complication in hospitalized and critically ill patients, highlighted by a rapid decline of kidney function occurring a few hours or days after sepsis onset. Systemic inflammation elicited by microbial infections is believed to lead to kidney damage under immunocompromised conditions. However, although AKI has been recognized as a disease with long-term sequelae, partly because of the associated higher risk of chronic kidney disease (CKD), the understanding of kidney pathophysiology at the molecular level and the global view of dynamic regulations in situ after S-AKI, including the transition to CKD, remains limited. Existing studies of S-AKI mainly focus on deriving sepsis biomarkers from body fluids. In the present study, we constructed a mid-severity septic murine model using cecal ligation and puncture (CLP), and examined the temporal changes to the kidney proteome and phosphoproteome at day 2 and day 7 after CLP surgery, corresponding to S-AKI and the transition to CKD, respectively, by employing an ultrafast and economical filter-based sample processing method combined with the label-free quantitation approach. Collectively, we identified 2,119 proteins and 2950 phosphosites through multi-proteomics analyses. Among them, we identified an array of highly promising candidate marker proteins indicative of disease onset and progression accompanied by immunoblot validations, and further denoted the pathways that are specifically responsive to S-AKI and its transition to CKD, which include regulation of cell metabolism regulation, oxidative stress, and energy consumption in the diseased kidneys. Our data can serve as an enriched resource for the identification of mechanisms and biomarkers for sepsis-induced kidney diseases.
Acute kidney injury (AKI) is characterized by a rapid decline of renal function resulting in elevated serum creatinine levels with or without decreased urine output (1). It is estimated that the incidence of AKI is about 10–15% of all patients admitted to the hospital, and its frequent occurrence with other organ dysfunction together causes high mortality (2). AKI has been recognized as a disease with long-term sequelae because survivors of AKI have a high risk of developing chronic kidney disease (CKD), which constitutes another high healthcare burden worldwide (3). In the clinic, the leading cause of AKI is sepsis, which is a systemic inflammatory response elicited by microbial infections (4). In the intensive care unit (ICU), sepsis accounts for up to 50% of AKI cases (5), and sepsis-induced AKI (S-AKI) has 6–8 fold higher risk of death compared with AKI caused by other etiologies (6).
Current understanding of the pathogenesis of S-AKI mainly includes alterations in renal microvascular circulation, adaptive immune responses to exacerbated inflammation, and reprogramming of metabolic pathways (6, 7). First, altered microvascular blood flow impairs tissue oxygenation, which in turn leads to multiple organs' dysfunction. In the kidney tissue, glomerular shunting and constriction of the efferent arteriole result in decreased intraglomerular pressure and subsequently reduced glomerular filtration rate and urine output (4, 6, 8). Increased vascular permeability and leakage can also cause interstitial edema and increase oxygen diffusion distant to the tubular cells. Endothelial activation promotes rolling and adhesion of leukocytes and platelets, resulting in an increased risk of thrombi formation and flow continuity alterations. Second, tubular endothelial cells are equipped with pattern recognition receptors (such as Toll-like receptors, TLRs), which can receive signals from sepsis-induced inflammation, pathogen-associated molecular patterns (PAMPs), or damage-associated molecular patterns (DAMPs). These result in a cascade of downstream signals and increase the synthesis of proinflammatory cytokines, reactive oxygen species, and oxidative stress that damage renal tubular cells (4, 8, 9). Lastly, the metabolism and energy state of renal tissue is reprogrammed during sepsis to conserve and reprioritize energy spending for the tubular cells to recover from damage (8, 10, 11). However, the global changes of the diseased kidneys after S-AKI at the molecular level remain largely unknown. In this regard, we constructed an S-AKI mouse model using the cecal ligation and puncture (CLP) procedure. CLP is a gold standard clinical model (12) as it allows the release of fecal components into the peritoneal cavity and circulatory system that induces exacerbated inflammatory responses. This procedure mimics gut injury-triggered severe sepsis in humans. Hence, it is more clinically relevant when compared with the endotoxic model (13, 14). In the current study, we analyzed the global proteome and phosphoproteome level changes of kidney tissues at day 2 and day 7 after CLP. With the results we obtained, we can better characterize the molecular mechanisms underlying the development of S-AKI and its transition to CKD.
EXPERIMENTAL PROCEDURES
Ethics Statement, Mouse Models, and CLP Surgery
Male C57BL/6J mice 6–8 weeks old and weighing about 20–25 g were obtained from the Jackson Laboratories (Bar Harbor, ME). Cecal ligation and puncture procedure was performed by using an established protocol described previously (13). Briefly, the abdomen of the mice was shaved and disinfected after anesthetizing the mice. Under aseptic conditions, a 1–2 cm midline laparotomy was performed to expose the cecum with the adjoining intestine. The cecum was tightly ligated with a 6.0 silk suture at its base below the ileo-cecal valve (1 cm distance from the distal end of the cecum to ligation point) and was perforated twice with a 19 gauge needle on the same side of the cecum. The cecum was then gently squeezed to extrude a small amount of feces from the perforation sites. The cecum was returned to the peritoneal cavity and peritoneum and skin were closed. Mice were sacrificed at 0, 2, or 7 days after the procedure. Kidney tissues were collected for various analyses. All the above animal experiments were approved by the Institutional Animal Care and Use Committee at the University of Pittsburgh.
Determination of Serum Creatinine
Blood was collected from infected mice at day 2 and day 7 respectively before being sacrificed. The serum creatinine level was determined using the QuantiChrom creatinine assay kit according to the protocols specified by the manufacturer (BioAssay Systems, Hayward, CA). The level of serum creatinine was expressed as milligrams per 100 ml (dl) as described previously (15).
Histology Staining, Western Blot, and ELISA Analysis
Paraffin-embedded mouse kidney sections (∼3 μm thickness) were prepared and then stained with Periodic acid–Schiff (PAS) reagents following standard protocols (16). For immunoassays, kidney tissues were lysed with radioimmunoprecipitation assay (RIPA) buffer containing 1% Nonidet P-40, 0.1% SDS, 100 μg/ml PMSF, 1% protease inhibitor mixture, and 1% phosphatase I and II inhibitor mixture (Sigma) in PBS on ice. The supernatants were collected after centrifugation at 13,000 × g at 4 °C for 15 min. Protein expression was analyzed by Western blot analysis as described previously (17). Based on our known knowledge of AKI and proteomic quantitation significance, confidence and antibody availability, we selected the following primary antibodies in our study: anti-FADD (05-486; Millipore, Billerica, MA); anti-NGAL (ab63929), anti-TNFα (ab1793; Abcam, Cambridge, MA); anti-Bad (#9268; Cell Signaling Technology, Danvers, MA); anti-Gasdermin D (sc-81868; Santa Cruz Biotechnology, Santa Cruz, CA); anti-α-tubulin (T9026; Sigma, St. Louis, MO), anti-Caspase-1 (#93709S; Cell Signaling Technology, Danvers, MA); anti-p-MAPK (#9101S; Cell Signaling Technology, Danvers, MA); anti-MAPK (#9102S; Cell Signaling Technology, Danvers, MA); anti-ASC (#67824T; Cell Signaling Technology, Danvers, MA); anti-p-ASC(Y144) (AP5631; ECM Biosciences, Versailles, KY); anti-HMGCS2 (#20940S; Cell Signaling Technology, Danvers, MA); anti-Chil3 (#H1165; Sino Biological, Beijing, China); anti-S100a8 (#15792-1-AP; ProteinTech, Rosemont, IL); anti-S100a9 (#26992-1-AP; ProteinTech, Rosemont, IL); anti-TLR-4 (#19811-1-AP; ProteinTech, Rosemont, IL); and anti-β-actin (#60008-1-Ig; ProteinTech, Rosemont, IL). For ELISA analysis, kidney samples were processed for TNFα expression using the Mouse TNFα Quantikine ELISA Kit (R&D Systems MTA00B). Samples were diluted 1:5 in reagent diluent prior to plating. Assay was performed according to manufacturer's instructions.
Global Proteomic and Phosphoproteomic Sample Preparation
Mouse kidneys were homogenized in PBS with HALT protease inhibitor mixture (Pierce) in a bead beater. After collecting soluble homogenate, the tissue debris was re-suspended in 2 × SED lysis buffer (4% SDS, 50 mm EDTA, 20 mm DTT, 2% Tween 20, 100 mm Tris-HCl, pH 8.0) and sonicated to further extract membrane-bound proteins. The homogenate from both PBS and SED treatment were combined (∼500 μg proteins) and digested using the Suspension Trapping (STrap) approach with in-house packed glass fiber filters (18). Around 50 μg digested peptides were directly desalted using spinnable StageTip protocol (19). To analyze the phosphoproteome of the kidney tissue, ∼450 μg digested peptides were processed with phosphorylation enrichment using TiO2 beads as described previously (20). Peptides were desalted using StageTip as described, lyophilized and stored in −80 °C until further analysis (21).
LC–MS/MS Analysis
The LC–MS/MS analysis was carried out using an Ultimate 3000 nanoLC coupled to Q Exactive mass spectrometer (Thermo Scientific). Peptides were first loaded onto a trap column (PepMap C18, 2 cm × 100 μm × I.D.; Thermo Scientific) and then separated by an in-house packed analytical column (C18 ReproSil, 3.0 μm, Dr. Maisch GmbH; 19 cm × 75 μm I.D.) using a binary buffer system (buffer A: 0.1% formic acid in water; buffer B: 0.1% formic acid in acetonitrile) with a 220-min gradient (2–35% buffer B over 180 min; 35–80% buffer B over 10 min; back to 2% B in 5 min for equilibration after staying on 80% B for 5 min). MS data were acquired in a data-dependent top-10 method with a maximum injection time of 20 ms, a scan range of 350–1700 Da, and an AGC target of 1e6. MS/MS was performed via higher energy collisional dissociation fragmentation with a target value of 5e5 and a maximum injection time of 100 ms. Full MS and MS/MS scans were acquired at a resolution of 70,000 and 17,500, respectively. Dynamic exclusion was set to 20 s.
Protein Identification and Quantitation
Protein identification and quantitation were performed using the MaxQuant-Andromeda software suite (version 1.6.3.4) with most of the default parameters (22). A mouse database (17,038 sequences; Reviewed only; version March 2019) downloaded from UniProt Knowledgebase (https://www.uniprot.org/) was used for the database search. For global proteome analysis, the following parameters were applied: 10 ppm and 20 ppm mass tolerances for precursor and fragments, respectively; trypsin as enzyme with two missed cleavage sites; protein N-terminal acetylation and methionine oxidation as variable modifications; cysteine carbamidomethylation as a fixed modification; peptide length with at least 7 amino acids. False discovery rate (FDR) was set at 1% for both proteins and peptides. For global proteome quantitation, the MaxQuant output result (proteinGroups.txt) was first filtered to exclude those 'Only identified by site, 'Potential contaminant', and 'Reversed' hits, and then log2-transformed. Downstream data analyses such as missing value imputation, Hierarchical clustering, Principal Component Analysis (PCA), t-tests, correlation, and volcano plots were performed in the Perseus environment using default parameters (version 1.6.1.3). For phosphoproteome analysis, phosphorylation at serine, threonine, and tyrosine was set as an additional variable modification during MaxQuant database search. The cutoff of phosphosite probability estimated by MaxQuant was required to be 0.75 or higher. The MaxQuant output file (Phospho (STY) Sites.txt) was used for further bioinformatics analyses, and were performed in Perseus environment as well.
For microbial protein identification, several different metaproteome database searches were tested in Proteome Discoverer platform (version 2.3, Thermo Scientific) using mouse gut microbiome and human gut and oral microbiomes obtained from China National GeneBank (https://db.cngb.org/microbiome/), NIH Human Microbiome Project (https://hmpdacc.org/hmp/HMRGD/), and eHOMD (http://www.homd.org/), respectively. Search parameters including enzyme, modifications, mass tolerance and FDR were set similarly to MaxQuant software.
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses were performed using DAVID Bioinformatics Resources 6.8 (https://david.ncifcrf.gov/home.jsp). For protein network analysis, the StringApp was employed in the Cytoscape environment (version 3.8.0) (23). The interaction score was set to 0.9, the highest confidence cutoff, to retrieve potential interactions. The 'Load Enrichment Data' option was enabled during this process to retrieve functional enrichment (minimum significance threshold FDR 0.05) for the STRING network.
Experimental Design and Statistical Rationale
The mouse CLP experiments and biospecimens at each time point were collected in at least six biological replicates. Western blot and biochemical assays were performed for all the collected samples. Proteomic and phosphoproteomic experiments were performed for six samples of each time point. All tissues were lysed and digested in parallel. For global quantitation of all proteomics and phosphoproteomics data, the files were processed in MaxQuant software in the same batch as well. Multi-sample variation test was determined by ANOVA in the Perseus environment corrected by the Permutation FDR 0.05. Pair-wise Student's t test was performed following a similar procedure, and the p value were corrected as well with Permutation FDR 0.05. Unsupervised clustering analyses used Euclidean as distance and average as linkage for both column and row clustering.
RESULTS
Characterization of Kidney Injury after S-AKI
Given that S-AKI is the most common complication associated with sepsis in the clinic, we aimed to explore its pathogenesis in detail. We created a moderate S-AKI mouse model in which the mice would survive to progress to chronic kidney disease. CLP surgery was performed by placing a 6.0 silk ligature at 1 cm from the cecal tip as described previously (Fig. 1A), with mice sacrificed at day 2 or day 7 (12). Only 1 out of 20 mice that went through the procedure died before the entire experiments were finished, indicating we did not induce severe sepsis. Next, we examined if the CLP procedure successfully induced AKI using rapid elevation of serum creatinine (SCr) as an established biomarker of AKI (24). We observed that SCr was significantly increased to an average of 0.33 mg/dl at 2 days after the CLP procedure, a nearly 3-fold increase compared with the sham control group. By 7 days after CLP, SCr returned to a relatively low level (Fig. 1B). We also investigated the expression of neutrophil gelatinase-associated lipocalin protein (NGAL; also known as lipocalin 2, Lcn2), a validated predictor for kidney damage. Under ischemic, septic, or post-transplant AKI, NGAL's expression in kidney tubules is often rapidly increased (25). NGAL expression increased dramatically in renal tissues at day 2 after CLP compared with the sham control, with a reduction by day 7 (Fig. 1C and 1D), indicating that CLP caused a definite injury to the kidneys. Consistent with this, periodic acid-Schiff (PAS) staining illustrated cell vacuolation, necrosis, and cellular debris present near blood vessels in the diseased kidneys at day 2 after CLP (Fig. 1E), which are consistent with previous observations of septic kidneys (26). Little to no differences in histological changes were observed between day 2 and day 7 after CLP. In contrast to S-AKI, ischemia-reperfusion associated AKI usually causes more profound histological changes, as reported by our previous study (17).
Fig. 1.
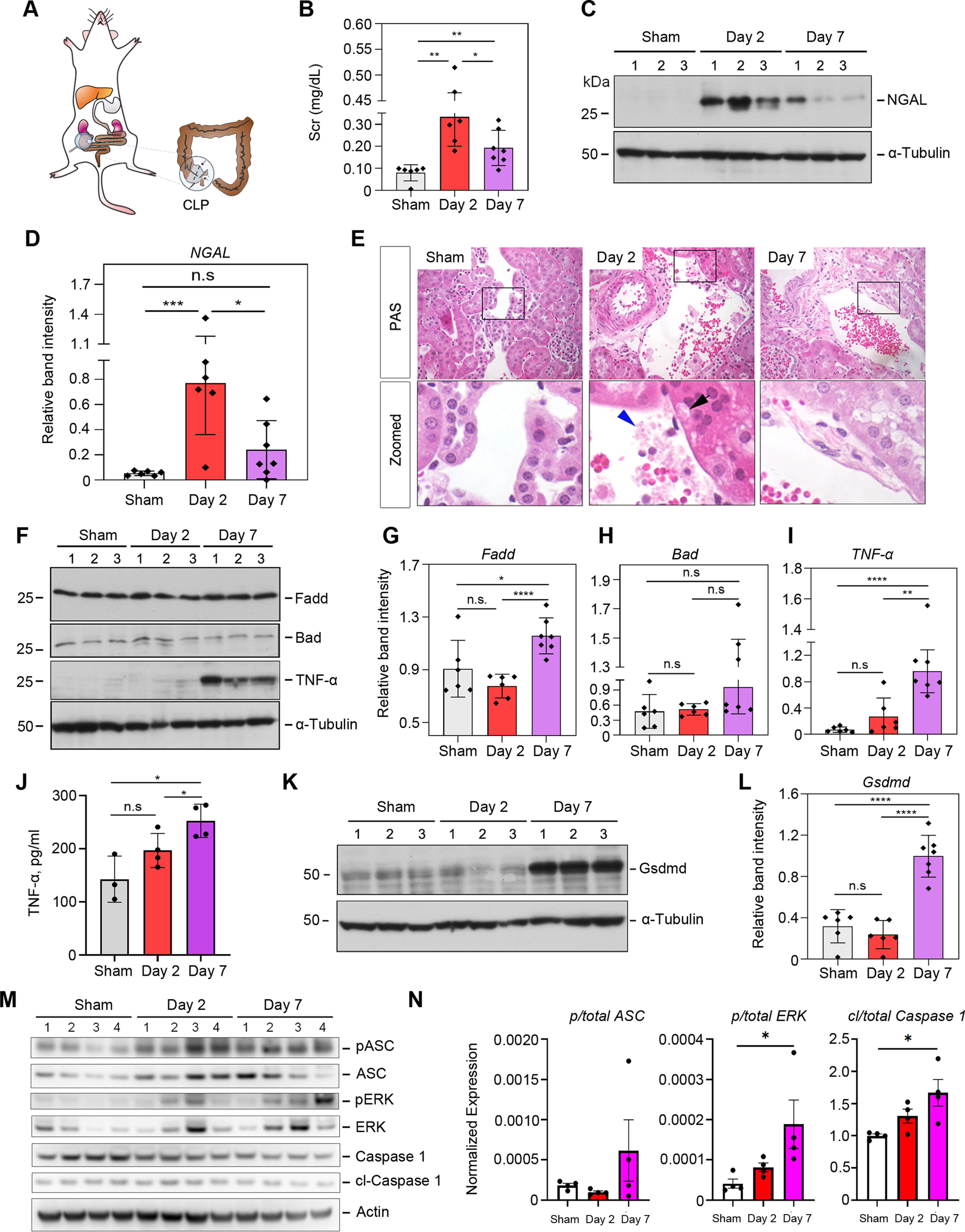
Cecal ligation leads to sepsis, kidney inflammation and pyroptosis in a time dependent manner. A, Schematic diagram illustrating the construction of the moderate CLP mouse model. B, Measurement of serum creatinine (SCr) levels from control mice (sham), day 2, and day 7 post-CLP surgery (n = 6–7). C, Western blot analyses of renal expression of NGAL protein in sham control and injured kidneys after CLP at day 2 and 7. Numbers (1–3) indicate each animal in a given group. D, Quantitative plots of (C) with more data points included. E, Representative micrographs showing kidney morphology at day-2 and day-7 after CLP. In the enlarged boxed areas, the black arrow indicates typical tubular cell vacuolation after CLP, and the blue arrowhead indicates cellular debris and possible migrated microbial. Scale bar, 50 μm. F, Western blot analyses of FADD, Bad, and TNFα protein abundance in the kidneys at day 2 and 7 after CLP compared with the sham control. Numbers (1-3) indicate each animal in a given group. G–I, Quantitative data of (F) with more data points. J, ELISA results of TNFα level in the kidneys at day-2 and day-7 after CLP. K, Western blot analyses of Gasdermin D abundance in the kidneys at day 2 and 7 after CLP compared with the sham control. L, Quantitative data of (K) with more data points. M, Western blot of phosphorylated ASC and ERK proteins and their respective total proteins, and cleaved Casp1 and the total uncleaved form. β-Actin was used as the loading control. N, Densitometry quantitation of (M). n.s, not significant; *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
It has been known that cell apoptosis and inflammatory cell infiltration are two major cellular processes in the development and progression of AKI (27). To investigate this, we examined two cell apoptosis-related proteins, Bcl2-associated agonist of cell death protein (Bad) and FAS-associated death domain protein (Fadd) (27, 28). Bad appeared to remain unchanged (p > 0.05) across the three time points, whereas Fadd showed mild increase at day 7 compared with sham control (Fig. 1F through 1H). Consistently, the expression of tumor necrosis factor-α (TNFα) displayed a similar trend to Fadd in the S-AKI kidneys by Western blot (Fig. 1I) and ELISA (ELISA) (Fig. 1J). As areas of necrosis were observed in our histological analysis (Fig. 1E), we tested the effector proteins of pyroptosis (29), whose activity has recently been associated with acute kidney injury (30). Gasdermin D, the effector molecule that forms the membrane pore and mediates pyroptosis (29), was significantly increased in the injured kidneys at day 7 after CLP (Fig. 1K and 1L). Cleaved caspase-1, which mediates Gsdmd cleavage and activation, was also significantly increased, along with phosphorylated ASC (apoptosis-associated speck-like protein containing a CARD; also known as Pycard), which regulates caspase-1 activation (Fig. 1M and 1N). In addition, phosphorylation of ERK, whose activation mediates LPS-induced inflammasome formation (31) was significantly increased as well (Fig. 1M and 1N). Collectively, our data strongly suggest canonical pyroptosis activation and inflammasome formation, and that a delayed biological response to septic injury in kidney dominates the transition from AKI to CKD.
The Global Proteomic View of the Septic Kidneys
To gain an unbiased understanding of the underlying molecular determinants that modulate S-AKI, we analyzed the global profile of the renal tissue proteomes of mice at day 0, 2 and 7 post-CLP following a label-free quantitative approach (Fig. 2A). The global kidney proteome was analyzed using a 220-min single-run LC–MS/MS approach to balance the throughput and proteomic depth. Collectively, 2,119 proteins were quantified with a less than 1% false discovery rate (FDR), and nearly 90% of them were detected across all three groups (supplemental Table S1). The correlation between biological replicates (i.e. different mice) within the same group was high (R2 = 0.95 ± 0.02, n = 45) (Fig. 2B), which validated the reliability of our CLP model and the reproducibility of the sample processing approach. We also noticed that the correlation between different groups was relatively high as well (R2 = 0.93 ± 0.02, n = 108). These data indicate that the CLP procedure induced low- to mid-grade sepsis, likely only allowing a minimal amount of feces to extrude from the perforation site. Interestingly, the overall unfiltered proteomics data enabled the unsupervised classification of the kidneys according to their global profiles, which clustered day-2 mice tightly together whereas control and day-7 samples were intermixed (supplemental Fig. S1). The data suggested that the kidney global proteome has undergone systematic changes after moderate septic injury, and may have reshaped back closer to healthy kidneys a week after CLP.
Fig. 2.
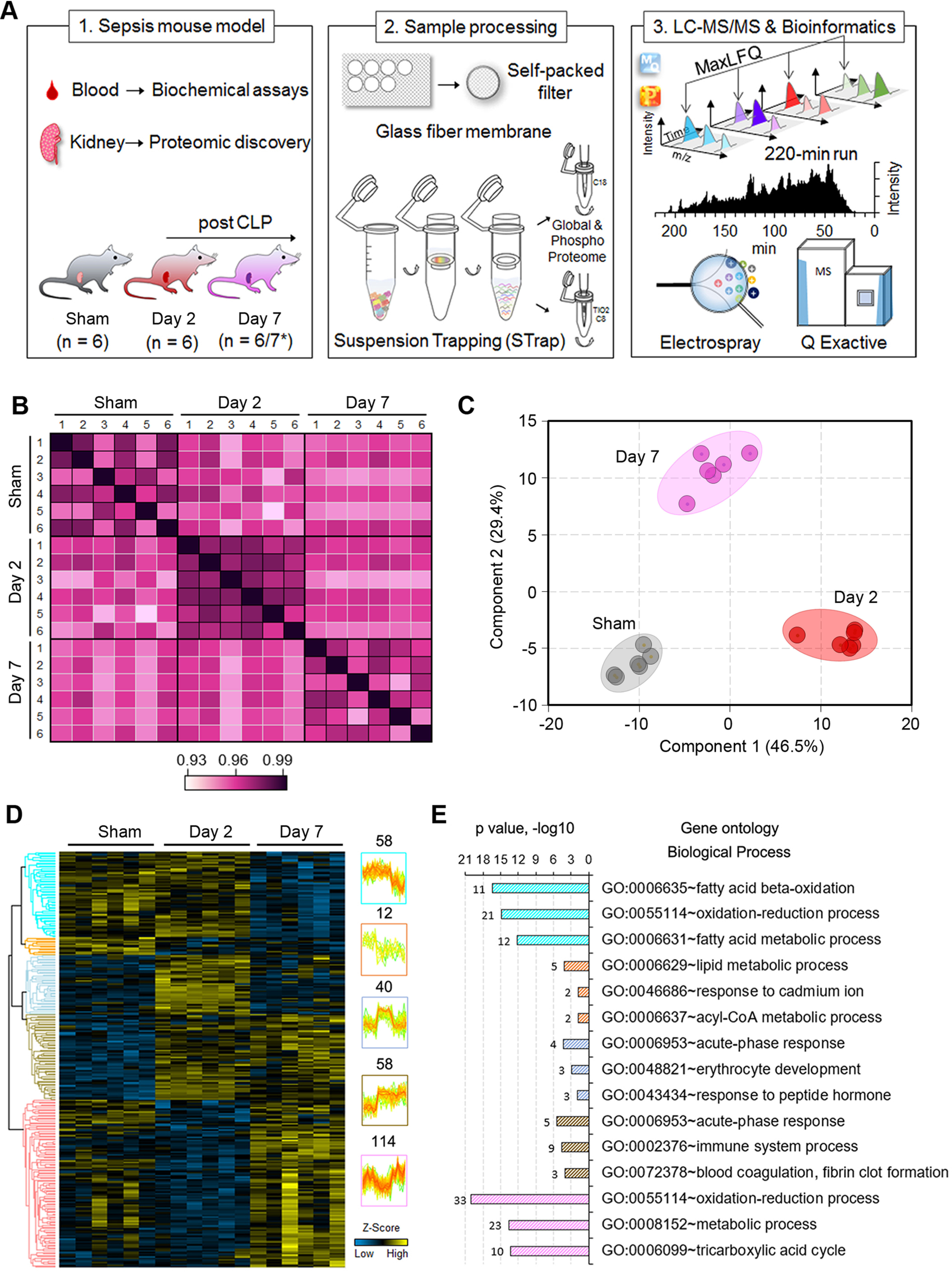
Differential proteomic profiles define acute versus chronic kidney injury. A, Experimental workflow of the study. * Seven mice were used in some of biochemical assays as indicated in the plots in Figure 1. B, Correlation of kidney proteome profiles between different groups of mice. Color scale represents R2 values. C, and D, PCA lot and heatmap of ANOVA significant proteins (Permutation FDR 0.05) among the three groups. LFQ intensity of represented proteins was z-scored and plotted according to the color bar. Five clusters of proteins with different patterns of abundance profile are highlighted in the heatmap. The numbers indicate the number of proteins in each cluster. E, Top three enriched GO biological process terms in each cluster of proteins are plotted with their names and significance (p value). The number of matched proteins in each category is listed next to the bar graph.
To define the proteins that were differentially regulated in response to septic injury, and determine underlying modulatory pathways, we performed a multiple-variations test (ANOVA). We first filtered the overall proteome to require that proteins be quantified in at least 4 out of 6 replicates in one of the groups, which led to 1,520 high quality proteins. The ANOVA analysis resulted in 282 significant proteins among the three groups (Permutation FDR 0.05) (Fig. 2D, supplemental Table S2), which was further verified by their distinct segregation in a PCA plot (Fig. 2C). Gene Ontology (GO) enrichment analysis indicated that these proteins were generally involved in the oxidation-reduction process, metabolic process, and fatty acid beta-oxidation. Although acute-phase response proteins and proteins that are associated with the immune system process were largely increased upon septic injury, oxidation-reduction, as well as metabolic process proteins, showed varied profiles (Fig. 2E). These data highlighted the complex and dynamic host response to the septic infection. By examining the subcellular localization of the ANOVA significant proteins, we found that nearly 44% of them (124 out of 282) were enriched in “mitochondrion” (p = 8.14 × 10−57) (supplemental Fig. S2). Of note, around 21% of mitochondrial-associated proteins were identified in a global kidney proteome (32), and less than 10% in the overall mouse proteome (17,034 Reviewed UniProt proteins). Among the mitochondrial proteins, 46 (37%) of them were annotated as mitochondrial inner membrane proteins, such as ATP synthase (e.g. Atp5j, Atp5l, and Atp5o), NADH dehydrogenase (e.g. Ndufb1/3/4/7/9/12/13) and cytochrome c oxidase (e.g. Cox2 and Cox4i1). This finding is consistent with the biological process enrichment result, and suggests that mitochondrial dysfunction is strongly associated with the decline of kidney integrity and hemostasis (33), and in our case, during sepsis (34).
Septic Injury Induces Dynamic Changes in Kidney Phosphoproteome
To gain more insights into the temporal regulation and functional changes of kidney proteins during S-AKI, we performed phosphoproteome analysis to the same set of tissue samples used for the global proteome analysis (as shown in Fig. 2A). In total, 4,745 phosphosites were identified from our study, of which 2,952 were unambiguously localized with high confidence (localization score > 0.75), corresponding to 1,279 nonredundant phosphoproteins (supplemental Table S3 and S8), which were used for downstream analysis. Of note, phosphorylation of Erk-1/Mapk3 (at Tyr205) and ASC (at Ser193), as described in the previous section, were both seen in our phosphoproteome data set. However, phosphoproteomic data did not correlate quite well with immunoblot assays (supplemental Fig. S3), which was likely because of the low abundance of these phosphoproteins. Majority (64%) of the 1279 proteins were exclusively identified from the phosphoproteomics experiments (supplemental Fig. S4A), implying the specificity of the TiO2-based phosphorylation enrichment process. In the context of the reproducibility of phosphoproteome measurements, a much lower correlation was seen both between biological replicates (average R2 = 0.82) and different groups (average R2 = 0.79) (supplemental Fig. S4B) as compared with those of global proteomes. These data suggest that different phosphorylation patterns exist, as indicated by a previous tissue phosphoproteome study (35), and temporal phosphoproteomic changes in response to microbial infection are more dynamic than global proteomic changes.
To base our analyses on high quality data, we filtered the phosphorylation data set to exclude samples that contained less than 1000 quantifiable phosphosites, and phosphosites that were quantified in less than 3 replicates, which resulted in 1870 phosphosites. Subsequent ANOVA analysis resulted in 370 significant phosphosites among the three groups (p < 0.05) (supplemental Table S4), and further p-value correction (Permutation FDR 0.05) resulted in 23 phosphosites, as presented in Fig. 3A. Overall, the majority of the kidney phosphorylation events drastically decreased at day 2 post CLP and gradually returned to basal level at day 7 (supplemental Fig. S5), indicating a declined energy state of the renal tissue upon acute infection and dynamic host response to septic injury. Enrichment analysis indicated that phosphoproteins associated with RNA splicing and mRNA processing were significantly over-represented (p < 10−15) (Fig. 3B). These include RNA binding motif (RBM) protein families (e.g. Rbm10/17/25/39/xl1), serine/arginine-rich splicing factors (e.g. Srsf1) and serine/arginine repetitive matrix proteins (e.g. Srrm1/2). Alternative RNA splicing regulates host immune response in a variety of viral and bacterial infection conditions (36), and RBM proteins have been found to modulate apoptosis during infection in addition to splicing (37). As an example, the phosphorylation of Srrm2 has been shown to modulate HIV (HIV-1) replication and release (38). Srrm2 is highly phosphorylated, with over 250 known phosphorylation sites listed in the UniProt Knowledgebase. In our study, we identified 78 of them, like a previous study (39). About 20 sites were differentially phosphorylated upon sepsis injury, including a highly significant one at serine 1229/1230 (ANOVA p < 0.0001). Its modulatory role in polymicrobial infection deserves further investigation.
Fig. 3.
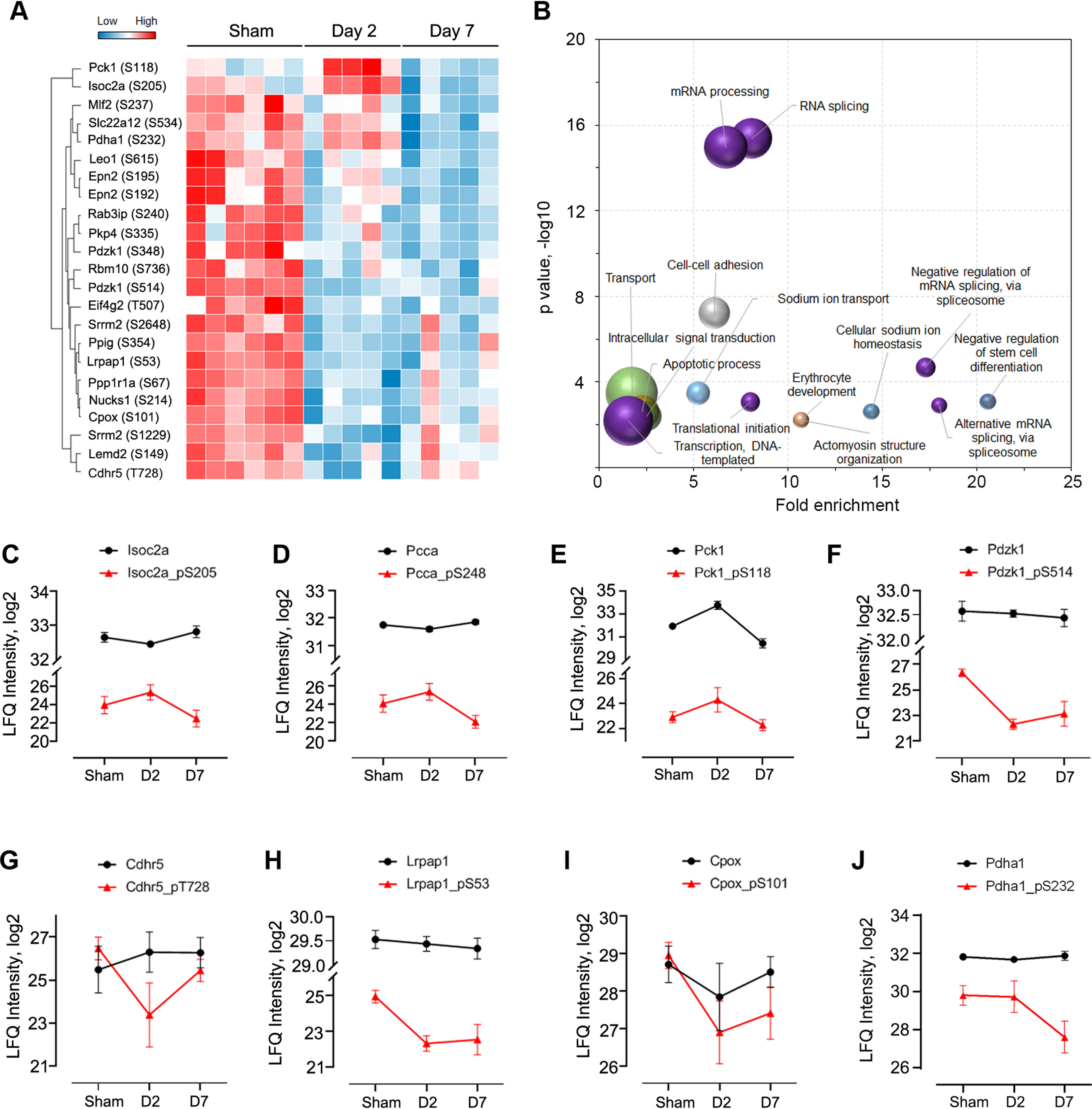
Quantitative phosphoproteome analysis of kidneys after septic injury. A, Heatmap of representative phosphosites significantly altered among the three groups (ANOVA, Permutation FDR 0.05). B, Enrichment analysis of biological process of ANOVA significant phosphoproteins. The top-15 terms are plotted. Bubble size depicts the number of proteins in the category, ranging from 3 to 42. C–J, Intensity plots of representative phosphosites (red line) and their unmodified forms (black line). Ctl, control; D2, day 2; D7, day 7.
Although most of the phosphorylation events were reduced after acute septic injury after global proteome and phosphoproteome normalization, a small number of them remained high at day 2 (Fig. 3C–3E). These include isochorismatase domain-containing protein 2A (Isoc2a, S205), phosphoenolpyruvate carboxykinase (Pck1, S118), and mitochondrial propionyl-CoA carboxylase alpha chain (Pcca, S248). The identified phosphorylation at S205 in Isoc2a is novel, therefore its role in S-AKI needs further investigation. Interestingly, the expression levels of both Isoc2a and Pcca remained stable among the three groups (p > 0.05), whereas their phosphorylated forms changed significantly (up-regulated at day 2 and down-regulated at day 7; p < 0.05), indicating a true up-regulation of their phosphorylation at day 2. Pcca is a mitochondrial enzyme involved in amino acid catabolism (40) and mitochondrial oxidative phosphorylation. In C. elegans, its mutation significantly reduced mitochondrial energy metabolism and increased oxidative stress (41), and previous studies have demonstrated that it is strongly linked to the genetic disease propionic acidemia (41). Phosphorylation of Pcca at site S248 has been identified by proteomic screening from various mouse tissues previously (42, 43), however, the phosphorylation/dephosphorylation of this site in response to polymicrobial infections and its association with mitochondrial metabolism in kidneys is unknown.
Another interesting class of proteins that was identified in our analyses is the solute carrier (SLC) proteins, a superfamily of membrane-bound transporters with nearly 400 members (44). They regulate ion transport, waste removal, and many other essential physiological functions, and have increasingly been targeted for therapeutic invention (45). However, their involvement in septic injury has not been well studied. In our global and phosphoproteomic studies, 72 SLC proteins as well as 113 phosphosites were identified (supplemental Table S1 and S3). The majority of SLC proteins identified in our study did not show significant differences upon septic injury, and even if changed, the fold differences were minimal (Fig. 4B). The significantly (ANOVA, p < 0.05) changed SLC proteins and/or their phosphorylated forms are shown in Fig. 4A. The Band 3 anion transport protein (Slc4a1) increased dramatically (FC > 4) during acute septic infection at day 2, whereas its phosphorylated form (pS18) did not change (Fig. 4C). On the other hand, the unmodified form of the solute carrier family 22 member 12 (Slc22a12, also known as Urat1) did not show significant alterations, but its phosphorylation at S534 was maintained at a similar level at day 2 and then down-regulated at day 7 (Fig. 4D). Phosphorylation of Slc22a12 has been shown to assist in the reabsorption of uric acid via other organic anion transporters (46, 47). Lastly, both the unmodified and phosphorylated forms of the solute carrier family 22 member 2 (Slc22a2) were changed in a similar trend after septic injury (Fig. 4E). These data indicate that the homeostasis of ions and small molecules in the kidney are broadly altered after sepsis induction.
Fig. 4.
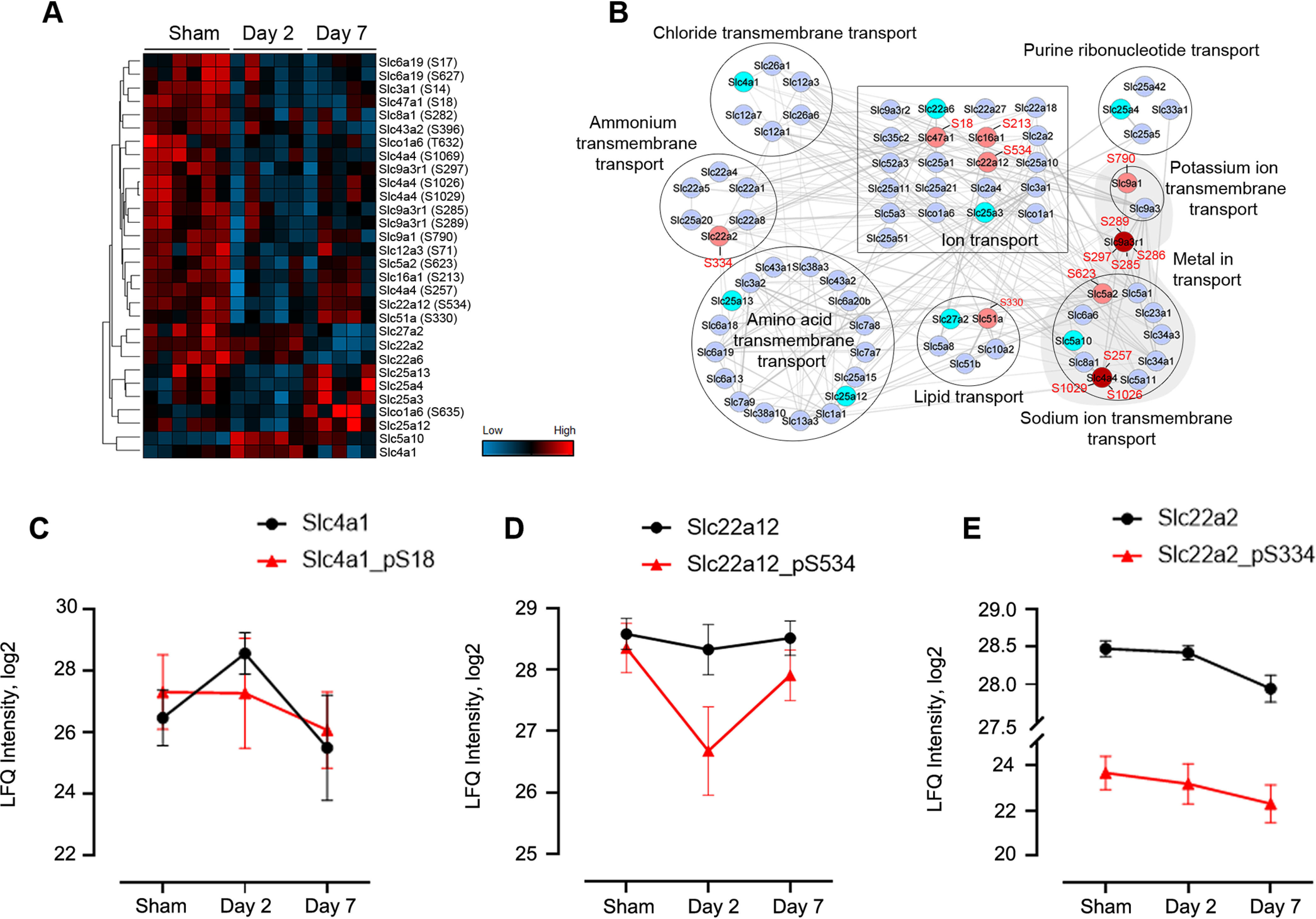
Quantitative analysis of SLC proteins. A, Heatmap of ANOVA significant (p < 0.05) proteins and phosphosites. B, Protein network and functional enrichment clusters. The network was built based on all the 72 identified SLC proteins as input using the String App in Cytoscape software. Nodes colored in light blue are nondifferential (FC < 1.5) proteins; in cyan are significantly different proteins; in pink and red are differential phosphoproteins. The protein clusters were annotated based on the Enrichment Data, an embedded function in Cytoscape. C–E Profiles of representative SLC proteins across the three groups with phosphosite intensities in red line and unmodified forms in black.
Discovery-Based Studies Recapitulate Signature Proteins for S-AKI Prognosis and Disease Progression
Next, we sought to examine signature proteins that correlate with the severity of S-AKI. We performed pairwise comparisons between control and septic kidneys at day 2 or day 7 to highlight significant marker proteins (Fig. 5A–5B; supplemental Table S5). In our data, the known AKI marker protein NGAL dramatically increased (> 64-fold; p < 0.001) at day 2 and gradually lowered down to near basal level (p > 0.05; Fig. 5C). This finding was consistent with the immunoblot analysis (Fig. 1C–1D), highlighting the value of our MS-based approach for kidney disease biomarker discovery. In addition to NGAL, we also identified other marker proteins and proteins that have not been previously associated with kidney injury. Hydroxymethylglutaryl-CoA synthase 2 (Hmgcs2), a key enzyme of ketogenesis mediating energy generation from lipids when carbohydrates are deprived, was increased in day-2 kidneys by more than 80-fold (Fig. 5D). These data were confirmed by immunoblot analysis (Fig. 5M and 5N). Hmgcs2 has been indicated to be up-regulated in both Type II diabetic kidneys and Type I diabetic heart (48, 49), suggesting enhanced ketone body production and activated ketogenesis in these diseased organs. Serine protease inhibitor A3N (Serpina3n) was also significantly increased at day 2 by nearly 60-fold (Fig. 5E). Serine protease inhibitors are usually found to be increased during acute inflammation and act by modulating protease activities (50, 51). Recently, Serpina3n was found to be elevated in the urine of rats with early AKI-to-CKD transition and relocated from cytoplasm to apical tubular cell membranes (52). In our immunoblot analysis, we found one of these serine protease inhibitors, α-1-antitrypsin, to be significantly increased at day 2 (Fig. 5M and 5N). Other proteins such as serum amyloid A-1 and A-2 protein (Saa1 and Saa2) and serum amyloid P-component (ApcS), proteins that were shown to be increased in diabetic kidneys and chronic renal failure (53, 54), were also markedly increased in day-2 post-CLP kidneys (Fig. 5F-5H).
Fig. 5.
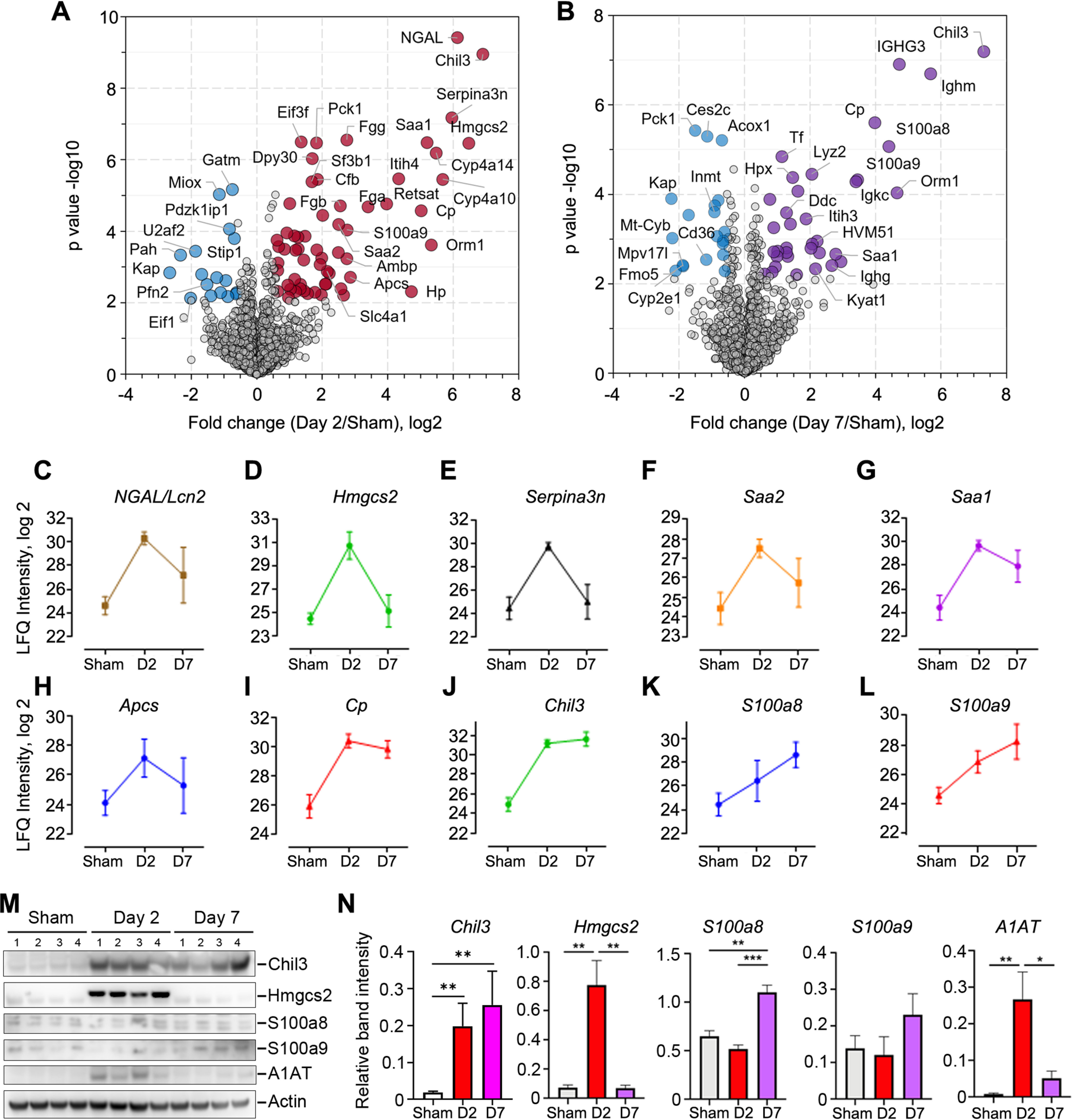
Kinetic changes of sepsis-induced kidney injury markers. A–B, Volcano plot of pairwise comparison between the kidney proteomes of day 2 or day 7 post-CLP versus control. Student t test (Permutation FDR 0.05) was used for the analyses. Up- and down-regulated proteins by 1.5-fold were colored in red/purple and blue, respectively. Gene name of represented proteins were indicated in the plots. C–L, Abundance plots of representative proteins in response to septic injury. Mean values and error bars from six replicates of each group were shown. M–N, Western blot validation and densitometry quantitation of representative marker proteins. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Of note, several proteins showed consistent up-regulation at both day 2 and day 7 post-CLP. Ceruloplasmin (Cp) is a copper-containing ferroxidase that oxidizes ferrous iron (Fe2+) to its nontoxic ferric iron (Fe3+) form. Its expression was shown to be increased in aging mice or mice consuming high-calorie diet (55), suggesting the increased amount of oxidative damage in these mice required enhancement for its antioxidant activity. The urinary Cp level was suggested to be a biomarker for CKD in sickle cell disease patients (56). Cp levels in our day-2 and day-7 mice post-CLP were elevated by 32- and 15-fold, respectively (Fig. 5I), suggesting increased oxidative stress in kidneys post-sepsis. Meanwhile, S100a8 and S100a9, DAMPs molecules that mediate pro-inflammatory responses via TLR4 binding (57), were previously indicated to be increased in plasma and associated with septic shock (58). In our proteome analysis, the increase was significant at both time points for S100a9, whereas it was significant only at day 7 for S100a8 (Fig. 5K–5L), suggesting the formation of the active dimerized calprotectin was gradually increased in kidneys after septic injury (57). Regarding the validation assays, Western blot confirmed the differential expression pattern of S100a8, whereas contradicted S100a9 expression (Fig. 5M–5N). The role of the latter in septic kidney would require further investigation.
Among the proteins that were significantly increased post-CLP, we found the up-regulation of chitinase-like protein 3 (Chil3) to be most striking, by almost 120-fold at day 2 and more than 150-fold at day 7 (Fig. 5J). These differences were validated with our immunoblot analysis (Fig. 5M and 5N). Chil3 is a member of the mammalian chitinase family that lacks the chitinase activity though it is highly secreted by macrophages and neutrophils (59, 60). Similar to the function of S100 proteins, chitinase-like proteins have also been shown to be mediators of inflammation and antimicrobial responses in multiple models of disease such as asthma and pulmonary fibrosis as well as involved in tissue remodeling and wound healing (61–63). In previous work in kidney disease models, a urinary proteomic study has shown that chitinases-like proteins could be candidate biomarkers for S-AKI (63). Recently, it has been demonstrated to be largely increased in urine from clinical AKI patients (64). Another study also showed renal Chil3 to be up-regulated at 6 h and 24 h after intraperitoneal injection of LPS into mice (65). Taken together, our study recapitulated previously described sepsis-related modulator proteins and provided a valuable resource of candidate targets to future studies of sepsis diagnosis and therapeutics.
Molecular-Level Understanding of AKI-CKD Transition
As described above, our data have shown distinct pathology of the septic kidneys at the acute and chronic stages by analyzing them at day 2 and day 7 post-septic injury, respectively. Therefore, a direct comparison between these two phases would lead to insights into how the disease progresses after acute septic injury. Following a similar t test (FC ≥ 1.5; Permutation FDR 0.05), we identified 38 and 56 up- and down-regulated proteins, respectively, from day-7 mice compared with day-2 mice (supplemental Table S6). Interestingly, although the functional enrichment analysis did not suggest particular biological process terms enriched among the up-regulated proteins, likely because of the low number of input proteins, we noticed several immunoglobulin proteins (such as Ighm, Ighg, Igkc, IGHG3, and HVM51) were significantly increased. This suggested that while recovering from the acute phase of septic-induced kidney injury, there may be an initiation of an adaptive immune response in renal tissue progressing toward chronic kidney disease. Indeed, a switch from innate to adaptive immunity has been reported to mediate the transition from acute septic kidney injury to prolonged chronic disease (66, 67), and some of these Igs can be used to develop biomarkers for CKD in the future (68).
To obtain a broader view of the dysregulated pathways during S-AKI-CKD transition, we slightly lowered the fold-change threshold (≥ 1.2) to capture more differential proteins (202 in total; Fig. 6A) and build a signaling network (see details under “Experimental Procedures”). The analysis with the highest confidence score (cutoff = 0.9) resulted in 396 interactions and 126 nodes (Fig. 6B), the majority (56%) of which were associated with small-molecule metabolic processes (FDR = 3.35e-48), such as lipids, nucleotides, alcohol, and other fatty acids. Abnormal lipid metabolism has been associated with diabetic nephropathy and other kidney dysfunctions (69, 70); lipid deposition contributes to atherosclerosis and cardiovascular diseases commonly seen in CKD patients (71). The network analysis also indicated several interesting protein clusters such as oxidoreductase complex, peroxisome, and cytochrome P450 (CYP) family proteins (Fig. 6B), which may play important roles in the AKI-CKD transition. Peroxisomes and oxidoreductases are involved in regulating oxidative stress caused by bacterial and/or viral infections (72). In our data, 18 of the 21 proteins annotated as peroxisome proteins showed down-regulation in day-7 kidneys. In contrast, all the 12 oxidoreductase complex associated proteins were up-regulated in day-7 kidneys. The role of oxidoreductases in the progression of the disease stages of septic kidneys remains to be identified. CYP proteins are known to metabolize a wide array of small molecules such as fatty acids, alkanes, and xenobiotic drugs. Recent studies have shown their involvement in microbial infections (73, 74). In our analysis, five CYP members (2A4, 2A5, 2E1, 4A10, and 4A14) were found to be significantly down-regulated in day-7 kidneys compared with day-2. For example, the expression of Cyp4a10 was dramatically increased after acute infection (at day 2) and declined back to the basal level at chronic stage (at day 7) (Fig. 6C). It has been noted that CYP enzymes can be selectively regulated in different states of inflammation by a multitude of cytokines released after infection or inflammation (73). In hepatic and extrahepatic tissues, chronic inflammation is usually associated with down-regulation of CYP enzymes (73). Hence, our quantitative data of CYP proteins were in line with previous studies and suggest that they could also be used as signatures for kidney disease severity and/or classification. Notably, carboxylesterase 1C (Ces1c), which aids in detoxification of xenobiotics and is responsible for poor pharmacokinetics of the anti-virus drug remdesivir (75), was also mapped in our cluster analysis. Ces1c has been shown to be significantly decreased in macrophages after LPS treatment (76), which was consistent with its changes in our data of septic kidneys (Fig. 6D). Together, our proteomic data provides an unbiased view of how kidney tissues dynamically modulate oxidative stress caused by sepsis, and shed light on future exploration for potential diagnostics and therapeutics.
Fig. 6.

Metabolic modifications in kidneys occur upon CLP-induced sepsis. A, Volcano plot shows the p value (y axis) and fold change (x axis) of proteins identified in both groups of kidneys. Significant proteins are highlighted in red and purple for up- and down-regulated proteins, respectively (Permutation FDR 0.05; FC ≥ 1.2). B, Interaction network of the significant proteins as shown in A. The network and enrichment results were derived from String App in Cytoscape software. The score cutoff for interaction confidence was set to 0.9. Shaded clusters are involved in metabolic processes. The color coding is in accordance to fold changes between day 7 and day 2. Diamond shape depicts mitochondrion associated proteins. C–D, Intensity plot of two representative proteins associated with the oxidation-reduction process.
DISCUSSION
In the present study, we have profiled the septic kidney proteome and phosphoproteome using a CLP mouse model established in our laboratory. CLP was performed in six mice (biological replicates) in each of three groups to better control reproducibility. The tissue samples were processed following an STrap approach developed by Zougman and colleagues (77). The STrap filters were in-house packed using bulk glass fiber membranes, which are nearly cost-free and as effective as commercial filters (18). The sample was analyzed in a 'single-shot' injection with label-free quantitation based on MaxLFQ algorithm. Our data showed minimal technical variations, thus offering critical insights into the molecular level alterations of renal proteins upon septic injury. Our CLP procedure induced only low to mid-grade severity and nearly all the day-7 mice survived from the acute sepsis and progressed to the chronic stage. Hence, the CLP model allowed us to investigate not only the acute host response (at day 2) but also the “acute-to-chronic” transition even after moderate kidney injury, the latter of which has not been well defined molecularly (78, 79). This transition from the acute to chronic stage reflects the refractoriness of septic kidney diseases in the clinical setting. Our study provides solid molecular evidence for the first time derived directly from septic kidneys.
S-AKI can occur in the absence of overt signs of hypoperfusion or clinical signs of hemodynamic instability. Therefore, early biomarkers are urgently needed to permit timely diagnosis and predict injury severity (80). However, sepsis is usually associated with multiorgan injuries, and traditional markers of S-AKI originated from plasma and urine (e.g. creatinine, urinary concentrating ability) are susceptible to interference from nonrenal factors, thus are not sensitive and specific (80). Our global proteome and phosphoproteome data broadly agree with previous urinary discoveries in the context of many known AKI markers such as NGAL. In addition, we validated several new and/or less-well studied marker proteins for S-AKI including Hmgcs2, Serpin, S100a8, and Chil3. The comprehensive global analyses revealed that at the AKI stage, immune-related proteins were up-regulated whereas metabolism-related proteins were repressed in septic kidneys. Our data suggests that multiple immune pathways could be activated in kidneys after septic injury, including adaptive DAMPs responses, CYP-mediated lipid metabolism, peroxisome-mediated oxidative stress, and likely the inflammasome-mediated pyroptosis pathway. Effector molecules involved in the canonical inflammasome formation and pyroptosis activation (81–83), including GSDMD, cleaved caspase-1, and phosphorylated-ASC, were all found to be significantly increased post-sepsis. Gsdmd-dependent pyroptosis cell death has been revealed for the first time in nonseptic AKI in a very recent study (30). Our data indicated that Gsdmd expression remained at a low level at the acute stage and drastically increased at the chronic phase. Adding another time point midway through this transition (for instance, at day 4 or day 5) would likely reveal the onset of pyroptosis, which can be considered in the future studies. Nevertheless, our findings implied the potential role of pyroptosis in initiating tubular cell damage, thus deteriorating CKD, or alternatively, plasma membrane repair can be initiated to counterbalance pyroptosis (for instance, through ESCRT (84, 85)). These questions are certainly of great interest for future investigations.
Among the responsive proteins to septic injury, Chil3 was found to be of particular interest. Chil3 (also known as YKL-40 in humans and BRP-39 in mice) does not contain known chitinase activity, though it has been implicated in host defense, inflammation, and remodeling processes (61, 62). Chil3 has been reported as a biomarker for skin wound infection (86), cardiovascular disease and diabetes (87), and cancer (60). A handful of studies have shown that Chil3 has critical functional relevance to kidney ischemic injury (88) and renal fibrosis (89). There is also evidence showing its potential in predicting the onset and recovery of acute kidney injuries (63, 89, 90). However, most of these studies investigated chitinase secretion into the urine and used the urinary form as an indicator of disease status, which might be complicated by other nonkidney diseases such as urothelial carcinoma (19). Emerging studies have also started to demonstrate the mechanism of Chil3 in mediating immunity, including its role in IL-17 production in γδ T cells (91), and recruitment of macrophages and neutrophils in colorectal cancer (92). A recent study showed that Chil3 interacts with interleukin-13 receptor α2 and activates pathways that regulate apoptosis, pyroptosis, inflammasome formation, and antibacterial responses (93). Our data showed that Chil3 has a strong correlation with S-AKI disease onset and severity. Chil3 was elevated almost 120-fold at day 2 and more than 150-fold at day-7 after S-AKI, implying that it was involved in the progression of septic kidney injures. It remains unclear, however, whether the expression Chil3 is linked to the direct microbial migration from gut to kidney.
Furthermore, plenty of metabolism-related proteins were significantly changed during the acute septic injury stage, which is consistent with previous findings that changes in metabolism and mitochondrial energetics occurred in response to renal damage (94, 95). Of note, the general reduction of phosphorylation after septic injury indicates that energy usage has been reprioritized in renal tissue. The drastic increase of Hmgcs2 at day 2 suggests that ketogenesis is highly up-regulated during acute kidney injury. Similar up-regulation of Hmgc2 in diabetic kidney and heart encourages us to speculate that the energy-producing pathway in kidneys during S-AKI may be ketogenesis. Therefore, we can imagine that our CLP procedure in this study merely generated a moderate S-AKI model, and it remains intriguing to see if, for example, during a severe S-AKI scenario, or at an earlier time point post-CLP, more drastic changes of these marker proteins will be seen.
Because cecal perforation breaks intestinal barrier, and leads to the translocation of fecal contents from gut lumina into the peritoneum and subsequently blood stream (96), we have attempted to detect gut microbial species, if they might have translocated to distal organs such as kidney. Metaproteomic database searches were performed to identify potential bacterial proteins in addition to the mouse host proteins. Although not totally unexpected, we did not obtain highly confident protein hits, as they were higher than the 1% FDR threshold and also majority matched with single peptides (supplemental Table S7). We did not intent to over interpret the data as our study used a mid-severity CLP murine model, gut microbes might be cleared already by the first line of host immunity before gaining access to distal tissues or organs, thus minimizing the chance of identifying them in the complex background of kidney tissue proteins. In addition, the histological data of kidney tissue did not show clear evidence of microbial presence neither (Fig. 1E). Indeed, gut bacterial DNA fragments have been detected in the blood of CKD patients (97, 98). Gut-originated uropathogenic Escherichia coli has also been reported to be able to replicate intracellularly in bladder urothelium, causing recurrent urinary tract infections and cystitis (99, 100). In addition, emerging evidence suggested that respiratory tract extracellular pathogen Streptococcus pneumoniae could translocate to heart tissue and replicate intracellularly, exacerbating community-acquired pneumonia by inducing cardiac microlesions (101, 102). Currently, a prevailing paradigm in renal physiology is the assumption that the kidney is sterile (103). Whether gut microbes can invade and colonize kidney is still an unanswered question. Our data from the current study did not provide strong evidence of microbial presence in kidney, however, we hypothesize that gut microbes, once release to circulation system, may gain access to other tissues, form local bacterial colonization, then amplify existing injury and lead to persistent injuries. Therefore, further investigations (for instance, using severe CLP model) are desired to pinpoint the potential role of gut microbes in the chronic kidney diseases.
Our study is not flawless. One of the apparent limitations is the relatively low coverage of the kidney proteome and phosphoproteome. With further fractionation prior to LC–MS/MS analysis, the sample complexity would be reduced and the chance to identify marker proteins that have low abundance would be increased. We have decided to demonstrate that the single-shot proteomics along with the extremely economical sample preparation approach would be a great alternative to biomarker discovery and pathology mechanism investigation. In summary, our study provides the first characterization of how moderate gut injury triggered sepsis promotes S-AKI and the molecular determinant alterations in situ. The findings from our study will facilitate our understandings of septic kidney. The comprehensive data set will serve as a valuable resource for sepsis biomarker discovery in the future.
DATA AVAILABILITY
All MS raw data associated with the study, along with detailed tables of MaxQuant output files reporting m/z, charge, identification, quantitation and phosphosite scores, and other details for all peptide identifications, have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifier PXD017045, and MassIVE with the data set identifier MSV000086126.
Supplementary Material
Acknowledgments
We would like to acknowledge JCVI internal startup funds to Y.Y. and G.J.N. D.Z. was supported by the National Institutes of Health (NIH) grant K01DK116816. We would like to thank Rodrigo Vargas Eguez for help with sample processing, and Dr. Karen E. Nelson for manuscript reviewing and useful discussions.
Footnotes
This article contains supplemental data.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
Abbreviations—The abbreviations used are:
- AKI
- Acute kidney injury
- BAD
- Bcl2-associated agonist of cell death protein
- CKD
- Chronic kidney disease
- CLP
- Cecal ligation and puncture
- DAMPs
- Damage-associated molecular patterns
- ELISA
- Enzyme-linked immunosorbent assay
- ESCRT
- Endosomal sorting complexes required for transport
- FADD
- FAS-associated death domain protein
- FC
- Fold change
- FDR
- False discovery rate
- GO
- Gene Ontology
- Gsdmd
- Gasdermin D
- ICU
- Intensive care unit
- KEGG
- Kyoto Encyclopedia of Genes and Genomes
- LFQ
- Label-free quantitation
- NGAL
- Neutrophil gelatinase-associated lipocalin protein
- PAMPs
- Pathogen-associated molecular patterns
- PAS
- Periodic acid-Schiff
- PCA
- Principle component analysis
- S-AKI
- Sepsis-induced acute kidney injury
- SLC
- Solute carrier proteins
- STrap
- Suspension Trapping
- TLRs
- Toll-like receptors.
REFERENCES
- 1. Ronco C., Bellomo R., and Kellum J. A. (2019) Acute kidney injury. Lancet 394, 1949–1964 [DOI] [PubMed] [Google Scholar]
- 2. Mercado M. G., Smith D. K., and Guard E. L. (2019) Acute Kidney Injury: Diagnosis and Management. Am. Fam. Physician. 100, 687–694 [PubMed] [Google Scholar]
- 3. Fraser S. D. S., and Roderick P. J. (2019) Kidney disease in the Global Burden of Disease Study 2017. Nat. Rev. Nephrol. 15, 193–194 [DOI] [PubMed] [Google Scholar]
- 4. Zarbock A., Gomez H., and Kellum J. A. (2014) Sepsis-induced acute kidney injury revisited: pathophysiology, prevention and future therapies. Curr. Opin. Crit. Care. 20, 588–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zarjou A., and Agarwal A. (2011) Sepsis and Acute Kidney Injury. J. Am. Soc. Nephrol. 22, 999–1006 [DOI] [PubMed] [Google Scholar]
- 6. Gómez H., and Kellum J. A. (2016) Sepsis-induced acute kidney injury. Curr. Opin. Crit. Care. 22, 546–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Peerapornratana S., Manrique-Caballero C. L., Gómez H., and Kellum J. A. (2019) Acute kidney injury from sepsis: current concepts, epidemiology, pathophysiology, prevention and treatment. Kidney Int. 96, 1083–1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gomez H., Ince C., De Backer D., Pickkers P., Payen D., Hotchkiss J., and Kellum J. A. (2014) A unified theory of sepsis-induced acute kidney injury: inflammation, microcirculatory dysfunction, bioenergetics, and the tubular cell adaptation to injury. Shock 41, 3–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kellum J. A., Wen X., de Caestecker M. P., and Hukriede N. A. (2019) Sepsis-associated acute kidney injury: a problem deserving of new solutions. Nephron 143, 174–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yang Q-h., Liu D-W., Long Y., Liu H-Z., Chai W-Z., and Wang X.-T. (2009) Acute renal failure during sepsis: Potential role of cell cycle regulation. J. Infect. 58, 459–464 [DOI] [PubMed] [Google Scholar]
- 11. Carré J. E., and Singer M. (2008) Cellular energetic metabolism in sepsis: The need for a systems approach. Biochim. Biophys. Acta 1777, 763–771 [DOI] [PubMed] [Google Scholar]
- 12. Ruiz S., Vardon-Bounes F., Merlet-Dupuy V., Conil J.-M., Buléon M., Fourcade O., Tack I., and Minville V. (2016) Sepsis modeling in mice: ligation length is a major severity factor in cecal ligation and puncture. Intensive care Med. Exp. 4, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Toscano M. G., Ganea D., and Gamero A. M. (2011) Cecal ligation puncture procedure. J. Vis. Exp. 7, 2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dejager L., Pinheiro I., Dejonckheere E., and Libert C. (2011) Cecal ligation and puncture: the gold standard model for polymicrobial sepsis?. Trends Microbiol. 19, 198–208 [DOI] [PubMed] [Google Scholar]
- 15. Fu H., Zhou D., Zhu H., Liao J., Lin L., Hong X., Hou F. F., and Liu Y. (2019) Matrix metalloproteinase-7 protects against acute kidney injury by priming renal tubules for survival and regeneration. Kidney Int. 95, 1167–1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhou D., Fu H., Han Y., Zhang L., Liu S., Lin L., Stolz D. B., and Liu Y. (2019) Sonic hedgehog connects podocyte injury to mesangial activation and glomerulosclerosis. JCI Insight 4, e130515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhou D., Fu H., Liu S., Zhang L., Xiao L., Bastacky S. I., and Liu Y. (2019) Early activation of fibroblasts is required for kidney repair and regeneration after injury. FASEB J. 33, 12576–12587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lin Y.-H., Eguez R. V., Torralba M. G., Singh H., Golusinski P., Golusinski W., Masternak M., Nelson K. E., Freire M., and Yu Y. (2019) Self-assembled STrap for global proteomics and salivary biomarker discovery. J. Proteome Res. 18, 1907–1915 [DOI] [PubMed] [Google Scholar]
- 19. Lee Y.-E., Chan T.-C., Tian Y.-F., Liang P.-I., Shiue Y.-L., Chen Y.-S., and He H.-L. (2019) High expression of Chitinase 3-like-1 is an unfavorable prognostic factor in urothelial carcinoma of upper urinary tract and urinary bladder. Urol. Oncol. 37, 299.e2–299.e18 [DOI] [PubMed] [Google Scholar]
- 20. Humphrey S. J., Azimifar S. B., and Mann M. (2015) High-throughput phosphoproteomics reveals in vivo insulin signaling dynamics. Nat. Biotechnol. 33, 990–995 [DOI] [PubMed] [Google Scholar]
- 21. Yu Y., and Pieper R. (2019) Using proteomics to identify inflammation during urinary tract infection. Methods Mol. Biol. 2021, 259–272 [DOI] [PubMed] [Google Scholar]
- 22. Yu Y., and Pieper R. (2015) Urinary pellet sample preparation for shotgun proteomic analysis of microbial infection and host-pathogen interactions. Methods Mol. Biol. 1295, 65–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Doncheva N. T., Morris J. H., Gorodkin J., and Jensen L. J. (2019) Cytoscape StringApp: network analysis and visualization of proteomics data. J. Proteome Res. 18, 623–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Waikar S. S., and Bonventre J. V. (2009) Creatinine kinetics and the definition of acute kidney injury. J. Am. Soc. Nephrol. 20, 672–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bennett M., Dent C. L., Ma Q., Dastrala S., Grenier F., Workman R., Syed H., Ali S., Barasch J., and Devarajan P. (2008) Urine NGAL predicts severity of acute kidney injury after cardiac surgery: a prospective study. Clin. J. Am. Soc. Nephrol. 3, 665–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Takasu O., Gaut J. P., Watanabe E., To K., Fagley R. E., Sato B., Jarman S., Efimov I. R., Janks D. L., Srivastava A., Bhayani S. B., Drewry A., Swanson P. E., and Hotchkiss R. S. (2013) Mechanisms of cardiac and renal dysfunction in patients dying of sepsis. Am. J. Respir. Crit. Care Med. 187, 509–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Havasi A., and Borkan S. C. (2011) Apoptosis and acute kidney injury. Kidney Int. 80, 29–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sanz A. B., Santamaría B., Ruiz-Ortega M., Egido J., and Ortiz A. (2008) Mechanisms of renal apoptosis in health and disease. J. Am. Soc. Nephrol. 19, 1634–1642 [DOI] [PubMed] [Google Scholar]
- 29. Gaidt M. M., and Hornung V. (2016) Pore formation by GSDMD is the effector mechanism of pyroptosis. EMBO J. 35, 2167–2169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Miao N., Yin F., Xie H., Wang Y., Xu Y., Shen Y., Xu D., Yin J., Wang B., Zhou Z., Cheng Q., Chen P., Xue H., Zhou L., Liu J., Wang X., Zhang W., and Lu L. (2019) The cleavage of gasdermin D by caspase-11 promotes tubular epithelial cell pyroptosis and urinary IL-18 excretion in acute kidney injury. Kidney Int. 96, 1105–1120 [DOI] [PubMed] [Google Scholar]
- 31. Ghonime M. G., Shamaa O. R., Das S., Eldomany R. A., Fernandes-Alnemri T., Alnemri E. S., Gavrilin M. A., and Wewers M. D. (2014) Inflammasome priming by lipopolysaccharide is dependent upon ERK signaling and proteasome function. J. Immunol. 192, 3881–3888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Geiger T., Velic A., Macek B., Lundberg E., Kampf C., Nagaraj N., Uhlen M., Cox J., and Mann M. (2013) Initial quantitative proteomic map of 28 mouse tissues using the SILAC mouse. Mol. Cell. Proteomics 12, 1709–1722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Funk J. A., and Schnellmann R. G. (2012) Persistent disruption of mitochondrial homeostasis after acute kidney injury. Am. J. Physiol. Renal Physiol. 302, F853–F864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Langley R. J., Tsalik E. L., Velkinburgh J. C. V., Glickman S. W., Rice B. J., Wang C., Chen B., Carin L., Suarez A., Mohney R. P., Freeman D. H., Wang M., You J., Wulff J., Thompson J. W., Moseley M. A., Reisinger S., Edmonds B. T., Grinnell B., Nelson D. R., Dinwiddie D. L., Miller N. A., Saunders C. J., Soden S. S., Rogers A. J., Gazourian L., Fredenburgh L. E., Massaro A. F., Baron R. M., Choi A. M. K., Corey G. R., Ginsburg G. S., Cairns C. B., Otero R. M., Fowler V. G., Rivers E. P., Woods C. W., and Kingsmore S. F. (2013) An integrated clinico-metabolomic model improves prediction of death in sepsis. Sci. Transl. Med. 5, 195ra195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sacco F., Seelig A., Humphrey S. J., Krahmer N., Volta F., Reggio A., Marchetti P., Gerdes J., and Mann M. (2019) Phosphoproteomics reveals the GSK3-PDX1 axis as a key pathogenic signaling node in diabetic islets. Cell Metab. 29, 1422–1432.e3 [DOI] [PubMed] [Google Scholar]
- 36. Chauhan K., Kalam H., Dutt R., and Kumar D. (2019) RNA splicing: a new paradigm in host-pathogen interactions. J. Mol. Biol. 431, 1565–1575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sutherland L. C., Rintala-Maki N. D., White R. D., and Morin C. D. (2005) RNA binding motif (RBM) proteins: a novel family of apoptosis modulators?. J. Cell. Biochem. 94, 5–24 [DOI] [PubMed] [Google Scholar]
- 38. Soderholm S., Kainov D. E., Ohman T., Denisova O. V., Schepens B., Kulesskiy E., Imanishi S. Y., Corthals G., Hintsanen P., Aittokallio T., Saelens X., Matikainen S., and Nyman T. A. (2016) Phosphoproteomics to characterize host response during influenza A virus infection of human macrophages. Mol. Cell. Proteomics 15, 3203–3219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rinschen M. M., Wu X., Konig T., Pisitkun T., Hagmann H., Pahmeyer C., Lamkemeyer T., Kohli P., Schnell N., Schermer B., Dryer S., Brooks B. R., Beltrao P., Krueger M., Brinkkoetter P. T., and Benzing T. (2014) Phosphoproteomic analysis reveals regulatory mechanisms at the kidney filtration barrier. J. Am. Soc. Nephrol. 25, 1509–1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wongkittichote P., Ah Mew N., and Chapman K. A. (2017) Propionyl-CoA carboxylase – A review. Mol. Genet. Metab. 122, 145–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Guenzel A. J., Hofherr S. E., Hillestad M., Barry M., Weaver E., Venezia S., Kraus J. P., Matern D., and Barry M. A. (2013) Generation of a hypomorphic model of propionic acidemia amenable to gene therapy testing. Mol. Ther. 21, 1316–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Huttlin E. L., Jedrychowski M. P., Elias J. E., Goswami T., Rad R., Beausoleil S. A., Villén J., Haas W., Sowa M. E., and Gygi S. P. (2010) A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Villen J., Beausoleil S. A., Gerber S. A., and Gygi S. P. (2007) Large-scale phosphorylation analysis of mouse liver. Proc. Natl. Acad. Sci. U S A. 104, 1488–1493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lin L., Yee S. W., Kim R. B., and Giacomini K. M. (2015) SLC transporters as therapeutic targets: emerging opportunities. Nat. Rev. Drug Discov. 14, 543–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. César-Razquin A., Snijder B., Frappier-Brinton T., Isserlin R., Gyimesi G., Bai X., Reithmeier R. A., Hepworth D., Hediger M. A., Edwards A. M., and Superti-Furga G. (2015) A call for systematic research on solute carriers. Cell 162, 478–487 [DOI] [PubMed] [Google Scholar]
- 46. Vadakedath S., and Kandi V. (2018) Probable potential role of urate transporter genes in the development of metabolic disorders. Cureus e2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Enomoto A., Kimura H., Chairoungdua A., Shigeta Y., Jutabha P., Ho Cha S., Hosoyamada M., Takeda M., Sekine T., Igarashi T., Matsuo H., Kikuchi Y., Oda T., Ichida K., Hosoya T., Shimokata K., Niwa T., Kanai Y., and Endou H. (2002) Molecular identification of a renal urate–anion exchanger that regulates blood urate levels. Nature 417, 447–452 [DOI] [PubMed] [Google Scholar]
- 48. Zhang D., Yang H., Kong X., Wang K., Mao X., Yan X., Wang Y., Liu S., Zhang X., Li J., Chen L., Wu J., Wei M., Yang J., and Guan Y. (2011) Proteomics analysis reveals diabetic kidney as a ketogenic organ in type 2 diabetes. Am. J. Physiol. Endocrinol. Metab. 300, E287–E295 [DOI] [PubMed] [Google Scholar]
- 49. Shukla S. K., Liu W., Sikder K., Addya S., Sarkar A., Wei Y., and Rafiq K. (2017) HMGCS2 is a key ketogenic enzyme potentially involved in type 1 diabetes with high cardiovascular risk. Sci. Rep 7, 4590–4590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chelbi S. T., Wilson M. L., Veillard A.-C., Ingles S. A., Zhang J., Mondon F., Gascoin-Lachambre G., Doridot L., Mignot T.-M., Rebourcet R., Carbonne B., Concordet J.-P., Barbaux S., and Vaiman D. (2012) Genetic and epigenetic mechanisms collaborate to control SERPINA3 expression and its association with placental diseases. Hum. Mol. Genet. 21, 1968–1978 [DOI] [PubMed] [Google Scholar]
- 51. Vicuña L., Strochlic D. E., Latremoliere A., Bali K. K., Simonetti M., Husainie D., Prokosch S., Riva P., Griffin R. S., Njoo C., Gehrig S., Mall M. A., Arnold B., Devor M., Woolf C. J., Liberles S. D., Costigan M., and Kuner R. (2015) The serine protease inhibitor SerpinA3N attenuates neuropathic pain by inhibiting T cell–derived leukocyte elastase. Nat. Med. 21, 518–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sánchez-Navarro A., Mejía-Vilet J. M., Pérez-Villalva R., Carrillo-Pérez D. L., Marquina-Castillo B., Gamba G., and Bobadilla N. A. (2019) SerpinA3 in the early recognition of acute kidney injury to chronic kidney disease (CKD) transition in the rat and its potentiality in the recognition of patients with CKD. Sci. Rep 9, 10350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Anderberg R. J., Meek R. L., Hudkins K. L., Cooney S. K., Alpers C. E., Leboeuf R. C., and Tuttle K. R. (2015) Serum amyloid A and inflammation in diabetic kidney disease and podocytes. Lab. Invest. 95, 250–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nelson S. R., Tennent G. A., Sethi D., Gower P. E., Ballardie F. W., Amatayakul-Chantler S., and Pepys M. B. (1991) Serum amyloid P component in chronic renal failure and dialysis. Clinica Chimica Acta 200, 191–199 [DOI] [PubMed] [Google Scholar]
- 55. Wiggins J. E., Goyal M., Wharram B. L., and Wiggins R. C. (2006) Antioxidant ceruloplasmin is expressed by glomerular parietal epithelial cells and secreted into urine in association with glomerular aging and high-calorie diet. JASN. 17, 1382–1387 [DOI] [PubMed] [Google Scholar]
- 56. Saraf S. L., Lin X., Lee G., Adjei E. A., Kumari N., Gordeuk V. R., Jerebtsova M., and Nekhai S. A. (2016) Urinary ceruloplasmin concentration predicts development of kidney disease in sickle cell disease patients. Blood 128, 4865–4865 [Google Scholar]
- 57. Wang S., Song R., Wang Z., Jing Z., Wang S., and Ma J. (2018) S100A8/A9 in Inflammation. Front. Immunol. 9, 1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Dubois C., Payen D., Simon S., Junot C., Fenaille F., Morel N., and Becher F. (2020) Top-down and bottom-up proteomics of circulating S100A8/S100A9 in plasma of septic shock patients. J. Proteome Res. 19, 914–925 [DOI] [PubMed] [Google Scholar]
- 59. Elias J. A., Homer R. J., Hamid Q., and Lee C. G. (2005) Chitinases and chitinase-like proteins in TH2 inflammation and asthma. J. Allergy Clin. Immunol. 116, 497–500 [DOI] [PubMed] [Google Scholar]
- 60. Kzhyshkowska J., Gratchev A., and Goerdt S. (2007) Human chitinases and chitinase-like proteins as indicators for inflammation and cancer. Biomarker Insights. 2, 117727190700200. [PMC free article] [PubMed] [Google Scholar]
- 61. Sutherland T. E. (2018) Chitinase-like proteins as regulators of innate immunity and tissue repair: helpful lessons for asthma?. Biochem. Soc. Trans. 46, 141–151 [DOI] [PubMed] [Google Scholar]
- 62. Lee C. G., Da Silva C. A., Dela Cruz C. S., Ahangari F., Ma B., Kang M. J., He C. H., Takyar S., and Elias J. A. (2011) Role of chitin and chitinase/chitinase-like proteins in inflammation, tissue remodeling, and injury. Annu. Rev. Physiol. 73, 479–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Maddens B., Ghesquière B., Vanholder R., Demon D., Vanmassenhove J., Gevaert K., and Meyer E. (2012) Chitinase-like proteins are candidate biomarkers for sepsis-induced acute kidney injury. Mol. Cell. Proteomics 11, M111.013094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Albeltagy E. S., Abdul-Mohymen A. M., and Taha D. R. A. (2020) Early diagnosis of acute kidney injury by urinary YKL-40 in critically ill patients in ICU: a pilot study. Int. Urol. Nephrol. 52, 351–361 [DOI] [PubMed] [Google Scholar]
- 65. Róka B., Tod P., Kaucsár T., Vizovišek M., Vidmar R., Turk B., Fonović M., Szénási G., and Hamar P. (2019) The acute phase response is a prominent renal proteome change in sepsis in mice. IJMS. 21, 200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kato S., Chmielewski M., Honda H., Pecoits-Filho R., Matsuo S., Yuzawa Y., Tranaeus A., Stenvinkel P., and Lindholm B. (2008) Aspects of immune dysfunction in end-stage renal disease. Clin. J. Am. Soc. Nephrol. 3, 1526–1533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Oleinika K., Mauri C., and Salama A. D. (2019) Effector and regulatory B cells in immune-mediated kidney disease. Nat. Rev. Nephrol. 15, 11–26 [DOI] [PubMed] [Google Scholar]
- 68. Hsu H.-M., Chu C.-M., Chang Y.-J., Yu J.-C., Chen C.-T., Jian C.-E., Lee C.-Y., Chiang Y.-T., Chang C.-W., and Chang Y.-T. (2019) Six novel immunoglobulin genes as biomarkers for better prognosis in triple-negative breast cancer by gene co-expression network analysis. Sci. Rep. 9, 4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Herman-Edelstein M., Scherzer P., Tobar A., Levi M., and Gafter U. (2014) Altered renal lipid metabolism and renal lipid accumulation in human diabetic nephropathy. J. Lipid Res. 55, 561–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Bobulescu I. A. (2010) Renal lipid metabolism and lipotoxicity. Curr. Opin. Nephrol. Hypertens 19, 393–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bulbul M. C., Dagel T., Afsar B., Ulusu N. N., Kuwabara M., Covic A., and Kanbay M. (2018) Disorders of lipid metabolism in chronic kidney disease. Blood Purif. 46, 144–152 [DOI] [PubMed] [Google Scholar]
- 72. Vasko R. (2016) Peroxisomes and kidney injury. Antioxid. Redox Signal. 25, 217–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Stavropoulou E., Pircalabioru G. G., and Bezirtzoglou E. (2018) The role of cytochromes P450 in infection. Front. Immunol. 9, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Van Bogaert I. N. A., Groeneboer S., Saerens K., and Soetaert W. (2011) The role of cytochrome P450 monooxygenases in microbial fatty acid metabolism. FEBS J. 278, 206–221 [DOI] [PubMed] [Google Scholar]
- 75. Sheahan T. P., Sims A. C., Leist S. R., Schäfer A., Won J., Brown A. J., Montgomery S. A., Hogg A., Babusis D., Clarke M. O., Spahn J. E., Bauer L., Sellers S., Porter D., Feng J. Y., Cihlar T., Jordan R., Denison M. R., and Baric R. S. (2020) Comparative therapeutic efficacy of remdesivir and combination lopinavir, ritonavir, and interferon beta against MERS-CoV. Nat. Commun. 11, 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Jones R. D., Taylor A. M., Tong E. Y., and Repa J. J. (2013) Carboxylesterases are uniquely expressed among tissues and regulated by nuclear hormone receptors in the mouse. Drug Metab. Dispos. 41, 40–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Zougman A., Selby P. J., and Banks R. E. (2014) Suspension trapping (STrap) sample preparation method for bottom-up proteomics analysis. Proteomics 14, 1006–1010 [DOI] [PubMed] [Google Scholar]
- 78. Liu J., Kumar S., Dolzhenko E., Alvarado G. F., Guo J., Lu C., Chen Y., Li M., Dessing M. C., Parvez R. K., Cippa P. E., Krautzberger A. M., Saribekyan G., Smith A. D., and McMahon A. P. (2017) Molecular characterization of the transition from acute to chronic kidney injury following ischemia/reperfusion. JCI Insight 2, e94716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Guzzi F., Cirillo L., Roperto R. M., Romagnani P., and Lazzeri E. (2019) Molecular mechanisms of the acute kidney injury to chronic kidney disease transition: an updated view. Int. J. Mol. Sci. 20, 4941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Vaidya V. S., Ferguson M. A., and Bonventre J. V. (2008) Biomarkers of acute kidney injury. Annu. Rev. Pharmacol. Toxicol. 48, 463–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Hara H., Tsuchiya K., Kawamura I., Fang R., Hernandez-Cuellar E., Shen Y., Mizuguchi J., Schweighoffer E., Tybulewicz V., and Mitsuyama M. (2013) Phosphorylation of the adaptor ASC acts as a molecular switch that controls the formation of speck-like aggregates and inflammasome activity. Nat. Immunol. 14, 1247–1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Yang J., Liu Z., and Xiao T. S. (2017) Post-translational regulation of inflammasomes. Cell. Mol. Immunol. 14, 65–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Martin B. N., Wang C., Willette-Brown J., Herjan T., Gulen M. F., Zhou H., Bulek K., Franchi L., Sato T., Alnemri E. S., Narla G., Zhong X. P., Thomas J., Klinman D., Fitzgerald K. A., Karin M., Nuñez G., Dubyak G., Hu Y., and Li X. (2014) IKKα negatively regulates ASC-dependent inflammasome activation. Nat. Commun. 5, 4977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Tonnus W., and Linkermann A. (2019) Gasdermin D and pyroptosis in acute kidney injury. Kidney Int. 96, 1061–1063 [DOI] [PubMed] [Google Scholar]
- 85. Ruhl S., Shkarina K., Demarco B., Heilig R., Santos J. C., and Broz P. (2018) ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science 362, 956–960 [DOI] [PubMed] [Google Scholar]
- 86. Murase T., Yamamoto T., Koide A., Yagi Y., Kagawa S., Tsuruya S., Abe Y., Umehara T., and Ikematsu K. (2017) Temporal expression of chitinase-like 3 in wounded murine skin. Int. J. Legal Med. 131, 1623–1631 [DOI] [PubMed] [Google Scholar]
- 87. Rathcke C. N., and Vestergaard H. (2009) YKL-40–an emerging biomarker in cardiovascular disease and diabetes. Cardiovasc. Diabetol. 8, 61–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Schmidt I. M., Hall I. E., Kale S., Lee S., He C. H., Lee Y., Chupp G. L., Moeckel G. W., Lee C. G., Elias J. A., Parikh C. R., and Cantley L. G. (2013) Chitinase-like protein Brp-39/YKL-40 modulates the renal response to ischemic injury and predicts delayed allograft function. J. Am. Soc. Nephrol. 24, 309–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Montgomery T. A., Xu L., Mason S., Chinnadurai A., Lee C. G., Elias J. A., and Cantley L. G. (2017) Breast regression protein-39/chitinase 3-like 1 promotes renal fibrosis after kidney injury via activation of myofibroblasts. J. Am. Soc. Nephrol. 28, 3218–3226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Puthumana J., Hall I. E., Reese P. P., Schroppel B., Weng F. L., Thiessen-Philbrook H., Doshi M. D., Rao V., Lee C. G., Elias J. A., Cantley L. G., and Parikh C. R. (2017) YKL-40 associates with renal recovery in deceased donor kidney transplantation. JASN. 28, 661–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Sutherland T. E., Logan N., Rückerl D., Humbles A. A., Allan S. M., Papayannopoulos V., Stockinger B., Maizels R. M., and Allen J. E. (2014) Chitinase-like proteins promote IL-17-mediated neutrophilia in a tradeoff between nematode killing and host damage. Nat. Immunol. 15, 1116–1125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Kawada M., Seno H., Kanda K., Nakanishi Y., Akitake R., Komekado H., Kawada K., Sakai Y., Mizoguchi E., and Chiba T. (2012) Chitinase 3-like 1 promotes macrophage recruitment and angiogenesis in colorectal cancer. Oncogene 31, 3111–3123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. He C. H., Lee C. G., Dela Cruz C. S., Lee C.-M., Zhou Y., Ahangari F., Ma B., Herzog E. L., Rosenberg S. A., Li Y., Nour A. M., Parikh C. R., Schmidt I., Modis Y., Cantley L., and Elias J. A. (2013) Chitinase 3-like 1 Regulates Cellular and Tissue Responses via IL-13 Receptor α2. Cell Rep. 4, 830–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Liu J. X., Yang C., Zhang W. H., Su H. Y., Liu Z. J., Pan Q., and Liu H. F. (2019) Disturbance of mitochondrial dynamics and mitophagy in sepsis-induced acute kidney injury. Life Sci. 235, 116828. [DOI] [PubMed] [Google Scholar]
- 95. Bhargava P., and Schnellmann R. G. (2017) Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 13, 629–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Doi K., Leelahavanichkul A., Yuen P. S. T., and Star R. A. (2009) Animal models of sepsis and sepsis-induced kidney injury. J. Clin. Invest. 119, 2868–2878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Wang F., Jiang H., Shi K., Ren Y., Zhang P., and Cheng S. (2012) Gut bacterial translocation is associated with microinflammation in end-stage renal disease patients. Nephrology (Carlton) 17, 733–738 [DOI] [PubMed] [Google Scholar]
- 98. Shi K., Wang F., Jiang H., Liu H., Wei M., Wang Z., and Xie L. (2014) Gut bacterial translocation may aggravate microinflammation in hemodialysis patients. Dig. Dis. Sci. 59, 2109–2117 [DOI] [PubMed] [Google Scholar]
- 99. Rosen D. A., Hooton T. M., Stamm W. E., Humphrey P. A., and Hultgren S. J. (2007) Detection of intracellular bacterial communities in human urinary tract infection. PLoS Med. 4, e329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Terlizzi M. E., Gribaudo G., and Maffei M. E. (2017) UroPathogenic Escherichia coli (UPEC) infections: virulence factors, bladder responses, antibiotic, and non-antibiotic antimicrobial strategies. Front. Microbiol. 8, 1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Shenoy A. T., Brissac T., Gilley R. P., Kumar N., Wang Y., Gonzalez-Juarbe N., Hinkle W. S., Daugherty S. C., Shetty A. C., Ott S., Tallon L. J., Deshane J., Tettelin H., and Orihuela C. J. (2017) Streptococcus pneumoniae in the heart subvert the host response through biofilm-mediated resident macrophage killing. PLoS Pathog. 13, e1006582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Brown A. O., Mann B., Gao G., Hankins J. S., Humann J., Giardina J., Faverio P., Restrepo M. I., Halade G. V., Mortensen E. M., Lindsey M. L., Hanes M., Happel K. I., Nelson S., Bagby G. J., Lorent J. A., Cardinal P., Granados R., Esteban A., LeSaux C. J., Tuomanen E. I., and Orihuela C. J. (2014) Streptococcus pneumoniae translocates into the myocardium and forms unique microlesions that disrupt cardiac function. PLoS Pathog. 10, e1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Abraham S. N., and Miao Y. (2015) The nature of immune responses to urinary tract infections. Nat. Rev. Immunol. 15, 655–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All MS raw data associated with the study, along with detailed tables of MaxQuant output files reporting m/z, charge, identification, quantitation and phosphosite scores, and other details for all peptide identifications, have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the data set identifier PXD017045, and MassIVE with the data set identifier MSV000086126.