

Abstract
Patients with lymphangioleiomyomatosis (LAM) develop pulmonary cysts associated with neoplastic, smooth muscle–like cells that feature neuroendocrine cell markers. The disease preferentially affects premenopausal women. Existing therapeutics do not cure LAM. As gp100 is a diagnostic marker expressed by LAM lesions, we proposed to target this immunogenic glycoprotein using TCR transgenic T cells. To reproduce the genetic mutations underlying LAM, we cultured Tsc2−/− kidney tumor cells from aged Tsc2 heterozygous mice and generated a stable gp100-expressing cell line by lentiviral transduction. T cells were isolated from major histocompatibility complex–matched TCR transgenic pmel-1 mice to measure cytotoxicity in vitro, and 80% cytotoxicity was observed within 48 hours. Antigen-specific cytotoxicity was likewise observed using pmel-1 TCR-transduced mouse T cells, suggesting that transgenic T cells may likewise be useful to treat LAM in vivo. On intravenous injection, slow-growing gp100+ LAM-like cells formed lung nodules that were readily detectable in severe combined immunodeficient/beige mice. Adoptive transfer of gp100-reactive but not wild-type T cells into mice significantly shrunk established lung tumors, even in the absence of anti–PD-1 therapy. These results demonstrate the treatment potential of adoptively transferred T cells to eliminate pulmonary lesions in LAM.
Keywords: adoptive T cell transfer, T cell receptor, lymphangioleiomyomatosis, gp100
Lymphangioleiomyomatosis (LAM) represents a benign neoplasm. Patients present with cystic lung disease, renal angiomyolipomas, retroperitoneal lymphangiomas, and chylous fluid collections (1). LAM cells exhibit biallelic mutations or epigenetic modifications to the TSC2 (or TSC1) gene (2). Resulting tumors are metastatic, as pulmonary tumors are frequently accompanied by renal tumors that harbor the same mutation (3). The disease almost exclusively affects premenopausal women. It can develop de novo in otherwise healthy women (so-called “sporadic LAM”) or in women with tuberous sclerosis complex who carry a germline mutated allele of one of the TSC genes. Patients with TSC mutations can acquire a second mutation or display disease features in cells with only a single affected TSC allele (4). Thus, LAM is relatively common among patients with TSC mutations (“TSC LAM”). LAM cells express elevated mTORC1 activity. Rapamycin slows the decline of lung function in women with LAM and stabilizes the disease but is not curative (5). Further treatments might specifically target affected LAM cells and mediate cytotoxic responses (6, 7). The expression of immune checkpoint programmed death-ligand 1 (PD-L1) in LAM (8) combined with preclinical data for checkpoint inhibition monotherapy suggests that anti–CTLA-4 and especially anti–PD-1 might be further supported by antigen-specific immunotherapy for LAM (9, 10). Interestingly, checkpoint inhibition may be effective even in parallel with rapamycin therapy (11).
LAM is a monogenic disorder (12), and personalized neoantigen vaccines (13) may not benefit patients with LAM. However, LAM tumors exhibit a relatively predictable genotype and phenotype, providing an opportunity to develop therapeutics that can benefit a wide range of patients. We have focused on targeting immunogenic proteins that are a hallmark of LAM development (14, 15). These are normally commonly found in melanosomes of neural crest–derived melanocytes and melanoma cells. Our group found consistent expression of melanosomal proteins, including glycoprotein 100 (gp100) (or pmel17), in LAM (15). Immunostaining with the gp100-reactive antibody human melanoma black 45 (HMB45) is in fact diagnostic for LAM (16). Their expression in LAM suggests a possible neuronal origin of LAM cells. However, other aspects of LAM cells might not support this, and their derivation from pluripotent stem cells is more likely. A breakthrough in understanding their neural stem cell–like characteristics has come from identifying Notch homolog 1 (Notch1) activation of Ras homolog enriched in brain (Rheb) (17).
Although CD68+ macrophages are abundant in LAM lesions (15), expression of immunogenic proteins is not otherwise accompanied by immune infiltration. This is surprising, as gp100 is an immunogenic target protein for T cells in melanoma, and LAM cells are susceptible to T cell–mediated cytotoxicity (15). We have similarly identified gp100 as a target molecule for T cells in vitiligo, and have isolated, cloned, and characterized a TCR with remarkable reactivity to human melanoma when expressed in human T cells and introduced via adoptive transfer in preclinical disease models (18). We expect this skin infiltrating lymphocyte v44 (SILv44) TCR to hold therapeutic value for LAM as well.
Here we focus on adoptive transfer of gp100 antigen-specific T cells as a possible treatment for LAM (19). The data underscore the importance of antigen specificity when developing immunotherapeutic options for LAM. Antigen-specific immunotherapy might find broad application for the treatment of benign tumors. These present with a relatively consistent genotype and a more predictable phenotype than hypermutated, aggressive tumors with powerful immune escape mechanisms, as in, for example, melanoma (20). The expression of gp100 by normal melanocytes of the skin, ears, and eyes would suggest that depigmentation could be associated with aggressive treatment.
We generated a Tsc2 knockout, gp100+ cell line to test the cytotoxicity of TCR-transduced gp100 reactive T cells in vitro and challenged severe combined immunodeficient (SCID)/beige mice with these cells, followed by adoptive transfer of gp100 reactive cells from pmel-1 mice. This strain is transgenic for a TCR reactive with gp100, resulting in robust T-cell responses against the glycoprotein (21). We also included a luciferase marker in the LAM-like cell line and treated mice with a combination of gp100-reactive T cells and anti–PD-1 in vivo. The experiments shed light on the treatment potential of antigen-specific T cells applied toward the treatment of benign tumors in LAM.
Methods
Generating LAM-like Cells
To reproduce properties of human LAM cells, we generated cells from tumors of Tsc2+/− mice and introduced gp100 expression. Cells were derived from a 21-month-old male Tsc2+/− mouse and transduced with a lentiviral vector encoding gp100. Resulting cells were maintained in selective medium and passaged repeatedly. Cells were injected and reisolated from immunodeficient mice to enhance tumorigenicity, and some were retrovirally transduced to express luciferase and sorted for injection and in vivo imaging system (IVIS) monitoring. A detailed description is provided in the data supplement.
Characterizing LAM-like Cells
To demonstrate that LAM-like cells exhibit mTOR hyperaction and expression of the gp100 antigen, cells were characterized in vitro. This involved amino acid deprivation, homogenization, and Western blotting to detect a lack of TSC expression, constitutive mTOR activation, and gp100 expression, as described in the data supplement.
Generating gp100-Reactive Mouse T Cells
To examine whether LAM-like cells can be targeted by gp100 reactive T cells, we used the TCR sequence expressed by CD8 T cells in pmel-1 mice. The TCR sequence was cloned into a retroviral vector to transduce wild-type (WT) mouse T cells that were sorted and Dynabead activated before repeated transduction procedures. Resulting cells were amplified in the presence of Dynabeads, and successful transduction was confirmed and quantified by FACS analysis, as described in the data supplement.
Co-culture Experiments
In vitro experiments served to establish that pmel-1 TCR-expressing T cells respond to LAM-like cells. Transduced or untransduced T cells were combined with Tsc2− kidney tumor cells with or without gp100 expression at different effector:target ratios. To establish successful transduction, responses to target cells pulsed with peptide were measured by enzyme-linked immunosorbent assay (ELISA). Cytotoxicity was estimated by counting remaining target cells, as described in the data supplement.
Adoptive T-Cell Transfer and Tumor Monitoring
Immunodeficient mice served to evaluate the anti-LAM activity of gp100-reactive T cells in vivo. Mice <8 weeks old were challenged with 106 LAM-like cells introduced subcutaneously or by intravenous injection. As relevant, luciferase was measured over time by IVIS or measured with calipers. On tumor growth, gp100-reactive or WT T cells were injected twice, 1 week apart, and maintained under low-dose IL-2. In one experiment, hamster anti-mouse PD-1 antibody was repeatedly administered to some animals. For IVIS monitoring, mice were injected with D-luciferin just before imaging, as described in the data supplement. Relevant tissues were harvested when mice were killed.
Immunostaining
To locate tumors and infiltrating immune cells, immunohistology was performed. Relevant tissues were snap frozen. For hematoxylin and eosin staining, sections were prefixed in neutral buffered formalin; otherwise, acetone-fixed cryosections were subjected to direct or indirect immunostaining using antibodies described in the data supplement. Microscopic images were subjected to Adobe Photoshop evaluation to quantify stained cells.
Statistics
Significant differences were determined by applying statistical software, namely Prism software (GraphPad), as described in the data supplement. For all tests, a value less than 0.05 was considered significant.
Study Approval
Before executing in vivo studies, all animal studies were approved by the Institutional Animal Care and Use Committee of Northwestern University and followed the institutional guidelines.
Results
LAM-like Target Cell Validation
Tsc2−/− mouse tumor cells were generated from kidney tumors of an aging mouse. Cell homogenates were subjected to Western blotting to evaluate Tsc2 expression. SV40 large T-expressing human embryonic kidney 293 (HEK293T) cells served as a positive control. The antibody used recognizes both mouse and human TSC2 proteins. In Figure 1A, expression of TSC2 is detected in HEK293 cells, but not in kidney tumor cells. No band was observed at 250 kD or a smaller molecular size, indicating that the tumor cells carry mutations in both alleles of the Tsc2 gene. These data support loss of expression of Tsc2 by kidney tumor cells. In Figure 1B, expression of gp100 is confirmed in LAM-like tumor cells, demonstrating that the transfection procedure was successful. The lack of expression of gp100 by mock-transduced cells indicates that the loss of Tsc2 alone is insufficient to prompt gp100 expression. Next, it is shown that loss of Tsc2 and gain of gp100 expression are maintained in vivo. In Figure 1C, intense expression of gp100 is observed in mouse Tsc2− cells transfected to express the protein, but not in mock-transfected cells. Expression appears more intense toward the border of the tumor. In Figure 1D, a hematoxylin and eosin stain in conjunction with a bright-field image helps to localize tumor cells. These display intense gp100 expression and coexpress phosphorylated S6 protein (pS6) as an mTOR activation marker, indicating that gp100 expression co-localizes with, and is restricted to, Tsc2− tumor cells. Finally, in Figure 1E, LAM-like gp100+, Tsc2− cells exhibit greater phosphorylation of S6 kinase (S6K) and S6 protein, as confirmed by quantification of Western blot band intensities in Figures 1F and 1G. The combined data support an LAM-like physiology for gp100-expressing, Tsc2-knockout “target” cells, henceforth named “LAM-like cells.”
Figure 1.
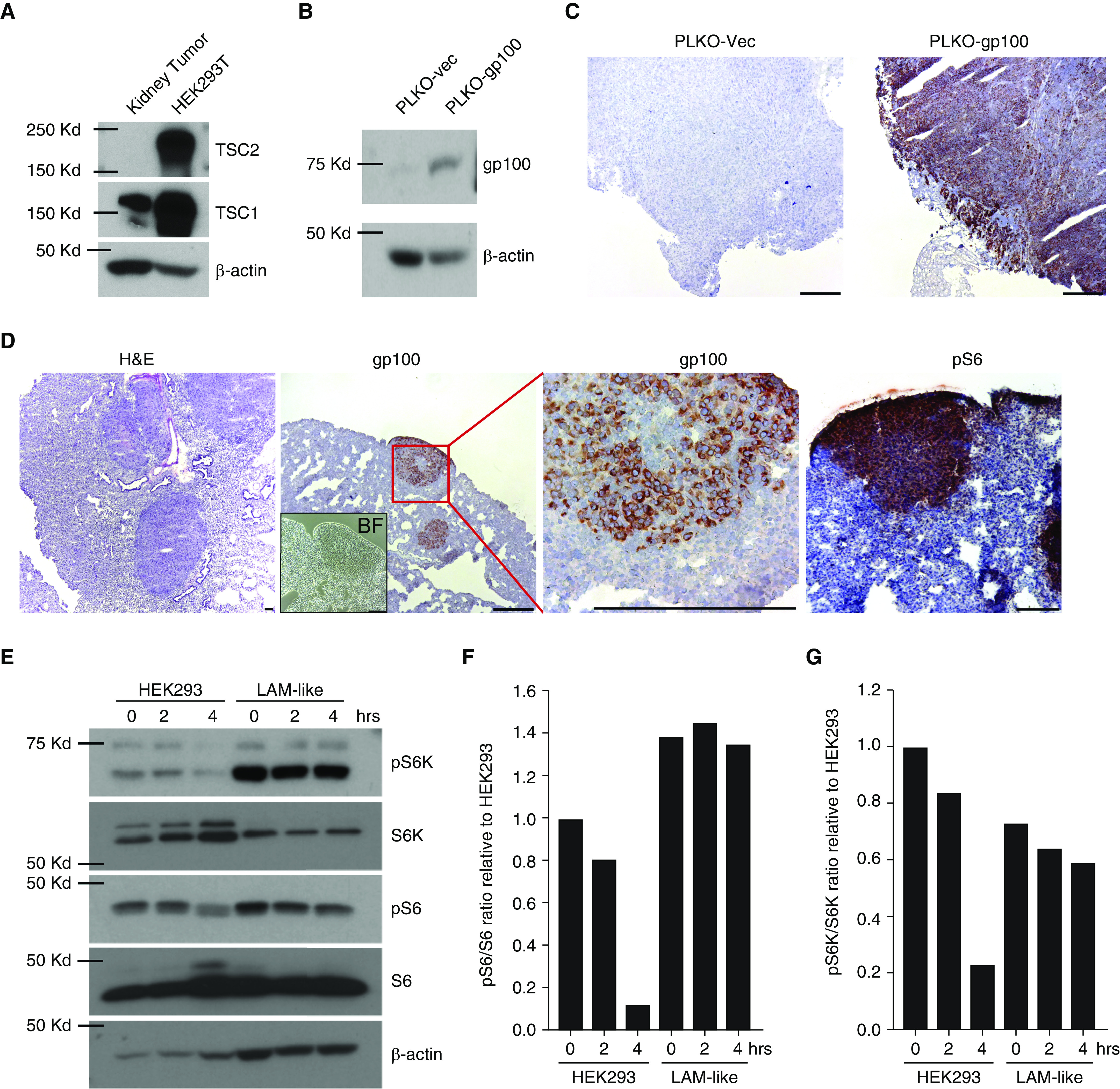
Generation and validation of lymphangioleiomyomatosis (LAM)-like cells. This figure displays the LAM-like features of target cells used for experiments throughout the article. (A) TSC1 and TSC2 expression is shown as found in kidney tumor cells isolated from 21-month-old Tsc2+/− mice. Human embryonic kidney (HEK) 293T cells served as a positive control. (B) Lack of gp100 expression by (PLKO-vec) control-transfected, and apparent gp100 expression by mouse gp100-transfected, Tsc2− kidney tumor cells. (C) Absence of gp100 expression in subcutaneous tumors from SCID-bg mice injected with Tsc2− kidney tumor cells, versus expression by gp100-transfected cells in vivo. (D) LAM-like cells from gp100+Tsc2− subcutaneous tumors and reinjected into SCID-bg mice via tail vein formed pulmonary tumors in 1 month; sections were stained with hematoxylin and eosin (H&E, left panel) and imaged in bright field (inset), and stained for gp100 (middle panels) or pS6 expression (right panel). (E) To identify constitutive mTOR activation measured as phosphorylation under nutrient deprivation in LAM-like cells, HEK293 and LAM-like cells were treated with normal- and amino acid–deprived FBS for 2 and 4 hours. Lysates were analyzed by Western blot using antibodies against indicated proteins. pS6 and pS6K bands from E were quantified and normalized to S6 and S6K, respectively, using ImageJ software. Relative ratios of quantified band intensity of (F) pS6 to S6 and (G) pS6K to S6K are shown in bar graphs. Scale bar: 100 μm. BF = bright-field; bg = beige; gp100 = glycoprotein 100; pS6 = phospho-S6; pS6K = phospho-S6 kinase; SCID = severe combined immunodeficiency; vec = vector.
TCR-transduced T Cells Are Specifically Reactive toward LAM-like Target Cells
Transgenic pmel-1 TCR expression can be detected by TCR-Vβ13–reactive antibodies (Figure 2A). We observed no T-cell staining without adding the primary antibody and minimal natural expression of this subunit among mock-transduced T cells. Subtracting natural expression, we observed ∼4% pBABE-pmel-1 TCR-transduced T cells, compared with 21% naturally occurring gp100-reactive T cells detected among cells from the pmel-1 transgenic mouse. In Figure 2B, T cell–target clustering, a sign of T-cell activation, is observed only in the combination of pmel-1–transduced T cells and gp100+ targets. More than 65% of target cells are eliminated by gp100-reactive T cells in vitro, which is evidence of cytotoxicity even at the low 1:1 effector:target ratio. In Figure 2C, killing efficiency by transduced T cells is shown in vitro. Here, only the pmel-1 TCR–transduced T cells were cytotoxic toward LAM-like target cells and also exhibited some cytotoxicity toward target cells pulsed with gp100 peptide. T-cell transductions were achieved at higher efficiency when using an alternative construct with pmel-1 in pMMLV (Figure E2A in the data supplement), and transduced T cells demonstrated targeted killing in co-culture assays (Figures E2B–E2D), providing an opportunity for more translational applications in the future.
Figure 2.
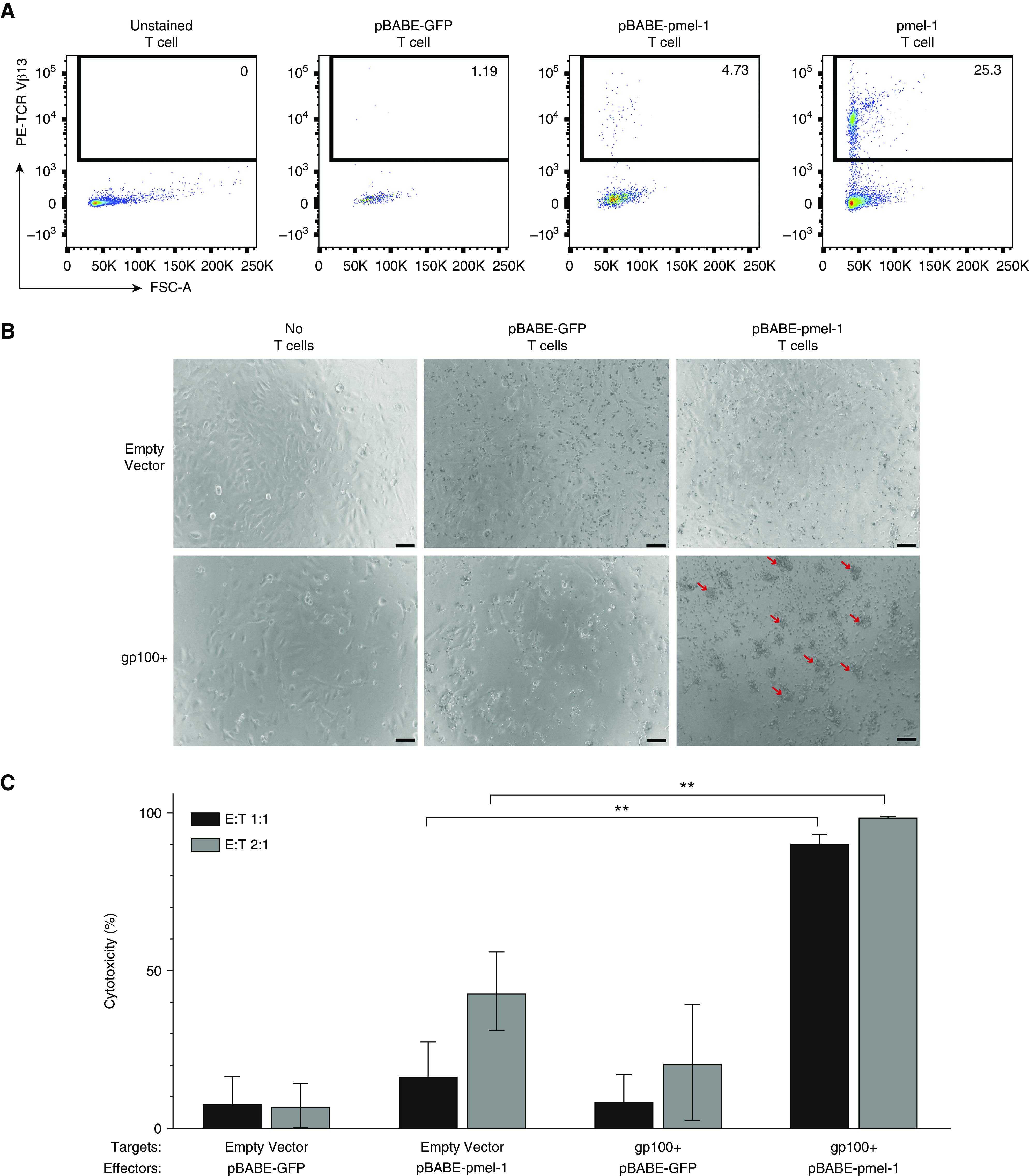
pmel-1–transduced T cells showed reactivity toward LAM-like cells in vitro. The expression and activity of a gp100-reactive TCR was analyzed in vitro. (A) Flow analysis of TCRVβ13 expression for the pmel-1 TCR in unstained (−control), pBABE-GFP vector-transduced, and pBABE-pmel-1–transduced mouse T cells, and T cells from pmel-1 transgenic mice (+control), showing TCR expression only in the two latter groups. (B) Vector control Tsc2− kidney tumor cells and gp100+ Tsc2− LAM-like cells pulsed with additional gp100 peptide were co-cultured with pBABE-GFP vector and pBABE-pmel-1–transduced T cells at 1:1 ratio. Bright-field images taken 48 hours after co-culture. Red arrows: clusters are visible only in combinations of gp100-reactive T cells and gp100+ target cells, indicative of ongoing cytotoxicity. (C) Cytotoxicity quantified: target cells remaining after 48 hours were quantified relative to target cell monocultures. With some background cytotoxicity observed, pmel-1–transduced T cells showed significant cytotoxicity toward gp100+ targets at 1:1 and 2:1 effector:target (E:T) ratios (represented as mean ± SD). N = 3. **P ≤ 0.01 (two-way ANOVA with Tukey’s multiple comparisons test). Scale bars: 50 μm. FSC-A = forward scatter area.
Containing LAM-like Tumor Growth In Vivo
To assess the treatment potential of transgenic T cells for LAM, we adoptively transferred pmel-1 T cells into SCID-beige mice previously challenged with LAM-like tumor cells. Mice (four per group) were challenged with 300,000 tumor cells by tail vein injection. One and 2 weeks later, two groups of mice received 1,000,000 T cells from either pmel-1 or C57BL/6 mice, while in another group the tumors grew unopposed, as outlined in Figure 3A. Another 3 weeks later, mice were killed and lung nodules counted. As shown in Figure 3B, the number of lung nodules (and the lung area covered by LAM-like nodules) was significantly lower in animals adoptively transferred with pmel-1, gp100-reactive T cells than with WT, polyclonal T cells, supporting the treatment potential of adoptive T cell transfer by gp100-reactive T cells. In Figure 3C, representative resected lung samples from each group are shown. Thus, robust antitumor responses by gp100-reactive T cells significantly reduced the number and size of LAM-like tumors.
Figure 3.
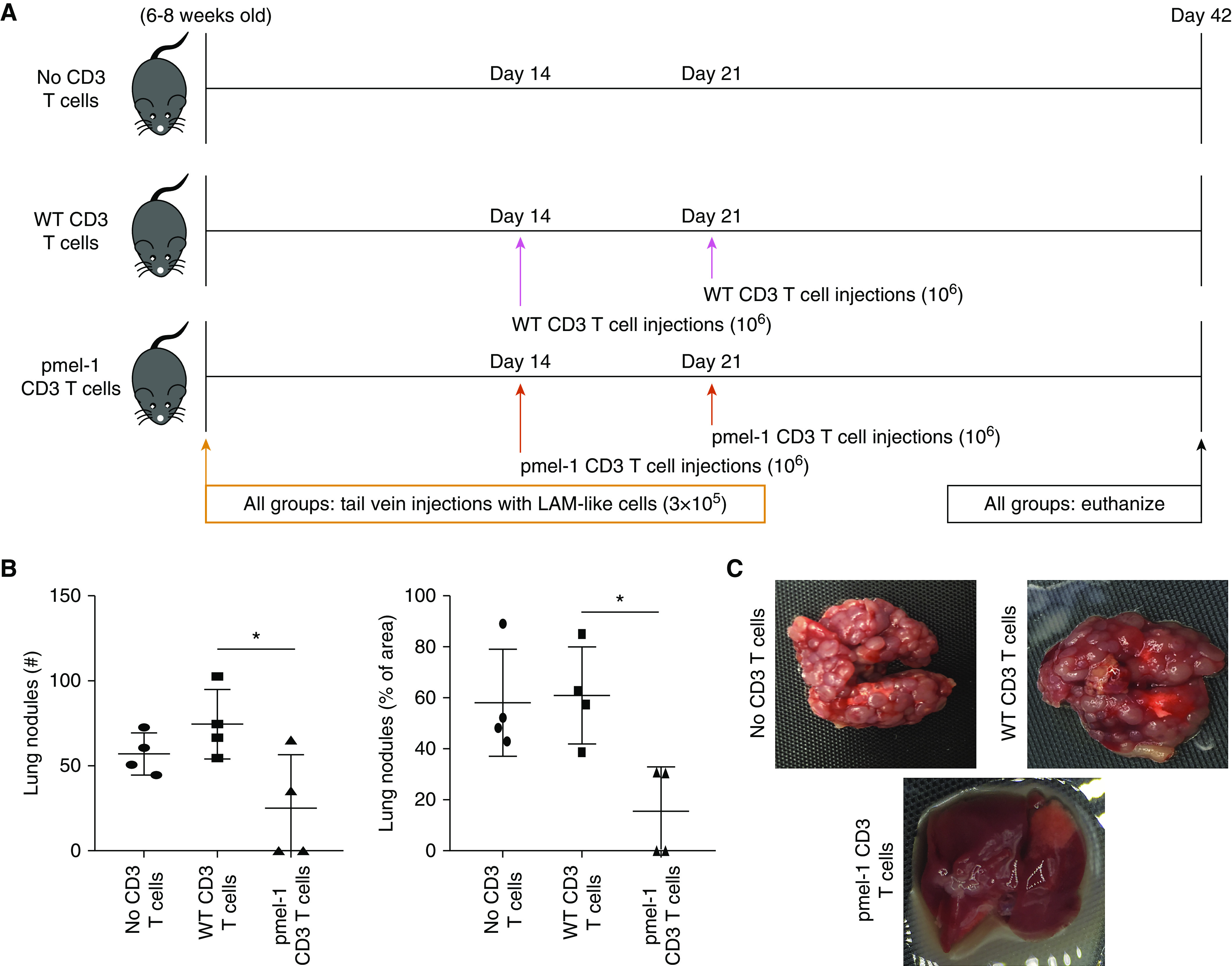
gp100-reactive T cells reduce LAM-like lung tumors in mice. The number and size of tumor nodules developing in the presence or absence of gp100-reactive T cells were measured in vivo. (A) The experimental design for a LAM-like tumor challenge and adoptive transfer of transgenic CD3 T cells is shown. Mice were killed after 42 days. (B) Surface lung nodules were counted and additionally quantified as percentage of lung area (represented as mean ± SD), and (C) representative lung images are shown for each treatment group. N = 4. *P < 0.05 (Kruskall-Wallis test with Dunn’s multiple comparisons test). WT = wild type.
Supporting Anti-Tumor Responses with Anti–PD-1 Treatment
Some animals challenged with LAM-like tumor cells and treated with gp100-reactive T cells still contained tumors at endpoint. As antitumor reactivity might be limited by T-cell exhaustion, expression of checkpoint proteins PD-1 and PD-L1 was assessed in the tumor tissue. A trend toward greater T-cell abundance was found in lung tumors from pmel-1 compared with WT T cell–treated animals (Figures E1A–E1C), suggesting that pmel-1 T cells have become activated through contact with LAM-like cells. Indeed, among T cells, PD-1 expression was greater on lung-infiltrating pmel-1 T cells than on WT T cells, as shown in Figures 4A and 4B and quantified in Figure 4C. Meanwhile, no overall differences in PD-L1 expression were found among treatment groups. But when tumor-containing areas for all tissues were combined for Figure 5A, lung sections analyzed for gp100 and PD-L1 expression showed that the abundance of PD-L1 within tumors is in fact inversely related to tumor presence (Figure 5B). As most PD-L1 expression observed was found in surrounding normal tissue, tumor clearance by infiltrating pmel-1 T cells is probably not limited by PD-L1–expressing tumor cells. To better understand if there might still be a treatment benefit to including anti–PD-1 therapy with adoptive transfer of gp100-reactive T cells, mice received the combination treatment in vivo, as outlined in Figure 6A. In addition to T cells, three mice within each treatment group received three anti–PD-1 injections per week. Four weeks after adoptive transfer, tumors became undetectable in pmel-1 T cell–treated mice, regardless of anti–PD-1 treatment. However, some therapeutic effects of anti–PD-1 treatment alone (WT T cells) are observed in IVIS imaging of the control (WT T cells) group, comparing the bars at 4 weeks after treatment (Figure 6B) and the associated quantification (Figure 6C). The overarching effect of transgenic T-cell treatment compared with WT T-cell treatment is illustrated in images of representative lungs after termination (Figure 6D).
Figure 4.

PD-1 expression is more frequently found on gp100-reactive T cells. To evaluate differential T-cell engagement as it relates to expression of a gp100-reactive TCR, PD-1 expression by (CD3+) T cells was measured in lung tissues from mice that received T cells from (A) WT or (B) pmel-1 mice, revealing (C) greater PD-1 expression among the latter and suggesting that expression represents T-cell activation rather than exhaustion (represented as mean ± SD). Arrows show examples of double-stained cells. Scale bars: 50 μm. *P < 0.05 (two-tailed, unpaired t test with Welch’s correction). N = 3.
Figure 5.

Pulmonary PD-L1 expression is inversely related to tumor size. The contribution of tumor or tissue cells to PD-1 engagement on T cells was evaluated by lung immunohistology. PD-L1 and gp100 co-expression was quantified in tissues with differing tumor burden after LAM-like tumor challenge and adoptive transfer of CD3+ T cells. (A) Example staining showing abundant expression of PD-L1 in sites without a tumor, whereas PD-L1 expression is absent from tumor cells. (B) When quantified relative to tumor burden, PD-L1 expression was inversely related (represented as mean ± SD). Thus, PD-1 expression by T cells may not represent PD-L1–induced T-cell exhaustion. Quantifications were calculated in a given field of view where N = 12, 10, 2, and 11 for large, intermediate, small, and no tumor areas, respectively. Scale bars: 100 μm. ***P ≤ 0.001 and ****P ≤ 0.0001 (Kruskell-Wallis test with Dunn’s multiple comparisons test).
Figure 6.
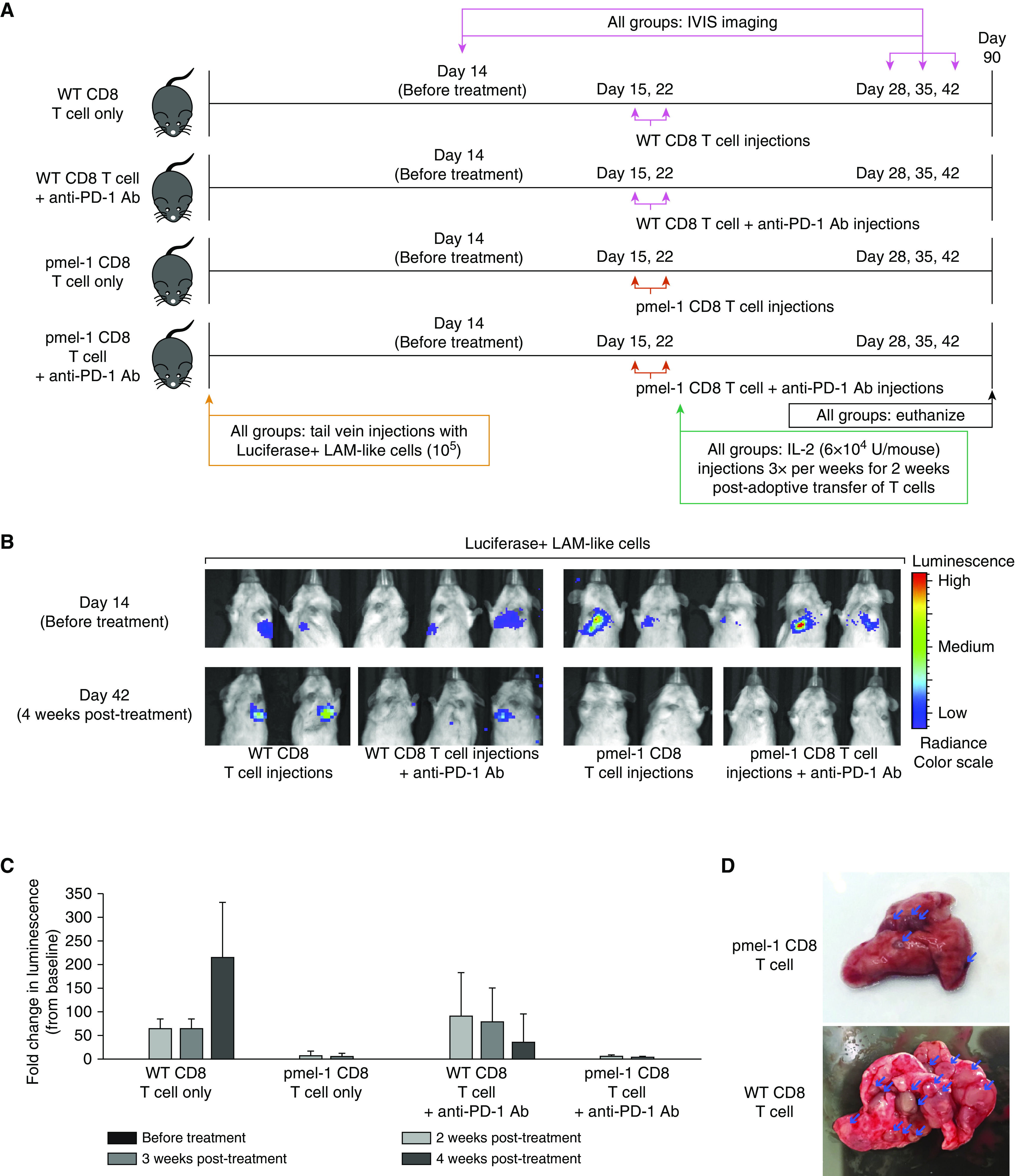
Transgenic T cells provide potent tumor control independent of immune checkpoint inhibition. A supportive role for checkpoint inhibition by anti–PD-1 to adoptively transferred, LAM-reactive T cells was tested in vivo. (A) The experimental outline is shown, comparing adoptive transfer of WT or pmel-1 transgenic CD8 T cells with or without anti–PD-1 antibody in SCID-bg mice. (B) IVIS imaging of luciferase+ tumors before and after treatment shows complete responses to T-cell transfer independent of checkpoint inhibition. (C) Relative luminescence intensities measured as ROI readings (represented as mean ± SD) suggest some treatment benefit to anti–PD-1 in the absence of gp100-reactive T cells. (D) Representative lungs are shown after treatment with pmel-1 or WT CD8 T cells. Group sizes were N = 2 for mice that received WT CD8 T cells or pmel-1 CD8 T cells and N = 3 for mice that received WT CD8 T cells with anti–PD-1 antibody or pmel-1 CD8 T cells with anti–PD-1 antibody. Arrows show examples of double-stained cells. IVIS = in vivo imaging system; ROI = region of interest.
An Immunocompetent Model to Test Immunotherapeutic Strategies for LAM
As LAM-like cells originated from Tsc2+/− mice, the cells are expected to grow in genetically identical littermates unless transgene expression precludes outgrowth of the cells in this setting. Mice were challenged with gp100+ LAM-like cells by subcutaneous and, in smaller numbers, intravenous injection. When subcutaneous necrotic tumors appeared after 3 months, mice had not developed pulmonary tumors. Subcutaneous tumors (Figure 7A) were screened for maintenance of LAM-like features. In Figures 7B–7D, expression of gp100 (Figure 7B) and phospho-S6 (Figure 7C) and an absence of Tsc2 expression (Figure 7D) were observed throughout the tumor, whereas in Figure 7E, a significant portion of CD3+ infiltrating T cells express PD-1. Infiltration by immunosuppressive elements, including F4/80+ macrophages (Figure 7F) and FoxP3+ regulatory T cells (Figure 7G), was also observed. Thus in an immuno-replete environment, as found in WT or Tsc2 heterozygote mice, as well as in actual patients with LAM, adoptively transferred T cells may fail to compete for nutrients and may require supportive treatment by checkpoint inhibitors to be efficacious. Under these conditions there may thus well be merit to combining adoptive T-cell transfer with anti–PD-1 therapy.
Figure 7.
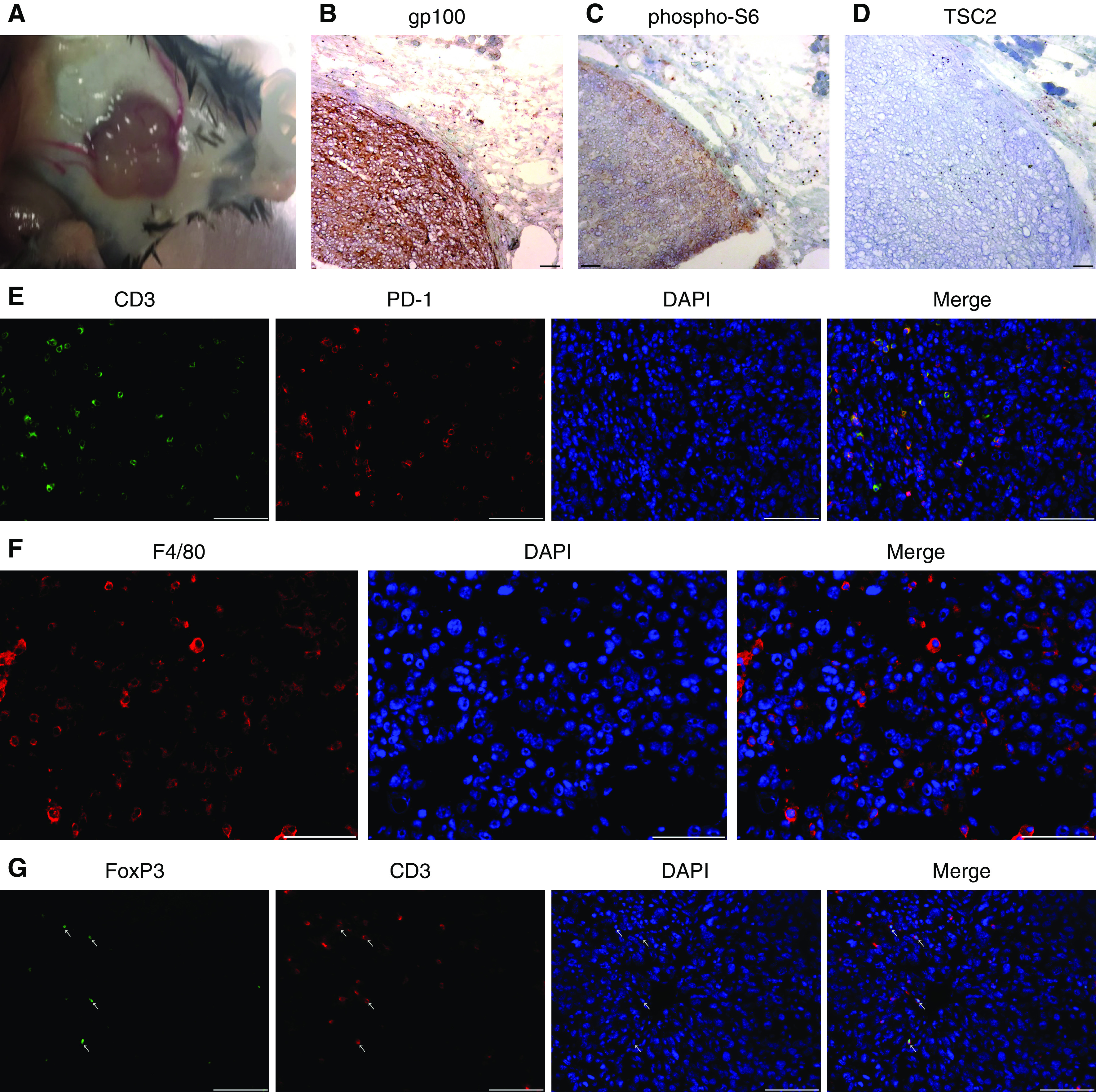
LAM-like tumor growth in immunocompetent mice. We tested the characteristics of LAM-like cells hosted by immunocompetent mice by subcutaneous and i.v. challenge of C57BL/6 mice with LAM-like cells. (A) Subcutaneous tumor growth was observed 2 months after subcutaneous challenge. Representative staining is shown for (B) gp100, (C) pS6, and (D) Tsc2, revealing the LAM-like identity of tumor cells. Immune infiltration was observed by detecting (E) CD3 and PD-1 co-localization, (F) the presence of F4/80+ macrophages, and (G) FoxP3 and CD3 co-localization indicative of regulatory T cells in an LAM-like subcutaneous tumor. Scale bars: 100 μm.
Discussion
Recognition by antibody HMB45 (14) used in LAM diagnosis illustrates disease properties shared with malignant melanoma, suggesting that LAM might likewise be amenable to antigen-specific immunotherapy. The HMB45 antibody responds to gp100, encoded by the Pmel-17 gene. This early melanosomal glycoprotein contributes to melanosomal striation, providing the fibrillar structures onto which melanin is deposited in pigmented cells (22). However, premelanosomal structures observed in LAM cells do not fully differentiate or exhibit pigmentation, and expression of tyrosinase, the rate-limiting enzyme in pigment formation, is lacking (23).
In the early 1990s, the gp100 antigen was defined as a target for melanoma-infiltrating T cells (24). This solidified the concept that expression of gp100 would leave LAM cells vulnerable to gp100-reactive, cytotoxic T cells. Indeed, when we generated primary cell cultures from LAM lung, resulting target cells were sensitive to human leukocyte antigen (HLA)-matched, gp100-reactive cytotoxic T cells (15). This interaction induced antigen- and major histocompatibility complex (MHC)-restricted cytokine release by gp100-reactive and HLA-A2–restricted T cells. Many TCRs identified are HLA-A2 restricted. Although MHC haplotyping has not been performed to reveal a possible association between LAM and HLA, we have several HLA-A2+ tissues in our inventory from patients who might benefit from treatment by HLA-A2–restricted and gp100-reactive T cells. In the absence of any known HLA association in LAM, we expect a prevalence of ∼50% for HLA-A*0201 among white patients, the same as in the healthy population (25).
Other patients might benefit from other means of enhancing immune responses to melanosomal antigens, for example by vaccines that newly elicit T-cell responses to MART-1 (melanoma antigen recognized by T cells) or to gp100 (26), or otherwise from tyrosinase-related protein-1 (TRP-1) vaccination to elicit humoral responses (27). Existing antibody responses to the TRP-1 antigen have not been measured in patients with LAM, and T-cell responses are generally considered more powerful. However, the therapeutic value of anti–TRP-1 antibodies has been documented in melanoma, and these antibodies may be beneficial to patients with LAM as well (28).
LAM pulmonary tissues are not infiltrated by T cells, despite marked overexpression of gp100, suggesting that patients do not mount a spontaneous response to the antigen. Such lack of infiltration might be assigned to immunosuppressive elements attracted to tumor sites by, for example, overexpression of MCP-1 (monocyte chemotactic protein-1) (29). Activation of mTORC supports M2 polarization (30), and lesional cells overexpressing the chemokine MCP-1 might subsequently attract M2 macrophages, which impair T-cell reactivity and support humoral responses instead. Moreover, expression of PD-1 ligands has been reported for LAM (31). Elevated secretion of soluble natural killer group 2D (NKG2D) ligands may contribute to the observed failure to activate T cells and mount effective T-cell responses in LAM (32, 33). These microenvironmental factors suggest that checkpoint inhibition alone may not suffice to mount a response to LAM lesional cells, but such treatment can well be supportive of adoptive T-cell therapy.
Thus, it will be important to support antigen-specific responses, by making use of existing tools that target melanoma. We identified and cloned a gp100-reactive TCR from human vitiligo skin, a disease where autoimmune T cells exhibit reactivity to self-antigens, including gp100 (19). Indeed, when compared with TCRs from patients with melanoma, the SILv44 gp100-reactive TCR exhibited superior antimelanoma reactivity in mice (18). Adoptive transfer of transgenic T cells expressing the SILv44 TCR might thus be therapeutic and truly eliminate the disease in affected MHC-matched, HLA-A2+ patients. Given the known expression of PD-1 ligands in LAM, such treatment might best be combined with anti–PD-1, although a patient with melanoma treated under a phase I clinical trial using tyrosinase-reactive T cells did not benefit from supportive anti–PD-1 treatment (34). The use of anti–PD-1 is currently approved primarily for immunogenic tumor types with a high mutation burden and resulting neoantigen expression, which are more likely to benefit from treatment (35). By contrast, LAM is driven by mutations in a single gene (12). Yet, opportunities remain to follow transgenic T-cell treatment by anti–PD-1 administration. It is possible that a supportive effect of anti–PD-1 therapy is better demonstrated when experiments are performed with large sample sizes. Another limitation to our study is the restriction of the Pmel-1 receptor that is limited to mice. However, the known in vivo antitumor efficacy of (human) SILv44-transduced T cells can partially compensate for this limitation (36).
The patient described above, who did not benefit from anti–PD-1 treatment, originally harbored stage 4 melanoma tumors and has now benefitted from adoptive T-cell therapy for several years (34). The persistence of transgenic T cells in treated patients can provide long-term immunosurveillance. It will be important to consider a potential for tissue damage in patients with LAM with extensive disease. A suicide gene can then be included for safety (37). Moreover, Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR-associated protein 9 (CRISPR/CAS9) technology can be used to avoid off-target cytotoxicity by TCR subunit mispairing in transduced cells, which might elicit unpredicted specificities (36).
We recently reported the in vivo effects of newly transduced, gp100-reactive T cells toward gp100-expressing human melanoma tumors (18). Taken together, there are compelling arguments supporting an initial application of gp100-reactive T cells in a setting of early disease, where the size of the lesions will not yet be limiting to treatment efficacy and where concerns about eliciting inflammation or tissue damage can be largely avoided. The actual therapeutic outcomes of adoptive T-cell transfer in LAM and its interactions with selected follow-up therapies are ultimately best gauged in actual patients.
Meanwhile, our data provide proof of principle for the use of adoptively transferred, gp100-reactive T cells to treat LAM. These data provide a rationale and in vivo support for our quest to bring LAM immunotherapy to the clinic.
Supplementary Material
Acknowledgments
Acknowledgment
The authors thank patients with LAM who have donated tissues to further our understanding of LAM. They also thank Dr. Ancy Thomas and Rohan S. Shivde (Lurie Comprehensive Cancer Center, Northwestern University, Chicago, IL), as well as Dr. Irina V. Balyasnikova and Dr. Markella Zannikou (Department of Neurological Surgery, Feinberg School of Medicine, Northwestern University, Chicago, IL) for their technical help.
Footnotes
Supported by The LAM Foundation through an Established Investigator award given to I.C.L.P.
Author Contributions: Experimental design: F.H. and I.C.L.P. Data collection and analyses: F.H., E.R.D., L.W.B., C.C., S.W.H., C.M.A., D.J., and A.Y. Manuscript writing: F.H., V.P.K., D.F.D., and I.C.L.P. Manuscript formatting: E.R.D. Preparation of figures: F.H. and E.R.D. All authors have read and approved the manuscript.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2019-0117OC on February 20, 2020
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Johnson SR, Taveira-DaSilva AM, Moss J. Lymphangioleiomyomatosis. Clin Chest Med. 2016;37:389–403. doi: 10.1016/j.ccm.2016.04.002. [DOI] [PubMed] [Google Scholar]
- 2.Taveira-DaSilva AM, Moss J. Epidemiology, pathogenesis and diagnosis of lymphangioleiomyomatosis. Expert Opin Orphan Drugs. 2016;4:369–378. doi: 10.1517/21678707.2016.1148597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carsillo T, Astrinidis A, Henske EP. Mutations in the tuberous sclerosis complex gene TSC2 are a cause of sporadic pulmonary lymphangioleiomyomatosis. Proc Natl Acad Sci USA. 2000;97:6085–6090. doi: 10.1073/pnas.97.11.6085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Julian LM, Delaney SP, Wang Y, Goldberg AA, Doré C, Yockell-Lelièvre J, et al. Human pluripotent stem cell-derived TSC2-haploinsufficient smooth muscle cells recapitulate features of lymphangioleiomyomatosis. Cancer Res. 2017;77:5491–5502. doi: 10.1158/0008-5472.CAN-17-0925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gao N, Zhang T, Ji J, Xu KF, Tian X. The efficacy and adverse events of mTOR inhibitors in lymphangioleiomyomatosis: systematic review and meta-analysis. Orphanet J Rare Dis. 2018;13:134. doi: 10.1186/s13023-018-0874-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pietrobon A, Delaney SP, Stanford WL. Could immunotherapy sink its teeth into lymphangioleiomyomatosis? Am J Respir Cell Mol Biol. 2018;59:663–665. doi: 10.1165/rcmb.2018-0251ED. [DOI] [PubMed] [Google Scholar]
- 7.Dilling DF, Gilbert ER, Picken MM, Eby JM, Love RB, Le Poole IC. A current viewpoint of lymphangioleiomyomatosis supporting immunotherapeutic treatment options. Am J Respir Cell Mol Biol. 2012;46:1–5. doi: 10.1165/rcmb.2011-0215TR. [DOI] [PubMed] [Google Scholar]
- 8.Maisel K, Merrilees MJ, Atochina-Vasserman EN, Lian L, Obraztsova K, Rue R, et al. Immune checkpoint ligand PD-L1 is upregulated in pulmonary lymphangioleiomyomatosis. Am J Respir Cell Mol Biol. 2018;59:723–732. doi: 10.1165/rcmb.2018-0123OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu HJ, Lizotte PH, Du H, Speranza MC, Lam HC, Vaughan S, et al. TSC2-deficient tumors have evidence of T cell exhaustion and respond to anti-PD-1/anti-CTLA-4 immunotherapy. JCI Insight. 2018;3:98674. doi: 10.1172/jci.insight.98674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu HJ, Krymskaya VP, Henske EP. Immunotherapy for lymphangioleiomyomatosis and tuberous sclerosis: progress and future directions. Chest. 2019;156:1062–1067. doi: 10.1016/j.chest.2019.08.005. [DOI] [PubMed] [Google Scholar]
- 11.Pluvy J, Brosseau S, Stelianides S, Danel C, Nguenang M, Khalil A, et al. Safe and effective use of nivolumab for treating lung adenocarcinoma associated with sporadic lymphangioleiomyomatosis: a rare case report. BMC Pulm Med. 2019;19:12. doi: 10.1186/s12890-018-0775-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krymskaya VP, McCormack FX. Lymphangioleiomyomatosis: a monogenic model of malignancy. Annu Rev Med. 2017;68:69–83. doi: 10.1146/annurev-med-050715-104245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee CH, Yelensky R, Jooss K, Chan TA. Update on tumor neoantigens and their utility: why it is good to be different. Trends Immunol. 2018;39:536–548. doi: 10.1016/j.it.2018.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matsumoto Y, Horiba K, Usuki J, Chu SC, Ferrans VJ, Moss J. Markers of cell proliferation and expression of melanosomal antigen in lymphangioleiomyomatosis. Am J Respir Cell Mol Biol. 1999;21:327–336. doi: 10.1165/ajrcmb.21.3.3693. [DOI] [PubMed] [Google Scholar]
- 15.Klarquist J, Barfuss A, Kandala S, Reust MJ, Braun RK, Hu J, et al. Melanoma-associated antigen expression in lymphangioleiomyomatosis renders tumor cells susceptible to cytotoxic T cells. Am J Pathol. 2009;175:2463–2472. doi: 10.2353/ajpath.2009.090525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grzegorek I, Lenze D, Chabowski M, Janczak D, Szolkowska M, Langfort R, et al. Immunohistochemical evaluation of pulmonary lymphangioleiomyomatosis. Anticancer Res. 2015;35:3353–3360. [PubMed] [Google Scholar]
- 17.Cho JH, Patel B, Bonala S, Manne S, Zhou Y, Vadrevu SK, et al. Notch transactivates Rheb to maintain the multipotency of TSC-null cells. Nat Commun. 2017;8:1848. doi: 10.1038/s41467-017-01845-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eby JM, Smith AR, Riley TP, Cosgrove C, Ankney CM, Henning SW, et al. Molecular properties of gp100-reactive T-cell receptors drive the cytokine profile and antitumor efficacy of transgenic host T cells. Pigment Cell Melanoma Res. 2019;32:68–78. doi: 10.1111/pcmr.12724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klarquist J, Eby JM, Henning SW, Li M, Wainwright DA, Westerhof W, et al. Functional cloning of a gp100-reactive T-cell receptor from vitiligo patient skin. Pigment Cell Melanoma Res. 2016;29:379–384. doi: 10.1111/pcmr.12458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karpanen T, Olweus J. The potential of donor T-cell repertoires in neoantigen-targeted cancer immunotherapy. Front Immunol. 2017;8:1718. doi: 10.3389/fimmu.2017.01718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Finkelstein SE, Heimann DM, Klebanoff CA, Antony PA, Gattinoni L, Hinrichs CS, et al. Bedside to bench and back again: how animal models are guiding the development of new immunotherapies for cancer. J Leukoc Biol. 2004;76:333–337. doi: 10.1189/jlb.0304120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoashi T, Muller J, Vieira WD, Rouzaud F, Kikuchi K, Tamaki K, et al. The repeat domain of the melanosomal matrix protein PMEL17/GP100 is required for the formation of organellar fibers. J Biol Chem. 2006;281:21198–21208. doi: 10.1074/jbc.M601643200. [DOI] [PubMed] [Google Scholar]
- 23.Svajdler M, Bohus P, Goc V, Tkácová V. Perivascular epithelioid cell tumor (PEComa) of the liver: a case report and review of the literature [in Czech] Cesk Patol. 2007;43:18–22. [PubMed] [Google Scholar]
- 24.Kawakami Y, Eliyahu S, Delgado CH, Robbins PF, Sakaguchi K, Appella E, et al. Identification of a human melanoma antigen recognized by tumor-infiltrating lymphocytes associated with in vivo tumor rejection. Proc Natl Acad Sci USA. 1994;91:6458–6462. doi: 10.1073/pnas.91.14.6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Middleton D, Menchaca L, Rood H, Komerofsky R. doi: 10.1034/j.1399-0039.2003.00062.x. New allele frequency database: http://www.allelefrequencies.net. Tissue Antigens 2003;61:403–407. [DOI] [PubMed]
- 26.Steele JC, Rao A, Marsden JR, Armstrong CJ, Berhane S, Billingham LJ, et al. Phase I/II trial of a dendritic cell vaccine transfected with DNA encoding melan A and gp100 for patients with metastatic melanoma. Gene Ther. 2011;18:584–593. doi: 10.1038/gt.2011.1. [DOI] [PubMed] [Google Scholar]
- 27.Trcka J, Moroi Y, Clynes RA, Goldberg SM, Bergtold A, Perales MA, et al. Redundant and alternative roles for activating Fc receptors and complement in an antibody-dependent model of autoimmune vitiligo. Immunity. 2002;16:861–868. doi: 10.1016/s1074-7613(02)00327-8. [DOI] [PubMed] [Google Scholar]
- 28.Khalil DN, Postow MA, Ibrahim N, Ludwig DL, Cosaert J, Kambhampati SR, et al. An open-label, dose-escalation phase I study of anti-TYRP1 monoclonal antibody IMC-20D7S for patients with relapsed or refractory melanoma. Clin Cancer Res. 2016;22:5204–5210. doi: 10.1158/1078-0432.CCR-16-1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li S, Takeuchi F, Wang JA, Fuller C, Pacheco-Rodriguez G, Moss J, et al. MCP-1 overexpressed in tuberous sclerosis lesions acts as a paracrine factor for tumor development. J Exp Med. 2005;202:617–624. doi: 10.1084/jem.20042469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Byles V, Covarrubias AJ, Ben-Sahra I, Lamming DW, Sabatini DM, Manning BD, et al. The TSC-mTOR pathway regulates macrophage polarization. Nat Commun. 2013;4:2834. doi: 10.1038/ncomms3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boorjian SA, Sheinin Y, Crispen PL, Lohse CM, Leibovich BC, Kwon ED. T-cell co-regulatory molecule expression in renal angiomyolipoma and pulmonary lymphangioleiomyomatosis. Urology. 2009;74:1359–1364. doi: 10.1016/j.urology.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Osterburg AR, Nelson RL, Yaniv BZ, Foot R, Donica WR, Nashu MA, et al. NK cell activating receptor ligand expression in lymphangioleiomyomatosis is associated with lung function decline. JCI Insight. 2016;1:e87270. doi: 10.1172/jci.insight.87270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Prajapati K, Perez C, Rojas LBP, Burke B, Guevara-Patino JA. Functions of NKG2D in CD8+ T cells: an opportunity for immunotherapy. Cell Mol Immunol. 2018;15:470–479. doi: 10.1038/cmi.2017.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moore T, Wagner CR, Scurti GM, Hutchens KA, Godellas C, Clark AL, et al. Clinical and immunologic evaluation of three metastatic melanoma patients treated with autologous melanoma-reactive TCR-transduced T cells. Cancer Immunol Immunother. 2018;67:311–325. doi: 10.1007/s00262-017-2073-0. [Published erratum appears in Cancer Immunol Immunother 67:327.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372:2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Legut M, Dolton G, Mian AA, Ottmann OG, Sewell AK. CRISPR-mediated TCR replacement generates superior anticancer transgenic T cells. Blood. 2018;131:311–322. doi: 10.1182/blood-2017-05-787598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou X, Brenner MK. Improving the safety of T-Cell therapies using an inducible caspase-9 gene. Exp Hematol. 2016;44:1013–1019. doi: 10.1016/j.exphem.2016.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.