

Abstract
The disease, COVID-19, is caused by the severe acute respiratory coronavirus 2 (SARS-CoV-2) for which there is currently no treatment. The SARS-CoV-2 main protease (Mpro) is an important enzyme for viral replication. Small molecules that inhibit this protease could lead to an effective COVID-19 treatment. The 2-pyridone scaffold was previously identified as a possible key pharmacophore to inhibit SARS-CoV-2 Mpro. A search for natural, antimicrobial products with the 2-pyridone moiety was undertaken herein, and their calculated potency as inhibitors of SARS-CoV-2 Mpro was investigated. Thirty-three natural products containing the 2-pyridone scaffold were identified from the literature. An in silico methodology using AutoDock was employed to predict the binding energies and inhibition constants (Ki values) for each 2-pyridone-containing compound with SARS-CoV-2 Mpro. This consisted of molecular optimization of the 2-pyridone compound, docking of the compound with a crystal structure of SARS-CoV-2 Mpro, and evaluation of the predicted interactions and ligand-enzyme conformations. All compounds investigated bound to the active site of SARS-CoV-2 Mpro, close to the catalytic dyad (His-41 and Cys-145). Thirteen molecules had predicted Ki values <1 μM. Glu-166 formed a key hydrogen bond in the majority of the predicted complexes, while Met-165 had some involvement in the complex binding as a close contact to the ligand. Prominent 2-pyridone compounds were further evaluated for their ADMET properties. This work has identified 2-pyridone natural products with calculated potent inhibitory activity against SARS-CoV-2 Mpro and with desirable drug-like properties, which may lead to the rapid discovery of a treatment for COVID-19.
Keywords: Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), COVID-19, Main protease (Mpro), 2-Pyridone, In silico molecular modelling, AutoDock
Abbreviations: ACE2, Angiotensin-converting enzyme 2; ADMET, Absorption, distribution, metabolism, excretion, toxicity; clogD, Calculated distribution coefficient; clogP, Calculated partition coefficient; F(20%), Oral bioavailability score to achieve 20% systemic concentration; ΔGcalc, Gibbs free energy, calculated; H-bonds, Hydrogen bonds; HBD, Hydrogen bond donors; IC50, Half maximal inhibitory concentration; Ki, Inhibition constant; LD50, acute oral toxicity median lethal dose; logPapp, Calculated Caco-2 permeability; logS, Calculated aqueous solubility; MPO, Multi-parametric optimization; Mpro, Main protease; PDB, Protein databank; pKa, Acid dissociation constant; PLpro, Papain-like protease; QM, quantum mechanics; R, gas constant; RdRp, RNA-dependent RNA polymerase; RMSD, root mean square deviation; SARS-CoV, Severe acute respiratory syndrome coronavirus; SARS-CoV-2, Severe acute respiratory syndrome coronavirus 2; T, Absolute temperature; TPSA, Total polar surface area
1. Introduction
In 2019, a novel human coronavirus, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2; NC_045512) [1], spread rapidly throughout the global population with suspected human-to-human transmission via respiratory droplets [2,3]. Most SARS-CoV-2-positive individuals developed mild symptoms including upper respiratory tract illness. However, many people with concomitant medical conditions, including pulmonary and cardiovascular disease, hypertension, diabetes mellitus, as well as older adults, have developed severe symptoms or succumbed to the virus [4,5]. The disease, COVID-19, caused by this virus was declared a pandemic by the World Health Organization in March 2020. Over 34.4 million individuals have been infected globally so far, with a case-fatality ratio of 2.98% [6].
SARS-CoV-2 is a 29,903 bp single-stranded RNA coronavirus [7]. It shares 82% nucleotide identity with the related, human severe acute respiratory syndrome coronavirus (SARS-CoV), and both of these viruses belong to clade b of the genus betacoronavirus [2,3]. Both SARS-CoV and SARS-CoV-2 have structural spike proteins anchored to the viral envelope, which recognize and bind to the host cell's membrane protease, angiotensin-converting enzyme 2 (ACE2) [3,8,9]. ACE2 activates the viral spike protein, allowing the virus to penetrate into the host cell [3,10,11] and it is expressed on the ciliated epithelial cells of the human lungs and other tissues [12]. The binding affinity of spike-ACE2 was found to be 10-20-fold higher in SARS-CoV-2 than in SARS-CoV [13], which may be attributed to differences in the receptor-binding domain of the S1 region of the spike protein in both viruses [7]. Once inside the host cell, viral RNA is translated by the host ribosome into two polyproteins, which are proteolytically processed by coronavirus main protease (Mpro) and papain-like protease (PLpro) [14,15], for assembly of new viral particles [16]. In order to replicate the RNA genome, the virus encodes an RNA-dependent RNA polymerase (RdRp). Spike, Mpro, PLpro and RdRP are essential proteins for coronaviruses to infect and replicate, thus making them suitable therapeutic targets. As such, Mpro has been characterized as one of the most attractive targets for the design of coronavirus inhibitors, by using small molecules to block enzyme activity and prevent viral replication [17].
Mpro has a unique cleavage specificity, Leu-Gln↓(Ser,Ala,Gly), that no other human proteases possess, safeguarding potential Mpro inhibitors from being harmful to humans [18]. The crystal structure of SARS-CoV-2 Mpro reveals a dimer that shares 96% amino acid identity with SARS-CoV Mpro [18]. SARS-CoV-2 Mpro has chymotrypsin- and picoronavirus 3C protease-like domains I and II that are six stranded anti-parallel β-barrels containing the substrate binding site between them [19]. Domain III of Mpro regulates dimerization by a salt-bridge interaction between Glu-290 of one protomer and Arg-4 of the other protomer [20]. Dimerization is important for the catalytic activity as it shapes the pocket of the substrate-binding site [21]. Catalytic efficiency of Mpro was shown to be slightly increased in SARS-CoV-2 than SARS-CoV [18], while the maximal accessible volume of the active site is decreased from SARS-CoV to SARS-CoV-2 [22]. For this reason, inhibitors for SARS-CoV might not be as effective for SARS-CoV-2.
There are several crystal structures that have been determined for SARS-CoV-2 Mpro. The x-ray crystallographic structure of ligand-free SARS-CoV-2 Mpro at room temperature (293K) revealed structural plasticity when compared to the x-ray crystallographic structure at low-temperature (100K) [23]. This work also revealed a water molecule deep in the active site, close to His-41, that aids in the catalytic activity of this enzyme [23]. The crystal structure of SARS-CoV-2 Mpro (PDB: 6WQF) is depicted in Fig. 1 with its catalytic dyad and the water molecule. This crystal structure has been regarded as the most appropriate model for performing docking studies as it is more representative of physiological conditions [22,23].
Fig. 1.

The crystal structure of the SARS-CoV-2 main protease (Mpro) monomer (PDB: 6WQF) is comprised of three domains (I–III). The domains, amino acid residues of the catalytic site, His-41 (yellow), Cys-145 (skyblue) and embedded catalytic water molecule (red/white spheres) as suggested by Kneller et al. [23], are depicted. Figure was developed using PyMOL [43].
Previously, α-ketoamides have been synthesized to inhibit the Mpro of beta- and alpha-coronaviruses, although, the half-life and solubility of these compounds in plasma was unsatisfactory [19]. To increase the half-life, solubility and anti-viral activity, α-ketoamides were modified to incorporate a pyridone ring that prevents cleavage from cellular proteases [18]. These new compounds were observed to increase the plasma half-life in mice 3-fold with significantly improved kinetic plasma and thermodynamic solubilities. To identify potential binding sites, the crystal structure of SARS-CoV-2 Mpro was complexed with the pyridone-containing compound (PDB: 6Y2F) [18]. The 2-pyridone compound was bound to the shallow substrate-binding site of SARS-CoV-2 Mpro containing the catalytic dyad (His-41 and Cys-145) and inhibited viral activity [18]. Favourable ADMET and pharmacokinetic properties (e.g. lung tropism) and tolerability when administered subcutaneously and/or via inhalation, gave further credence for the development of 2-pyridone compounds as inhibitors of SARS-CoV-2 [18].
Natural products and their analogues have played a significant role in the development of medicines to treat a wide variety of conditions, including infections [24]. The 2-pyridone moiety is observed in a wide variety of simple and complex organic molecules in nature. Many organisms retain the ability to synthesize, degrade, or utilize molecules containing 2-pyridone rings [25,26]. In humans, 2-pyridone compounds have been identified as metabolites of regular cell function [[27], [28], [29]]. A number of 2-pyridone-containing natural products have antimicrobial, -viral, -inflammatory, -proliferative, -diabetic and -cancer properties making them attractive molecules for drug development [[30], [31], [32], [33], [34]]. The chemical properties of 2-pyridones and their presence in many bioactive compounds make them a valuable resource for drug design [33,34]. Furthermore, the 2-pyridone moiety has been shown to restrict degrees of freedom within chemical structures, while mimicking a peptide backbone [18].
The present work was undertaken to identify potential new inhibitors of SARS-CoV-2 Mpro from the trove of natural products containing a 2-pyridone scaffold. The intention of the present study is to allow for a streamlined and rapid approach in developing therapeutics towards COVID-19.
2. Materials and methods
2.1. Identification of 2-pyridone natural products
The natural products containing a 2-pyridone moiety assessed in the present study were selected through a literature search of 2-pyridone alkaloids discovered from the 1960s to present. The Chemical Abstract Service database, SciFinder (https://scifinder.cas.org), was utilized for conducting structure and keyword (“bioactive 2-pyridones”, “2-pyridone alkaloids”, “antimicrobial 2-pyridones”, “2-pyridone natural products”, “antibacterial 2-pyridones” and “antifungal 2-pyridones”) searches. An initial investigation provided a review from Jessen and Gademann from which a list of notable, bioactive compounds was developed [35]. Additional compounds, discovered following the publication of this review, were identified through further literature searches on compounds containing a 2-pyridone moiety. The 2-pyridone natural products chosen for the current study were selected based on their putative antimicrobial activity. Natural products with a 2-pyridone scaffold, as well as their putative biological actions, are summarized in Appendix A Supplementary Data (Table A1 ).
Table 1.
Calculated binding energies, inhibition constants (Ki values) <1 μM and all-atom RMSD identified for the interaction between 2-pyridone natural products and SARS-CoV-2 Mpro.
| Compound Name [Reference] | Structure (Compound Number) | SARS-CoV-2 Mpro |
||
|---|---|---|---|---|
| Calculated Binding Energy (ΔGcalc; kJ/mol) | Calculated Ki (μM) | RMSD (Å) | ||
| Fusapyridon A-2 [67] |
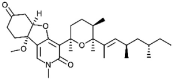 1 |
−43.85 | 0.02086 | 2.00 (Cluster: 13/50) |
| Ilicicolin H [68] |
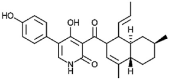 2 |
−43.39 | 0.02498 | 1.98 (Cluster: 11/50) |
| Fusapyridon B-2 [67] |
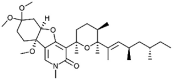 3 |
−42.72 | 0.03258 | 2.00 (Cluster: 12/50) |
| Leporin A [69] |
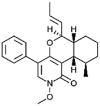 4 |
−39.58 | 0.11584 | 1.02 (Cluster: 29/50) |
| Fusapyridon A-1 [67] |
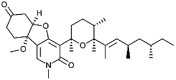 5 |
−38.53 | 0.17601 | 1.69 (Cluster: 11/50) |
| Leporin B [70] |
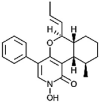 6 |
−37.91 | 0.22961 | 0.26 (Cluster: 40/50) |
| (+) Epipyridone [71] |
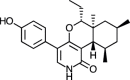 7 |
−37.61 | 0.25692 | 0.19 (Cluster: 37/50) |
| (+) Apiosporamide [72] |
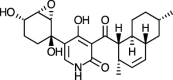 8 |
−36.65 | 0.38145 | 1.12 (Cluster 7/50) |
| Torrubiellone E [73] |
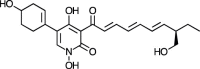 9 |
−36.48 | 0.40795 | 1.60 (Cluster: 8/50) |
| Farinosone A [74] |
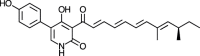 10 |
−36.28 | 0.44335 | 0.85 (Cluster: 5/50) |
| Farinosone B [75] |
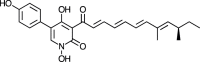 11 |
−35.82 | 0.5296 | 0.60 (Cluster: 10/50) |
| YM-215343 [76] |
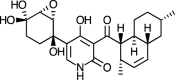 12 |
−34.85 | 0.78177 | 1.23 (Cluster: 13/50) |
| Cordypyridone D [77] |
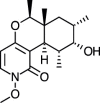 13 |
−34.43 | 0.9259 | 0.17 (Cluster: 31/50) |
2.2. In silico binding studies
Due to the recently identified conformational flexibility of the SARS-CoV-2 Mpro active site and the revelation of a water molecule aiding in catalysis, the room temperature, ligand-free Mpro structure with an embedded water molecule has been suggested to be the most physiologically relevant crystal structure for performing molecular docking (PDB: 6WQF) [23]. Therefore, this structure was selected for in silico binding studies. The calculated binding energies and inhibition constants (K i values) for the selected 2-pyridone natural products with SARS-CoV-2 Mpro were obtained using a molecular modelling procedure. This process consisted of optimization of the molecular geometry of each natural product using GaussView 5.0 [36] and docking each minimized molecular structure with a crystal structure of SARS-CoV-2 Mpro (PDB: 6WQF) using AutoDock 4.2.6 software equipped with AutoDock Tools (Scripps Research Institute) [37].
2.2.1. Molecular geometry optimization of 2-pyridone natural products
In order to minimize the molecular geometry of compounds assessed, each 2-pyridone-containing natural product was constructed and optimized using a quantum mechanics (QM) calculation (Hartree-Fock using the 3-21G basis set) within GaussView 5.0 [36]. The minimized structure was saved as a .pdb file and was subsequently used for docking procedures.
2.2.2. Docking of minimized natural products with crystal structure of SARS-CoV-2 Mpro
To estimate the potential interaction and conformation of the minimized 2-pyridone natural product and SARS-CoV-2 Mpro complex, a published molecular modeling procedure using AutoDock 4.2 Software was conducted [38,39]. The published SARS-CoV-2 Mpro crystal structure (PDB code: 6WQF) was obtained from the protein databank (http://www.rcsb.org/) and uploaded to UCSF Chimera (https://www.cgl.ucsf.edu/chimera/). Water molecules were removed from this structure, except for the oxygen atom located 2.8 Å from His-41, as suggested by Kneller et al. [23]. Hydrogen atoms were added to the oxygen to complete valency of the embedded water, and the structure was saved as a .pdb file. The structure was then uploaded into AutoDock 4.2.6 and each non-polar hydrogen was merged with its bonding partner. Gasteiger partial charges for all atoms within the structure were computed and added to the enzyme. The resulting file was saved as a .pdb file.
The data from the geometry optimization of the 2-pyridone natural product was uploaded into AutoDock 4.2.6. Immediately, all non-polar hydrogen atoms were merged with their bonding partner, aromatic carbon atoms were identified, Gasteiger partial charges for all the atoms within the 2-pyridone natural product were computed and added to the molecule and bonds that could rotate were identified. The data was saved as a .pdbqt file.
Both .pdbqt files were uploaded to AutoDock and the grid maps were produced around the active site of SARS-CoV-2 Mpro. The nucleophilic sulfur atom in Cys-145 (X: 16.67 Å, Y: 3.63 Å, Z: 12.50 Å) was located and used as the center of the grid map. The grid map size was 120 grid points × 120 grid points × 120 grid points with a grid-point spacing of 0.375 Å. The docking calculation was subsequently performed using the Lamarckian genetic algorithm [38], and each 2-pyridone natural product was evaluated over 50 runs. The initial population size of each run was 150. During each run, the maximum number of generations simulated was 27,000, while the maximum number of energy evaluations to identify the fitness of the 2-pyridone : SARS-CoV-2 Mpro interaction was 2,500,000. All predicted binding energies were calculated using a semiempirical free energy force field [38].
The most probable docking location and the lowest binding energy predicted (ΔGcalc, Table 1) between each 2-pyridone : SARS-CoV-2 Mpro complex were visualized within AutoDock. In order to quantify the performance of the docking calculation, a known procedure was used wherein each of the 50 docking runs were evaluated for repetition in the location and orientation of the ligand and placed into groups (clusters) within an all-atom root mean square deviation (RMSD) of ≤2 Å [40,41]. The cluster data produced from each run was placed on a scatter graph to demonstrate the accuracy of the calculation for each 2-pyridone assessed (Appendix A, Supplementary Data, Figure A1). The best cluster, i.e. the cluster containing the highest number of docking runs or the lowest binding energy (where clusters contained the same number of runs) was selected to represent the 2-pyridone : SARS-CoV-2 Mpro complex [42]. The complex with the lowest binding energy from the best cluster was saved as a .pdbqt file and evaluated using the open source visualization software, PyMOL [43]. The amino acid residues that interact with the 2-pyridone natural product, produce hydrogen bonds (H-bonds) [44,45] or surround the docked 2-pyridone compound were identified (Appendix A, Supplementary Data, Table A1). The calculated K i values were obtained using ΔGcalc = RTlnK i; where R is the gas constant, 8.314 J K−1mol−1, and T is the absolute temperature (298.15 K), based on the temperature for enzyme crystallography (Table 1, Table 2, Table 3, Table 4 ) [46,47].
Table 2.
Calculated binding energies, inhibition constants (Ki values) 1–10 μM and all-atom RMSD identified for the interaction between 2-pyridone natural products and SARS-CoV-2 Mpro.
| Compound Name [Reference] | Structure (Compound Number) | SARS-CoV-2 Mpro |
||
|---|---|---|---|---|
| Calculated Binding Energy (ΔGcalc; kJ/mol) | Calculated Ki (μM) | RMSD (Å) | ||
| Cordypyridone C (N-methoxyPF1140) [77] |
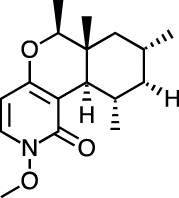 14 |
−33.05 | 1.62 | 0.12 (Cluster: 35/50) |
| Pyridovericin [[78], [79]] |
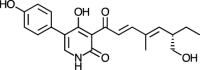 15 |
−31.55 | 2.99 | 1.15 (Cluster: 7/50) |
| Fusapyridon B-1 [67] |
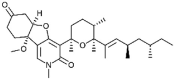 16 |
−30.79 | 4.04 | 1.99 (Cluster: 5/50) |
| Akanthomycin [80] |
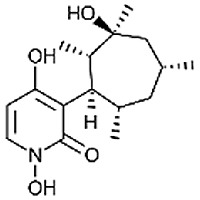 17 |
−30.25 | 5.03 | 0.51 (Cluster: 19/50) |
| Aurodox [81] |
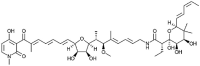 18 |
−29.87 | 5.87 | 0.00 (Cluster: 1/50) |
| N-deoxyakanthomycin [82] |
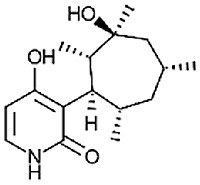 19 |
−29.29 | 7.38 | 1.15 (Cluster: 20/50) |
| PF1140 [83] |
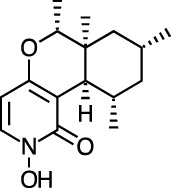 20 |
−28.74 | 9.20 | 0.25 (Cluster: 41/50) |
| Pyridoxatin [84] |
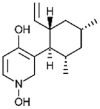 21 |
−28.70 | 9.40 | 0.17 (Cluster: 18/50) |
Table 3.
Calculated binding energies, inhibition constants (Ki values) 10–40 μM and all-atom RMSD identified for the interaction between 2-pyridone natural products and SARS-CoV-2 Mpro.
| Compound Name [Reference] | Structure (Compound Number) | SARS-CoV-2 Mpro |
||
|---|---|---|---|---|
| Calculated Binding Energy (ΔGcalc; kJ/mol) | Calculated Ki (μM) | RMSD (Å) | ||
| Cordypyridone B (8-methylpyridoxatin) [77] |
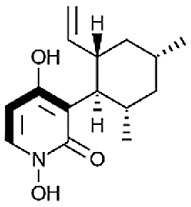 22 |
−28.33 | 10.87 | 0.53 (Cluster: 28/50) |
| Cordypyridone A (8-methylpyridoxatin) [77] |
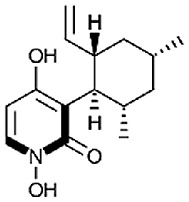 23 |
−28.16 | 11.73 | 0.63 (Cluster: 27/50) |
| Harzianopyridone [[85], [86]] |
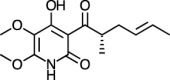 24 |
−28.03 | 12.25 | 1.54 (Cluster: 7/50) |
| (R) Flavipucine [87] |
 25 |
−27.91 | 12.96 | 0.15 (Cluster: 32/50) |
| Atpenin A4 [88] |
 26 |
−27.45 | 15.56 | 1.17 (Cluster: 5/50) |
| (S) Flavipucine [87] |
 27 |
−27.45 | 15.64 | 0.12 (Cluster: 15/50) |
| Torrubiellone A [89] |
 28 |
−27.11 | 17.90 | 0.39 (Cluster: 3/50) |
| Atpenin A5 [88] |
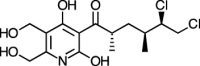 29 |
−25.77 | 30.32 | 1.60 (Cluster: 4/50) |
Table 4.
Calculated binding energies, inhibition constants (Ki values) 40–245 μM and all-atom RMSD identified for the interaction between 2-pyridone natural products and SARS-CoV-2 Mpro.
| Compound Name [Reference] | Structure (Compound Number) | SARS-CoV-2 Mpro |
||
|---|---|---|---|---|
| Calculated Binding Energy (ΔGcalc; kJ/mol) | Calculated Ki (μM) | RMSD (Å) | ||
| Kirromycin [90] |
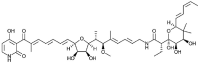 30 |
−24.85 | 43.91 | 0.00 (Cluster: 1/50) |
| Factumycin [91] |
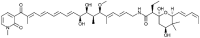 31 |
−24.56 | 49.44 | 0.00 (Cluster: 1/50) |
| Atpenin B [88] |
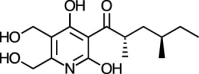 32 |
−23.72 | 69.99 | 1.23 (Cluster: 5/50) |
| Ricinine [[34], [92]] |
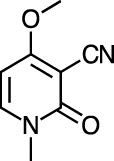 33 |
−20.62 | 244.7 | 0.09 (Cluster: 46/50) |
2.3. Calculation of ADMET pharmacokinetic properties
To quantify the suitability of the most favourable 2-pyridone inhibitors as drugs for SARS-CoV-2 Mpro, several absorption, distribution, metabolism, excretion, and toxicity (ADMET) characteristics were calculated. The multi-parameter optimization (MPO) algorithm, was used to score the most favourable 2-pyridone compounds with the lowest calculated K i values (1–10) and estimate their theoretical ability to be used as a drug. MPO is based on six pharmacokinetic parameters: calculated partition coefficient (clogP), calculated distribution coefficient (clogD), total polar surface area (TPSA), number of H-bonding donors (HBD), acid dissociation constant (pK a), and molecular weight [48]. These chemical properties were calculated using their corresponding plugin and default settings in MarvinSketch (ChemAxon, https://www.chemaxon.com) [49]. pK a values were likewise determined using the default settings for macro mode and static acid/base prefix at room-temperature (298 K). Each 2-pyridone compound was scored for a total MPO value from 0.0 to 6.0 (Table 5 ), where values >3.0 are considered acceptable for compounds possessing drug-like properties [48]. Theoretical aqueous solubility for compounds 1–10 were calculated using MarvinSketch, where values >10 μg/mL are considered optimal [49,50]. Additional ADMET properties (i.e. oral bioavailability F(20%), acute oral toxicity (LD50), and Caco-2 permeability) were generated using the ADMETLab open-source web calculator (https://admet.scbdd.com/) [51,52]. Oral bioavailability is expressed as the fraction of drug required to achieve 20% in systemic circulation (>20% optimal) [53,54]. Toxicity is expressed as mg/kg where values >500 mg/kg are considered optimal [55,56]. Membrane permeability expressed as logPapp values (m/s) for the human colorectal cell line, Caco-2 (>-5.15 cm/s is optimal) [57]. The theoretical subcellular localization of compounds 1–10, were also calculated using the admetSAR2.0 open-source web calculator (https://lmmd.educ.cn/admetsar2.0/) [58,59].
Table 5.
ADMET properties of the ten 2-pyridone natural products with Ki values, <1 μM when bound to SARS-CoV-2 Mpro.
| Compound | MPO | TPSA (Å) | HBD | pKa | clogP | clogD at pH 7.40 | logS (μg/mL) | F(20%) | LD50 (mg/kg) | logPapp (m/s) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2.00 | 65.1 | 0 | 18.8 | 5.07 | 5.07 | 0.361 | 0.386 | 185 | −0.0473 |
| 2 | 2.27 | 86.6 | 3 | 8.93 | 4.84 | 4.83 | 0.0119 | 0.465 | 342 | −0.0529 |
| 3 | 2.00 | 66.5 | 0 | 27.2 | 5.71 | 5.71 | 0.367 | 0.312 | 176 | −0.0476 |
| 4 | 3.15 | 38.8 | 0 | 23.8 | 4.53 | 4.53 | 0.123 | 0.619 | 552 | −0.0461 |
| 5 | 2.00 | 65.1 | 0 | 18.8 | 5.07 | 5.07 | 0.361 | 0.415 | 161 | −0.0473 |
| 6 | 4.70 | 49.8 | 1 | 6.38 | 4.15 | 3.11 | 0.323 | 0.632 | 150 | −0.0492 |
| 7 | 3.23 | 58.6 | 2 | 9.18 | 4.28 | 4.27 | 0.103 | 0.602 | 234 | −0.0506 |
| 8 | 3.10 | 119 | 4 | 8.87 | 1.84 | 1.83 | 0.349 | 0.424 | 229 | −0.0558 |
| 9 | 3.78 | 118 | 4 | −1.84 | 1.60 | 0.39 | 171 | 0.473 | 444 | −0.0533 |
| 10 | 2.48 | 86.6 | 3 | 8.86 | 4.89 | 4.88 | 0.00128 | 0.499 | 867 | −0.0523 |
2.4. Comparison of top 2-pyridones natural products with known drugs
To display the relevance and drug-suitability of the top 2-pyridone-containing compounds investigated herein, calculated binding energies and MPO scores of the 2-pyridone compounds (K i values ≤1 μM) were compared with that of a selection of known drugs suggested for repurposing/repositioning against SARS-CoV-2 Mpro (Table 6 ). The drugs chosen for comparison were selected based on their consideration in several COVID-19-related in silico and drug repurposing studies [[60], [61], [62], [63]]. These known compounds offer a variety of therapeutic classes, from antivirals to neuroleptic drugs.
Table 6.
Comparison of calculated binding energies and MPO scores from select 2-pyridone natural products with potential drugs targeting SARS-CoV-2 Mpro suggested in the literature for possible treatment of COVID-19.
| 2-Pyridone Natural Products | Binding Energy (kJ/mol) | MPO | Repurposed Drugs Against SARS-CoV-2 Mpro | Binding Energy (kJ/mol) | MPO |
|---|---|---|---|---|---|
| 1 | −43.85 | 2.00 | Darunavir | −48.10 [61] | 2.76 |
| 2 | −43.39 | 2.27 | Lopinavir | −40.58 [62] | 2.50 |
| 3 | −42.72 | 2.00 | Saquinavir | −36.30 [61] | 2.90 |
| 4 | −39.58 | 3.15 | Chlorpromazine | −34.73 [63] | 2.58 |
| 5 | −38.53 | 2.00 | Festinavir | −29.92 [62] | 4.53 |
| 6 | −37.91 | 4.70 | Oseltamivir | −29.33 [62] | 5.48 |
| 7 | −37.61 | 3.23 | Remdesivir | −28.33 [62] | 3.00 |
| 8 | −36.65 | 3.10 | Ribavirin | −26.36 [60] | 4.00 |
| 9 | −36.48 | 3.78 | Azithromycin | −25.69 [62] | 2.38 |
| 10 | −36.28 | 2.48 | Favipiravir | −20.00 [62] | 4.50 |
3. Results
3.1. Search for natural products with the 2-pyridone moiety
From the literature search, a total of 61 natural products containing the 2-pyridone moiety were identified. The putative biological actions of many of these compounds were antimicrobial (anti-bacterial, -viral and -fungal), anti-cancer, and/or anti-proliferative or cytotoxic. Given the objective of the present study to assess potentially efficacious inhibitors of SARS-CoV-2 Mpro with limited side-effects, only compounds that possessed anti-microbial properties were evaluated herein. For the putative mechanism of action of the selected compounds, see Appendix A, Supplementary Data Table A1.
3.2. Evaluation and validation of docking poses
To validate the AutoDock procedure utilized in the present work, a cluster plot was generated summarizing the docking data for each of the 33 compounds assessed (Appendix A, Supplementary Data, Figure A1). The cluster plots reveal that most of the 2-pyridone natural products (30 out of 33 compounds) produced their largest cluster with a similarly low binding energy to that of their most favourable (lowest ΔG) run. Furthermore, 27 of the 2-pyridones evaluated showed RMSD values ≤2 Å, indicating good correlation within the best cluster result. The data sets for Aurodox 18, Kirromycin 30, and Factumycin 31 did not generate any clusters greater than a single run. Within the best cluster for each compound, the conformation showing the lowest binding energy was further analyzed in PyMOL.
3.3. Calculated binding energies and Ki values
A total of 33 natural products containing the 2-pyridone moiety were docked to SARS-CoV-2 Mpro using AutoDock [38,39]. Calculated binding energies and K i values for each top-ranked 2-pyridone natural product complexed with SARS-CoV-2 Mpro are summarized in Table 1, Table 2, Table 3, Table 4. The binding energies ranged from −43.85 to −20.62 kJ/mol and the K i values ranged from 0.021 μM–244.70 μM. Compound 1 had the lowest binding energy (−43.85 kJ/mol) and calculated K i value (0.021 μM). Compounds 2–13 (Table 1) also had strong binding energies (−43.39 to −34.43 kJ/mol), with small calculated K i values (0.025 to 0.926 μM). The stereochemistry of the chiral C-3 methyl substituent in the tetrahydropyran ring of fusapyridon A (1 and 5) and fusapyridon B (3 and 16) was not indicated in the literature; therefore, both configurations of the methyl substituent (axial and equatorial) were subjected to the docking procedure.
Compounds 14–21 had affinity for SARS-CoV-2 Mpro with binding energies ranging from −33.05 to −28.70 kJ/mol and the calculated K i values ranging from 1.62 to 9.40 μM (Table 2). Compounds 22–29 had binding energies ranging from −28.33 to −25.77 kJ/mol, and larger calculated K i values ranging from 10.87 to 30.32 μM (Table 3); while compounds 30–33 had the weakest binding energies (−24.85 to −20.62 kJ/mol) and the largest calculated K i values (43.91–244.70 μM) (Table 4).
3.4. Amino acid interactions
The 2-pyridone natural products investigated in this study exhibited very similar interactions with amino acid residues in the SARS-CoV-2 Mpro active site. All 33 compounds showed binding inside of the active site gorge. Only compound 9 showed no H-bonding. The recurrent H-bonding amino acid residues were: Glu-166 (22/33), Asn-142 (11/33), Gln-189 (9/33), His-163 (7/33), Gln-192 (7/33), Thr-190 (7/33). The prominent amino acid residues in close contact to the bound 2-pyridone compounds were: Met-165 (33/33), Met-49 (32/33), His-41 (30/33), Cys-145 (29/33), His-164 (28/33), Leu-167 (24/33), Phe-140 (23/33), Gln-189 (22/33), Leu-141 (20/33), Gly-143 (19/33), Pro-168 (19/33), His-163 (18/33), Arg-188 (18/33), Ser-144 (17/33), Asn-142 (16/33), His-172 (13/33), Thr-190 (13/33), Gln-192 (12/33), Ala-191 (10/33), Glu-166 (9/33). The amino acid residues that were most common for both H-bonding and/or as close contacts were: Met-165 (33/33), His-41 (32/33), Met-49 (32/33), His-164 (31/33). Glu-166 (31/33), Gln-189 (31/33). Almost all compounds exhibited bonding or close contact interactions with the catalytic dyad (His-41 and Cys-145). For the compounds with the most favourable binding energies and K i values (1–13), residues Gln-189 (12/13), His-164 (12/13), and Arg-188 (10/13) were recurrent. The details regarding amino acid interactions for each of the 2-pyridone compounds are summarized in Appendix A, Supplementary Data, Table A1. The amino acid residues in the SARS-CoV-2 Mpro active site that interact with the four most potent compounds assessed in this study are depicted in Fig. 2 .
Fig. 2.

The most potent 2-pyridone natural products, compounds 1–4 (A–D), are docked with SARS-CoV-2 main protease (Mpro) (PDB: 6WQF). The structures, binding energies and inhibition constants (Ki values) for 1–4 are summarized in Table 1, as well as their ADMET properties in Table 5. The H-bonding (dashed black lines) and close contact amino acid residues for 1–4 are depicted in their corresponding panel, as well as the catalytic dyad, His-41 (skyblue), Cys-145 (yellow), and embedded catalytic water (red/white spheres) as suggested by Kneller et al. [23]. Figures were generated using PyMOL [43].
The observed H-bonding interactions primarily involved the 2-pyridone ring portion of the natural product molecules. Frequently, H-bonding occurred between the previously highlighted amino acid residues and the 2-pyridone carbonyl (19/33), the C-4 hydroxy/oxygen group (18/33), or the pyridone amine/oxime (8/33). The 13 compounds that exhibited the most favourable calculated K i values (<1 μM) were mostly ring-based structures containing little or no chains (Tables 1 and A1). These compounds also tended to possess carbonyl, hydroxy, or ether groups at C-3 and a 6 membered-ring functional group off C-5. Interestingly, compound 6 (leporin B) showed no H-bonding despite favourable binding affinity. H-bond interactions are shown in Fig. 2.
3.5. Drug suitability
The ten 2-pyridone natural products (1–10) that produced the lowest K i values when complexed with SARS-CoV-2 Mpro were evaluated for their fitness as potential drugs to treat COVID-19. The ADMET properties identified through the MPO score (clogP, clogD, TPSA, HBD, pKa, and molecular weight) as well as theoretical aqueous solubility, oral bioavailability F(20%), LD50, subcellular localization, and membrane permeability were calculated and are shown in Table 5. Only compounds 4, 6, 7, 8, and 9 produced MPO scores ≥3.0. Leporin B (6) had the largest MPO (4.70). All compounds except 9 showed low theoretical aqueous solubility. The oral bioavailability values were all favourably >20%, with Leporin A (4) and B (6) exhibiting the greatest F(20%) values (0.632 and 0.619). All compounds displayed very similar membrane permeability values ranging from −5.578 to −4.614 cm/s, with compounds 1, and 3–7 showing optimal values. Most compounds were indicated to possess mild-toxicity; however, compounds 4 and 10 were in the low-toxicity range. All compounds showed subcellular localization to the mitochondria, except for compound 6 which was suggested to further localize to the plasma membrane.
When compared with drugs currently under repurpose for the inhibition of SARS-CoV-2 Mpro, compounds 1–10 exhibited similar, if not, slightly greater binding energies (Table 6). A comparable range of MPO values were identified for the ten 2-pyridone compounds (2.00 to 4.70) and the repurposed drugs targeting SARS-CoV-2 Mpro (2.38 to 5.48).
4. Discussion
The novel coronavirus, SARS-CoV-2, has resulted in the global pandemic, COVID-19 [[1], [2], [3]]. This virus is highly contagious [2,3], more so than other coronaviruses [64] and can cause serious and potentially fatal respiratory conditions [4,5]. Currently, there are no vaccines or treatments available to combat SARS-CoV-2. As a result, a global effort has been initiated in search of a cure by repurposing or repositioning existing drugs [[65], [66]].
While a number of unique SARS-CoV-2 proteins are being evaluated for drug development to treat COVID-19, Mpro has been identified as an appropriate target and a number of inhibitors have been evaluated not only for SARS-CoV-2 but for SARS-CoV as well [17]. In the literature, 2-pyridone molecules have shown promise for the inhibition of SARS-CoV-2 Mpro. Thus, in the present work, natural products that contain the 2-pyridone scaffold were evaluated through in silico studies. These molecules, most of which are of fungal origin, have shown putative antimicrobial properties against bacterial and fungal strains [35]. Given the urgency of discovering a cure for COVID-19, these natural products show promise for further evaluation as therapeutic agents for SARS-CoV-2.
Using the in silico AutoDock methodology, selected compounds from the literature were docked and evaluated for their potential inhibitory properties against SARS-CoV-2 Mpro through calculation of binding energies and K i values. To validate the in silico binding procedure, each 2-pyridone natural product was docked with SARS-CoV-2 Mpro to produce a cluster plot. The cluster plots showed convergence of docking poses to the lowest possible binding energy, while the docking conformations within the best cluster had acceptable RMSD values (≤2 Å) (Appendix A, Supplementary Data, Figure A1). In this study, 33 2-pyridone molecules were identified from the literature for investigation, 13 of which proved to have calculated K i <1 μM (compounds 1–13; Table 1), indicating their promise as inhibitors of SARS-CoV-2 Mpro.
Based on the bonding interactions of the 2-pyridone natural products investigated, it is evident that the 2-pyridone ring is a key scaffold that aids in inhibitor binding to SARS-CoV-2 Mpro. A previous investigation of α-ketoamide inhibitors of Mpro highlighted the potential importance of the 2-pyridone moiety in inhibitor design [18]. In the present work, the 2-pyridone carbonyl and nitrogen were shown to be important atoms in the H-bonding interactions at the active site gorge among all compounds studied. In addition, compounds also exhibited prominent interactions with a hydroxy moiety at the C-4 carbon of the 2-pyridone ring. This suggests that, specifically, 4-hydroxy-2-pyridones may be successful inhibitors of SARS-CoV-2. Moreover, these results suggest that greater affinity for SARS-CoV-2 Mpro is achieved with the presence of multiple ring structures (e.g. compound 1, K i value of 0.021 μM) rather than large portions of carbon chains (e.g. compound 30, K i value of 43.91 μM) (Table 1, Table 4). As well, compounds 1–13 were among the most appropriately sized compounds for binding to and filling the active site gorge of Mpro. The weakest binding 2-pyridone was ricinine (33), which was far too small to fill the active site and to engage with the key amino acids previously identified to be important for enzymatic activity. Similarly, compounds 18, 30, and 31 were the largest compounds assessed. Their cluster results indicated poor docking with the SARS-CoV-2 Mpro active site, likely due to the large, bulky carbon chains of these compounds.
It has been suggested that the active site of SARS-CoV-2 Mpro contains a catalytic dyad, His-41 (base) and Cys-145 (nucleophile), with an embedded water molecule (Fig. 1) that enables the enzyme to catalyse the hydrolysis of polyproteins and generate the replicase/transcriptase complex for SARS-CoV-2 replication [[14], [15], [16],23]. The results of the current study show that Glu-166 was present in almost all of the 2-pyridone binding studies conducted, either as a close contact residue or as a participant in H-bonding, as shown previously [18]. This residue may therefore act as a proton donor in hydrolysis, an alternative to the embedded water molecule, to provide increased drug potency. In addition, the frequent occurrence of ligand interactions with Met-49 and Met-165 and for compounds 1–33 and His-164, Arg-188, and Gln-189 for compounds 1–13, also suggests that these residues may be important mediators in inhibitor binding to Mpro. As well, almost all 2-pyridone natural products investigated in this study engaged in interactions with the catalytic dyad as proposed.
The ten top 2-pyridone-containing compounds (K i values ≤1.0 μM) were further evaluated on their suitability for future research as potential drugs to treat COVID-19. ADMET properties including clogD, clogP, HBD, pK a, TPSA, aqueous solubility, oral bioavailability, acute oral toxicity, subcellular localization, and membrane permeability were calculated. The ADMET properties summarize several pharmacokinetic parameters that are considered to provide a basis on the drug-like capacity of a theoretical pharmaceutical [[53], [54], [55], [56], [57], [58], [59]]. Other assessments that include ADMET parameters, such as the MPO calculation have been developed to help theorize the drug-suitability of any given compound [48]. Of the top ten 2-pyridone natural products, compounds 4, 6, 7, 8, and 9 produced favourable MPO values, with leporin B (6) producing the largest MPO value (4.70). Further ADMET analysis showed that all compounds had low, but suitable aqueous solubility [50], favourable oral bioavailability values [53,54], and mild to low toxicity [55,56]. Based on the data for all ADMET parameters (Table 5), only compound (4) was in the optimal range of all parameters, suggesting leporin A (4) may be the most suitable compound for further drug evaluation as an inhibitor of SARS-CoV-2 Mpro.
Additionally, a comparison of the pharmacokinetic properties of the top ten 2-pyridone natural products (K i <1.0 μM) with ten similar active compounds (i.e. darunavir, lopinavir, saquinavir, chlorpromazine, festinavir, oseltamivir, remdesivir, ribavirin, azithromycin, and favipiravir) tested against SARS-CoV-2 Mpro [[60], [61], [62], [63]] showed a similar range of binding energies and MPO scores. This suggests that several of the 2-pyridone-containing compounds highlighted in this study, in addition to favourable calculated inhibitory properties against SARS-CoV-2 Mpro, also exhibit comparable pharmacokinetic and drug-like profiles to those of approved therapeutics, particularly leporin A (4). These results provide further credence for the potential therapeutic value of the natural products explored herein against COVID-19, and emphasizes the further need for pre-clinical and clinical evaluation of these 2-pyridone natural products.
In summary, 33 natural products containing the 2-pyridone moiety were studied using in silico docking techniques against SARS-CoV-2 Mpro. Mpro is a key protein involved in the infection and replication processes of SARS-CoV-2, making it a valuable drug target. All 33 natural product 2-pyridones docked to Mpro, bound to the active site gorge and exhibited promising binding energies and calculated Ki values in the nanomolar to micromolar range. These 2-pyridones indicated key interactions with amino acid residues of the SARS-CoV-2 Mpro active site, such as the catalytic dyad His-41 and Cys-145. Other key residues such as Met-49, His-164, Met-165, Glu-166, and Gln-189 suggest these amino acids may have importance in the inhibitor-SARS-CoV-2 Mpro binding properties to the active site. These in silico findings, and those described previously [18,19], suggest that 2-pyridone natural products may act as potent inhibitors of SARS-CoV-2 Mpro. Five of these 2-pyridone compounds (K i values ≤1.0 μM) also exhibited pharmacokinetic parameters favourable for drug design, where leporin A (4) showed the most promise for future therapeutic use. This in silico study provides a solid foundation for the discovery and development of other effective therapies to combat SARS-CoV-2. Further investigation using in vivo and in vitro experimentation is required to confirm the effectiveness of 2-pyridone natural products as possible treatments for COVID-19.
5. Conclusion
Natural products are a rich source of compounds that can be utilized as therapeutics. In the context of COVID-19, natural products that are inhibitors of SARS-CoV-2 Mpro may provide another avenue for combating the disease. Thirteen 2-pyridone natural products were identified to have favourable predicted inhibitory properties against the virus, five of which also exhibited favourable pharmacokinetic profiles, especially leporin A (4). The present work indicates that natural products containing a 2-pyridone scaffold have the potential to be used as therapeutics against the novel coronavirus SARS-CoV-2 to treat COVID-19.
CRediT authorship contribution statement
Katrina L. Forrestall: Formal analysis, Investigation, Methodology, Validation, Visualization, Writing - original draft, Writing - review & editing. Darcy E. Burley: Formal analysis, Investigation, Validation, Visualization, Writing - original draft, Writing - review & editing. Meghan K. Cash: Writing - original draft, Writing - review & editing. Ian R. Pottie: Conceptualization, Formal analysis, Funding acquisition, Investigation, Methodology, Resources, Supervision, Validation, Visualization, Writing - original draft, Writing - review & editing. Sultan Darvesh: Conceptualization, Formal analysis, Funding acquisition, Project administration, Resources, Supervision, Writing - original draft, Writing - review & editing.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported in part by the Canadian Institutes of Health Research (PJT – 153319), the Dalhousie Medical Research Foundation (DMRF Chemists and The Durland Breakthrough Fund), the Canadian Foundation for Innovation (37854),and the Dalhousie Medical Research Foundation Irene MacDonald Sobey Endowed Chair in Curative Approaches to Alzheimer's Disease.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jclepro.2020.125216.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Gorbalenya B.S., Baric R.S., de Groot R.J., Drosten C., Gulyaeva A.A., Haagmans B.L., Lauber C., Leontovich A.M., Neuman B.W., Penzar D., Perlman S., Poon L.L.M., Samborskiy D.V., Sidorov I.A., Sola I., Ziebuhr J. The species Severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020;5:536–544. doi: 10.1038/s41564-020-0695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu F., Zhao S., Yu B., Chen Y.M., Wang W., Song Z.G., Hu Y., Tao Z.W., Tian J.H., Pei Y.Y., Yuan M.L., Zhang Y.L., Dai F.H., Liu Y., Wang Q.M., Zheng J.J., Xu L., Holmes E.C., Zhang Y.Z. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhou P., Yang X.L., Wang X.G., Hu B., Zhang L., Zhang W., Si H.R., Zhu Y., Li B., Huang C.L., Chen H.D., Chen J., Luo Y., Guo H., Jiang R.D., Liu M.Q., Chen Y., Shen X.R., Wang X., Zheng X.S., Zhao K., Chen Q.J., Deng F., Liu L.L., Yan B., Zhan F.X., Wang Y.Y., Xiao G.F., Shi Z.L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang J., Zheng Y., Gou X., Pu K., Chen Z., Guo Q., Ji R., Wang H., Wang Y., Zhou Y. Prevalence of comorbidities and its effects in coronavirus disease 2019 patients: a systematic review and meta-analysis. Int. J. Infect. Dis. 2020;94:91–95. doi: 10.1016/j.ijid.2020.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang J.J., Dong X., Cao Y.Y., Yuan Y.D., Yang Y.B., Yan Y.Q., Akdis C.A., Gao Y.D. Vol. 75. Allergy; 2020. Clinical characteristics of 140 patients infected with SARS-CoV-2 in Wuhan, China; pp. 1730–1741. [DOI] [PubMed] [Google Scholar]
- 6.Johns Hopkins University, Coronavirus Resource Centre, Johns Hopkins University, United States, 2020.
- 7.Baig A.M., Khaleeq A., Ali U., Syeda H. Evidence of the COVID-19 virus targeting the CNS: tissue distribution, host-virus interaction, and proposed neurotropic mechanisms. ACS Chem. Neurosci. 2020;11:995–998. doi: 10.1021/acschemneuro.0c00122. [DOI] [PubMed] [Google Scholar]
- 8.Li W., Moore M.J., Vasilieva N., Sui J., Wong S.K., Berne M.A., Somasundaran M., Sullivan J.L., Luzuriaga K., Greenough T.C., Choe H., Farzan M. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Netland J., Meyerholz D.K., Moore S., Cassell M., Perlman S. Severe acute respiratory syndrome coronavirus infection causes neuronal death in the absence of encephalitis in mice transgenic for human ACE2. J. Virol. 2008;82:7264–7275. doi: 10.1128/jvi.00737-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li F., Li W., Farzan M., Harrison S.C. Structure of SARS coronavirus spike receptor-binding domain complexed with receptor. Science. 2005;309:1864–1868. doi: 10.1126/science.1116480. [DOI] [PubMed] [Google Scholar]
- 11.Letko M., Marzi A., Munster V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 2020;5:562–569. doi: 10.1038/s41564-020-0688-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Palasca O., Santos A., Stolte C., Gorodkin J., Jensen L.J. Database; Oxford): 2018. TISSUES 2.0: an Integrative Web Resource on Mammalian Tissue Expression. 2018 bay003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wrapp D., Wang N., Corbett K.S., Goldsmith J.A., Hsieh C.L., Abiona O., Graham B.S., McLellan J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367:1260–1263. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ziebuhr J., Snijder E.J., Gorbalenya A.E. Virus-encoded proteinases and proteolytic processing in the Nidovirales. J. Gen. Virol. 2000;81:853–879. doi: 10.1099/0022-1317-81-4-853. [DOI] [PubMed] [Google Scholar]
- 15.Hilgenfeld R. From SARS to MERS: crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J. 2014;281:4085–4096. doi: 10.1111/febs.12936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baranov P.V., Henderson C.M., Anderson C.B., Gesteland R.F., Atkins J.F., Howard M.T. Programmed ribosomal frameshifting in decoding the SARS-CoV genome. Virology. 2005;332:498–510. doi: 10.1016/j.virol.2004.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 18.Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science. 2020;368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang L., Lin D., Kusov Y., Nian Y., Ma Q., Wang J., von Brunn A., Leyssen P., Lanko K., Neyts J., de Wilde A., Snijder E.J., Liu H., Hilgenfeld R. Alpha-ketoamides as broad-spectrum inhibitors of coronavirus and enterovirus replication: structure-based design, synthesis, and activity assessment. J. Med. Chem. 2020;63:4562–4578. doi: 10.1021/acs.jmedchem.9b01828. [DOI] [PubMed] [Google Scholar]
- 20.Shi J., Song J. The catalysis of the SARS 3C-like protease is under extensive regulation by its extra domain. FEBS J. 2006;273:1035–1045. doi: 10.1111/j.1742-4658.2006.05130.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anand K., Palm G.J., Mesters J.R., Siddell S.G., Ziebuhr J., Hilgenfeld R. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra α-helical domain. EMBO J. 2002;21:3213–3224. doi: 10.1093/emboj/cdf327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bzowka M., Mitusinska K., Raczynska A., Samol A., Tuszynski J.A., Gora A. Structural and evolutionary analysis indicate that the SARS-CoV-2 Mpro is a challenging target for small-molecule inhibitor design. Int. J. Mol. Sci. 2020;21:3099. doi: 10.3390/ijms21093099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kneller D.W., Phillips G., O’Neill H.M., Jedrzejczak R., Stols L., Langan P., Joachimiak A., Coates L., Kovalevsky A. Structural plasticity of SARS-CoV-2 3CL Mpro active site cavity revealed by room temperature X-ray crystallography. Nat. Commun. 2020;11 doi: 10.1038/s41467-020-16954-7. 3202 (2020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dias D.A., Urban S., Roessner U. A historical overview of natural products in drug discovery. Metabolites. 2012;2:303–336. doi: 10.3390/metabo2020303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sims G.K., Lee L.E. Degradation of pyridine derivatives in soil. J. Environ. Qual. 1985;14:580–584. doi: 10.2134/jeq1985.00472425001400040022x. [DOI] [Google Scholar]
- 26.Chen G.Y., Zhong W., Zhou Z., Zhang Q. Simultaneous determination of tryptophan and its 31 catabolites in mouse tissues by polarity switching UHPLC-SRM-MS. Anal. Chim. Acta. 2018;1037:200–210. doi: 10.1016/j.aca.2018.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grant R.S., Coggan S.E., Smythe G.A. The physiological action of picolinic Acid in the human brain. Int. J. Tryptophan Res. 2009;2:71–79. doi: 10.4137/IJTR.S2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lenglet A., Liabeuf S., Bodeau S., Louvet L., Mary A., Boullier A., Lemaire-Hurtel A.S., Jonet A., Sonnet P., Kamel S., Massy Z.A. N-methyl-2-pyridone-5-carboxamide (2PY)-Major metabolite of nicotinamide: an update on an old uremic toxin. Toxins (Basel) 2016;8:339. doi: 10.3390/toxins8110339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Trammell S.A., Schmidt M.S., Weidemann B.J., Redpath P., Jaksch F., Dellinger R.W., Li Z., Abel E.D., Migaud M.E., Brenner C. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat. Commun. 2016;7:12948. doi: 10.1038/ncomms12948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hamama W.S., Waly M., El-Hawary I., Zoorob H.H. Developments in the chemistry of 2-pyridone. Synth. Commun. 2014;44:1730–1759. doi: 10.1080/00397911.2013.862836. [DOI] [Google Scholar]
- 31.Sośnicki J.G., Idzik T.J. Pyridones – powerful precursors for the synthesis of alkaloids, their derivatives. and alkaloid-inspired compounds Synthesis. 2019;51:3369–3396. doi: 10.1055/s-0037-1611844. [DOI] [Google Scholar]
- 32.Bao J., Zhai H., Zhu K., Yu J.H., Zhang Y., Wang Y., Jiang C.S., Zhang X., Zhang H. vol. 16. Mar Drugs; 2018. (Bioactive Pyridone Alkaloids from a Deep-Sea-Derived Fungus Arthrinium Sp. UJNMF0008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li L.N., Wang L., Cheng Y.N., Cao Z.Q., Zhang X.K., Guo X.L. Discovery and characterization of 4-Hydroxy-2-pyridone derivative sambutoxin as a potent and promising anticancer drug candidate: activity and molecular mechanism. Mol. Pharm. 2018;15:4898–4911. doi: 10.1021/acs.molpharmaceut.8b00525. [DOI] [PubMed] [Google Scholar]
- 34.Tan Y.-N., Zeng J., Zhang S.-N., Ma R.-J., Pan Z.-H., Tan Q.-G. Pyridone alkaloids from the leaves of Ricinus communis and their inhibitory effect against protein tyrosine phosphatase 1B. Chem. Nat. Compd. 2019;55:395–397. doi: 10.1007/s10600-019-02702-x. [DOI] [Google Scholar]
- 35.Jessen H.J., Gademann K. 4-Hydroxy-2-pyridone alkaloids: structures and synthetic approaches. Nat. Prod. Rep. 2010;27:1168–1185. doi: 10.1039/B911516C. [DOI] [PubMed] [Google Scholar]
- 36.Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R., Scalmani G., Barone V., Petersson G.A., Nakatsuji H., Li X., Caricato M., Marenich A., Bloino J., Janesko B.G., Gomperts R., Mennucci B., Hratchian H.P., Ortiz J.V., Izmaylov A.F., Sonnenberg J.L., Williams-Young D., Ding F., Lipparini F., Egidi F., Goings J., Peng B., Petrone A., Henderson T., Ranasinghe D., Zakrzewski V.G., Gao J., Rega N., Zheng G., Liang W., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Throssell K., Montgomery J.A., Peralta J.E., Ogliaro F., Bearpark M., Heyd J.J., Brothers E., Kudin K.N., Staroverov V.N., Keith T., Kobayashi R., Normand J., Raghavachari K., Rendell A., Burant J.C., Iyengar S.S., Tomasi J., Cossi M., Millam J.M., Klene M., Adamo C., Cammi R., Ochterski J.W., Martin R.L., Morokuma K., Farkas O., Foresman J.B., Fox D.J. Gaussian, Inc.; Wallingford, CT: 2016. Gaussian 09. [Google Scholar]
- 37.Sanner M.F. Python: a programming language for software integration and development. J. Mol. Graph. Model. 1999;17:57–61. [PubMed] [Google Scholar]
- 38.Morris G.M., Goodsell D.S., Halliday R.S., Huey R., Hart W.E., Belew R.K., Olson A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998;19:1639–1662. doi: 10.1002/(SICI)1096-987X(19981115)19:14<1639::AID-JCC10>3.0.CO;2-B. [DOI] [Google Scholar]
- 39.Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vieth M., Hirst J.D., Kolinski A., Brooks C.L., III Assessing energy functions for flexible docking. J. Comput. Chem. 1998;19:1612–1622. doi: 10.1002/(SICI)1096-987X(19981115)19:14<1612::AID-JCC7>3.0.CO;2-M. [DOI] [Google Scholar]
- 41.Bursulaya B.D., Totrov M., Abagyan R., Brooks C.L. Comparative study of several algorithms for flexible ligand docking. J. Comput. Aided Mol. Des. 2003;17:755–763. doi: 10.1023/b:jcam.0000017496.76572.6f. [DOI] [PubMed] [Google Scholar]
- 42.Ballante F., Marshall G.R. An automated strategy for binding-pose selection and docking assessment in structure-based drug design. J. Chem. Inf. Model. 2016;56:54–72. doi: 10.1021/acs.jcim.5b00603. [DOI] [PubMed] [Google Scholar]
- 43.The PyMOL Molecular Graphics System, Schrödinger, LLC.
- 44.Jeffrey G.A. Oxford University Press; New York and Oxford: 1997. An Introduction to Hydrogen Bonding. [Google Scholar]
- 45.Pinney M.M., Natarajan A., Yabukarski F., Sanchez D.M., Liu F., Liang R., Doukov T., Schwans J.P., Martinez T.J., Herschlag D. Structural coupling throughout the active site hydrogen bond networks of ketosteroid isomerase and photoactive yellow protein. J. Am. Chem. Soc. 2018;140:9827–9843. doi: 10.1021/jacs.8b01596. [DOI] [PubMed] [Google Scholar]
- 46.Phosrithong N., Ungwitayatorn J. Molecular docking study on anticancer activity of plant-derived natural products. Med. Chem. Res. 2010;19:817–835. doi: 10.1007/s00044-009-9233-5. [DOI] [Google Scholar]
- 47.Thongnum K., Chanthai S. Inhibitory reactivity of capsaicin with α-amylase and α-glucosidase related to antidiabetes using molecular docking and quantum calculation methods. Orient. J. Chem. 2018;34:2211–2228. doi: 10.13005/ojc/340501. [DOI] [Google Scholar]
- 48.Wager T.T, Hou X., Verhoest P.R., Villalobos A. Moving beyond rules: the development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem. Neurosci. 2010;1:435–449. doi: 10.1021/cn100008c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hou T.J., Xia K., Zhang W., Xu X.J. ADME Evaluation in Drug Discovery. 4. Prediction of Aqueous Solubility Based on Atom Contribution Approach. J. Chem. Inf. Comput. Sci. 2004;44:266–275. doi: 10.1021/ci034184n. [DOI] [PubMed] [Google Scholar]
- 50.Lipinski C.A., Lombardo F., Dominy B.W., Feeney P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001;46:3–26. doi: 10.1016/S0169-409X(96)00423-1. [DOI] [PubMed] [Google Scholar]
- 51.Dong J., Wang N.-N., Yao Z.-J., Zhang L., Cheng Y., Ouyang D., Lu A.-P., Cao D.-S. ADMETlab: a platform for systematic ADMET evaluation based on a comprehensively collected ADMET database. J. Cheminform. 2018;10 doi: 10.1186/s13321-018-0283-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ferreira L.L.G., Andricopulo A.D. Vol. 24. Drug Discov. Today; 2019. ADMET modeling approaches in drug discovery; pp. 1157–1165. [DOI] [PubMed] [Google Scholar]
- 53.Ahmed S.S.S.J., Ramakrishnan V. Vol. 7. PLOS One; 2012. Systems biological approach of molecular descriptors connectivity: optimal descriptors for oral bioavailability prediction; p. e40654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Watanabe R., Esaki T., Kawashima H., Natsume-Kitatani Y., Nagao C., Ohashi R., Mizuguchi K. Predicting fraction unbound in human plasma from chemical structure: improved accuracy in the low value ranges. Mol. Pharm. 2018;15:5302–5311. doi: 10.1021/acs.molpharmaceut.8b00785. 1203. [DOI] [PubMed] [Google Scholar]
- 55.Zhu H., Martin T.M., Ye L., Sedykh A., Young D.M., Tropsha A. Quantitative structure−activity relationship modeling of rat acute toxicity by oral exposure. Chem. Res. Toxicol. 2009;22:1913–1921. doi: 10.1021/tx900189p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lei T., Li Y., Song Y., Li D., Sun H., Hou T. ADMET evaluation in drug discovery: 15. Accurate prediction of rat oral acute toxicity using relevance vector machine and consensus modeling. J. Cheminform. 2016;8:1–19. doi: 10.1186/s13321-016-0117-7. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang N.-N., Dong J., Deng Y.-H., Zhu M.-F., Wen M., Yao Z.-J., Lu A.-P., Wang J.-B., Cao D.-S. ADME properties evaluation in drug discovery: prediction of Caco-2 cell permeability using a combination of NSGA-II and boosting. J. Chem. Inform. Model. 2016;56:763–773. doi: 10.1021/acs.jcim.5b00642. [DOI] [PubMed] [Google Scholar]
- 58.Yang H., Lou C., Sun L., Li J., Cai Y., Wang Z., Li W., Liu G., Tang Y. admetSAR 2.0: web-service for prediction and optimization of chemical ADMET properties. Bioinformatics. 2018;35:1067–1069. doi: 10.1093/bioinformatics/bty707. [DOI] [PubMed] [Google Scholar]
- 59.Yang H., Sun L., Wang Z., Li W., Liu G., Tang Y. ADMETopt: a web server for ADMET optimization in drug design via scaffold hopping. J. Chem. Inform. Model. 2018;58:2051–2056. doi: 10.1021/acs.jcim.8b00532. [DOI] [PubMed] [Google Scholar]
- 60.Deshpande R.R., Tiwari A.P., Nyayanit N., Modak M. In silico molecular docking analysis for repurposing therapeutics against multiple proteins from SARS-CoV-2. Eur. J. Pharmacol. 2020;886:173430. doi: 10.1016/j.ejphar.2020.173430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Khan S.A., Zia K., Ashraf S., Uddin R., Ul-Haq Z. Identification of chymotrypsin-like protease inhibitors of SARS-CoV-2 via integrated computational approach. J. Biomol. Struct. Dyn. 2020:1–10. doi: 10.1080/07391102.2020.1751298. [DOI] [PubMed] [Google Scholar]
- 62.Shivanika C., Deepak Kumar S., Venkataraghavan R., Pawan T., Sumitha A., Brindha Devi P. Molecular docking, validation, dynamics simulations, and pharmacokinetic prediction of natural compounds against the SARS-CoV-2 main-protease. J. Biomol. Struct. Dyn. 2012;78:1020–1023. doi: 10.1080/07391102.2020.1815584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Udrea A.-M., Avram S., Nistorescu S., Pascu M.-L., Romanitan M.O. Laser irradiated phenothiazines: New potential treatment for COVID-19 explored by molecular docking. J. Photochem. Photobiol. B. 2020;211:111997. doi: 10.1016/j.jphotobiol.2020.111997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.de Wit E., van Doremalen N., Falzarano D., Munster V.J. SARS and MERS: recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016;14:523–534. doi: 10.1038/nrmicro.2016.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sanders J.M., Monogue M.L., Jodlowski T.Z., Cutrell J.B. Pharmacologic Treatments for Coronavirus Disease 2019 (COVID-19): A Review. JAMA. 2020;323:1824–1836. doi: 10.1001/jama.2020.6019. [DOI] [PubMed] [Google Scholar]
- 66.Weston S., Coleman C.M., Haupt R., Logue J., Matthews K., Frieman M.B. Broad Anti-coronaviral Activity of FDA Approved Drugs against SARS-CoV-2 in Vitro and SARS-CoV in Vivo. bioRixv. 2020 doi: 10.1101/2020.03.25.008482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tsuchinari M., Shimanuki K., Hiramatsu F., Murayama T., Koseki T., Shioni Y. Fusapyridons A and B, Novel Pyridone Alkaloids from an Endophytic Fungus, Fusarium sp. YG-45. Z Naturforsch B J Chem Sci. 2007;62:1203–1207. doi: 10.1515/znb-2007-0916. [DOI] [Google Scholar]
- 68.Rotsaert F.A., Ding M.G., Trumpower B.L. Differential efficacy of inhibition of mitochondrial and bacterial cytochrome bc1 complexes by center N inhibitors antimycin, ilicicolin H and funiculosin. Biochim. Biophys. Acta. 2008;1777:211–219. doi: 10.1016/j.bbabio.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tao Q., Ding C., Auckloo B.N., Wu B. Bioactive metabolites from a hydrothermal vent fungus Aspergillus sp. YQ-13. Nat. Prod. Commun. 2018;13:571–573. doi: 10.1177/1934578X1801300514. [DOI] [Google Scholar]
- 70.Zhang C., Jin L., Mondie B., Mitchell S.S., Castelhano A.L., Cai W., Bergenhem N. Leporin B., A novel hexokinase II gene inducing agent from an unidentified fungus. Bioorg. Med. Chem. Lett. 2003;13:1433–1435. doi: 10.1016/S0960-894X(03)00153-7. [DOI] [PubMed] [Google Scholar]
- 71.Kemami Wangun H.V., Hertweck C. Epicoccarines A, B and epipyridone: tetramic acids and pyridone alkaloids from an Epicoccum sp. associated with the tree fungus Pholiota squarrosa. Org. Biomol. Chem. 2007;5:1702–1705. doi: 10.1039/B702378B. [DOI] [PubMed] [Google Scholar]
- 72.Zhang Y., Zhang Q., Bao J., Huang J., Zhang H. Apiosporamide, A 4-hydroxy-2-pyridone alkaloid, induces apoptosis via PI3K/akt signaling pathway in osteosarcoma cells. OncoTargets Ther. 2019;12:8611–8620. doi: 10.2147/OTT.S218692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Isaka M., Haritakun R., Intereya K., Thanakitpipattana D., Hywel-Jones N.L. Vol. 9. Nat. Prod. Commun; 2014. Torrubiellone E., An antimalarial N-hydroxypyridone alkaloid from the spider pathogenic fungus Torrubiella longissima BCC 2022; pp. 627–628. [DOI] [PubMed] [Google Scholar]
- 74.Cheng Y., Schneider B., Riese U., Schubert B., Li Z., Hamburger M. Farinosones A.-C., Neurotrophic alkaloidal metabolites from the entomogenous deuteromycete Paecilomyces farinosus. J. Nat. Prod. 2004;67:1854–1858. doi: 10.1021/np049761w. [DOI] [PubMed] [Google Scholar]
- 75.Talontsi F.M., Nwemeguela Kenla T.J., Dittrich B., Douanla-Meli C., Laatsch H. Paeciloside A., A new antimicrobial and cytotoxic polyketide from Paecilomyces sp. strain CAFT156. Planta Med. 2012;78:379–382. doi: 10.1055/s-0031-1298622. [DOI] [PubMed] [Google Scholar]
- 76.Shibazaki M., Taniguchi M., Yokoi T., Nagai K., Watanabe M., Suzuki K., Yamamoto T. YM-215343, a novel antifungal compound from Phoma sp. QN04621. J. Antibiot. 2004;57:379–382. doi: 10.7164/antibiotics.57.379. [DOI] [PubMed] [Google Scholar]
- 77.Isaka M., Tanticharoen M., Kongsaeree P., Thebtaranonth Y. Structures of cordypyridones A-D, antimalarial N-hydroxy- and N-methoxy-2-pyridones from the insect pathogenic fungus Cordyceps nipponica. J. Org. Chem. 2001;66:4803–4808. doi: 10.1021/jo0100906. [DOI] [PubMed] [Google Scholar]
- 78.Takahashi S., Kakinuma N., Uchida K., Hashimoto R., Yanagisawa T., Nakagawa A. Pyridovericin and pyridomacrolidin: novel metabolites from entomopathogenic fungi, Beauveria bassiana. J. Antibiot. 1998;51:596–598. doi: 10.7164/antibiotics.51.596. [DOI] [PubMed] [Google Scholar]
- 79.de Souza Santos M., Jonis Andrioli W., Freire de Morais Del Lama M.P., Kenupp Bastos J., Nanayakkara N.P., Zumstein Georgetto Naal R.M. In vitro anti-allergic activity of the fungal metabolite pyridovericin. Int. Immunopharm. 2013;15:532–538. doi: 10.1016/j.intimp.2013.01.017. [DOI] [PubMed] [Google Scholar]
- 80.Wagenaar M.M., Gibson D.M., Clardy J. Akanthomycin, a new antibiotic pyridone from the entomopathogenic fungus Akanthomyces gracilis. Org. Lett. 2002;4:671–673. doi: 10.1021/ol016737q. [DOI] [PubMed] [Google Scholar]
- 81.McHugh R.E., O’Boyle N., Connolly J.P.R., Hoskisson P.A., Roe A.J. Characterization of the mode of action of aurodox, a type III secretion system inhibitor from streptomyces goldiniensis. Infect. Immun. 2019;87(2):e00595–18. doi: 10.1128/IAI.00595-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.de Silva E.D., Geiermann A.S., Mitova M.I., Kuegler P., Blunt J.W., Cole A.L., Munro M.H. Isolation of 2-pyridone alkaloids from a New Zealand marine-derived penicillium species. J. Nat. Prod. 2009;72:477–479. doi: 10.1021/np800627f. [DOI] [PubMed] [Google Scholar]
- 83.Fujita Y., Oguri H., Oikawa H. Biosynthetic studies on the antibiotics PF1140: a novel pathway for a 2-pyridone framework. Tetrahedron Lett. 2005;46:5885–5888. doi: 10.1016/j.tetlet.2005.06.115. [DOI] [Google Scholar]
- 84.Dorsaz S., Snaka T., Favre-Godal Q., Maudens P., Boulens N., Furrer P., Ebrahimi S.N., Hamburger M., Allemann E., Gindro K., Queiroz E.F., Riezman H., Wolfender J.L., Sanglard D. Identification and mode of action of a plant natural product targeting human fungal pathogens. Antimicrob. Agents Chemother. 2017;61:e00829–17. doi: 10.1128/AAC.00829-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ióca L.P., Romminger S., Santos M.F.C., Bandeira K.F., Rodrigues F.T., Kossuga M.H., Nicacio K.J., Ferreira E.L.F., Morais-Urano R.P., Passos M.S., Kohn L.K., Arns C.W., Sette L.D., Berlinck R.G.S. A strategy for the rapid identification of fungal metabolites and the discovery of the antiviral activity of pyrenocine a and harzianopyridone. Quím. Nova. 2016;39:720–731. doi: 10.5935/0100-4042.20160092. [DOI] [Google Scholar]
- 86.Bat-Erdene U., Kanayama D., Tan D., Turner W.C., Houk K.N., Ohashi M., Tang Y. Iterative catalysis in the biosynthesis of mitochondrial complex II inhibitors harzianopyridone and atpenin B. J. Am. Chem. Soc. 2020;142:8550–8554. doi: 10.1021/jacs.0c03438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kusakabe Y., Mizutani S., Kamo S., Yoshimoto T., Tomoshige S., Kawasaki T., Takasawa R., Tsubaki K., Kuramochi K. Synthesis, antibacterial and cytotoxic evaluation of flavipucine and its derivatives. Bioorg. Med. Chem. Lett. 2019;29:1390–1394. doi: 10.1016/j.bmcl.2019.03.034. [DOI] [PubMed] [Google Scholar]
- 88.Miyadera H., Shiomi K., Ui H., Yamaguchi Y., Masuma R., Tomoda H., Miyoshi H., Osanai A., Kita K., Omura S. Atpenins, potent and specific inhibitors of mitochondrial complex II (succinateubiquinone oxidoreductase) Proc. Natl. Acad. Sci. U. S. A. 2003;100:473–477. doi: 10.1073/pnas.0237315100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Isaka M., Chinthanom P., Supothina S., Tobwor P., Hywel-Jones N.L. Pyridone and tetramic acid alkaloids from the spider pathogenic fungus Torrubiella sp. BCC 2165. J. Nat. Prod. 2010;73:2057–2060. doi: 10.1021/np100492j. [DOI] [PubMed] [Google Scholar]
- 90.Grubmuller H., Bock L.V. tRNA Dissociation from EF-Tu after GTP Hydrolysis: Primary Steps and Antibiotic Inhibition. Biophys. J. 2020;118:151–161. doi: 10.1016/j.bpj.2019.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kimura K., Iwatsuki M., Nagai T., Matsumoto A., Takahashi Y., Shiomi K., Omura S., Abe A. A small-molecule inhibitor of the bacterial type III secretion system protects against in vivo infection with Citrobacter rodentium. J. Antibiot. 2011;64:197–203. doi: 10.1038/ja.2010.155. [DOI] [PubMed] [Google Scholar]
- 92.Ferraz A.C., Anselmo-Franci J.A., Perosa S.R., de Castro-Neto E.F., Bellissimo M.I., de Oliveira B.H., Cavalheiro E.A., Naffah-Mazzacoratti Mda G., Da Cunha C. Amino acid and monoamine alterations in the cerebral cortex and hippocampus of mice submitted to ricinine-induced seizures. Pharmacol. Biochem. Behav. 2002;72:779–786. doi: 10.1016/S0091-3057(02)00750-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.