

Enoxacin mitigates obesity by inducing fat and muscle energy metabolism independently of its antibiotic function.
Abstract
MicroRNAs (miRNAs) have been implicated in oxidative metabolism and brown/beige adipocyte identity. Here, we tested whether widespread changes in miRNA expression promoted by treatment with the small-molecule enoxacin cause browning and prevent obesity. Enoxacin mitigated diet-induced obesity in mice, and this was associated with increased energy expenditure. Consistently, subcutaneous white and brown adipose tissues and skeletal muscle of enoxacin-treated mice had higher levels of markers associated with thermogenesis and oxidative metabolism. These effects were cell autonomous since they were recapitulated in vitro in murine and human cell models. In preadipocytes, enoxacin led to a reduction of miR-34a-5p expression and up-regulation of its target genes (e.g., Fgfr1, Klb, and Sirt1), thus increasing FGF21 signaling and promoting beige adipogenesis. Our data demonstrate that enoxacin counteracts obesity by promoting thermogenic signaling and inducing oxidative metabolism in adipose tissue and skeletal muscle in a mechanism that involves, at least in part, miRNA-mediated regulation.
INTRODUCTION
Obesity affects more than 600 million people worldwide and contributes to the onset of type 2 diabetes, cardiovascular disorders, and cancer (1). Obesity is the result of an imbalance between food intake and energy expenditure, leading to accumulation of excess energy in the form of lipids within adipocytes. Skeletal muscle and adipose are the main tissues where energy is expended in the body. To fuel oxidative metabolism and produce adenosine 5′-triphosphate (ATP), the skeletal muscle uses large amounts of substrates available from the diet or provided by the white adipose tissue (WAT), particularly during exercise. Heat is a by-product of these processes and can also be promoted by induction of uncoupled respiration and futile cycles, which, in turn, contribute to body temperature control in mammals along with brown adipose tissue (BAT)–induced thermogenesis. BAT is the most efficient thermogenic tissue in mammals, burning more calories per gram of tissue than any other tissue in the body (2). This occurs in part due to the high number of metabolically active mitochondria expressing uncoupling protein 1 (UCP1) (3, 4). A pool of adipocytes that express UCP1 is also found in specific WAT depots in response to stimuli such as cold or exercise (5, 6). These adipocytes are called beige adipocytes and the recruitment of these cells to WAT is often referred to as browning or beiging of WAT (3). When active, brown and beige adipocytes are important sites for glucose (7) and triglyceride (8) metabolism. Therefore, even a small increase in the activity or number of brown and beige adipocytes is envisioned with great promise in the treatment of obesity and its complications (9).
To date, the most potent and well-characterized interventions that induce energy expenditure in rodents and potentially in humans are β-adrenergic pathway stimulators (10) and peroxisome proliferator–activated receptor γ (PPARγ) agonists (11). These molecules trigger a variety of pathways that promote thermogenic gene expression; most of them are dependent on the activation of the transcriptional coactivator PGC1α (12). Another important inducer of energy expenditure is the fibroblast growth factor 21 (FGF21) (13). FGF21 is increased in response to cold or dietary restriction (14, 15) and acts to up-regulate Ppargc1a/PGC1α levels and function via SIRT1 stabilization (16). Despite having promising effects on browning and energy expenditure, β-adrenergic receptor agonists, certain PPARγ stimulators, and recombinant FGF21 display a number of notable side effects, such as tachycardia, gain of adiposity, and osteoporosis, respectively (17–19). Thus, finding previously unidentified ways to increase energy expenditure is a matter of considerable interest.
MicroRNAs (miRNAs) are small endogenous noncoding RNAs that target sequences within mRNAs to inhibit their translation and/or induce their degradation (20, 21). Emerging evidence has suggested that miRNAs play essential roles in brown/beige adipocyte determination and function. For example, adipocyte-specific knockout of DICER (22) or DiGeorge syndrome critical region protein 8 (23) results in whitening of the interscapular BAT and impaired mitochondrial function in mice (24). Among the miRNAs expressed in adipose tissue, many have been implicated in thermogenic gene expression and oxidative metabolism [reviewed in (25)]. Therefore, we hypothesized that widespread changes in miRNA abundance would affect adipose tissue browning and energy expenditure in vivo.
Enoxacin is a broad-spectrum antibacterial fluoroquinolone that has been shown to interfere with miRNA processing in eukaryotic cells (26, 27). In cancer cells, enoxacin appears to facilitate the interaction between double-stranded RNA structures and TAR RNA binding protein (TRBP)—a molecule that recruits DICER to the AGO2 complex—thus promoting small interfering RNA (siRNA) and miRNA processing and RNA-induced silencing complex (RISC) activation (27, 28). TRBP association with DICER has also been proposed to play a role in strand selection during pre-miRNA processing and AGO2 recruitment (29).
When given to mice, enoxacin may inhibit tumor growth (28, 30, 31) and reduces amyotrophic lateral sclerosis symptoms (31) by interfering with miRNA biogenesis. Enoxacin and other fluoroquinolones have also been described as glucose-lowering agents potentially by acting in ATP-sensitive K+ channels in pancreatic β cells (33, 34). Enoxacin is relatively safe to administer to humans and has been previously approved by the U.S. Food and Drug Administration for the treatment of bacterial urinary tract infections and gonorrhea, although its clinical use has been replaced by other more potent fluoroquinolones.
Here, we show that enoxacin mitigates diet-induced obesity in mice by increasing energy expenditure and promoting oxidative metabolism and uncoupled respiration in adipocytes and myotubes. This mechanism was further explored in adipocytes, where it was associated with increased FGF21 signaling and increased UCP1 and Ppargc1a expression, the latter mediated by down-regulation of miR-34a-5p. These observations suggest a potential new class of anti-obesity drugs and serve as proof of principle for studies targeting miRNA biogenesis in metabolic diseases.
RESULTS
Acute treatment with enoxacin induces markers of oxidative metabolism and thermogenesis
Enoxacin has pleiotropic effects in vivo in eukaryotic cells, and some of these effects are attributed to its activity over the miRNA processing pathway (28, 30, 32). Since miRNAs have been involved in energy metabolism, we tested whether enoxacin treatment influences key regulators of thermogenesis and oxidative phosphorylation in vivo. To begin to uncover it, we treated C57BL/6J mice raised at room temperature with daily intraperitoneal injections of enoxacin for up to 10 days, at a dose that had been previously described as effective and safe in mice, i.e., 10 mg/kg body weight per day (35). Enoxacin led to up-regulation of Ucp1 (Fig. 1A) and Ppargc1a gene expression in BAT (Fig. 1B) as early as 3 days after the first injection. The increase of UCP1 was confirmed at the protein level after 10 days of daily injections with enoxacin (Fig. 1, C and D). Ucp1 was also induced by enoxacin in subcutaneous inguinal WAT (scWAT) as early as 24 hours after the first injection (fig. S1A), whereas Ppargc1a was not affected during the 10-day time course (fig. S1B).
Fig. 1. Effect of acute enoxacin treatment in murine BAT of chow-fed mice.
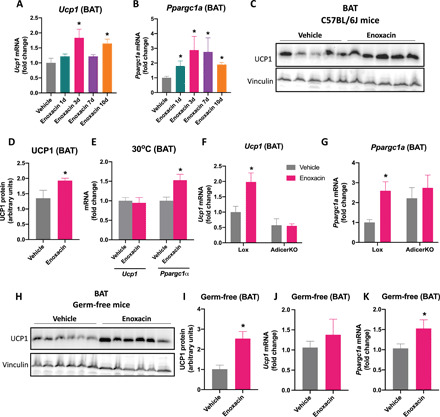
C57BL/6J mice were injected daily with enoxacin for the time indicated, and thermogenic markers were measured in BAT. Time course of (A) Ucp1 and (B) Ppargc1a (n = 4 to 6 animals per group). (C) Representative UCP1 blot and (D) quantification in BAT of mice injected with enoxacin (or vehicle) every day for 10 days (n = 5 animals per group). (E) Ucp1 and Ppargc1a in BAT of mice maintained in thermoneutrality and injected with enoxacin (or vehicle) every day for 3 days (n = 9 animals per group). *P < 0.05 versus vehicle (Mann-Whitney U test). (F) Ucp1 and (G) Ppargc1a in 8-week-old AdicerKO and floxed control (Lox) mice injected with enoxacin (or vehicle) every day for 3 days (n = 3 and 4 animals/group). *P < 0.05 versus vehicle [two-way analysis of variance (ANOVA) with Tukey post hoc test]. (H to K) Germ-free mice were treated with enoxacin (or vehicle) every day for 3 days. (H) Representative UCP1 blot and (I) quantification (n = 6 animals per group). (J) Ucp1 and (K) Ppargc1a (n = 6 and 7 per group). Values are the means ± SEM. *P < 0.05 versus vehicle (Mann-Whitney U test).
We also measured the expression of genes involved in oxidative metabolism and UCP1-independent thermogenesis in BAT, scWAT and muscle after 10 days of daily injections with enoxacin. Enoxacin increased uncoupling protein 3 mRNA (Ucp3) levels in all tissues (fig. S1, C to E) and had mild or no effect on genes related to the creatine biosynthetic (fig. S1, F to H) and calcium cycling (fig. S1, I to K) pathways. We did not observe alterations in heart rate (fig. S1L), systolic blood pressure (fig. S1M), or double product (i.e., systolic blood pressure multiplied by the heart rate used as an index of myocardial oxygen consumption) (fig. S1N) (36) in mice treated with enoxacin. In summary, enoxacin may increase genes involved in thermogenesis and oxidative metabolism without inducing cardiac output.
Next, we focused on BAT and investigated whether the effect of enoxacin remained at thermoneutrality (30°C), when β-adrenergic and other endogenous thermogenic stimuli are negligible (37). While Ucp1 levels were not affected by enoxacin in mice at thermoneutrality, Ppargc1a expression was still up-regulated in BAT (Fig. 1E), suggesting that enoxacin induces Ppargc1a, but not Ucp1, independent of cold-induced triggers. Moreover, young (3-month-old) adipocyte-specific Dicer knockout (AdicerKO) mice did not show up-regulation of Ucp1 (Fig. 1F) and Ppargc1a (Fig. 1G) expression in BAT in response to enoxacin, pointing to a role for adipose tissue miRNAs in the induction of these genes independently of the lipodystrophic phenotype that AdicerKO mice manifest when they are older than 6 months of age or are fed a high-fat diet (HFD) (18).
Given the antibiotic nature of enoxacin, we tested whether the gut microbiome was affected by the drug treatment and whether it played a role in enoxacin’s effects. We found no significant differences between vehicle- and enoxacin-treated animals in terms of microbiome composition, as analyzed by Bray-Curtis dissimilarities or abundance of specific taxa [(Analysis of composition of microbiomes (ANCOM) test] (fig. S2, A to C). Consistent with a role for enoxacin independent of the microbiota, the protein level of UCP1 was up-regulated by 2.5-fold in BAT of enoxacin-treated germ-free mice (Fig. 1, H and I). However, enoxacin failed to up-regulate Ucp1 in BAT of both antibiotic mix–treated (fig. S2E) and germ-free mice (Fig. 1J). In contrast, enoxacin led to increases in Ppargc1a mRNA levels in BAT of antibiotic mix–treated mice (fig. S2E) and germ-free mice (Fig. 1K), although in the former, the difference did reach statistical significance (P = 0.2284). These results indicate that enoxacin treatment does not change the composition of the gut microbiome and leads to up-regulation of UCP1 and Ppargc1a in BAT independently of its function as an antibiotic.
Enoxacin mitigates diet-induced obesity
To determine the effect of enoxacin treatment on obesity and energy balance, we fed C57BL/6 mice with HFD for 4 weeks and then treated these mice with enoxacin for 10 weeks (10 mg/kg body weight per day, 5 intraperitoneal injections/week) in combination with the diet. Mice treated with enoxacin gained 31% less weight on HFD in comparison to vehicle-treated mice (Fig. 2A). This was accompanied by reduced WAT mass and no changes in BAT mass (Fig. 2B). Consistent with reduced adiposity, enoxacin treatment decreased fasting glucose levels (Fig. 2C), improved glucose tolerance (Fig. 2, D and E), and increased insulin sensitivity (Fig. 2, F and G), although no differences were observed in insulin levels (fig. S3A). Enoxacin led to a nonsignificant reduction in blood total cholesterol levels (P = 0.0636) (fig. S3B), but it did not affect HDL levels (fig. S3C) or altered accumulation of liver triglycerides (fig. S3D). Moreover, enoxacin treatment did not affect the content of bacteria or bacterial phyla distribution in the feces (fig. S3, E to G), and it did not modify fecal energy content (fig. S3H). Food intake was also not changed during enoxacin treatment (Fig. 2H). Together, these results indicate that enoxacin mitigates diet-induced obesity and insulin resistance by a mechanism that does not involve changes in energy intake, energy loss, or depletion of the gut microbiota.
Fig. 2. Enoxacin treatment mitigates diet-induced obesity by increasing energy expenditure.
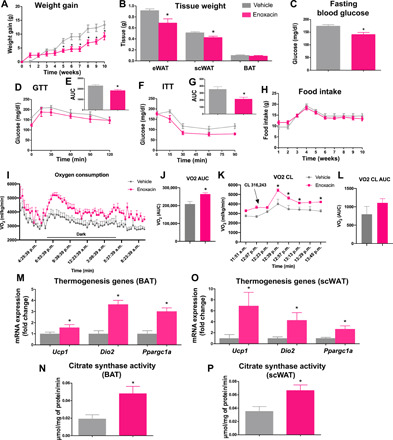
HFD-fed C57BL/6 mice were treated for 10 weeks with enoxacin (or vehicle). (A) Weight gain (g); (B) adipose tissue weight (eWAT, epididymal adipose tissue; scWAT, subcutaneous inguinal adipose tissue; BAT, intrascapular brown adipose tissue) (n = 5 to 8 animals per group); (C) fasting blood glucose levels; (D) glucose tolerance test (GTT), expressed also as (E) the area under the curve (AUC); (F) insulin tolerance test (ITT), expressed also as (G) AUC (n = 4 to 5 per group); (H) food intake (g); (I) O2 consumption, also expressed as (J) AUC (n = 5 to 6 animals per group). (K) Mice were intraperitoneally injected with CL-316,243 (1.0 mg/kg), and O2 consumption was measured and expressed also as (L) AUC. In this case, the baseline was established on the basis of the level of O2 consumption per group before injection. (M and O) Gene expression (n = 5 to 7 per group) and (N and P) citrate synthase activity were measured in BAT and scWAT (n = 5 per group), respectively. Values are the means ± SEM. *P < 0.05 versus vehicle (Mann-Whitney U test).
Enoxacin promotes energy expenditure and stimulates a thermogenic gene program in scWATs and BATs in HFD-fed mice
We next examined whether enoxacin counteracts obesity by increasing energy expenditure. Using Comprehensive Lab Animal Monitoring System, we found higher oxygen consumption and increased carbon dioxide production in animals treated with enoxacin (Fig. 2, I and J, and fig. S3, I and J). Respiratory exchange ratio (RER) was also increased in enoxacin-treated mice, indicating a preference for carbohydrate utilization (fig. S3, K and L). Heat production trended upward in animals treated with enoxacin, but this increase was only statistically significant in the dark cycle (fig. S3, M to O). No differences were observed in spontaneous activity (fig. S3, P and Q), suggesting that the increase in energy expenditure is due to increased metabolic rate.
To estimate the contribution of brown/beige adipocyte–mediated energy expenditure, we acutely activated these cells by giving mice a single dose of CL-316,243—a β3-adrenergic receptor agonist—and measured the increase in VO2. CL-316,243 injection increased VO2 levels in both vehicle- and enoxacin-treated mice, although this increase trended to be more prominent in the latter, exacerbating the changes between the two groups of animals (Fig. 2, K and L). This suggests, at least in part, increased potential for brown/beige adipocyte stimulation. Consistent with this phenotype, markers associated with the increased oxidative and thermogenic capacity of brown/beige adipocytes, such as Ucp1, Dio2, and Ppargc1a gene expression (Fig. 2M) and citrate synthase activity (Fig. 2N), were all up-regulated in BAT of enoxacin-treated mice. These markers were also increased in scWAT of mice treated with enoxacin (Fig. 2, O and P). Mitochondrial DNA was not significantly affected by enoxacin in this tissue (fig. S3R). These data show that enoxacin increases BAT thermogenic capacity and induces browning of WAT.
To investigate the effect of enoxacin on thermogenesis, we exposed mice to cold. Enoxacin-treated mice were more thermotolerant than the vehicle group in both acute (within 6 hours of cold exposure; when thermogenesis is largely sustained by activation of preexisting brown and beige adipocytes; Fig. 3A) and chronic (24 to 72 hours of cold exposure; when new beige adipocytes are recruited; Fig. 3B) challenges. BAT of enoxacin-treated mice was browner (Fig. 3C) and expressed higher levels of thermogenic genes (i.e., Ucp1, Dio2, and Ppargc1a) after 72 hours of cold exposure (Fig. 3D). These genes were also up-regulated by enoxacin in scWAT, some of them by over 10-fold (Fig. 3E). Histologically, enoxacin reversed HFD-induced BAT “whitening” and decreased the size of fat cells in scWAT (Fig. 3F). Liver triglyceride levels were reduced in enoxacin-treated mice after cold exposure (fig. S3S). Together, these results demonstrate that enoxacin potentiates the effect of cold to promote browning and activate thermogenesis.
Fig. 3. Enoxacin induces browning and oxidative metabolism.
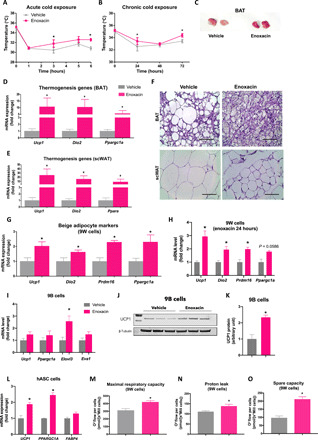
HFD-fed mice treated for 10 weeks with enoxacin and subjected to 6°C. (A and B) Rectal temperature during cold exposure (n = 6 to 8 per group). (C) Representative image of BAT after 72 hours of cold exposure. Photo credit: Andrea L. Rocha, Photographer Institution, University of Campinas. Gene expression in (D) BAT and (E) scWAT (n = 6 per group). (F) Representative hematoxylin and eosin images of BAT and scWAT. Gene expression in (G) 9W preadipocytes treated with enoxacin during differentiation (n = 4 independent pools per group; representative of three experiments). Scale bar 100 µm (40x magnification) or (H) for 24 hours after differentiation (n = 3 independent pools per group; representative of two experiments). 9B preadipocytes treated with enoxacin during differentiation. (I) Gene expression (n = 5 to 7 independent pools per group; representative of three experiments). (J) Representative UCP1 blot and (K) quantification (n = 6 independent pools per group; representative of two experiments). (L) Gene expression in human adipose–derived stem cells (hASC) differentiated into adipocytes and treated with enoxacin for 10 days (n = 3 independent pools per group; representative of two experiments). (M to O) Respiratory parameters in 9W cells treated with enoxacin during differentiation (n = 3 to 5 independent pools per group; representative of three experiments). *P < 0.05 versus vehicle (Mann-Whitney U test or unpaired t test with Welch’s correction when n = 3).
Enoxacin induces oxidative metabolism and uncoupled respiration in a cell autonomous manner
To test whether enoxacin acts cell-autonomously to induce browning and promote oxidative metabolism, we used a mouse subcutaneous preadipocyte cell line (9W) (22). First, we established that treatment with enoxacin up to 100 μM did not cause significant toxicity to 9W cells (fig. S4, A to C). We then chose to study the effect of 50 μM enoxacin during differentiation of 9W cells. When differentiated under probrowning conditions, i.e., stimulated by the classical adipocyte differentiation cocktail plus T3, indomethacin, and rosiglitazone, these cells differentiated into bona fide beige adipocytes (Figs. 4 to 6 and figs. S4 to S9). When exposed to enoxacin during differentiation, they expressed even more of the brown/beige fat markers (Ucp1, Dio2, Prdm16, and Ppargc1a mRNAs) (Fig. 3G). This occurred despite any changes in Fasn, Pparg, and Fabp4 mRNA (fig. S5, A to C), suggesting that overall adipogenesis was not affected. Ucp1, Dio2, Prdm16, and Ppargc1a mRNAs were also up-regulated when fully differentiated beige adipocytes were given enoxacin for 24 hours, although the latter did not reach statistical significance (P = 0.0586) (Fig. 3H). We also investigated the effects of enoxacin treatment during differentiation of murine brown preadipocytes (9B cells). Enoxacin treatment did not alter Ucp1, Ppargc1a, and Eva1 mRNA expression, but it did increase the expression of Elovl3—a marker of brown adipocyte activation (Fig. 3I). UCP1 protein levels were increased by over twofold upon enoxacin treatment (Fig. 3, J and K).
Fig. 4. Enoxacin induces energy turnover in beige adipocytes.
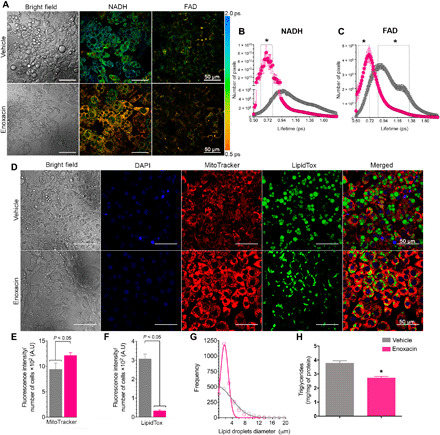
9W cells were treated with enoxacin (50 μM) or vehicle during beige adipocyte differentiation. (A) Representative images and (B) quantification of mean fluorescence lifetime (τm) of NADH (two-photon excitation at 730 nm and emission at 430 nm) and (C) FAD in picoseconds (excitation 730 nm/emission 515 nm) (n = 8 to 12 independent pools per group; representative of three experiments). (D) Representative images and (E) quantification of MitoTracker Red and (F) LipidTox. (G) Lipid droplet frequency per diameter was quantified from confocal images (n = 6 to 8 independent pools per group; representative of three experiments). (H) Quantification of the triglyceride content of differentiated adipocytes (n = 4 to 6 independent pools per group; representative of two experiments). Values are the means ± SEM. *P < 0.05 versus vehicle (Mann-Whitney U test). A.U., arbitrary units.
We also tested the effect of enoxacin on murine 3T3-F442A differentiated white adipocytes. Ucp1 was measured in these cells after differentiation with the probrowning cocktail, but its expression was undetected under all conditions, consistent with the strong commitment of these cells to the white adipocyte lineage. Nonetheless, treatment with enoxacin during differentiation significantly increased genes associated with oxidative metabolism and the brown/beige adipocyte phenotype, including Ppargc1a and Eva1 (fig. S5, D and E). In contrast, Fabp4 expression was not changed, indicating no effect on overall adipocyte differentiation (fig. S5F).
We also tested whether enoxacin promotes browning in a human model of beige adipocyte differentiation. In human adipose–derived stem cells (hASCs) predifferentiated into mature adipocytes, 10-day enoxacin treatment led to 1.9- and 2.5-fold increases in UCP1 and PPARGC1A mRNAs, respectively (Fig. 3L). Again, no changes in FABP4 expression were seen (Fig. 3L). Together, these data confirm that enoxacin induces genes associated with oxidative metabolism and promotes beige adipogenesis in mice and humans in a cell autonomous manner.
To explore the functional implications of enoxacin treatment, we measured cell respiration in differentiated 9W cells using the Oroboros apparatus. Treatment with enoxacin during adipocyte differentiation resulted in increased maximum respiration [Carbonyl cyanide m-chlorophenyl hydrazone (CCCP)–induced respiration in Fig. 3M], higher proton leak (Fig. 3N], and improved mitochondrial spare capacity (measured as respiration above basal after uncoupling with CCCP) (Fig. 3O). Furthermore, we observed that cells treated with enoxacin had an increase in mitochondrial and cytoplasmic reactive oxygen species (ROS) levels (fig. S5, G and H), denoting higher oxidative metabolism. Given the role of ROS in controlling uncoupled respiration, we tested whether the increase in ROS was the cause of higher uncoupling in enoxacin-treated cells. However, incubation with the antioxidant N-acetyl-l-cysteine (NAC) during differentiation or in mature adipocytes (i.e., NAC, 24 hours) did not completely abrogate the capacity of enoxacin to induce maximal respiration, proton leak, and mitochondrial spare capacity in adipocytes (fig. S5, I to K), suggesting that increased ROS production is a consequence and not a cause of increased uncoupling and improved oxidative metabolism in enoxacin-treated cells.
To study the metabolic effects of enoxacin at the single-cell level, we used fluorescence lifetime imaging microscopy (FLIM) to measure the reduced form of nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FAD) turnover rate. Consistent with accelerated metabolism, the overall half-life of NADH and FAD was shorter in enoxacin-treated cells (Fig. 4, A to C). Enoxacin-treated cells also had increased mitochondrial mass/membrane potential as determined by MitoTracker Red FM (Fig. 4, D and E) and decreased lipid content (Fig. 4F) and increased frequency of smaller lipid droplets (Fig. 4G). In agreement, enoxacin led to a reduction in triglyceride accumulation (Fig. 4H). This was not due to an inhibition of adipocyte differentiation since general adipocyte markers such as Pparg and Fabp4 were not affected by enoxacin treatment (fig. S5, B and C). Increased NADH and FAD turnover, higher mitochondrial content, and multilocular distribution of lipid droplets are hallmarks of beige adipocytes, thus confirming that enoxacin is a potent inducer of beige adipogenesis.
Knowing that enoxacin treatment in vivo up-regulates Ucp3 in skeletal muscle, we tested whether the drug could also promote oxidative metabolism and uncoupled respiration in C2C12 myotubes. Consistent with this hypothesis, enoxacin up-regulated Ppargc1a and several PPAR target genes related to oxidative metabolism (fig. S6A). In addition, enoxacin treatment increased maximum mitochondrial respiration (fig. S6B), induced proton leak (fig. S6C), and enhanced mitochondrial spare capacity (fig. S6D), demonstrating that the effects of enoxacin to induce oxidative metabolism and uncoupled respiration go beyond the adipose tissue and include the skeletal muscle.
Enoxacin effects on adipose tissue are partially dependent on miRNAs
To determine to what degree the metabolic effects of enoxacin depend on the regulation of adipose tissue miRNAs in vivo, we took advantage of the AdicerKO mice. These mice and their littermate wild-type controls (Lox) were subjected to the previously described protocol of HFD following enoxacin or vehicle treatment. Again, enoxacin treatment resulted in reduced adiposity in wild-type mice, but these effects were blunted in AdicerKO mice (Fig. 5, A to C, and fig. S7A). No statistical differences were found in the insulin levels or liver weight of these animals (fig. S7, B and C). Ucp1 and Ppargc1a were up-regulated by enoxacin in BAT of wild-type mice but not in AdicerKO mice (Fig. 5, D and E). Similarly, enoxacin trended to induce UCP1 protein abundance in BAT of wild-type (80% increase; P = 0.1358) but not AdicerKO mice (Fig. 5, F and G). The same overall pattern of gene expression was observed in scWAT for Ppargc1a but not Ucp1 mRNA, which was still up-regulated by enoxacin in AdicerKO mice (Fig. 5, H and I). Despite the reduced UCP1 and Ppargc1a levels in adipose tissues of AdicerKO mice, enoxacin treatment could still improve thermotolerance in these mice (fig. S7D), indicating that enoxacin-induced thermogenesis does not entirely depend on adipose tissue miRNAs or UCP1 induction.
Fig. 5. Enoxacin effects are partially dependent on miRNA expression in adipocytes.
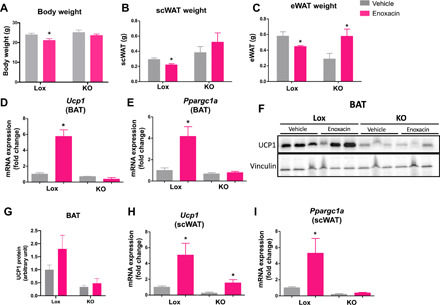
AdicerKO (KO) mice or floxed control (Lox) mice were fed with HFD and treated with enoxacin or vehicle. (A) Body weight and (B and C) adipose tissue weight (n = 4 to 7 per group). Expression of genes involved in (D and E) thermogenesis and (F and G) representative Western blot and (G) quantification of UCP1 BAT (n = 5 per group; P = 0.1358 Lox enoxacin versus Lox vehicle). (H) Expression of Ucp1 and (I) Ppargc1a in subcutaneous adipose tissue (scWAT) (n = 4 to 5 per group). Values are the means ± SEM. *P < 0.05 versus vehicle (two-way ANOVA with Bonferroni’s multiple comparison posttest).
To confirm the requirement of miRNAs to the effects of enoxacin in adipocytes, we used C3H10T1/2 mouse mesenchymal cells transduced with virus carrying shRNA targeting Dicer (shDicer) or green fluorescent protein (shGFP). These cells were differentiated into adipocytes using the probrowning cocktail and differentiated normally even with a >50% down-regulation of Dicer in the shDicer cells (fig. S8, A and B). Consistent with a role for miRNAs in Ppargc1a regulation, Ppargc1a was up-regulated in a dose-dependent manner by enoxacin in shGFP but not in shDicer cells (fig. S8C). Ucp1, on the other hand, was not affected by enoxacin in these cells (fig. S8D). Moreover, enoxacin led to a reduction in triglyceride accumulation in a Dicer-dependent manner (fig. S8E), but no changes in Fabp4 mRNA expression (fig. S8F). Together with the AdicerKO mouse data, these results demonstrate that enoxacin requires miRNAs in adipocytes to increase Ppargc1a and reduce lipid accumulation cell autonomously.
Enoxacin acts via miR-34a-5p to regulate FGF21 signaling and Ppargc1a expression
Next, we explored how enoxacin controls Ppargc1a via miRNA regulation. Consistent with the role of enoxacin in miRNA biogenesis, DICER processing activity was increased in 9W beige cells treated with enoxacin during differentiation (fig. S8G). However, of the 720 miRNAs detected in these cells by small RNA sequencing (i.e., at least one generated read), 105 (14%) and 93 (13%) miRNAs had at least a twofold decrease or increase, respectively, upon enoxacin treatment (Fig. 6A and table S1). Six miRNAs were selected for validation using quantitative reverse transcription polymerase chain reaction (RT-qPCR) and four of them confirmed the pattern found in the small RNA sequencing data (Fig. 6C and fig. S8, H to M).
Fig. 6. Enoxacin controls FGF21 signaling and Ppargc1a expression via down-regulation of miR-34a-5p.
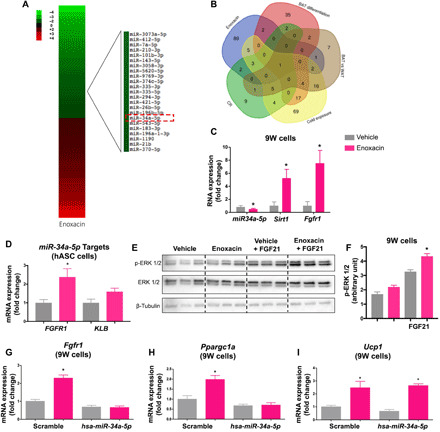
(A) Heatmap of differentially expressed miRNAs (fold change < 2 or > 2) in 9W cells upon treatment with enoxacin or vehicle during differentiation. Red, up-regulated; green, down-regulated. (B) Venn’s diagram showing consistent changes in miRNA expression after enoxacin treatment, BAT differentiation, BAT from cold exposed mice, BAT versus WAT, and scWAT from caloric restriction mice. miR-34a-5p and/or miR34a-5p target genes in (C) 9W (n = 4 to 6 independent pools per group; representative of three experiments) and (D) hASC adipocytes (n = 3 independent pools per group; representative of two experiments). (E) Phosphorylation state of p44/42 MAPK in differentiated 9W cells treated with enoxacin or vehicle during differentiation and stimulated with 50 nM FGF21 or vehicle for 15 min (n = 3 independent pools per group; performed once). (F) Blot quantification. (G to I) Effects of enoxacin and/or the miR-34a-5p mimic on miR-34a-5p target genes in 9W adipocytes (n = 4 independent pools per group; performed once). Values are the means ± SEM. *P < 0.05 versus vehicle. Two-way ANOVA (F to I), Mann-Whitney U test (C), and unpaired t test with Welch’s correction (D).
To select the miRNAs potentially involved in Ppargc1a regulation among the ones changed by enoxacin (Fig. 6A), we compared our dataset with publicly available datasets of miRNAs expressed in adipose tissue and affected by brown adipocyte differentiation (38), cold exposure (39), and caloric restriction (40) or increased in BAT versus WAT (41). The result of this analysis revealed only one miRNA commonly down-regulated under all conditions—miR-34a-5p (Fig. 6B). The down-regulation of miR-34a-5p by enoxacin was confirmed by RT-qPCR in 9W adipocytes (Fig. 6C), C3H10T1/2 adipocytes (fig. S8M), and scWAT (fig. S9A). miR-34a-5p was rapidly down-regulated in 9W cells upon enoxacin exposure (within 30 min), then increased at 60 min, and went back down after 3 hours, while miR-34a-3p stayed mostly unchanged until 60 min, when it also went up and then down after 3 hours (fig. S9B). We also traced the expression of miR-34a-5p during differentiation and observed that this miRNA remained unchanged during the course of adipogenesis in the vehicle control group, while it went progressively down in the enoxacin-treated cells (fig. S9C). These effects were mirrored by the up-regulation of Ucp1 mRNA at the end of the differentiation (fig. S9D). miR-34a-3p was not affected by the enoxacin treatment and neither was its target Frat1 (fig. S9, E and F). Moreover, P53—a master regulator of pri-miR-34a transcription (42)—was not changed by enoxacin (fig. S9, G to I). These results suggest that enoxacin primarily controls the levels of the 5p strand of miR-34a.
To link miR-34a-5p down-regulation to metabolic changes in adipocytes, we looked for validated target genes of this miRNA among components of the FGF21 signaling pathway, which is known to induce PGC1α and promote oxidative metabolism. Consistent with the down-regulation of miR-34a-5p, enoxacin treatment increased the expression of the known miR-34a-5p targets Fgfr1, Klb, and Sirt1 (42) in murine 9W beige adipocytes (Fig. 6C), human adipose–differentiated hASC beige adipocytes (Fig. 6D), murine C3H10T1/2 adipocytes (fig. S8, N and O), and mouse scWAT (fig. S9, J to L). In the last two models, the effects of enoxacin were entirely dependent on Dicer (fig. S8, C and M to Q). Enoxacin-treated 9W beige adipocytes were also more sensitive to FGF21 (Fig. 6, E and F), confirming that enoxacin promotes FGF21 signaling. Moreover, enoxacin treatment induces a fourfold increase in Fgf21 mRNA in 9W beige adipocytes (fig. S9M).
Last, to determine the requirement of miR-34a-5p to enoxacin-induced Ppargc1a up-regulation, we transfected 9W cells with a miR-34a-5p mimic (or the scramble control), and after 24 hours, we induced beige differentiation in the presence or absence of enoxacin. Once again, enoxacin treatment significantly increased the levels of Fgfr1 and Ppargc1a in the control group (Fig. 6, G and H); however, these effects were blunted in cells overexpressing miR-34a-5p (Fig. 6, G and H). Ucp1, on the other hand, was increased by enoxacin regardless of the overexpression of miR-34a-5p (Fig. 6I). Together, these results demonstrate that enoxacin controls the synthesis of miR-34a-5p and therefore the levels of components of the FGF21 signaling pathway including Ppargc1a, whereas it up-regulates Ucp1 through a different mechanism.
DISCUSSION
Here, we provide proof-of-principle, preclinical evidence for a new class of drugs capable of promoting oxidative metabolism and inducing uncoupled respiration in adipose tissues and skeletal muscle, at least in part, by interfering with miRNA biogenesis. We used the small-molecule enoxacin to demonstrate these effects since this molecule has been previously shown to modulate the miRNA processing pathway (27) and is safe to be administered to humans (NDA 19-616). Our study shows that enoxacin induces energy expenditure in mice and counteracts diet-induced obesity. These effects are related to the activation of BAT, induction of browning in scWAT and up-regulation of oxidative metabolism in skeletal muscle. Browning induction is also observed in human cells. Part of these effects is mediated by miRNAs, particularly a miR-34a-5p–mediated up-regulation of FGF21 signaling and induction of Ppargc1a expression.
While studies have reported a role for enoxacin inducing miRNA biogenesis (26, 27), the few studies that actually measured global levels of miRNAs in enoxacin-treated cells failed to observe an overall induction of miRNA expression. We (this study) and others (27, 28) found that most miRNAs are not affected by enoxacin, although in the latter studies, the miRNAs that were affected by the drug were mostly up-regulated. Here, we show that in adipocytes, about the same percentage of miRNAs is increased and decreased by the drug. We found a similar pattern in Caenorhabditis elegans treated with enoxacin (44), and in both adipocyte and C. elegans, miR-34-5p appears down-regulated by enoxacin, suggesting an evolutionarily conserved mechanism. In worms, while enoxacin inhibits cel-miR-34-5p, it does not affect the levels of cel-miR-34-3p (44). Likewise, we found that mmu-miR-34a-5p expression decreases within the first 30 min upon enoxacin treatment and remains down-regulated after adipocyte differentiation in the presence of the drug, while the levels of mmu-miR-34a-3p are similar to the vehicle group in differentiated cells. Considering the selective down-regulation of the 5p strand, we propose a model where enoxacin interferes with strand stability during pre-miRNA processing. The mechanisms of arm selection controlling the synthesis and stability of each miRNA strand are a matter of continuous study. Double-stranded RNA binding proteins that partner with DICER such as TRBP and PACT have been proposed to determine, at least partially, which strain in a miRNA duplex is stabilized and loaded into the RISC and which strand is discarded (29, 45). Previous reports have demonstrated a direct interaction between enoxacin and TRBP (28). Whereas the model suggested by the authors was that enoxacin favors miRNA biogenesis by stabilizing pre-miRNA/TRBP/DICER interaction, we propose that for some miRNAs like miR-34a-5p, this stabilization may also affect strand bias and result in miRNA down-regulation. Pre-miR-674 has been previously identified as one of the miRNA precursors for which strand selection is markedly altered in the absence of TRBP and PACT (29), and its 5p strand is the most down-regulated miRNA upon enoxacin treatment (174-fold, table S1), while its 3p strand is barely changed by the drug (15% decrease, table S1) in our study. On the other hand, the levels of miR-451—a miRNA processed by AGO2 using a DICER/TRBP-independent mechanism (46, 47)—is not affected by enoxacin.
Our data also show that enoxacin acts via miR-34a-5p to control the expression of several members of the FGF21 pathway, namely, Fgfr1, Sirt1, and Ppargc1a. Consequently, enoxacin increases FGF21 sensitivity. FGF21 is a major regulator of metabolism in mammals and acts in different organs to promote oxidative metabolism (16). One important function of FGF21 is promoting browning and increasing thermogenesis in UCP1-dependent and UCP1-independent manners (15). These effects are, at least in part, mediated by an increase in Ppargc1a expression in adipose tissue (13). Here, we show that Ppargc1a is up-regulated by enoxacin in various fat depots and cell lines, this up-regulation occurs in a manner that is cell autonomous and independent of the ambient temperature and is entirely dependent on miR-34a-5p.
In agreement with our data, other studies have implicated miR-34a-5p in the regulation of FGF21 signaling, browning, and metabolism (43, 48). For example, Fu et al. (43) demonstrated an inhibition of browning by miR-34a-5p during obesity and linked FGF21 signaling and PGC-1α regulation to this process. They also demonstrated that lentivirus-mediated down-regulation of miR-34a-5p in mice reduces adiposity and increases adipose tissue oxidative metabolism. Likewise, whole-body knockout of Mir34a results in increased PGC-1α levels in many tissues, including fat depots (49). miR-34a-5p also controls lipogenesis, fat accumulation, inflammation, and endoplasmic reticulum stress in liver via down-regulation of nicotinamide phosphoribosyltransferase and changes in NAD+ turnover (50). Together, these results confirm the importance of miR-34a-5p in controlling oxidative metabolism.
Nonetheless, not all the metabolic effects of enoxacin depend on miR-34a-5p. Ucp1 expression, for example, is up-regulated by enoxacin in 9W beige adipocytes regardless of the levels of miR-34a-5p. Moreover, enoxacin does not require miRNAs in scWAT mature adipocytes to induce Ucp1, as observed in the AdicerKO mice. This is in sharp contrast with the results found in BAT, where enoxacin-induced up-regulation of UCP1 depends entirely on Dicer expression in adipocytes. These results may be interpreted in two different ways. One possibility is that in beige cells, enoxacin-induced up-regulation of Ucp1 does not need miRNAs, differently from brown adipocytes. Another possibility is that enoxacin may still recruit newly formed, UCP1+ beige adipocytes in scWAT of AdicerKO mice given that the adiponectin promoter is up-regulated later during differentiation and hence knockout occurs only in mature adipocytes. In BAT, on the other hand, the turnover of adipocytes is low and up-regulation of Ucp1 by enoxacin is likely taking place in mature brown adipocytes, in which miRNAs appear to be required for the phenotype.
The up-regulation of UCP1+ beige adipocytes in scWAT of AdicerKO mice may also explain, at least in part, why enoxacin treatment improves cold tolerance in these mice despite the reduced UCP1 levels in BAT. BAT paucity may lead to compensatory beige adipocyte activation via increased sympathetic input to WAT, and this is sufficient to maintain normal temperature homeostasis in mice (51). UCP1-independent mechanisms and nonadipose-related increases in energy expenditure could also explain the thermogenic effect of enoxacin in AdicerKO mice, although Pm20d1 or genes related to the creatine and calcium futile cycles (52) are not consistently affected by enoxacin in BAT, scWAT, or skeletal muscle. Nonetheless, we found up-regulation of Ucp3 in all three tissues in response to the drug. Moreover, enoxacin promotes oxidative metabolism and increases uncoupled respiration in C2C12 myotubes. Hence, enoxacin induces energy expenditure and thermogenesis by a combination of mechanisms that involve UCP1 up-regulation in BAT, beige adipocyte recruitment in scWAT, and overall increases in oxidative metabolism in adipose tissues and muscle. These mechanisms are likely additive and redundant, which might explain the amplitude of induction of energy expenditure by enoxacin at the whole-body level.
Another potential nonadipose effect of enoxacin is its predicted antibiotic function in the microbiota. However, perhaps because of the route of administration (i.e., intraperitoneal), enoxacin treatment does not deplete fecal bacterial content or change bacterial diversity. Consistent with these results, the up-regulation of UCP1 protein and Ppargc1a mRNA in BAT is still observed in germ-free mice treated with enoxacin. We thus conclude that intraperitoneal injections with enoxacin do not alter the composition of the gut microbiome. Furthermore, enoxacin does not act through the microbiome to enhance UCP1 and Ppargc1a levels in BAT.
Fluoroquinolones have also been reported to have many different effects on eukaryotic cells, and it is unlikely that all of these effects involve miRNAs. For instance, fluoroquinolones affect mitochondrial metabolism in pancreatic β cells and contribute to hyperglycemic episodes (53). The effect of fluoroquinolones over mitochondria may also lead to increases in oxidative stress markers, such as ROS, lipid peroxidation, and oxidized glutathione (54). Continuous use of some fluoroquinolones may cause rare, disabling side effects in individuals (55). One of them is insomnia (56), although, in our study, mice chronically treated with enoxacin display the same ambulatory activity during daytime and nighttime than vehicle-treated mice, suggesting that they do not suffer from alterations in sleep/wake cycle. Another common undesired effect among thermogenic drugs is increased heart rate and elevated systolic blood pressure, although we found no increases in these parameters in enoxacin-treated mice. Despite the potential side effects, fluoroquinolones are relatively safe to use in humans, and the fact that enoxacin also induces browning in human cells raises the possibility of repurposing and developing a new class of drugs to treat obesity complications in humans, ideally with no antibiotic effect.
As recently reported, approximately 32 million of fluoroquinolone prescriptions are registered annually in the United States, making these drugs one of the most popular classes of antibiotics (55). Here, we demonstrate that chronic treatment of preclinical models with the fluoroquinolone enoxacin induces oxidative metabolism, increases energy expenditure, and protects from diet-induced obesity through a mechanism that involves, at least in part, miRNA regulation in adipose tissue. This study contributes to the elucidation of the metabolic effects of fluoroquinolones and serves as proof of principle for the development of therapies targeting the miRNA processing pathway in the treatment of obesity and its comorbidities.
METHODS
Animal procedures
Twelve-week-old male C57BL/6 and AdicerKO and their floxed littermates (Lox) were obtained from the Centro de Desenvolvimento de Modelos Experimentais para Medicina e Biologia at Federal University of São Paulo (São Paulo, Brazil) or the Multidisciplinary Center for Biological Research at University of Campinas (Campinas, Brazil). AdicerKO were generated by breeding Dicerlox/lox mice with mice carrying the Cre recombinase driven by the adiponectin promoter (22). Experiments with germ-free mice animals were performed in the Department of Microbiology, Institute of Biological Science of the Federal University of Minas Gerais. Male germ-free Swiss mice were derived from germ-free nucleus (Taconic Farms, Germantown, NY) and maintained in flexible plastic isolators (Standard Safety Equipment, Marietta, OH) using classical gnotobiology techniques. All experimental procedures using germ-free were conducted under aseptic conditions. Animal procedures were performed in accordance with standards established by the Ethics Committee on Animal Use of the Federal University of São Paulo (CEUA UNIFESP, 1932250915/2016) and University of Campinas (CEUA UNICAMP, 4749-1/2017 and 5183-1/2019). Mice were maintained on a 12-hour/12-hour light/dark cycle, at 22° ± 2°C, with ad libitum access to tap water (reverse osmosis purified) and chow diet (Nuvilab CR1 Radiated diet) unless stated otherwise. Enoxacin (100 mg/ml; Sigma-Aldrich) was initially dissolved in 1 M NaOH (Sigma-Aldrich) solution to a concentration of 312 mM and then diluted 100× in phosphate-buffered saline (PBS) for injection. Vehicle was 10 mM NaOH in PBS.
For the acute enoxacin treatment, 16-week-old male C57BL/6JUnib, germ-free Swiss mice and AdicerKO mice (and their littermate floxed controls) were injected for up to 10 consecutive days with 10 mg/kg body weight per day of enoxacin or vehicle. For the thermoneutrality experiment, mice were adapted to 30°C in a climate chamber (HPP750life) for 5 days before enoxacin or vehicle injections (injected intraperitoneally every day for 3 days) and were maintained at this temperature during the whole procedure. For gut microbiome depletion, antibiotics were administered via gavage every day for 5 days. In the last 3 days of the treatment with the antibiotic mix, enoxacin or vehicle were injected intraperitoneally every day in parallel with the gavage. The antibiotic cocktail contained neomycin (50 mg/ml), gentamicin (50 mg/ml), ampicillin (50 mg/ml), metronidazole (50 mg/ml), and vancomycin (25 mg/ml). For the HFD studies, 12-week-old mice were given a 60% HFD (PragSoluções) until the day of sacrifice. After 4 weeks on HFD, animals were treated daily (five intraperitoneal injections/week) with enoxacin at a dose of 10 mg/kg body weight for 10 weeks. Body weight and food intake were measured weekly. Fecal energy content (calories/g) was measured by calorimetry at the Labcontrol facility (Labcontrol, São Paulo, Brazil). Feces were collected daily, pooled, and dried at 50°C before analysis. At sacrifice, tissues and feces were collected, snap-frozen in N2, and stored at −80°C.
Insulin sensitivity and glucose tolerance
Glucose and insulin tolerance tests were performed 1 week before euthanasia and followed the protocols described elsewhere (22). Briefly, mice were injected intraperitoneally with glucose (1 g/kg body weight) after overnight fasting or insulin (0.75 U/kg body weight; Humalog, Eli Lilly) after 2-hour fasting. Blood glucose was measured at the indicated time points using a digital glucometer.
Serum assays
Total cholesterol and HDL cholesterol were measured using commercial colorimetric kits obtained from Labtest (Lagoa Santa, MG, Brazil). Insulin levels were quantified using Rat/Mouse Insulin ELISA Millipore Sigma (ref. EZRMI-13K). The protocols followed the manufacturer’s instructions.
Metabolic rate
Following procedures previously described in (22), we used the Oxymax OPTO-M3 system (Columbus Instruments) to assess basal metabolic rate in mice treated for 9 weeks with enoxacin. Rates of oxygen consumption (VO2), CO2 production (VCO2), RER, heat, and spontaneous activity were determined. For β3-adrenergic receptor stimulation in vivo, mice were injected intraperitoneally with CL-316,243 (1 mg/kg; Sigma-Aldrich). For cold exposure, mice were housed in a cold room at 6°C, and the temperature was measured at the indicated time points using a RET-3 rectal probe (Physitemp).
Tail cuff
Mouse CODA tail-cuff system (Kent Scientific Corp., Torrington, CT) was used to estimate systolic blood pressure values. Mouse tail was placed in a cuff to occlude blood flow, and a pressure sensor was used to measure systolic blood pressure and heart rate.
Citrate synthase activity
Subcutaneous and BAT samples were homogenized in 0.1 M potassium phosphate buffer with 5 mM EDTA using a Dounce grinder. The homogenate was centrifuged at 10,000g for 10 min at 4°C to remove debris and excessive fat, and the aqueous phase was used for protein quantification (BCA kit, Pierce) and determination of citrate synthase activity as previously described (20) using 5 to 10 μg of total homogenate.
Histological analyses
Tissues were freshly fixed in 10% paraformaldehyde-PBS, dehydrated by increasing ethanol concentrations, washed in xylene, and then embedded in paraffin. Paraffin blocks were sectioned and stained with hematoxylin and eosin.
Cell culture and adipocyte differentiation
Mouse cells
Murine 3T3-F442A, C3H10T1/2, white subcutaneous (9W), and brown (9B) preadipocytes (18) were cultured until confluence and differentiated with Dulbecco’s modified Eagle’s medium (DMEM; Sigma-Aldrich) containing 20 nM insulin, 1 nM triiodothyronine, 0.5 mM isobutyl methylxanthine, 1 μM dexamethasone, and 0.125 mM indomethacin according to the protocol published elsewhere (57, 58). Cells were treated with vehicle (NaOH) or enoxacin (50 μM) throughout the differentiation process (days 2 to 8) or during 24 hours after the last day of differentiation (day 8). C2C12 undifferentiated myoblasts were cultivated in DMEM (high glucose) containing fetal bovine serum (FBS) (10%). At 70 to 90% of confluency, the medium was switched to DMEM (high glucose) containing horse serum (2%) to induce differentiation. Enoxacin at 100 μM or vehicle (NaOH) was directly added into the differentiation medium. Differentiation medium containing the compound or vehicle was changed every 2 days until the formation of myotubes, after 4 days.
Human cells
For hASC isolation and adipocyte differentiation, we followed procedures previously described in (59). Briefly, the cells were obtained from human subcutaneous adipose tissue collected from the abdominal region of a female patient undergoing elective surgery at Sahlgrenska University Hospital in Gothenburg, Sweden. The study subject gave written informed consent for the use of the tissue, and the study was approved by The Regional Ethical Review Board in Gothenburg, Sweden. After becoming confluent, cells were differentiated in basal medium (Zenbio BM-1) with 3% FBS Gold (PAA-A15-104), 1% penicillin/streptomycin, 0.5 mM IBMX (Sigma-Aldrich), 0.1 μM dexamethasone (Sigma-Aldrich), 20 nM insulin (Sigma-Aldrich), 1 nM T3 (3,3″,5-triiodo-l-thyronine sodium, Sigma-Aldrich), and BMP4 (10 ng/ml; R&D systems). After 7 days, the medium was replaced with the same medium without IBMX. On day 10, cells were treated for an additional 10 days with either vehicle (NaOH) or enoxacin (50 μM) in the same medium.
Gain or loss of function
For Dicer knockdown, C3H10T1/2 cells were transduced with pLKO.1 lentivirus carrying a short hairpin targeting Dicer, or GFP as control (Dharmacon), and selected with puromycin. We transfected the hsa-miR-34a-5p mimic or the scramble control (MISSION miRNA Mimic, Sigma-Aldrich) in 9W white preadipocytes using the HiPerFect Transfection Reagent (Qiagen) following the manufacturer’s instructions. After 24 hours, the culture medium (DMEM, 10% FBS, and 1% penicillin/streptomycin) was changed, and preadipocytes were subjected to differentiation.
Triglyceride quantification
Differentiated adipocytes were washed once with PBS, lysed with 10% NP-40 solution in deionized water, and frozen. For the reaction, 200 μl of the reagent TG (Labtest Diagnostica) was added to 2 μl of lysate, and the absorbance of 505 nm was measured after 30 min of incubation at 37°C. Triglyceride concentration was determined using a standard and normalized by protein concentration as measured by the BCA kit.
Cell viability
Cells were treated with enoxacin in different concentrations. Twenty-four hours after treatment, cells were incubated for 4 hours in culture medium containing the 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reagent (0.5 mg/ml; Sigma-Aldrich). After 4 hours, the medium was removed followed by addition of dimethyl sulfoxide to lyse the cells and release the formazan product. After 30 min at 37°C, the absorbance was read at 570 nm. Cell viability was also assessed by flow cytometry. After enoxacin treatment, the supernatant was collected, and cells were treated with trypsin. Cells were centrifuged at 300g for 5 min, resuspended in 200 μl of PBS + 2% FBS, and transferred to a 96-well plate followed by a new centrifugation step. The supernatant was removed, and the cells were resuspended in 25 μl of the fluorescent dye Zombie AquaTM Fixable Viability Kit (BioLegend) diluted 1:1000 in PBS and incubated for 20 min at 4°C. Cells were then washed with 100 μl of PBS + 2% FBS and resuspended in 200 μl of PBS + 2% FBS. Unstained cells were used as a negative control. Cells were counted using the BD FACSVerse flow cytometer (BD Biosciences) and quantified using FlowJo software.
Staining and microscopy
For mitochondrial staining, 9W cells were resuspended in culture medium and stained with MitoTracker Deep Red FM (50 nM) (Thermo Fisher Scientific) at 37°C for 30 min. After washing with PBS, the cells were then stained with Hoechst 33258 for 5 min and visualized by confocal laser scanning microscopy (LSM 780 NLO, Carl Zeiss). For lipid droplet staining, 9W cells were fixed in 4% paraformaldehyde in PBS for 30 min at room temperature. Cells were then rinsed twice with PBS, incubated for 30 min in 1∶200 dilution with PBS of LipidTOX Green (Invitrogen), then rinsed twice with distilled water, and visualized by confocal laser scanning microscopy. FLIM was performed according to protocol previously described (60) and using a LSM780 NLO confocal microscopy equipped with a femtosecond pulsed laser and a time-correlated single photon counting system (Becker & Hickel).
Oxygen consumption
For oxygen consumption measurement, 9W or C2C12 cells were trypsinized, and 1 × 106 cells were resuspended in Krebs buffer [130 mM NaCl, 4.7 mM KCl, 1.24 mM MgSO4, 2.5 mM CaCl2, 10 mM Hepes, 2.5 mM NaH2PO4, 5 mM d-glucose, and 2% bovine serum albumin (pH 7.4)] or differentiation medium supplemented with Hepes 20 mM, respectively. After the acquisition of the initial reading, the Oroboros chamber was closed and oxygen consumption (oxygen flow) was monitored, followed by the addition of oligomycin (1 μM), CCCP (2 μM) for 9W, or carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP) (2 μM) for C2C12 and rotenone/antimycin (1 μM). Data were acquired and analyzed by DatLab 4 software (Oroboros Instruments). ATP-linked respiration was calculated by subtracting the uncoupled (after the addition of oligomycin) from the basal oxygen consumption rate (OCR). Maximum respiratory capacity was determined after CCCP or FCCP addition, and proton leak was determined by subtracting the nonmitochondrial respiration from the uncoupled respiration (after oligomycin addition). Spare capacity was determined by subtracting basal from the CCCP- or FCCP-induced OCR.
ROS measurements
Cytosolic and mitochondrial ROS were detected using a dihydroethidium (Thermo Fisher Scientific) for the detection of superoxide and MitoSOX Red (Thermo Fisher Scientific) for the detection of mitochondrial superoxide. Staining was performed according to the manufacturer.
miRNA sequencing
miRNAs were sequenced as described in Pinto et al. (44). Sequencing data were analyzed using CLC Genomics Workbench 6.0.1 software (CLC Bio). Briefly, adapter sequences were trimmed, and the remaining sequences were mapped against the mouse miRNA database, miRBase (release 21), allowing no more than one mismatch. Known mature miRNAs were annotated according to miRBase. Normalization was performed as follows: for a given sample, the number of sequences of each mature miRNA was divided by the total number of sequences mapped to miRNAs and multiplied by one million (counts per million).
Dicer enzymatic assays
To monitor DICER catalytic activity, in vitro assays were performed as described by Davies and Arenz (61) using 20 mM tris-HCl (pH 7.4), 12 mM NaCl, 2.5 mM MgCl2. As a substrate for the reaction, we used 100 nM of a pre-miRNA probe linked to a fluorophore moiety (FAM-EX-5-GGCAAAUUGAGGUAGUAGGUUGUAUAGUAGUAAUUA) at the 5′-end and a nonfluorescent quencher moiety (phosphate CACAUCAUACUAUACAAUGUGCUAGCUUUCUUUGCU-DABCYL) at the 3′-end. Fluorescence was detected real-time upon DICER-mediated cleavage. As a DICER source, we used the extract obtained from 9W cells differentiated in the presence of 50 μM enoxacin or vehicle. Cells were harvested in SDS-free radioimmunoprecipitation assay buffer, protein concentration was determined by BCA (Thermo Fisher Scientific), and the chosen concentration used in the assays was determined on the basis of the results of the enzymatic assay after performing serial dilutions of the cell extract. Cell extracts were added to the reaction and incubated for 30 min at room temperature. The increase in fluorescence was accompanied every minute for 4 hours.
Western blotting and RT-qPCR
Western blotting and RT-qPCR were performed as previously described (24). Antibodies were as follows: Erk (9102), phospho-Erk (9101), p53 (9282), phospho-p53 (Ser15, 9284), and β-tubulin (2146) from Cell Signaling; Vinculin (ab18058) and Dicer (ab13502) from Abcam; and UCP1 from Abcam (ab10983) and Cell Signaling (14670S). Primer sequences are available upon request. For miRNA quantification, we used TRIzol reagent (Life Technologies) to isolate RNA and the miScript II RT kit to generate cDNA (Qiagen). For that, we followed the manufacturer’s instructions and used the HiSpec buffer and 400 ng of total RNA. After cDNA synthesis, miRNA expression was quantified by qPCR using the corresponding miScript primer assay (Qiagen).
Mitochondrial DNA quantification
For DNA extraction, we used the QIAamp DNA Micro Kit (Qiagen), following the manufacturer’s instructions. qPCR analysis was used to measure mitochondrial gene mtCox1 and nuclear gene nNdufs as previously described (24).
Microbiota quantification
For stool bacterial load quantification, total DNA was extracted using the QIAamp DNA Stool Mini Kit (Qiagen), according to the manufacturer’s protocol and subjected to qPCR using primers targeting 16S ribosomal RNA universal primer (forward: AAACTCAAAKGAATTGACGG; reverse: CTCACRRCACGAGCTGAC), specific against Firmicutes (forward: TGAAACTYAAAGGAATTGACG; reverse: ACCATGCACCACCTGTC) or specific against Bacteroidetes (forward: CRAACAGGATTAGATACCCT; reverse: GGTAAGGTTCCTCGCGTAT).
Microbiome analyses were performed by Neoprospecta (https://neoprospecta.com). The V3/V4 regions of the 16S ribosomal gene were amplified by PCR using the primers 341F (CCTACGGGRSGCAGCAG) and 806R (GGACTACHVGGGTWTCTAAT) and sequenced using MiSeq Sequencing System (Illumina Inc., USA). A 305-nt single-end run was performed using a V2x300 sequencing kit. Read quality was evaluated using FastQC software (version 0.11.8) and imported into QIIME2 version 2020.2 (https://qiime2.org) for further analysis. Sequencing reads were denoised and clustered into amplicon sequence variants using DADA2. The sequence variants were taxonomically classified against SILVA version 138 reference database, normalized into relative abundance (%), and plotted using the QIIME2 taxa plugin. Relative abundance of Amplicon sequence variants (ASVs) at each taxonomic level was compared using ANCOM test. Beta diversity (Bray Curtis distance) was calculated at an even sampling depth of 20,000 sequences per sample. The significance of the differences between groups was tested by Permutational multivariate analysis of variance (PERMANOVA), with 999 permutations used to calculate P values. The data are deposited at National Center for Biotechnology Information (NCBI)’s BioProject database (PRJNA660801).
Statistics
Results are expressed as means ± SEM. We used nonparametric Mann-Whitney U test or unpaired t test with Welch’s correction to compare two independent groups depending on the number of samples per group. We used analysis of variance (ANOVA) to compare more than two groups. Two-way ANOVA was used when data had more than one categorical independent variable. Statistical significance was considered when P < 0.05.
Supplementary Material
Acknowledgments
We are grateful for the technical assistance of E. E. Saviani, R. G. Ludwig, V. N. Sato, B. B. Brandão, and S. P. da Silva. We thank J. Ferreira and L. A. Kiyuna for providing the substrate for the DICER activity assay. We are also thankful to A. S. Martins for performing pilot experiments and for generating preliminary data. Funding: This research was supported by grants from FAPESP (2017/01184-9, 2017/07975-8, 2016/12294-7, 2010/52557-0, 2016/24163-4, 2016/23142-3, 2018/15313-8, and 2015/01316-7), CNPq (474397/2011-4 and 305069/2015-2), CAPES (1431744/2014-2016), and the Ecole Polytechnique Fédérale de Lausanne. INFABiC is co-funded by FAPESP and CNPq. Author contributions: Conceptualization: A.L.R. and M.A.M. Methodology: A.L.R., T.I.L., G.P.S., R.O.C., D.L.F., B.R., C.L.-R., D.N., T.L.K., P.R.C., and M.F.F. Validation: A.L.R., G.P.S., and T.L.K. Formal analysis: A.L.R., T.I.L., G.P.S., D.L.F., B.R., C.-L.R., D.N., T.L.K., P.R.C., M.F.F., M.A.R.V., J.B., and M.A.M. Investigation: A.L.R., T.I.L., G.P.S., R.O.C., D.L.F., B.R., C.L.-R., D.N., T.L.K., P.R.C., M.A.R.V., J.B., and M.A.M. Resources: F.S.M., R.B.P., L.R.S., H.F.C., J.A., M.A.R.V., J.B., and M.A.M. Writing—original draft: A.L.R. and M.A.M. Writing—review and editing: All authors. Visualization: A.L.R., M.A.R.V., and M.A.M. Supervision: R.B.P., L.R.S., H.F.C., J.A., M.A.R.V., J.B., and M.A.M. Project administration: M.A.M. Funding acquisition: F.S.M., R.B.P., L.R.S., H.F.C., J.A., M.A.R.V., J.B., and M.A.M. Competing interests: M.A.M. and A.L.R. are inventors on a pending patent application related to this work filed by Instituto Nacional de Propriedade Intelectual (INPI) and deposited by Universidade Federal de São Paulo - UNIFESP (no. BR 10 2015 016473 4, filed 8 July 2015). The authors declare no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Metagenomics data are deposited at NCBI’s BioProject database (PRJNA660801). Raw data, primer sequences, reagents, and additional information related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/49/eabc6250/DC1
REFERENCES AND NOTES
- 1.World Health Organization. Obesity and Overweigh. Geneva: World Health Organization (2016). [Google Scholar]
- 2.Cypess A. M., Kahn C. R., The role and importance of brown adipose tissue in energy homeostasis. Curr. Opin. Pediatr. 22, 478–484 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu J., Boström P., Sparks L. M., Ye L., Choi J. H., Giang A. H., Khandekar M., Virtanen K. A., Nuutila P., Schaart G., Huang K., Tu H., van Marken Lichtenbelt W. D., Hoeks J., Enerbäck S., Schrauwen P., Spiegelman B. M., Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell 150, 366–376 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Golozoubova V., Cannon B., Nedergaard J., UCP1 is essential for adaptive adrenergic nonshivering thermogenesis. Am. J. Physiol. Endocrinol. Metab. 291, E350–E357 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Cinti S., Cancello R., Zingaretti M. C., Ceresi E., De Matteis R., Giordano A., Himms-Hagen J., Ricquier D., CL316,243 and cold stress induce heterogeneous expression of UCP1 mRNA and protein in rodent brown adipocytes. J. Histochem. Cytochem. 50, 21–31 (2002). [DOI] [PubMed] [Google Scholar]
- 6.Bostrom P., Wu J., Jedrychowski M. P., Korde A., Ye L., Lo J. C., Rasbach K. A., Boström E. A., Choi J. H., Long J. Z., Kajimura S., Zingaretti M. C., Vind B. F., Tu H., Cinti S., Højlund K., Gygi S. P., Spiegelman B. M., A PGC1-α-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 481, 463–468 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee P., Bova R., Schofield L., Bryant W., Dieckmann W., Slattery A., Govendir M. A., Emmett L., Greenfield J. R., Brown adipose tissue exhibits a glucose-responsive thermogenic biorhythm in humans. Cell Metab. 23, 602–609 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Bartelt A., Bruns O. T., Reimer R., Hohenberg H., Ittrich H., Peldschus K., Kaul M. G., Tromsdorf U. I., Weller H., Waurisch C., Eychmüller A., Gordts P. L. S. M., Rinninger F., Bruegelmann K., Freund B., Nielsen P., Merkel M., Heeren J., Brown adipose tissue activity controls triglyceride clearance. Nat. Med. 17, 200–205 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Wang C. H., Lundh M., Fu A., Kriszt R., Huang T. L., Lynes M. D., Leiria L. O., Shamsi F., Darcy J., Greenwood B. P., Narain N. R., Tolstikov V., Smith K. L., Emanuelli B., Chang Y.-T., Hagen S., Danial N. N., Kiebish M. A., Tseng Y.-H., CRISPR-engineered human brown-like adipocytes prevent diet-induced obesity and ameliorate metabolic syndrome in mice. Sci. Transl. Med. 12, eaaz8664 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cypess A. M., Weiner L. S., Roberts-Toler C., Elía E. F., Kessler S. H., Kahn P. A., English J., Chatman K., Trauger S. A., Doria A., Kolodny G. M., Activation of human brown adipose tissue by a β3-adrenergic receptor agonist. Cell Metab. 21, 33–38 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ohno H., Shinoda K., Spiegelman B. M., Kajimura S., PPARγ agonists induce a white-to-brown fat conversion through stabilization of PRDM16 protein. Cell Metab. 15, 395–404 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Puigserver P., Wu Z., Park C. W., Graves R., Wright M., Spiegelman B. M., A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 92, 829–839 (1998). [DOI] [PubMed] [Google Scholar]
- 13.Fisher F. M., Kleiner S., Douris N., Fox E. C., Mepani R. J., Verdeguer F., Wu J., Kharitonenkov A., Flier J. S., Maratos-Flier E., Spiegelman B. M., FGF21 regulates PGC-1α and browning of white adipose tissues in adaptive thermogenesis. Genes Dev. 26, 271–281 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hondares E., Iglesias R., Giralt A., Gonzalez F. J., Giralt M., Mampel T., Villarroya F., Thermogenic activation induces FGF21 expression and release in brown adipose tissue. J. Biol. Chem. 286, 12983–12990 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee P., Linderman J. D., Smith S., Brychta R. J., Wang J., Idelson C., Perron R. M., Werner C. D., Phan G. Q., Kammula U. S., Kebebew E., Pacak K., Chen K. Y., Celi F. S., Irisin and FGF21 are cold-induced endocrine activators of brown fat function in humans. Cell Metab. 19, 302–309 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chau M. D., Gao J., Yang Q., Wu Z., Gromada J., Fibroblast growth factor 21 regulates energy metabolism by activating the AMPK-SIRT1-PGC-1α pathway. Proc. Natl. Acad. Sci. U.S.A. 107, 12553–12558 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wei W., Dutchak P. A., Wang X., Ding X., Wang X., Bookout A. L., Goetz R., Mohammadi M., Gerard R. D., Dechow P. C., Mangelsdorf D. J., Kliewer S. A., Wan Y., Fibroblast growth factor 21 promotes bone loss by potentiating the effects of peroxisome proliferator-activated receptor γ. Proc. Natl. Acad. Sci. U.S.A. 109, 3143–3148 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moniotte S., Kobzik L., Feron O., Trochu J. N., Gauthier C., Balligand J. L., Upregulation of β3-adrenoceptors and altered contractile response to inotropic amines in human failing myocardium. Circulation 103, 1649–1655 (2001). [DOI] [PubMed] [Google Scholar]
- 19.Jones J. R., Barrick C., Kim K.-A., Lindner J., Blondeau B., Fujimoto Y., Shiota M., Kesterson R. A., Kahn B. B., Magnuson M. A., Deletion of PPARgamma in adipose tissues of mice protects against high fat diet-induced obesity and insulin resistance. Proc. Natl. Acad. Sci. U.S.A. 102, 6207–6212 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bartel D. P., MicroRNAs: Target recognition and regulatory functions. Cell 136, 215–233 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ha M., Kim V. N., Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 15, 509–524 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Mori M. A., Thomou T., Boucher J., Lee K. Y., Lallukka S., Kim J. K., Torriani M., Yki-Järvinen H., Grinspoon S. K., Cypess A. M., Kahn C. R., Altered miRNA processing disrupts brown/white adipocyte determination and associates with lipodystrophy. J. Clin. Invest. 124, 3339–3351 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim H. J., Cho H., Alexander R., Patterson H. C., Gu M., Lo K. A., Xu D., Goh V. J., Nguyen L. N., Chai X., Huang C. X., Kovalik J. P., Ghosh S., Trajkovski M., Silver D. L., Lodish H., Sun L., MicroRNAs are required for the feature maintenance and differentiation of brown adipocytes. Diabetes 63, 4045–4056 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reis F. C., Branquinho J. L. O., Brandão B. B., Guerra B. A., Silva I. D., Frontini A., Thomou T., Sartini L., Cinti S., Kahn C. R., Festuccia W. T., Kowaltowski A. J., Mori M. A., Fat-specific Dicer deficiency accelerates aging and mitigates several effects of dietary restriction in mice. Aging 8, 1201–1222 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brandão B. B., Guerra B. A., Mori M. A., Shortcuts to a functional adipose tissue: The role of small non-coding RNAs. Redox Biol. 12, 82–102 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Q., Zhang C., Xi Z., Enhancement of RNAi by a small molecule antibiotic enoxacin. Cell Res. 18, 1077–1079 (2008). [DOI] [PubMed] [Google Scholar]
- 27.Shan G., Li Y., Zhang J., Li W., Szulwach K. E., Duan R., Faghihi M. A., Khalil A. M., Lu L., Paroo Z., Chan A. W. S., Shi Z., Liu Q., Wahlestedt C., He C., Jin P., A small molecule enhances RNA interference and promotes microRNA processing. Nat. Biotechnol. 26, 933–940 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Melo S., Villanueva A., Moutinho C., Davalos V., Spizzo R., Ivan C., Rossi S., Setien F., Casanovas O., Simo-Riudalbas L., Carmona J., Carrere J., Vidal A., Aytes A., Puertas S., Ropero S., Kalluri R., Croce C. M., Calin G. A., Esteller M., Small molecule enoxacin is a cancer-specific growth inhibitor that acts by enhancing TAR RNA-binding protein 2-mediated microRNA processing. Proc. Natl. Acad. Sci. U.S.A. 108, 4394–4399 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wilson R. C., Tambe A., Kidwell M. A., Noland C. L., Schneider C. P., Doudna J. A., Dicer-TRBP complex formation ensures accurate mammalian microRNA biogenesis. Mol. Cell 57, 397–407 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sousa E., Graça I., Baptista T., Vieira F. Q., Palmeira C., Henrique R., Jerónimo C., Enoxacin inhibits growth of prostate cancer cells and effectively restores microRNA processing. Epigenetics 8, 548–558 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abell N. S., Mercado M., Caneque T., Rodriguez R., Xhemalce B., Click quantitative mass spectrometry identifies PIWIL3 as a mechanistic target of RNA interference activator enoxacin in cancer cells. J. Am. Chem. Soc. 139, 1400–1403 (2017). [DOI] [PubMed] [Google Scholar]
- 32.Emde A., Eitan C., Liou L. L., Libby R. T., Rivkin N., Magen I., Reichenstein I., Oppenheim H., Eilam R., Silvestroni A., Alajajian B., Ben-Dov I. Z., Aebischer J., Savidor A., Levin Y., Sons R., Hammond S. M., Ravits J. M., Möller T., Hornstein E., Dysregulated miRNA biogenesis downstream of cellular stress and ALS-causing mutations: A new mechanism for ALS. EMBO J. 34, 2633–2651 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kasuya F., Miwa Y., Kazumi M., Inoue H., Ohta H., Effect of enoxacin, felbinac, and sparfloxacin on fatty acid metabolism and glucose concentrations in rat tissues. Int. J. Toxicol. 30, 367–376 (2011). [DOI] [PubMed] [Google Scholar]
- 34.Hori S., Kizu J., Kawamura M., Effect of fluoroquinolones on plasma glucose levels in fasted and glucose-loaded mice. J. Infect. Chemother. 12, 109–111 (2006). [DOI] [PubMed] [Google Scholar]
- 35.Smalheiser N. R., Zhang H., Dwivedi Y., Enoxacin elevates microRNA levels in rat frontal cortex and prevents learned helplessness. Front. Psych. 5, 6 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nelson R. R., Gobel F. L., Jorgensen C. R., Wang K., Wang Y., Taylor H. L., Hemodynamic predictors of myocardial oxygen consumption during static and dynamic exercise. Circulation 50, 1179–1189 (1974). [DOI] [PubMed] [Google Scholar]
- 37.Fischer A. W., Cannon B., Nedergaard J., Optimal housing temperatures for mice to mimic the thermal environment of humans: An experimental study. Mol. Metab. 7, 161–170 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen Y., Siegel F., Kipschull S., Haas B., Fröhlich H., Meister G., Pfeifer A., miR-155 regulates differentiation of brown and beige adipocytes via a bistable circuit. Nat. Commun. 4, 1769 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Trajkovski M., Ahmed K., Esau C. C., Stoffel M., MyomiR-133 regulates brown fat differentiation through Prdm16. Nat. Cell Biol. 14, 1330–1335 (2012). [DOI] [PubMed] [Google Scholar]
- 40.Mori M. A., Raghavan P., Thomou T., Boucher J., Robida-Stubbs S., Macotela Y., Russell S. J., Kirkland J. L., Blackwell T. K., Kahn C. R., Role of microRNA processing in adipose tissue in stress defense and longevity. Cell Metab. 16, 336–347 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun L., Xie H., Mori M. A., Alexander R., Yuan B., Hattangadi S. M., Liu Q., Kahn C. R., Lodish H. F., Mir193b-365 is essential for brown fat differentiation. Nat. Cell Biol. 13, 958–965 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Raver-Shapira N., Marciano E., Meiri E., Spector Y., Rosenfeld N., Moskovits N., Bentwich Z., Oren M., Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol. Cell 26, 731–743 (2007). [DOI] [PubMed] [Google Scholar]
- 43.Fu T., Seok S., Choi S., Huang Z., Suino-Powell K., Xu H. E., Kemper B., Kemper J. K., MicroRNA 34a inhibits beige and brown fat formation in obesity in part by suppressing adipocyte fibroblast growth factor 21 signaling and SIRT1 function. Mol. Cell. Biol. 34, 4130–4142 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pinto S., Sato V. N., De-Souza E. A., Ferraz R. C., Camara H., Pinca A. P. F., Mazzotti D. R., Lovci M. T., Tonon G., Lopes-Ramos C. M., Parmigiani R. B., Wurtele M., Massirer K. B., Mori M. A., Enoxacin extends lifespan of C. elegans by inhibiting miR-34-5p and promoting mitohormesis. Redox Biol. 18, 84–92 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim Y., Yeo J., Lee J. H., Cho J., Seo D., Kim J.-S., Kim V. N., Deletion of human tarbp2 reveals cellular microRNA targets and cell-cycle function of TRBP. Cell Rep. 9, 1061–1074 (2014). [DOI] [PubMed] [Google Scholar]
- 46.Cifuentes D., Xue H., Taylor D. W., Patnode H., Mishima Y., Cheloufi S., Ma E., Mane S., Hannon G. J., Lawson N. D., Wolfe S. A., Giraldez A. J., A novel miRNA processing pathway independent of Dicer requires Argonaute2 catalytic activity. Science 328, 1694–1698 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cheloufi S., Dos Santos C. O., Chong M. M., Hannon G. J., A dicer-independent miRNA biogenesis pathway that requires Ago catalysis. Nature 465, 584–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Han H.-S., Choi B. H., Kim J. S., Kang G., Koo S.-H., Hepatic Crtc2 controls whole body energy metabolism via a miR-34a-Fgf21 axis. Nat. Commun. 8, 1878 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lavery C. A., Kurowska-Stolarska M., Holmes W. M., Donnelly I., Caslake M., Collier A., Baker A. H., Miller A. M., miR-34a−/− mice are susceptible to diet-induced obesity. Obesity 24, 1741–1751 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Choi S. E., Fu T., Seok S., Kim D. H., Yu E., Lee K. W., Kang Y., Li X., Kemper B., Kemper J. K., Elevated microRNA-34a in obesity reduces NAD+ levels and SIRT1 activity by directly targeting NAMPT. Aging Cell 12, 1062–1072 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schulz T. J., Huang P., Huang T. L., Xue R., McDougall L. E., Townsend K. L., Cypess A. M., Mishina Y., Gussoni E., Tseng Y.-H., Brown-fat paucity due to impaired BMP signalling induces compensatory browning of white fat. Nature 495, 379–383 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chang S.-H., Song N.-J., Choi J. H., Yun U. J., Park K. W., Mechanisms underlying UCP1 dependent and independent adipocyte thermogenesis. Obes. Rev. 20, 241–251 (2019). [DOI] [PubMed] [Google Scholar]
- 53.Ghaly H., Jörns A., Rustenbeck I., Effect of fluoroquinolones on mitochondrial function in pancreatic beta cells. Eur. J. Pharm. Sci. 52, 206–214 (2014). [DOI] [PubMed] [Google Scholar]
- 54.Kalghatgi S., Spina C. S., Costello J. C., Liesa M., Morones-Ramirez J. R., Slomovic S., Molina A., Shirihai O. S., Collins J. J., Bactericidal antibiotics induce mitochondrial dysfunction and oxidative damage in Mammalian cells. Sci. Transl. Med. 5, 192ra185 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Marchant J., When antibiotics turn toxic. Nature 555, 431–433 (2018). [DOI] [PubMed] [Google Scholar]
- 56.Kandasamy A., Srinath D., Levofloxacin-induced acute anxiety and insomnia. J. Neurosci. Rural Pract. 3, 212–214 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Marinovic M. P., Campeiro J. D., Lima S. C., Rocha A. L., Nering M. B., Oliveira E. B., Mori M. A., Hayashi M. A. F., Crotamine induces browning of adipose tissue and increases energy expenditure in mice. Sci. Rep. 8, 5057 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Macotela Y., Boucher J., Tran T. T., Kahn C. R., Sex and depot differences in adipocyte insulin sensitivity and glucose metabolism. Diabetes 58, 803–812 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Harms M. J., Li Q., Lee S., Zhang C., Kull B., Hallen S., Thorell A., Alexandersson I., Hagberg C. E., Peng X.-R., Mardinoglu A., Spalding K. L., Boucher J., Mature human white adipocytes cultured under membranes maintain identity, function, and can transdifferentiate into brown-like adipocytes. Cell Rep. 27, 213–225.e5 (2019). [DOI] [PubMed] [Google Scholar]
- 60.Borghi F., Silva C., da Silva P. C., Ferrucci D. L., Morais C. L., Conceição-Vertamatti A. G., Carvalho H. F., Fonseca M. d. C., Vieira A. S., Grassi-Kassisse D. M., The influence of hypertensive environment on adipose tissue remodeling measured by fluorescence lifetime imaging in spontaneously hypertensive rats. Mol. Cell. Endocrinol. 506, 110758 (2020). [DOI] [PubMed] [Google Scholar]
- 61.Davies B. P., Arenz C., A fluorescence probe for assaying micro RNA maturation. Bioorg. Med. Chem. 16, 49–55 (2008). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/49/eabc6250/DC1