

Functional annotations of a placental cell genome aid in identifying genes associated with preterm birth.
Abstract
While a genetic component of preterm birth (PTB) has long been recognized and recently mapped by genome-wide association studies (GWASs), the molecular determinants underlying PTB remain elusive. This stems in part from an incomplete availability of functional genomic annotations in human cell types relevant to pregnancy and PTB. We generated transcriptome (RNA-seq), epigenome (ChIP-seq of H3K27ac, H3K4me1, and H3K4me3 histone modifications), open chromatin (ATAC-seq), and chromatin interaction (promoter capture Hi-C) annotations of cultured primary decidua-derived mesenchymal stromal/stem cells and in vitro differentiated decidual stromal cells and developed a computational framework to integrate these functional annotations with results from a GWAS of gestational duration in 56,384 women. Using these resources, we uncovered additional loci associated with gestational duration and target genes of associated loci. Our strategy illustrates how functional annotations in pregnancy-relevant cell types aid in the experimental follow-up of GWAS for PTB and, likely, other pregnancy-related conditions.
INTRODUCTION
Spontaneous preterm birth (PTB), defined as spontaneous labor and birth before 37 weeks of gestation, is associated with considerable infant mortality and morbidity, as well as long-term health consequences into adulthood (1). A genetic component to PTB has long been recognized, but the significant role of environmental factors and the etiologic heterogeneity of birth before 37 weeks (2–4) have made it challenging to discover genetic associations and causal genes. For example, recent genome-wide association studies (GWASs) of gestational duration in 43,568 women (3331 with a preterm delivery) (5) and in 84,689 infants (4775 born preterm) (6) reported six and one genome-wide significant associations, respectively, with gestational duration considered as a continuous variable. Three loci were also associated with PTB (defined as a categorical variable of birth) in the maternal GWAS (5), but no loci were associated with PTB in the infant GWAS (6). These studies highlight the challenges of such complex and multifactorial phenotypes and the need for additional approaches to facilitate discovery of genes contributing to gestational duration and PTB.
Integrating GWAS that results with genomic and epigenomic annotations is a promising approach for assigning function to variants discovered by GWAS, as well as for identifying additional associations that do not reach stringent genome-wide significance threshold (7, 8). While large consortia [e.g., ENCODE (Encyclopedia of DNA Elements) (9), GTEx (Genotype-Tissue Expression Project) (10), and Roadmap Epigenomics (11)] have generated annotations of putative functional elements and genetic variants for many human cell types and tissues, there is a remarkable absence in these databases for the cell types and tissues that are relevant to pregnancy in general and to PTB in particular. Because the regulation of transcription has strong cell type–specific components and because annotations in disease-relevant tissues or cells tend to be most enriched among GWAS signals for those specific diseases (10, 12), follow-up studies of GWASs of pregnancy-associated conditions have been disadvantaged compared to most other complex diseases due to the paucity of functional annotations in cells relevant to pregnancy. To fill this gap in knowledge, we characterized the transcriptional and chromatin landscapes of cultured mesenchymal stromal/stem cells (MSCs) collected from human placental membranes and decidualized MSCs, also known as decidual stromal cells (DSCs). These cells play critical roles in promoting successful pregnancy, interfacing with fetal cells throughout pregnancy, and the timing of birth (13, 14). We then built a computational framework that integrated these decidua-derived stromal cell annotations with the results of a large GWAS of gestational duration to facilitate discovery of PTB genes.
This integrated analysis revealed a significant enrichment of heritability estimates for gestational duration in decidua-derived stromal cell genomic regions marked by open chromatin or histone marks. Leveraging those functional annotations in a Bayesian statistical framework, we discovered additional loci associated with gestational duration and improved fine mapping in regions associated with gestational duration. Last, using promoter capture Hi-C (pcHi-C), we linked functionally annotated gestational age-associated variants to their putative target genes. More generally, these functional annotations should prove a valuable resource for studying other pregnancy-related conditions, such as preeclampsia and recurrent miscarriage, as well as conditions associated with endometrial dysfunction, such as endometriosis and infertility.
RESULTS
Generation of transcriptome and epigenome maps of untreated (MSC) and in vitro differentiated DSCs
Decidualization is the process of transformation of endometrial MSCs into DSCs that is induced by progesterone production that begins during the luteal phase of the menstrual cycle and then increases throughout pregnancy when successful implantation occurs [reviewed in (15)]. Using progesterone and estrogen or cyclic adenosine 5′-monophosphate (cAMP) to induce decidualization of MSCs in culture has been used in cells derived from endometrial biopsies in nonpregnant women to characterize their transcriptomes and epigenomes and to identify genes and molecular pathways involved in this process (16–21).
Because obtaining endometrial cells in nonpregnant women through biopsies requires an invasive procedure that carries some risk and MSCs can also be obtained from human placentas (22–24), we isolated these cells from the decidua parietalis of three women who had delivered at term and established one primary MSC line from each to model the process of decidualization (see Materials and Methods). Briefly, cells were treated with medroxyprogesterone acetate (MPA) and cAMP for 48 hours, and a paired set of untreated samples was cultured in parallel for 48 hours. Three replicates of treated/untreated sets of each cell line were studied to assess experimental variability in the two conditions. Each of the 18 samples (3 individual lines × 3 replicates × 2 conditions) were assayed to generate transcriptome [RNA sequencing (RNA-seq)], open chromatin [assay for transposase-accessible chromatin sequencing (ATAC-seq)], and histone modification [chromatin immunoprecipitation sequencing (ChIP-seq)] maps. A summary of those data is shown in table S1, and a representative example of the full set of annotations for one primary cell line is shown in Fig. 1. The number of reads generated for each sample in each condition and other descriptive data are provided in data file S1.
Fig. 1. Schematic of RNA-seq, ATAC-seq, ChIP-seq, and pcHi-C maps centered on the prolactin (PRL) gene, as an example.
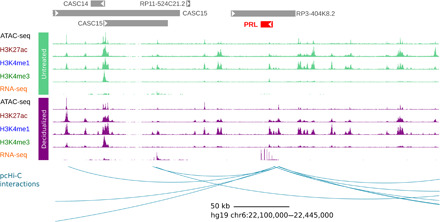
Each histone modification and RNA-seq track shows read counts per base pair for each experiment. The pcHi-C signal track shows the number of reads per MboI restriction fragment. Arcs in the pcHi-C interactions track show significant interactions between the promoter of the PRL gene and putative distal regulatory elements identified with pcHi-C. Pooled data (three replicates) for one cell line are shown for untreated cells (MSCs, in green) and decidualized cells (DSCs, in purple). pcHi-C data were generated in a fourth cell line that was decidualized.
Robust gene expression changes occur in decidualized stromal cells
Analysis of the RNA-seq data using DESeq2 (25) revealed 1135 differentially expressed genes after decidualization (table S1). Genes with decreased expression after 48 hours of treatment were highly enriched for cell cycle genes (data file S2), consistent with observations from endometrial biopsies from nonpregnant women that decidualization is associated with cell cycle arrest (19, 26). Genes with increased expression after treatment were enriched for insulin-related terms, also consistent with previous results from endometrial biopsies (26), and for glucose metabolism (18).
Identification of regulatory elements associated with decidualization
To identify putative regulatory elements in MSCs and DSCs, we assayed H3K27ac, H3K4me1, and H3K4me3 histone modifications, which are markers of active enhancers, poised enhancers, and active promoters, respectively [reviewed in (27)]. We also used ATAC-seq to identify open chromatin regions to complement ChIP-seq data. To identify regulatory regions that might be altered in response to, and potentially regulate decidualization, we compared read counts of ATAC-seq and ChIP-seq peaks in untreated and decidualized cells, revealing tens of thousands of regions that differed between untreated and treated samples (table S1). Most of the differential peaks were marked with H3K27Ac and H3K4me1, indicating that the epigenetic changes underlying alterations in gene expression during decidualization predominantly occur in distant regulatory elements, such as enhancers.
We observed a moderate degree of overlap between the differential peaks across ATAC-seq and ChIP-seq data, with the two enhancer marks, H3K27ac and H3K4me1, showing the most overlap (Fig. 2A). In addition, putative regulatory regions that showed chromatin changes in response to decidualization were associated with genes whose expression also changed in response to decidualization (Fig. 2B). Regulatory regions with increased read counts clustered around genes that were more highly expressed after decidualization, indicating increased chromatin accessibility or activation of enhancers of those genes. Conversely, genes that were more lowly expressed after decidualization were enriched for enhancers that became less accessible or active. These observations indicate that the differential peaks of open chromatin and histone marks observed after decidualization correspond to regulatory elements that become more or less active, resulting in correlated gene expression changes of the nearby genes.
Fig. 2. Differential histone modification and ATAC-seq peaks are associated with differential expression and enriched for transcription factors with roles in decidualization.
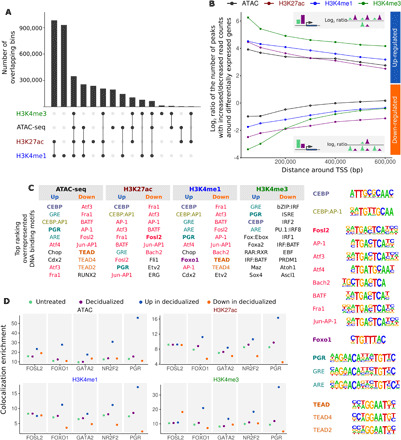
(A) Plot showing the overlap between the different histone modifications and ATAC-seq maps (intersection between annotations). Peaks were assigned to 100-bp bins to avoid ambiguity in overlap due to different peak borders. Black circles indicate overlap with other annotations; light gray circles indicate that the annotation does not overlap others. (B) Each data point shows the ratio between the number of increased/decreased differential peaks nearby genes that increase expression after decidualization (blue, positive log ratios; upper half of the figure) or decrease expression after decidualization (orange, negative log ratios; lower half of the figure). Genes that were more highly expressed in decidualized cells were flanked by a higher number of ChIP-seq and ATAC-seq peaks that displayed increased read counts in decidualized samples compared to peaks that displayed decreased read counts (top inset). Genes that were down-regulated in decidualized cells showed the opposite trend (bottom inset). All enrichments: P < 10−25. (C) DNA binding motifs of transcription factors relevant in decidualization are enriched in peaks that change following decidualization treatment. Motifs are color-coded by similarity. (D) Colocalization of PGR, FOSL2, FOXO1, GATA2, and NR2F2 with ATAC-seq and ChIP-seq peaks. Transcription factor binding sites co-occur with ATAC-seq and ChIP-seq peaks in both untreated (green) and decidualized (purple) cells more often than with random peaks. Enrichment of the co-occurrences of PGR, FOXO1, GATA2, and NR2F2 are higher when co-occurring with peaks that have increased read counts (navy blue) and lower with peaks that have decreased read counts (orange) in decidualized compared to untreated cells. Enrichment of co-occurrences with peak sets was calculated as the fold difference between the number of transcription factor peaks overlapping with ATAC-seq/ChIP-seq peaks and with a random set of peaks (see Materials and Methods).
Previous work identified transcription factors that play critical roles in decidualized stromal cells (28–32). Several of the DNA binding motifs that were enriched in peaks with increased or decreased read counts in our data correspond to transcription factors previously implicated in decidualization (Fig. 2C), such as CAAT-enhancer binding protein (CEBP) (33), progesterone receptor (PGR) (28) that shares the same motif with androgen response element, and glucocorticoid receptor, FOSL2 (Fos-related antigen 2) (28), that shares the same motif with Fra1 (Fos-related antigen 1), Atf3 (Activating transcription factor 3), and BATF (basic leucine zipper ATF-like transcription factor), and TEA (transcriptional enhancer factor) domain transcription factors (21, 34). Whereas CEBP and PGR were exclusively enriched in peaks with increased read counts in decidualized cells, the FOSL2 motif was present in peaks that both changed positively and negatively in decidualized cells.
To better understand the role of these transcription factors in decidualization, we obtained publicly available ChIP-seq data for PGR (28) and FOSL2 (28) from endometrial biopsies and analyzed the colocalization of their binding locations with the putative regulatory elements identified by ATAC-seq and ChIP-seq identified in our study (Fig. 2B). We additionally analyzed FOXO1 (Forkhead box O1) (29), NR2F2 (nuclear receptor subfamily 2 group F member 2) (30), and GATA2 (GATA binding protein 2) (31) ChIP-seq data because these transcription factors have also been implicated in decidualization (29–31). With the exception of FOSL2, the colocalization enrichments of PGR, FOXO1, GATA2, and NR2F2 with ATAC-seq and ChIP-seq peaks were higher (9 to 16 folds) among peaks that were increased in decidualized cells (more open chromatin or increased histone modification levels) compared to all peaks (7.5 to 12.8 folds) and to peaks that decreased in decidualized cells (2 to 5 folds). This observation supports the notion that these transcription factors are involved in regulation of decidualization (28–31, 35). Although FOSL2 has been reported as a positive coregulator of PGR (28), the presence of FOSL2 motifs in peaks that both increased and decreased in decidualized cells (Fig. 2C) and the lack of difference in the colocalization enrichment between these two sets of peaks (Fig. 2D) suggests that FOSL2 may have a dual role in decidualization.
Together, our results support a model of decidualization that involves changes in the regulatory landscape during the differentiation of MSCs into DSCs, including alterations in chromatin accessibility and in the activation levels of distant regulatory elements, accompanied by the differential binding of key transcription factors, resulting in increases or decreases in gene expression.
Chromatin interactions aid in the identification of target genes of distal regulatory elements
As shown in Fig. 2B, the surrounding regions of differentially expressed genes were enriched for differential ChIP-seq and ATAC-seq peaks that changed in the same direction as the genes in decidualized samples. Accordingly, when we paired differential peaks with the nearest expressed gene as its putative gene target, we observed that these pairs were more likely to have matching directions of change (i.e., both the peak and the gene have increased or decreased read counts in decidualized samples) than nonmatching directions when compared with pairs that were assigned randomly (Fig. 3A).
Fig. 3. pcHi-C connects predicted regulatory elements to their putative target genes.
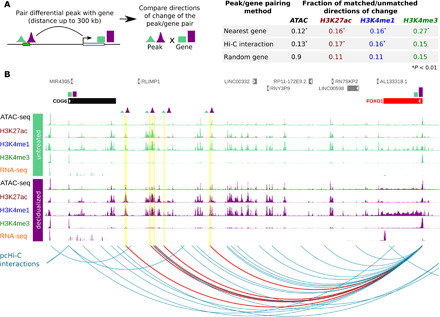
(A) Randomly assigning a gene to a peak (see Materials and Methods) resulted in fewer peaks that matched the direction of change with that of differentially expressed genes than when using pcHi-C interactions or the nearest gene to pair peaks to genes. (B) The FOXO1 gene is more highly expressed in decidualized samples (fourfold increase, P = 7 × 10−22) and its promoter physically interacts (red arcs) with distal regulatory elements (yellow highlights) that show increased activation in decidualized samples. The nearest expressed gene to these differential peaks is COG6.
In many cases, however, the target gene for a regulatory element is not the nearest gene (36), and therefore, information about distal chromatin interactions can be useful in prioritizing candidate gene targets of variants identified in GWAS. To this end, we generated a pcHi-C map of a decidualized cell line, thus enriching for the identification of long-range chromatin interactions between promoters and distant regulatory elements (37–39). We identified a total of 161,337 interactions, of which 53,211 were between promoters and distal regions of accessible chromatin assayed by ATAC-seq and ChIP-seq, suggestive of their regulatory role. We used the significant interactions between promoters and distal regions that we identified to pair differential peaks with putative target genes. As shown in Fig. 3A, using pcHi-C interactions as a pairing method resulted in enhanced identification of differential peak/differential target gene pairs that have matching directions of change compared to random assignment of gene-target pairs.
Whereas assigning peaks to the nearest expressed gene also led to enhanced assignment of differential peaks to target genes with matching directions of change (Fig. 3A), pcHi-C was helpful in identifying less obvious target genes, as shown in Fig. 3B. In this example, several pcHi-C interactions link distal regulatory elements up to 847 kb away that became more active in decidualized cells to the promoter of a gene (FOXO1) that was up-regulated in decidualized cells and is known to be involved in decidualization (32). The nearest expressed gene method assigned those differential peaks to COG6, a gene that does not change expression in decidualized samples and is therefore a less likely target.
In conclusion, by combining pcHi-C interactions with the epigenome maps and transcriptome data, we were able to identify genes and putative regulatory elements that respond to, or regulate, the decidualization process. We next used these functional genomic maps and datasets to fine map GWAS loci for gestational duration and identify new candidate genes with a potential role in PTB.
The heritability of gestational duration is enriched for functional annotations in DSCs
To identify candidate genes that may play a role in gestational duration and PTB, we used summary data from a GWAS of gestational duration based on a meta-analysis of a 23andMe GWAS (n = 42,121) (5) and the results from six European datasets (n = 14,263). A detailed description of the GWAS is in the Supplementary Materials and figs. S1 and S2. After filtering for single-nucleotide polymorphism (SNPs) that are present in the 1000 Genomes Project data and minor allele frequency of >0.01, we identified SNPs at six autosomal loci, defined as approximately independent blocks by LDetect (40), that were associated with gestational duration at genome-wide significance of P < 5 × 10−8 (table S2). We then created a computational pipeline to assess enrichment of GWAS signals in functional annotations that we generated in untreated (MSCs) and decidualized (DSCs) stromal cells to fine map GWAS loci and discover candidate causal genes and to potentially provide support for additional loci that did not reach genome-wide significance in the GWAS (Fig. 4A). Each step of this procedure is explained below and described in details in Materials and Methods.
Fig. 4. GWAS analysis pipeline and heritability enrichment in functional annotations.
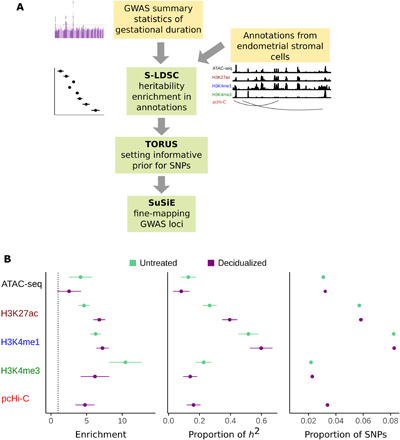
(A) Computational pipeline for analyzing GWAS of gestation duration. Yellow boxes (input data): GWAS summary statistics and functional annotations from endometrial stromal cells (in both untreated and decidualized cells). Green boxes: Stages of statistical analysis (see Materials and Methods). (B) Stratified LDSC heritability analysis of GWAS of gestational duration using functional annotations. Left: Fold enrichment of heritability in each annotation. Dashed line shows values at 1, i.e., no enrichment. Center: Proportion of heritability explained by each annotation. Right: Proportion of SNPs across the genome that fall within an annotation. For each annotation, enrichment (left) is the ratio of h2 proportion (center) divided by the SNP proportion (right). Error bars represent 95% confidence intervals.
We first used stratified linkage disequilibrium (LD) score regression (S-LDSC) (41) to assess enrichment of GWAS signals in functional annotations in endometrial stromal cells. S-LDSC takes as input GWAS summary statistics across the genome and functional annotations of SNPs, e.g., whether an SNP is in ATAC-seq peak, and returns as output heritability enrichment of each annotation. S-LDSC is a commonly used tool for estimating the proportion of heritability of complex phenotypes that is explained by variants in certain functional annotations. The heritability enrichment is defined by the proportion of heritability explained by annotations divided by the expected proportion, which is the percent of SNPs genome wide that are in these functional annotations. To account for possible systematic bias in this analysis, i.e., SNPs within annotations of interest may differ from background SNPs in systematic ways such as their LD structure and epigenomic properties, we included a range of baseline annotations (default S-LDSC setting), including LD-related annotations, deoxyribonuclease (DNase) hypersensitivity, enhancer annotation, H3K27ac, H3K4me1, and other histone marks (the union across cell types). Thus, if an annotation is shared by many cell types, then it would not show the enrichment in S-LDSC analysis (see Materials and Methods).
Using S-LDSC, we found 5- to 10-fold enrichments of GWAS heritability for gestational duration in our functional annotations compared to the baseline model of S-LDSC (Fig. 4). The enrichment of enhancer marks H3K27ac and H3K4me1 was higher in decidualized than in untreated cells, but the opposite pattern was observed for the promoter mark H3K4me3, which was more enriched in untreated (MSCs) than in decidualized (DSCs) cells. These findings are consistent with previous observations that enhancers are often more dynamic and condition- or tissue-specific than promoters (10). We observed weaker heritability enrichments of open chromatin regions defined by ATAC-seq and of interaction regions in pcHi-C. However, because we performed joint analysis of all annotations together, the enrichment of one annotation (e.g., ATAC-seq peaks) will be reduced if the enrichment is partially explained by other, overlapping annotations (e.g., H3K27ac). Although the promoter mark H3K4me3 in untreated cells showed the highest enrichment, the annotations that contributed most to the heritability of gestational duration were enhancers (Fig. 4) due to the much larger number of enhancer histone marks than promoters in the genome. Our results thus highlight the importance of functional annotations in endometrial stromal cells at GWAS loci for gestational duration.
Integrated analysis of GWAS and decidual cell functional annotations improves fine mapping of causal variants of gestational duration and identifies putative target genes
We next developed a computational procedure, based on fine mapping, to integrate the decidua stromal cell functional maps with a GWAS of gestational duration to identify putative causal variants (Fig. 4A). Because of extensive LD in the human genome, the causal variants driving the associations are unknown at most loci discovered by GWAS. Fine mapping is a Bayesian statistical procedure that takes as input GWAS summary statistics and patterns of LD at trait-associated loci and computes the probability of each variant at a locus to be a causal variant (7). These probabilities, known as posterior inclusion probabilities (PIPs), reflect our confidence of certain SNPs being causal variants. The PIP of a variant ranges from 0 to 1, with 1 indicating full confidence that the SNP is a causal variant. If a region contains a single causal variant, the PIPs of all SNPs in the region should approximately sum to 1.
While fine mapping has been commonly used in identifying putative causal variants from GWAS of complex traits (7), it is often difficult to narrow down causal signals to one or a small number of variants in most GWAS loci. Standard fine mapping treats all SNPs at a locus equally. Recent work suggests that incorporating Bayesian prior probabilities that favor functional SNPs improves fine mapping (8, 42). We posited that integrating functional annotations in pregnancy-relevant cells in a statistical fine-mapping framework would aid in (i) identifying candidate causal variants at each locus associated with gestation duration, (ii) linking those variants to their target genes, and (iii) discovering additional loci and genes associated with gestational duration that may have failed to reach the stringent threshold for significance in GWAS.
We first leveraged the enrichments of DSC annotations to create Bayesian prior probabilities for a variant being causal. On the basis of the results of S-LDSC, we chose H3K27ac, H3K4me1, and pcHi-C interactions from the decidualized cells, and H3K4me3 from untreated cells, as functional genomic annotations to create informative priors using TORUS (42). TORUS takes as input genome-wide summary statistics from GWAS and the functional annotations of SNPs and computes enrichment parameters of annotations, which reflect how much more likely an SNP is a causal variant than randomly chosen SNPs (table S3). SNPs associated with functional annotations are generally assigned higher prior probabilities. In addition, TORUS computes statistical evidence at the level of genomic blocks, defined as the probability that a block (determined by LD) contains at least one causal SNP. Without including any histone marks or chromatin accessibility annotations, TORUS implicated six autosomal blocks in the genome at false discovery rate (FDR) of < 0.05, including five of the six genome-wide significant autosomal loci identified in the GWAS (P < 5 × 10−8). One locus on chromosome 3 had an FDR = 0.11 and was therefore not identified by TORUS, and one locus on chromosome 9 that was not identified in the GWAS was implicated by TORUS (data file S3). By including the functional genomic annotations from endometrial stromal cells, the number of high confidence blocks increased to 10, including all 6 that were significant in the gestational duration GWAS and 4 that were not significant in the GWAS (data file S3).
We next performed computational fine mapping on these 10 blocks, with the informative priors learned by TORUS, using sum of single effects (SuSiE) regression (43). Conceptually, SuSiE is a Bayesian version of the stepwise regression analysis commonly used in GWAS (i.e., conditioning on one variant and testing if there is any remaining signal in a region). SuSiE accounts for the uncertainty of causal variants in each step and reports the results in the form of PIPs. Including the priors defined by TORUS using DSC functional annotations significantly improved fine mapping (Fig. 5A, table S3, and data file S4). For example, only one SNP reached PIP > 0.3 across all 10 blocks using the default setting under SuSiE (uniform prior, treating all SNPs in a block equally). This reflects the general uncertainty of pinpointing causal variants due to LD, e.g., a strong GWAS SNP in close LD with nine other SNPs would have PIP about 0.1. By using the annotation-informed priors, eight SNPs in six different blocks reached PIP > 0.3 (Fig. 5A). In some blocks, we were able to fine-map a single high-confidence SNP, e.g., the FOXL2 locus on chromosome 3, while in other blocks, we had considerable uncertainty of the causal variants, as shown by large credible sets, i.e., the minimum set of SNPs to include the causal SNP with 95% probability (Fig. 5B). Table 1 summarizes the most probable causal variants in eight blocks (fine mapping in the remaining two blocks produced large credible sets with no high-PIP SNPs) and their likely target genes based on promoter assignment or chromatin interactions from pcHi-C. We note that our results of the WNT4 locus identified rs3820282 as the likely causal variant. This is consistent with our previous results demonstrating experimentally that the T allele of this SNP disrupts the binding of estrogen receptor 1 (5). This SNP was among the three most likely SNPs in our fine-mapping study, with a PIP of 0.27 (Table 1).
Fig. 5. Fine-mapping GWAS loci of gestational duration.
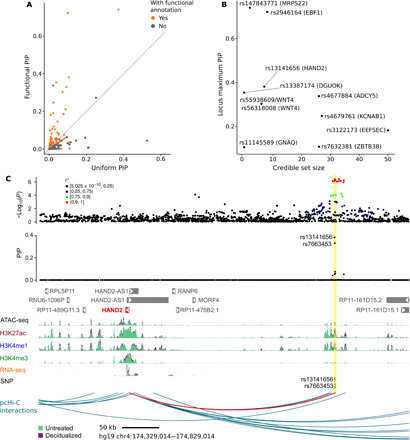
(A) PIPs of SNPs using uniform vs. functional priors in SuSiE (each dot is an SNP). The functional prior of an SNP is based on SNP annotations and is estimated using TORUS. (B) Summary of fine-mapping statistics of all 10 regions. X axis: The size (number of SNPs) of credible set. Y axis: The maximum PIP in a region. We label each region by its top SNP (by PIP) and the likely causal gene, according to Table 1 or the nearest gene of the top SNP. (C) Likely causal variants near HAND2 and their functional annotations. The top panel shows the significance of SNP association in the GWAS and the middle panel shows fine-mapping results (PIPs) in the region. The vertical yellow bar highlights the two SNPs with high PIPs. These SNPs are located in a region annotated with ATAC-seq, H3K27ac, H3K4me1, and H3K4me3 peaks (bottom). This putative enhancer also had increased ATAC-seq, H3K27ac, and H3K4me1 levels in decidualized samples and interacts with the HAND2 promoter (red arc).
Table 1. Most probable SNPs identified from computational fine mapping of regions associated with gestational duration.
Functional annotations are based on data from endometrial stromal cells. We list an annotation if the SNP is located in a sequence with that annotation in either untreated or decidualized condition. Functional prior is the prior probability of an SNP being a causal variant. For an SNP without any functional annotation, its prior probability is 3.6 × 10−6. We list the pcHi-C annotation if the SNP is within 1 kb of a region involved in a pcHi-C interaction. We call a gene the target of an SNP if (i) the SNP is located in the promoter (< 1 kb of transcription start site) of that gene or (ii) the promoter of that gene has a pcHi-C interaction with a region within 1 kb of the SNP. In the case of rs147843771 at the FOXL2 locus, the target was defined by literature evidence (69). The number of credible SNPs at each region is shown in Fig. 5B. SNPs in bold are discussed in the text. FOXL2 (69), forkhead box L2; GATA2, GATA-binding protein 2; HAND2, heart and neural crest derivatives expressed 2; KCNAB1, potassium voltage-gated channel subfamily A member regulatory beta subunit 1; WNT4, Wnt family member 4.
| SNP | Location (hg19) | GWAS P value | Functional prior | PIP |
Functional annotations |
Likely target |
| rs147843771 | chr3:138843356 | 3.8 × 10−8 | 8.3 × 10−5 | 0.74 | K4me1 | FOXL2 |
| rs17315501 | chr3:139029676 | 1.7 × 10−7 | 9.9 × 10−5 | 0.21 | K4me1, K4me3, ATAC, K27ac |
|
| rs2946164 | chr5:157884706 | 3.0 × 10−26 | 8.3 × 10−5 | 0.72 | K4me1 | |
| rs13141656 | chr4:174728703 | 3.9 × 10−7 | 5.1 × 10−4 | 0.38 | K4me1, K27ac, K4me3, ATAC, pcHi-C |
HAND2 (pcHi-C) |
| rs7663453 | chr4:174729014 | 4.5 × 10−7 | 5.1 × 10−4 | 0.33 | K4me1, K27ac, K4me3, pcHi-C |
HAND2 (pcHi-C) |
| rs13387174 | chr2:74206685 | 4.7 × 10−7 | 3.8 × 10−4 | 0.35 | pcHi-C, K4me1, K27ac | WBP1 (pcHi-C) |
| rs13390332 | chr2:74207357 | 2.0 × 10−7 | 9.9 × 10−5 | 0.18 | K4me1, K27ac | WBP1 (pcHi-C) |
| rs4677884 | chr3:123062970 | 4.1 × 10−9 | 8.3 × 10−5 | 0.34 | K4me1, ATAC | |
| rs56318008 | chr1:22470407 | 2.3 × 10−12 | 4.3 × 10−4 | 0.3 | K4me1, K4me3, ATAC, pcHi-C |
WNT4 (promoter) |
| rs55938609 | chr1:22470451 | 2.3 × 10−12 | 4.3 × 10−4 | 0.3 | K4me1, K4me3, ATAC, pcHi-C |
WNT4 (promoter) |
| rs3820282 | chr1:22468215 | 6.4 × 10−13 | 1.1 × 10−4 | 0.27 | K4me1, K4me3, ATAC |
WNT4 (promoter) |
| rs4679761 | chr3:155868039 | 5.0 × 10−9 | 9.9 × 10−5 | 0.24 | K4me1, K27ac | KCNAB1 (pcHi-C) |
| rs9882088 | chr3:155867092 | 5.5 × 10−9 | 8.3 × 10−5 | 0.19 | K4me1 | |
| rs3122173 | chr3:127889287 | 5.4 × 10−12 | 3.2 × 10−4 | 0.18 | K4me1, pcHi-C | GATA2 (pcHi-C) |
| rs2999048 | chr3:127878416 | 2.0 × 10−12 | 8.3 × 10−5 | 0.12 | K4me1, K27ac, pcHi-C |
GATA2 (pcHi-C) |
| rs1554535 | chr3:127895986 | 1.2 × 10−11 | 3.8 × 10−4 | 0.10 | K4me1, pcHi-C, K27ac |
GATA2 (pcHi-C) |
We highlight the results from two regions. In both cases, we were able to identify putative risk genes with relatively high confidence, and neither is the nearest gene of lead SNPs in GWAS. In the first case, two adjacent SNPs [311–base pair (bp) apart], rs13141656 and rs7663453, on chromosome 4q34 did not reach genome-wide significance in the GWAS (P = 3.9 × 10−7 and 4.5 × 10−7, respectively). After using functional annotations in decidua-derived stromal cells, the block containing these SNPs was highly significant (TORUS q = 0.02), suggesting the presence of at least one causal variant in this block. The two SNPs together explained most of the PIP signal in the block (PIP 0.38 and 0.33, respectively, Table 1). The two SNPs are located in a region of open chromatin in endometrial stromal cells, with enhancer activity marked by both H3K27ac and H3K4me1 (Fig. 5C). Only 9 of the 129 tissues from the Epigenome Roadmap (11) also had H3K27ac, H3K4me1, or H3K4me3 peaks spanning the rs13141656 locus and only 2 spanning the rs7663453 locus. In addition, this putative enhancer is bound by multiple transcription factors, including GATA2, FOXO1, NR2F2, and PGR, based on ChIP-seq data. The only physical interaction of this enhancer in the pcHi-C data in decidualized stromal cells is with the promoter of the HAND2 gene, located 277 kb away (Fig. 5C). Summing over the PIPs of all SNPs whose nearby sequences interact with HAND2 (heart and neural crest derivatives expressed 2) via chromatin looping gives an even higher probability, 0.89, suggesting that HAND2 is very likely to be the causal gene in this region (table S4). HAND2 is an important transcription factor that mediates the effect of progesterone on uterine epithelium (44). Thus, in this example, we identified a previously unknown locus, the likely causal variant(s), the enhancers they act on, and an outstanding candidate gene for gestational duration and PTB.
The second example focuses on the locus showing a strong GWAS association with gestational duration on chromosome 3q21. The lead SNP, rs144609957 (GWAS P = 4 × 10−13), is located upstream of the EEFSEC (eukaryotic elongation factor, selenocysteine-tRNA–specific) gene. There is considerable uncertainty of the causal variants in this region, with 50 SNPs in the credible set and the lead SNP explaining only a small fraction of signal (PIP = 0.02). Among all 12 SNPs with PIP > 0.01, 11 have functional annotations, most commonly H3K4me1 and pcHi-C interactions. For nine SNPs (first three shown in Table 1), the sequences in which they are located physically interact with the promoter of GATA2 in the pcHi-C data but not with any other promoters in the region (fig. S3). The PIPs of all SNPs in the genomic regions that likely target GATA2 through chromatin looping sum to 0.68 (table S5). Thus, despite uncertainty of causal variants in this region, our results implicate GATA2 as a candidate causal gene in endometrial stromal cells. GATA2 is a master regulator of embryonic development and differentiation of tissue-forming stem cells (45). As support for the possible role of GATA2 in pregnancy, GATA2 deficient mice show defects in embryo implantation and endometrial decidualization (35), making this another excellent candidate causal gene for gestational duration and PTB.
DISCUSSION
The molecular processes that signal the onset of parturition in human pregnancies, and how perturbation of those processes result in PTB, are largely unknown. Yet, understanding these processes would reveal important insights into the potential causes of adverse pregnancy outcomes, including spontaneous labor before 37 weeks’ gestation, and potentially lead to the identification of biomarkers and therapeutic targets for PTB. Although it is experimentally challenging to link decidualization processes directly to parturition in humans, it is well accepted that shallow implantation due to suboptimal decidualization is associated with poor pregnancy outcomes in general (46–48) and that the decidua is key in triggering parturition (13, 14). Thus far, however, specific genes that perturb decidualization processes and lead to PTB are poorly defined.
Unbiased GWASs do not require prior knowledge of molecular processes underlying disease phenotypes and have the potential to identify novel genes and pathways contributing to common diseases. However, the significant heterogeneity of most common diseases and small effects of most common disease-associated variants lead to the requirement for very large sample sizes (in the tens to hundreds of thousands of cases) to discover more than a handful of associated loci that meet stringent criteria for genome-wide significance. To address this limitation and provide orthogonal evidence for assessment of associations, we characterized the transcriptional and chromatin landscapes in decidua-derived stromal cells and integrated those functional annotations with a GWAS of gestational duration to discover novel loci and genes. The primary motivation for these studies was the notable paucity of genomic and epigenomic functional annotations in pregnancy-relevant primary cells among those studied by large consortia (9–11). Here, we filled a significant gap by providing maps in untreated and decidualized stromal cells and used these maps for annotating GWAS of pregnancy-related traits.
We chose to focus these studies on endometrial stromal cells because of their central importance in both the establishment and maintenance of pregnancy, as well as their intimate juxtaposition to fetal trophoblast cells throughout pregnancy. Of particular relevance are the roles that decidualized stromal cells play in regulating trophoblast invasion, modulating maternal immune and inflammatory responses at the maternal-fetal interface, and controlling remodeling of the endometrium (48). Defects in all of these processes have been considered a contributing factor to pregnancy disorders (48, 49). Moreover, we showed that the SNPs in regions with endometrial stromal cell functional annotations explained more of the heritability of gestational duration compared to just using baseline annotations. Among all annotations, enhancer marks H3K4me1 (in both decidualized and untreated stromal cells) and H3K27ac (in decidualized cells) were 8- to 10-folds enriched at GWAS loci after adjusting for the general annotations and accounted for 50 to 70% of the GWAS heritability. The lack of complete independence between these marks makes it difficult to delineate their individual effects but, nonetheless, highlights the importance of enhancers and of gene regulation in endometrial stromal cells in modulating the effects of GWAS variants on gestational duration. This is consistent with both the known tissue-specific roles of enhancers and the observation that more than 90% of GWAS loci reside outside of the coding portion of the genome and are enriched in regions of open chromatin and enhancers (12, 41).
Integrating transcriptional and chromatin annotations of gene regulation from MSCs and DSCs improved our ability to discover novel GWAS loci and identify likely causal SNPs and genes associated with gestational duration. We illustrate how our integrated platform identified a novel causal locus and candidate gene (HAND2) associated with gestational duration, as well as refined the annotation of loci that had been previously identified. Our data suggest that in endometrial stromal cells, GATA2 is likely the target gene of enhancers harboring SNPs associated with gestational duration. This does not exclude the possibility that the nearest gene to the associated SNPs, EEFSEC, may be a target gene in other cell types. Both HAND2 (50) and GATA2 (51) are involved in decidualization processes in humans, and perturbations in this process have been linked to poor pregnancy outcomes (46–48). Neither GATA2 nor HAND2 was identified as potential candidate genes in previous GWASs of gestational duration, or PTB supports our approach and the importance of using functional annotations from cell types relevant to pregnancy to fine map and identify candidate genes for the pregnancy-related traits. Overall, the integrated analyses performed in this study resulted in the identification of both novel GWAS loci and novel candidate genes for gestational duration, as well as maps of the regulatory architecture of these cells and their response to decidualization.
However, there are some limitations. Our results are based on cells from only three individuals, which may not fully capture the regulatory landscape of endometrial stromal cells. For pcHI-C, we used cells from a single individual to generate the chromatin interactions map. Another limitation is that we focused on only one cell type, albeit one that plays a central role in pregnancy and only one exposure (hormonal induction of decidualization) at one time point (48 hours). Furthermore, it is unclear how our model of in vitro decidualization mimics the endogenous decidualization of endometrial cells during pregnancy. While we chose decidualization as a perturbation to ascertain the dynamic features of functional genomic annotations, we fully anticipate that obtaining annotations in other cell types and in response to other relevant perturbations will improve the ability to identify novel loci, variants, and genes associated with PTB. Future studies that include fetal cells from the placenta and uterine or cervical myometrial cells could reveal additional processes that contribute to gestational duration and PTB, such as those related to fetal signaling and the regulation of labor, respectively. Inclusion of additional exposures, such as trophoblast conditioned media (52) and additional exposure times, may further reveal processes that are pregnancy specific. Second, to maximize power, we focused on a GWAS of gestational duration and not PTB per se. While previous GWAS have shown that all PTB loci were among the gestational age loci (5), we realize that some of the loci that we identified could be related to normal variation in gestational duration and not specifically to PTB. Nonetheless, our findings contribute to our understanding of potential mechanisms underlying the timing of human gestation, about which we still know little. Last, although our ChIP-seq results revealed an association between GATA2 binding and decidualization, confirming the role of this transcription factor in decidual cell biology (53, 54), and studies in murines support its role in endometrial processes (35), we do not yet have direct evidence showing that perturbations in the expression of GATA2, or any of the other target genes identified, influence the timing of parturition in humans. Future studies will be needed to directly implicate the expression of these genes in gestational duration or PTB. Our study highlights the importance of generating functional annotations in pregnancy-relevant cell types to inform GWASs of pregnancy-associated conditions. Our results suggest that the expression of two transcription factors, GATA2 and HAND2, in endometrial stromal cells may regulate transcriptional programs that influence the timing of parturition in humans, which could lead to the identification of biomarkers of or therapeutic targets for PTB.
MATERIALS AND METHODS
Ethics statement
This study was approved by the Institutional Review Boards at the University of Chicago, Northwestern University, and Duke University Medical School. Informed consent for the use of data collected via questionnaires and clinics was obtained from participants following the recommendations of the ALSPAC (Avon Longitudinal Study of Parents and Children) Ethics and Law Committee at the time. Informed consent for the use of genetic data in the other six GWASs used in this study was also obtained from participants. Details are available in the Supplementary Materials.
Sample collections
Placentas were collected from three African American women (≥18 years old) who delivered at term (≥37 weeks) following spontaneous labor; all were vaginal deliveries of singleton pregnancies. Within 1 hour of delivery, 5 cm by 5 cm pieces of the membranes were sampled from a distant location of the rupture site. Pieces were placed in Dulbecco’s modified Eagle’s medium (DMEM)-Ham’s F12 media containing 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. Samples were kept at 4°C and processed within 24 hours of tissue collection. This study was approved by the Institutional Review Boards at the University of Chicago, Northwestern University, and Duke University Medical School.
Isolation of mesenchymal stromal cells from human placental membranes
Third trimester placental tissue was enzymatically digested by a modification of previously described methods (55, 56). Decidua tissue was gently scraped from chorion, and tissue was enzymatically digested in a solution (1× Hanks’ balanced salt solution, 20 mM Hepes, 30 mM sodium bicarbonate, and 1% bovine serum albumin fraction V) containing collagenase type IV (200 U/ml; Sigma-Aldrich, C-5138), hyaluronidase type IS (1 mg/ml; Sigma-Aldrich, H-3506), and DNase type IV (0.45 KU/ml, Sigma-Aldrich, D-5025) at 37°C, until a single-cell suspension was obtained (usually three rounds of 30 min digestion using fresh digestion media each round). Epithelial cells were removed by filtering through a 75 μM nylon membrane and RPMI (Sigma-Aldrich) containing 10% FBS was added for enzyme inactivation. Dissociated cells were collected by centrifugation at 400g for 10 min and washed in RPMI/10% FBS. Erythrocytes were removed by cell pellet incubation with 1× red blood cell lysis buffer (Sigma-Aldrich) for 2.5 min at room temperature. The resulting cells were counted and resuspended in seeding media [1× phenol red-free high-glucose DMEM (Gibco)] supplemented with 10% FBS (Thermo Fisher Scientific), 2 mM l-glutamine (Life Technologies), 1 mM sodium pyruvate (Fisher Scientific), 1× insulin-transferrin-selenium (ITS; Thermo Fisher Scientific), 1% penicillin/streptomycin, and 1× antibiotic-antimycotic (Thermo Fisher Scientific). Dissociated cells were plated into a T75 flask and incubated at 37°C and 5% CO2 for 15 to 30 min (enrichment by attachment). The supernatant was carefully removed, and loosely attached cells were discarded. Plates were allowed to grow in fresh media containing 10% charcoal-stripped FBS (CS-FBS), and 1× antibiotic-antimycotic until the plate was 80% confluent. The antibiotic-antimycotic was removed from the culture media after 2 weeks of culture. We obtained >99% vimentin-positive cells after three passages (fig. S4). Cells were expanded, harvested in 0.05% trypsin, and cryopreserved in 10% dimethylsulfoxide culture media for subsequent use. Each cell line was defined as coming from a different sample collection (different pregnancy).
Decidualization of mesenchymal stromal cells in vitro
Cells were plated and grown for 2 days in cell culture media (1× phenol red-free high-glucose DMEM, 10% CS-FBS, 2 mM l-glutamine, 1 mM sodium pyruvate, and 1× ITS). After 2 days, cells were treated either with control media (1× phenol red-free high-glucose DMEM, 2% CS-FBS, 2 mM l-glutamine) or decidualization media (1× phenol red-free high-glucose DMEM, 2% CS-FBS, 2 mM % l-glutamine, 0.5 mM 8-Br-cAMP, and 1 μM MPA) for 48 hours. Cells were incubated at 37°C and 5% CO2 and harvested for ATAC-seq, ChIP-seq, and RNA-seq, and prolactin (PRL) and insulin-like growth factor-binding protein 1 (IGFBP1) mRNA were assessed by quantitative real-time polymerase chain reaction (PCR) before each downstream assay was performed.
RNA sequencing
Total RNA was extracted from approximately 1 million cells using the AllPrep DNA/RNA Kit (QIAGEN) according to manufacturer’s instructions. RNA quality (RNA integrity number) and concentration was assessed by Bioanalyzer 2100 (Agilent technology). RNA-seq libraries were generated by a TruSeq stranded total RNA library prep kit (Illumina) and TruSeq RNA CD Index Plate.
Chromatin immunoprecipitation sequencing
For ChIP experiments, cells were cross-linked by adding to the media 37% formaldehyde to a final concentration of 1%, gently mixed, incubated for 10 min, and quenched for 5 min with 2.5 M glycine for a final of 0.125 M per plate. Cells were washed using cold 1× phosphate-buffered saline and scraped in 15 ml of cold Farnham lysis buffer and protease inhibitor (Roche, 11836145001), and cell pellets were flash frozen and kept at −80°C. Thawed pellets were resuspended in radioimmunoprecipitation assay buffer on ice, aliquoted into 20 million cells per tube, and sonicated by Bioruptor (three 15-min rounds of 30 s ON, 30 s). ChIP was performed on 10 million cells using antibodies to H3K27ac, H3K4me3, and H3K4me1 histone marks (ab4729/lot no. GR274237, ab8580/lot no. GR273043, and ab8895/lot no. GR262515, respectively). M-280 sheep anti-rabbit immunoglobulin G Dynabeads (Invitrogen, 11203D) was used for chromatin immunoprecipitation. DNA was purified using the Qiagen MinElute PCR Purification Kit, quantified by Qubit, and prepared for sequencing using the Kapa Hyper Prep Kit. All libraries were pooled to 10 nM per sample before sequencing.
Assay for transposase-accessible chromatin sequencing
Approximately, 50,000 cells were harvested and used for ATAC-seq library preparation as described in the Fast-ATAC protocol (57). ATAC-seq libraries were uniquely indexed with Nextera PCR Primers and amplified with 9 to 12 cycles of PCR amplification. Amplified DNA fragments were purified with 0.8:1 ratio of Agencourt AMPure XP (Beckman Coulter) to sample. Libraries were quantified by Qubit, and size distribution was inspected by Bioanalyzer (Agilent Genomic DNA chip, Agilent Technologies). All libraries were pooled to 10 nM per sample before sequencing.
Promoter capture Hi-C
In situ Hi-C was performed as described previously (58). Briefly, 5 million decidualized cells were treated with formaldehyde 1% to cross-link interacting DNA loci. Cross-linked chromatin was treated with lysed and digested with MboI endonuclease (New England Biolabs). Subsequently, the restriction fragment overhangs were filled in and the DNA ends were marked with biotin-14-dATP (Life Technologies). The biotin-labeled DNA was sheared and pulled down using Dynabeads MyOne Stretavidin T1 beads (Life Technologies, 65602) and prepared for Illumina paired-end sequencing. The in situ Hi-C library was amplified directly off of the T1 beads with nine cycles of PCR using Illumina primers and protocol (Illumina, 2007). Promoter capture was performed as described previously (39). The Hi-C library was hybridized to 81,735 biotinylated 120-bp custom RNA oligomers (Custom Array) targeting promoter regions (four probes/RefSeq transcription start sites). After hybridization, postcapture PCR was performed on the DNA bound to the beads via biotinylated RNA.
Differential expression
Read counts per gene were calculated with Salmon (59) version 0.12.0 on transcripts from human Gencode release 19 (ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_19/gencode.v19.pc_transcripts.fa.gz and ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_19/gencode.v19.lncRNA_transcripts.fa.gz). Estimated counts were used in exploratory analysis (transformed with DESeq2’s rlog function) and in DESeq2 (25) version 1.24.0 to identify differentially expressed genes (adjusted P ≤ 0.05 and absolute fold change of ≥1.2). After observing that replicates for each cell lines clustered together, we pooled reads for each cell line, combining three decidualization experiments in each sample. We then performed a paired analysis to obtain genes that were differentially expressed between untreated and decidualized samples. The six samples clustered by treatment and by cell line and analysis with svaseq (60) showed that the two surrogate variables identified correlated with cell line, and therefore, a paired analysis was enough to correct the data.
Peak calling
ATAC-seq reads were trimmed with cutadapt and aligned with bowtie2 (61) version 2.3.4.1. Reads with mapping quality lower than 10 were discarded. ChIP-seq reads were also aligned with bowtie2. Peaks were called using MACS2 (62) version 2.1.2 with parameters --llocal 20000 --shift -100 --extsize 200 -q 0.05 for ATAC-seq and default parameters for ChIP-seq. Peaks overlapping coordinates blacklisted by Kundaje were excluded (http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeMapability/wgEncodeDacMapabilityConsensusExcludable.bed.gz).
Differential ATAC-seq and ChIP-seq peaks
Similarly to RNA-seq, we pooled reads from replicates for each cell line. We called peaks for each of the six samples using MACS2 and converted peak coordinates into 100-bp contiguous bins. Bins covered by less than 60% of their extension were excluded. To identify reproducible peaks, we only kept bins that were present in at least two of the three cell lines in each condition, allowing for condition-specific peaks. See table S7 for an assessment of the contribution of each cell line to the universe of peaks obtained. We then merged all adjacent bins, expanding them back into longer peaks. We counted the number of reads in all peaks and in all samples and compared the read counts using DESeq2 (adjusted P < 0.05 and absolute fold change >1.2).
Statistical analysis of the frequencies of differential peaks near differentially expressed genes
The P values in Fig. 2B were calculated with a chi-square test of the number of peaks with increased or decreased numbers of reads observed and an expected probability based on the number of peaks in each category for each dataset. Bonferroni correction was performed to correct for multiple testing.
Transcription factor ChIP-seq
ChIP-seq reads were downloaded from National Center for Biotechnology Information Gene Expression Omnibus and processed locally. HOMER 4.9 (63) was used to call peaks for the following samples: PGR (GSE94038); NR2F2 (GSE52008); FOSL2 (GSE94038), FOXO1 (GSE94037); and NR2F2 input (GSE52008); and FOXO1, PGR, and FOSL2 input (GSE94038).
Overlap between ATAC-seq and ChIP-seq peaks
Reproducible peaks were converted into 100 bp bins and those with >60% of their extension covered by a peak was retained. Common bins were counted, and the number of counts was plotted with UpSetR 1.4.0.
Motif enrichment
We used HOMER 4.9 to identify DNA binding motifs enriched in peaks with parameters -len 8,10,12 -size 200 -mask.
Enrichment of overlap between peaks
Enrichment was calculated as the observed number of overlapping peaks divided by the expected number of overlapping peaks using bedtools intersectBed with a 1 bp minimum. The expected number of overlapping peaks was obtained by averaging 100 random samples of peaks with bedtools shuffle excluding gaps annotated by the University of California, Santa Cruz Genome Browser (64). While shuffling peaks does not account for mapping and other biases that make peak locations nonuniform and may result in overestimation of enrichment, our results are limited to comparisons between enrichments, which should cancel any biases.
Hi-C interaction calling
We used HiCUP v0.5.9 (65) to align and filter Hi-C reads. HiCUP used bowtie2 version 2.2.3 to align reads. Unique reads were used as input by CHiCAGO (66) version 1.2.0, and significant interactions were called with default parameters. We only kept interactions identified by CHiCAGO that were in cis and with an end located at least 10 kb from a capture probe.
Pairing differential peaks with putative target genes
To pair peaks using pcHi-C, significant interactions identified by CHiCAGO that overlapped an ATAC-seq or ChIP-seq peak and were less than 300 kb away from a promoter were used. We chose 300 kb because the mean distance between interacting promoters and other regions was 280 kb (median, 200 kb). To pair peaks to the nearest gene, BEDTools closest -t first -d was used to find the gene closest to a peak, up to 300 kb away. To pair peaks to a random gene, all genes up to 300 kb from a peak were selected and one gene was randomly assigned to each peak. For each of these sets of pairs, we calculated the fraction of peak/gene pairs that had the same direction of change according to differential read count analysis with DESeq2, of the total number of peak/gene pairs. Only genes expressed at >1 transcript per million across all samples were used in the nearest and random gene assignments.
P values were calculated with a chi-square test comparing the number of cases in the matched and unmatched categories observed in the random set (average from 200 iterations) and in the two peak/gene pairing methods: nearest gene and pcHi-C interactions.
Gestational duration GWAS
The GWAS results used in this study was an extension of our previously published results (5). Like our previous study, we used summary results from 23andMe, which were obtained from GWAS of gestational duration in 42,121 mothers of European ancestry. In addition, we performed GWA analyses in 14,263 European mothers from six academic datasets. To increase the power of GWA discovery, we performed meta-analysis between the results from 23andMe and the results from the six datasets. See the Supplementary Materials for a full description of the GWAS.
GWAS enrichment analysis with S-LDSC
We assessed how much of the heritability of gestational duration is contained within ATAC-seq, H3K4me1, H3K4me3, H3K27ac, and pcHi-C peaks using S-LDSC (41). S-LDSC is a generalization of LD score regression, a method for estimating the heritability of a trait using SNP-level GWAS summary statistics and SNP-level estimates of the amount of genetic variation tagged at each variant, known as LD scores. Under the LD score regression model, the expected value of the GWAS summary statistic for a variant (specifically, the expected value of the χ2 statistic) is a linear function of the LD score at that site, and h2, the per-SNP heritability, and a an intercept parameter. Under the S-LDSC model, rather than estimating a single per-SNP heritability parameter, a parameter is estimated for each of several functional annotations. In a standard S-LDSC analysis, user-provided annotations are combined with a “baseline” set of genomic annotations from publicly available datasets. For this analysis, LD scores were calculated using the peaks identified as reproducible across either treated or untreated samples as annotations and the genotype data from the European individuals from phase 3 of the 1000 Genomes project (obtained from the Price Lab website: https://alkesgroup.broadinstitute.org/LDSCORE/) as a reference LD panel, using only the HapMap3 SNP list (also from the Price lab website). S-LDSC was performed on the gestational duration GWAS using the endometrial-tissue derived LD scores and the baseline LD scores contained in version 2.2 of the LD score regression baseline LD model. We include all annotations from the baseline LD model except those “flanking” annotations. This resulted in a total of 64 baseline annotations used in our S-LDSC analysis.
Fine-mapping GWAS loci associated with gestational length
Fine mapping proceeded in three stages. In the first stage, we partitioned the genome into 1703 regions approximately independent regions using breakpoints derived by Berisa et al. (40). Next, we constructed an SNP-level prior probability of being causal variant, informed by the functional genomic data that we collected. We used a Bayesian hierarchical model [TORUS (42)]. TORUS takes as input GWAS summary statistics and genomic annotations and estimates the extent to which SNPs with functional genomic annotations are likely to be causal for a trait of interest. Specifically, under TORUS, each SNP has a small prior probability of being a causal variant, which is a logistic function of the annotations of the SNP. Then, TORUS estimates the parameters of this logistic function using genome-wide summary statistics. Once these parameters are estimated, each SNP will have a prior causal probability based on its unique functional annotations. We ran TORUS with the gestational age GWAS summary statistics and the reproducible H3K27ac and H3K4me1 peaks from the treated samples along with the pcHi-C contact regions to obtain an SNP-level prior.
Last, fine mapping was performed using a summary statistics-based version of the “sum of single effects” model (43) using 1000 Genome as reference panel. SuSiE (as implemented in the R package “susieR”) was run on the 10 regions believed to have one or more causal variants with an FDR of 0.1 as estimated by TORUS. For each region, SuSiE was run with a uniform prior (default setting of SuSiE) and with an informed prior learned by TORUS. The parameter L of SuSiE (maximum number of causal variants) is set at 1 when running SuSiE (67, 68).
SNPs in Epigenome roadmap histone modification peaks
H3K27ac, H3K4me1, and H4K4me3 histone modification peak coordinates were downloaded from the Epigenome Roadmap data website, and bedtools intersect was used to find peaks that overlapped SNPs coordinates.
Supplementary Material
Acknowledgments
We acknowledge C. Billstrand, K. Naughton, M. Soliai, and R. Nicolae for assistance with sample processing; R. Nasim, S. Chinthala, and R. Loth for help with sample collection; R. Minhas for help with clinical questions and data entry; and B. Furner and S. Choi for designing and maintaining the sample tracking database. ALSPAC GWAS data: We are grateful to all the families who took part in this study, the midwives for help in recruiting them, and the whole ALSPAC team, which includes interviewers, computer and laboratory technicians, clerical workers, research scientists, volunteers, managers, receptionists, and nurses. Funding: The UChicago-Northwestern-Duke Prematurity Research Center was supported by a research grant from the March of Dimes to C.O., M.A.N., G.E.C., and J.K. This work was also supported by the March of Dimes Prematurity Research Center Ohio Collaborative and Bill and Melinda Gates Foundation (OPP1113966) to L.J.M. and G.Z. The UK Medical Research Council and Wellcome (grant reference 217065/Z/19/Z) and the University of Bristol provide core support for ALSPAC. A comprehensive list of grants funding is available on the ALSPAC website (http://www.bristol.ac.uk/alspac/external/documents/grant-acknowledgements.pdf). This research was specifically funded by Wellcome Trust WT088806 (Maternal genotype). This work was partially funded from grants from the National Institutes of Health, R01HL129735 (C.O.), 1R01MH110531 (X.H.), R01HL128075, R01119577, and R01DK114661 (M.A.N.). Ethics statement: This study was approved by the Institutional Review Boards at the University of Chicago, Northwestern University and Duke University Medical School. Informed consent for the use of data collected via questionnaires and clinics was obtained from participants following the recommendations of the ALSPAC Ethics and Law Committee at the time. Informed consent for the use of genetic data in the other six GWAS used in this study was also obtained from participants. Details are available in the Supplementary Materials. Author contributions: C.O., M.A.N., G.E.C., and X.H. designed and conceptualized the work. N.J.S., N.K., and J.M. analyzed data and interpreted results. I.A. coordinated all experiments. N.C., D.R.S., C.P., C.H., R.Z., H.K., and I.A. performed experiments. R.A. and S.R. facilitated sample collection. G.Z., B.J., M.H., K.T., and L.J.M. contributed the GWAS data. X.L., V.C.C., J.K., S.R., W.G., A.M., C.G., and I.A. contributed to discussions on study design. S.R., G.Z., L.J.M., V.J.L., G.E.C., C.O., X.H., and M.A.N. supervised aspects of the study. N.J.S., I.A., C.O., G.E.C., X.H., and M.A.N. wrote the manuscript. All authors read and commented on the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data generated or analyzed during this study are included in this published article and available at www.immport.org/shared/study/SDY1626. All the peaks sets, pcHi-C, read count, and TPM data are available. An expanded set of 2530 differentially expressed genes and sets of differential peaks called without statistically testing for fold change is also made available. Source code for the GWAS enrichment analyses can be found at https://github.com/CreRecombinase/ptb_workflowr. Blacklisted regions: http://mitra.stanford.edu/kundaje/akundaje/release/blacklists/hg19-human/. Price Lab website: https://data.broadinstitute.org/alkesgroup/LDSCORE/. Epigenome Roadmap data website: https://egg2.wustl.edu/roadmap/data/byFileType/peaks/consolidated/narrowPeak/ucsc_compatible/. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/49/eabc8696/DC1
REFERENCES AND NOTES
- 1.Saigal S., Doyle L. W., An overview of mortality and sequelae of preterm birth from infancy to adulthood. Lancet 371, 261–269 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Crider K. S., Whitehead N., Buus R. M., Genetic variation associated with preterm birth: A HuGE review. Genet. Med. 7, 593–604 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Pennell C. E., Jacobsson B., Williams S. M., Buus R. M., Muglia L. J., Dolan S. M., Morken N. H., Ozcelik H., Lye S. J.; PREBIC Genetics Working Group, Relton C., Genetic epidemiologic studies of preterm birth: Guidelines for research. Am. J. Obstet. Gynecol. 196, 107–118 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Varner M. W., Esplin M. S., Current understanding of genetic factors in preterm birth. BJOG 112 ( Suppl 1), 28–31 (2005). [DOI] [PubMed] [Google Scholar]
- 5.Zhang G., Feenstra B., Bacelis J., Liu X., Muglia L. M., Juodakis J., Miller D. E., Litterman N., Jiang P.-P., Russell L., Hinds D. A., Hu Y., Weirauch M. T., Chen X., Chavan A. R., Wagner G. P., Pavlicev M., Nnamani M. C., Maziarz J., Karjalainen M. K., Rämet M., Sengpiel V., Geller F., Boyd H. A., Palotie A., Momany A., Bedell B., Ryckman K. K., Huusko J. M., Forney C. R., Kottyan L. C., Hallman M., Teramo K., Nohr E. A., Smith G. D., Melbye M., Jacobsson B., Muglia L. J., Genetic associations with gestational duration and spontaneous preterm birth. N. Engl. J. Med. 377, 1156–1167 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu X., Helenius D., Skotte L., Beaumont R. N., Wielscher M., Geller F., Juodakis J., Mahajan A., Bradfield J. P., Lin F. T. J., Vogelezang S., Bustamante M., Ahluwalia T. S., Pitkanen N., Wang C. A., Bacelis J., Borges M. C., Zhang G., Bedell B. A., Rossi R. M., Skogstrand K., Peng S., Thompson W. K., Appadurai V., Lawlor D. A., Kalliala I., Power C., McCarthy M. I., Boyd H. A., Marazita M. L., Hakonarson H., Hayes M. G., Scholtens D. M., Rivadeneira F., Jaddoe V. W. V., Vinding R. K., Bisgaard H., Knight B. A., Pahkala K., Raitakari O., Helgeland Ø., Johansson S., Njølstad P. R., Fadista J., Schork A. J., Nudel R., Miller D. E., Chen X., Weirauch M. T., Mortensen P. B., Børglum A. D., Nordentoft M., Mors O., Hao K., Ryckman K. K., Hougaard D. M., Kottyan L. C., Pennell C. E., Lyytikainen L.-P., Bønnelykke K., Vrijheid M., Felix J. F., Lowe W. L. Jr., Grant S. F. A., Hyppönen E., Jacobsson B., Jarvelin M.-R., Muglia L. J., Murray J. C., Freathy R. M., Werge T. M., Melbye M., Buil A., Feenstra B., Variants in the fetal genome near pro-inflammatory cytokine genes on 2q13 associate with gestational duration. Nat. Commun. 10, 3927 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schaid D. J., Chen W., Larson N. B., From genome-wide associations to candidate causal variants by statistical fine-mapping. Nat. Rev. Genet. 19, 491–504 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pickrell J. K., Joint analysis of functional genomic data and genome-wide association studies of 18 human traits. Am. J. Hum. Genet. 94, 559–573 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.The ENCODE Project Consortium , A user’s guide to the encyclopedia of DNA elements (ENCODE). PLOS Biol. 9, e1001046 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.GTEx Consortium; Laboratory; Data Analysis &Coordinating Center (LDACC)—Analysis Working Group; Statistical Methods groups—Analysis Working Group; Enhancing GTEx (eGTEx) groups; NIH Common Fund; NIH/NCI; NIH/NHGRI; NIH/NIMH; NIH/NIDA; Biospecimen Collection Source Site—NDRI; Biospecimen Collection Source Site—RPCI; Biospecimen Core Resource—VARI; Brain Bank Repository—University of Miami Brain Endowment Bank; Leidos Biomedical—Project Management; ELSI Study; Genome Browser Data Integration &Visualization—EBI; Genome Browser Data Integration &Visualization—UCSC Genomics Institute, University of California Santa Cruz; Lead analysts; Laboratory, Data Analysis &Coordinating Center (LDACC); NIH program management; Biospecimen collection; Pathology; eQTL manuscript working group, Battle A., Brown C. D., Engelhardt B. E., Montgomery S. B., Genetic effects on gene expression across human tissues. Nature 550, 204–213 (2017).29022597 [Google Scholar]
- 11.Roadmap Epigenomics Consortium, Kundaje A., Meuleman W., Ernst J., Bilenky M., Yen A., Heravi-Moussavi A., Kheradpour P., Zhang Z., Wang J., Ziller M. J., Amin V., Whitaker J. W., Schultz M. D., Ward L. D., Sarkar A., Quon G., Sandstrom R. S., Eaton M. L., Wu Y.-C., Pfenning A. R., Wang X., Claussnitzer M., Liu Y., Coarfa C., Harris R. A., Shoresh N., Epstein C. B., Gjoneska E., Leung D., Xie W., Hawkins R. D., Lister R., Hong C., Gascard P., Mungall A. J., Moore R., Chuah E., Tam A., Canfield T. K., Hansen R. S., Kaul R., Sabo P. J., Bansal M. S., Carles A., Dixon J. R., Farh K.-H., Feizi S., Karlic R., Kim A.-R., Kulkarni A., Li D., Lowdon R., Elliott G., Mercer T. R., Neph S. J., Onuchic V., Polak P., Rajagopal N., Ray P., Sallari R. C., Siebenthall K. T., Sinnott-Armstrong N. A., Stevens M., Thurman R. E., Wu J., Zhang B., Zhou X., Beaudet A. E., Boyer L. A., De Jager P. L., Farnham P. J., Fisher S. J., Haussler D., Jones S. J., Li W., Marra M. A., McManus M. T., Sunyaev S., Thomson J. A., Tlsty T. D., Tsai L.-H., Wang W., Waterland R. A., Zhang M. Q., Chadwick L. H., Bernstein B. E., Costello J. F., Ecker J. R., Hirst M., Meissner A., Milosavljevic A., Ren B., Stamatoyannopoulos J. A., Wang T., Kellis M., Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maurano M. T., Humbert R., Rynes E., Thurman R. E., Haugen E., Wang H., Reynolds A. P., Sandstrom R., Qu H., Brody J., Shafer A., Neri F., Lee K., Kutyavin T., Stehling-Sun S., Johnson A. K., Canfield T. K., Giste E., Diegel M., Bates D., Hansen R. S., Neph S., Sabo P. J., Heimfeld S., Raubitschek A., Ziegler S., Cotsapas C., Sotoodehnia N., Glass I., Sunyaev S. R., Kaul R., Stamatoyannopoulos J. A., Systematic localization of common disease-associated variation in regulatory DNA. Science 337, 1190–1195 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Norwitz E. R., Bonney E. A., Snegovskikh V. V., Williams M. A., Phillippe M., Park J. S., Abrahams V. M., Molecular regulation of parturition: The role of the decidual clock. Cold Spring Harb. Perspect. Med. 5, a023143 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rinaldi S. F., Makieva S., Saunders P. T., Rossi A. G., Norman J. E., Immune cell and transcriptomic analysis of the human decidua in term and preterm parturition. Mol. Hum. Reprod. 23, 708–724 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gellersen B., Brosens J. J., Cyclic decidualization of the human endometrium in reproductive health and failure. Endocr. Rev. 35, 851–905 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Popovici R. M., Kao L. C., Giudice L. C., Discovery of new inducible genes in in vitro decidualized human endometrial stromal cells using microarray technology. Endocrinology 141, 3510–3513 (2000). [DOI] [PubMed] [Google Scholar]
- 17.Grimaldi G., Christian M., Steel J. H., Henriet P., Poutanen M., Brosens J. J., Down-regulation of the histone methyltransferase EZH2 contributes to the epigenetic programming of decidualizing human endometrial stromal cells. Mol. Endocrinol. 25, 1892–1903 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tamura I., Ohkawa Y., Sato T., Suyama M., Jozaki K., Okada M., Lee L., Maekawa R., Asada H., Sato S., Yamagata Y., Tamura H., Sugino N., Genome-wide analysis of histone modifications in human endometrial stromal cells. Mol. Endocrinol. 28, 1656–1669 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Katoh N., Kuroda K., Tomikawa J., Ogata-Kawata H., Ozaki R., Ochiai A., Kitade M., Takeda S., Nakabayashi K., Hata K., Reciprocal changes of H3K27ac and H3K27me3 at the promoter regions of the critical genes for endometrial decidualization. Epigenomics 10, 1243–1257 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Kin K., Nnamani M. C., Lynch V. J., Michaelides E., Wagner G. P., Cell-type phylogenetics and the origin of endometrial stromal cells. Cell Rep. 10, 1398–1409 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Vrljicak P., Lucas E. S., Lansdowne L., Lucciola R., Muter J., Dyer N. P., Brosens J. J., Ott S., Analysis of chromatin accessibility in decidualizing human endometrial stromal cells. FASEB J. 32, 2467–2477 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muñoz-Fernández R., De La Mata C., Requena F., Martin F., Fernandez-Rubio P., Llorca T., Ruiz-Magaña M. J., Ruiz-Ruiz C., Olivares E. G., Human predecidual stromal cells are mesenchymal stromal/stem cells and have a therapeutic effect in an immune-based mouse model of recurrent spontaneous abortion. Stem Cell Res. Ther. 10, 177 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Choi Y. S., Park Y.-B., Ha C.-W., Kim J. A., Heo J.-C., Han W.-J., Oh S.-Y., Choi S.-J., Different characteristics of mesenchymal stem cells isolated from different layers of full term placenta. PLOS ONE 12, e0172642 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hass R., Kasper C., Böhm S., Jacobs R., Different populations and sources of human mesenchymal stem cells (MSC): A comparison of adult and neonatal tissue-derived MSC. Cell Commun. Signal 9, 12 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaya H. S., Hantak A. M., Stubbs L. J., Taylor R. N., Bagchi I. C., Bagchi M. K., Roles of progesterone receptor A and B isoforms during human endometrial decidualization. Mol. Endocrinol. 29, 882–895 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sakabe N. J., Nobrega M. A., Beyond the ENCODE project: Using genomics and epigenomics strategies to study enhancer evolution. Philos. Trans. R. Soc. Lond. B Biol. Sci. 368, 20130022 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mazur E. C., Vasquez Y. M., Li X., Kommagani R., Jiang L., Chen R., Lanz R. B., Kovanci E., Gibbons W. E., DeMayo F. J., Progesterone receptor transcriptome and cistrome in decidualized human endometrial stromal cells. Endocrinology 156, 2239–2253 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vasquez Y. M., Mazur E. C., Li X., Kommagani R., Jiang L., Chen R., Lanz R. B., Kovanci E., Gibbons W. E., DeMayo F. J., FOXO1 is required for binding of PR on IRF4, novel transcriptional regulator of endometrial stromal decidualization. Mol. Endocrinol. 29, 421–433 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li X., Large M. J., Creighton C. J., Lanz R. B., Jeong J.-W., Young S. L., Lessey B. A., Palomino W. A., Tsai S. Y., Demayo F. J., COUP-TFII regulates human endometrial stromal genes involved in inflammation. Mol. Endocrinol. 27, 2041–2054 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mika K. M., Li X., DeMayo F. J., Lynch V. J., An ancient fecundability-associated polymorphism creates a GATA2 binding site in a distal enhancer of HLA-F. Am. J. Hum. Genet. 103, 509–521 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kajihara T., Brosens J. J., Ishihara O., The role of FOXO1 in the decidual transformation of the endometrium and early pregnancy. Med. Mol. Morphol. 46, 61–68 (2013). [DOI] [PubMed] [Google Scholar]
- 33.Ramathal C., Wang W., Hunt E., Bagchi I. C., Bagchi M. K., Transcription factor CCAAT enhancer-binding protein β (C/EBPβ) regulates the formation of a unique extracellular matrix that controls uterine stromal differentiation and embryo implantation. J. Biol. Chem. 286, 19860–19871 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kessler C. A., Bachurski C. J., Schroeder J., Stanek J., Handwerger S., TEAD1 inhibits prolactin gene expression in cultured human uterine decidual cells. Mol. Cell. Endocrinol. 295, 32–38 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rubel C. A., Wu S.-P., Lin L., Wang T., Lanz R. B., Li X., Kommagani R., Franco H. L., Camper S. A., Tong Q., Jeong J.-W., Lydon J. P., DeMayo F. J., A Gata2-dependent transcription network regulates uterine progesterone responsiveness and endometrial function. Cell Rep. 17, 1414–1425 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spitz F., Gene regulation at a distance: From remote enhancers to 3D regulatory ensembles. Semin. Cell Dev. Biol. 57, 57–67 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Schoenfelder S., Furlan-Magaril M., Mifsud B., Tavares-Cadete F., Sugar R., Javierre B.-M., Nagano T., Katsman Y., Sakthidevi M., Wingett S. W., Dimitrova E., Dimond A., Edelman L. B., Elderkin S., Tabbada K., Darbo E., Andrews S., Herman B., Higgs A., LeProust E., Osborne C. S., Mitchell J. A., Luscombe N. M., Fraser P., The pluripotent regulatory circuitry connecting promoters to their long-range interacting elements. Genome Res. 25, 582–597 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mifsud B., Tavares-Cadete F., Young A. N., Sugar R., Schoenfelder S., Ferreira L., Wingett S. W., Andrews S., Grey W., Ewels P. A., Herman B., Happe S., Higgs A., LeProust E., Follows G. A., Fraser P., Luscombe N. M., Osborne C. S., Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nat. Genet. 47, 598–606 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Montefiori L. E., Sobreira D. R., Sakabe N. J., Aneas I., Joslin A. C., Hansen G. T., Bozek G., Moskowitz I. P., McNally E. M., Nóbrega M. A., A promoter interaction map for cardiovascular disease genetics. eLife 7, e35788 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Berisa T., Pickrell J. K., Approximately independent linkage disequilibrium blocks in human populations. Bioinformatics 32, 283–285 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Finucane H. K., Bulik-Sullivan B., Gusev A., Trynka G., Reshef Y., Loh P.-R., Anttila V., Xu H., Zang C., Farh K., Ripke S., Day F. R.; ReproGen Consortium; Schizophrenia Working Group of the Psychiatric Genomics Consortium; RACI Consortium, Purcell S., Stahl E., Lindstrom S., Perry J. R., Okada Y., Raychaudhuri S., Daly M. J., Patterson N., Neale B. M., Price A. L., Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 47, 1228–1235 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wen X., Lee Y., Luca F., Pique-Regi R., Efficient integrative multi-SNP association analysis via deterministic approximation of posteriors. Am. J. Hum. Genet. 98, 1114–1129 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang G., Sarkar A., Carbonetto P., Stephens M., A simple new approach to variable selection in regression, with application to genetic fine-mapping. bioRxiv 10.1101/501114 , (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li Q., Kannan A., DeMayo F. J., Lydon J. P., Cooke P. S., Yamagishi H., Srivastava D., Bagchi M. K., Bagchi I. C., The antiproliferative action of progesterone in uterine epithelium is mediated by Hand2. Science 331, 912–916 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fujiwara T., GATA transcription factors: Basic principles and related human disorders. Tohoku J. Exp. Med. 242, 83–91 (2017). [DOI] [PubMed] [Google Scholar]
- 46.Ochoa-Bernal M. A., Fazleabas A. T., Physiologic events of embryo implantation and decidualization in human and non-human primates. Int. J. Mol. Sci. 21, 1973 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Garrido-Gomez T., Dominguez F., Quinonero A., Diaz-Gimeno P., Kapidzic M., Gormley M., Ona K., Padilla-Iserte P., McMaster M., Genbacev O., Perales A., Fisher S. J., Simón C., Defective decidualization during and after severe preeclampsia reveals a possible maternal contribution to the etiology. Proc. Natl. Acad. Sci. U.S.A. 114, E8468–E8477 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Norwitz E. R., Defective implantation and placentation: Laying the blueprint for pregnancy complications. Reprod. Biomed. Online 13, 591–599 (2006). [DOI] [PubMed] [Google Scholar]
- 49.Pavlicev M., Norwitz E. R., Human parturition: Nothing more than a delayed menstruation. Reprod. Sci. 25, 166–173 (2018). [DOI] [PubMed] [Google Scholar]
- 50.Cho H., Okada H., Tsuzuki T., Nishigaki A., Yasuda K., Kanzaki H., Progestin-induced heart and neural crest derivatives expressed transcript 2 is associated with fibulin-1 expression in human endometrial stromal cells. Fertil. Steril. 99, 248–255.e2 (2013). [DOI] [PubMed] [Google Scholar]
- 51.Kohlmeier A., Sison C. A. M., Yilmaz B. D., Coon V. J., Dyson M. T., Bulun S. E., GATA2 and Progesterone Receptor Interaction in Endometrial Stromal Cells Undergoing Decidualization. Endocrinology 161, bqaa070 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dowling P., Clynes M., Conditioned media from cell lines: A complementary model to clinical specimens for the discovery of disease-specific biomarkers. Proteomics 11, 794–804 (2011). [DOI] [PubMed] [Google Scholar]
- 53.Rubel C. A., Franco H. L., Jeong J. W., Lydon J. P., DeMayo F. J., GATA2 is expressed at critical times in the mouse uterus during pregnancy. Gene Expr. Patterns 12, 196–203 (2012). [DOI] [PubMed] [Google Scholar]
- 54.Rubel C. A., Franco H. L., Camper S. A., Lanz R. B., Jeong J. W., Lydon J. P., DeMayo F. J., Gata2 is a master regulator of endometrial function and progesterone signaling. Biol. Reprod. 85, 179 (2011).21471298 [Google Scholar]
- 55.Vince G. S., Starkey P. M., Jackson M. C., Sargent I. L., Redman C. W., Flow cytometric characterisation of cell populations in human pregnancy decidua and isolation of decidual macrophages. J. Immunol. Methods 132, 181–189 (1990). [DOI] [PubMed] [Google Scholar]
- 56.Narahara H., Kawano Y., Nasu K., Yoshimatsu J., Johnston J. M., Miyakawa I., Platelet-activating factor inhibits the secretion of platelet-activating factor acetylhydrolase by human decidual macrophages. J. Clin. Endocrinol. Metab. 88, 6029–6033 (2003). [DOI] [PubMed] [Google Scholar]
- 57.Corces M. R., Buenrostro J. D., Wu B., Greenside P. G., Chan S. M., Koenig J. L., Snyder M. P., Pritchard J. K., Kundaje A., Greenleaf W. J., Majeti R., Chang H. Y., Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat. Genet. 48, 1193–1203 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rao S. S., Huntley M. H., Durand N. C., Stamenova E. K., Bochkov I. D., Robinson J. T., Sanborn A. L., Machol I., Omer A. D., Lander E. S., Aiden E. L., A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Patro R., Duggal G., Love M. I., Irizarry R. A., Kingsford C., Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Leek J. T., svaseq: Removing batch effects and other unwanted noise from sequencing data. Nucleic Acids Res. 42, e161 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Langmead B., Salzberg S. L., Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang Y., Liu T., Meyer C. A., Eeckhoute J., Johnson D. S., Bernstein B. E., Nusbaum C., Myers R. M., Brown M., Li W., Liu X. S., Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Heinz S., Benner C., Spann N., Bertolino E., Lin Y. C., Laslo P., Cheng J. X., Murre C., Singh H., Glass C. K., Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kent W. J., Sugnet C. W., Furey T. S., Roskin K. M., Pringle T. H., Zahler A. M., Haussler D., The human genome browser at UCSC. Genome Res. 12, 996–1006 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wingett S., Ewels P., Furlan-Magaril M., Nagano T., Schoenfelder S., Fraser P., Andrews S., HiCUP: Pipeline for mapping and processing Hi-C data. F1000Res 4, 1310 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cairns J., Freire-Pritchett P., Wingett S. W., Varnái C., Dimond A., Plagnol V., Zerbino D., Schoenfelder S., Javierre B.-M., Osborne C., Fraser P., Spivakov M., CHiCAGO: Robust detection of DNA looping interactions in Capture Hi-C data. Genome Biol. 17, 127 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wakefield J., Bayes factors for genome-wide association studies: Comparison with P-values. Genet. Epidemiol. 33, 79–86 (2009). [DOI] [PubMed] [Google Scholar]
- 68.Servin B., Stephens M., Imputation-based analysis of association studies: Candidate regions and quantitative traits. PLOS Genet. 3, e114 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Elbaz M., Hadas R., Bilezikjian L. M., Gershon E., Uterine Foxl2 regulates the adherence of the Trophectoderm cells to the endometrial epithelium. Reprod. Biol. Endocrinol. 16, 12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Boyd A., Golding J., Macleod J., Lawlor D. A., Fraser A., Henderson J., Molloy L., Ness A., Ring S., Davey Smith G., Cohort profile: The ’children of the 90s’—The index offspring of the avon longitudinal study of parents and children. Int. J. Epidemiol. 42, 111–127 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fraser A., Macdonald-Wallis C., Tilling K., Boyd A., Golding J., Davey Smith G., Henderson J., Macleod J., Molloy L., Ness A., Ring S., Nelson S. M., Lawlor D. A., Cohort Profile: The Avon longitudinal study of parents and children: ALSPAC mothers cohort. Int. J. Epidemiol. 42, 97–110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Olsen J., Melbye M., Olsen S. F., Sørensen T. I., Aaby P., Andersen A. M., Taxbøl D., Hansen K. D., Juhl M., Schow T. B., Sorensen H. T., Andresen J., Mortensen E. L., Olesen A. W., Søndergaard C., The Danish national birth cohort—Its background, structure and aim. Scand. J. Public Health 29, 300–307 (2001). [DOI] [PubMed] [Google Scholar]
- 73.Magnus P., Birke C., Vejrup K., Haugan A., Alsaker E., Daltveit A. K., Handal M., Haugen M., Høiseth G., Knudsen G. P., Paltiel L., Schreuder P., Tambs K., Vold L., Stoltenberg C., Cohort profile update: The Norwegian mother and child cohort study (MoBa). Int. J. Epidemiol. 45, 382–388 (2016). [DOI] [PubMed] [Google Scholar]
- 74.Myking S., Boyd H. A., Myhre R., Feenstra B., Jugessur A., Devold Pay A. S., Ostensen I. H., Morken N.-H., Busch T., Ryckman K. K., Geller F., Magnus P., Gjessing H. K., Melbye M., Jacobsson B., Murray J. C., X-chromosomal maternal and fetal SNPs and the risk of spontaneous preterm delivery in a Danish/Norwegian genome-wide association study. PLOS ONE 8, e61781 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Plunkett J., Doniger S., Orabona G., Morgan T., Haataja R., Hallman M., Puttonen H., Menon R., Kuczynski E., Norwitz E., Snegovskikh V., Palotie A., Peltonen L., Fellman V., DeFranco E. A., Chaudhari B. P., McGregor T. L., McElroy J. J., Oetjens M. T., Teramo K., Borecki I., Fay J., Muglia L., An evolutionary genomic approach to identify genes involved in human birth timing. PLOS Genet. 7, e1001365 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Carvalho B., Bengtsson H., Speed T. P., Irizarry R. A., Exploration, normalization, and genotype calls of high-density oligonucleotide SNP array data. Biostatistics 8, 485–499 (2007). [DOI] [PubMed] [Google Scholar]
- 77.Scharpf R. B., Irizarry R. A., Ritchie M. E., Carvalho B., Ruczinski I., Using the R package crlmm for genotyping and copy number estimation. J. Stat. Softw. 40, 1–32 (2011). [PMC free article] [PubMed] [Google Scholar]
- 78.Delaneau O., Marchini J., Zagury J.-F., A linear complexity phasing method for thousands of genomes. Nat. Methods 9, 179–181 (2012). [DOI] [PubMed] [Google Scholar]
- 79.1000 Genomes Project Consortium, Abecasis G. R., Auton A., Brooks L. D., DePristo M. A., Durbin R. M., Handsaker R. E., Kang H. M., Marth G. T., McVean G. A., An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fuchsberger C., Abecasis G. R., Hinds D. A., minimac2: Faster genotype imputation. Bioinformatics 31, 782–784 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/49/eabc8696/DC1