

Abstract
Tumor genotyping is not routinely performed in localized non-small cell lung cancer (NSCLC) due to lack of associations of mutations with outcome. Here, we analyze 232 consecutive patients with localized NSCLC and demonstrate that KEAP1 and NFE2L2 mutations are predictive of high rates of local recurrence (LR) after radiotherapy but not surgery. Half of LRs occurred in KEAP1/NFE2L2 mutation tumors, indicating they are major molecular drivers of clinical radioresistance. Next, we functionally evaluate KEAP1/NFE2L2 mutations in our radiotherapy cohort and demonstrate that only pathogenic mutations are associated with radioresistance. Furthermore, expression of NFE2L2 target genes does not predict LR, underscoring the utility of tumor genotyping. Finally, we show that glutaminase inhibition preferentially radiosensitizes KEAP1 mutant cells via depletion of glutathione and increased radiation-induced DNA damage. Our findings suggest that genotyping for KEAP1/NFE2L2 mutations could facilitate treatment personalization and provide a potential strategy for overcoming radioresistance conferred by these mutations.
Introduction:
Over 40% of patients with lung cancer are diagnosed with localized disease and are potential candidates for curative treatment (1). For stage III NSCLC, concurrent chemoradiotherapy (CRT) with conventionally fractionated radiotherapy (RT) is a mainstay of treatment. Although distant recurrence is the most common pattern of failure, ~30% of patients experience LR which is associated with worse overall survival (OS) (2,3). For patients with stage I-II NSCLC who are not candidates for surgery, SABR is the preferred treatment and is associated with ~90% local control (4,5). For NSCLC treated by CRT or SABR, tumor burden is the only variable that has been repeatedly associated with risk of LR (6–10).
Although somatic mutations play a critical role in personalizing systemic therapy in advanced NSCLC, this is not the case for patients undergoing RT. Recently, mutations in several genes have been suggested to be associated with clinical LR, but these findings remain preliminary and not validated (11–13). Mutations leading to activation of the KEAP1-NFE2L2 pathway are particularly promising candidates for contributing to clinical radioresistance since NFE2L2 is a transcription factor that drives expression of free radical defense genes which could interfere with radiation-induced DNA damage. KEAP1 is an adaptor protein that targets NFE2L2 for ubiquitination and proteasomal destruction under normal homeostasis (14). Mutations in KEAP1 or NFE2L2 occur in approximately 20% of NSCLC (15,16), and lead to constitutive activation of the pathway. Previous studies have demonstrated that activation of the pathway in vitro (14,17–19) or in vivo promotes radioresistance (17). Furthermore, our group previously reported an association of KEAP1/NFE2L2 mutations with high risk of LR in a heterogeneous pilot cohort of patients with localized NSCLC treated with radiotherapy (17).
In the current study, we sought to identify recurrent mutations in localized NSCLC that are associated with LR after radiotherapy. We found that KEAP1/NFE2L2 mutations strongly increase the risk of LR after RT but not surgery. Functional classification of mutations using isogenic cell line systems to distinguish pathogenic and passenger mutations further strengthened the association of mutations with LR. Finally, we demonstrate that glutaminase inhibition is a potential approach for personalized radiosensitization of KEAP1/NFE2L2 mutant tumors.
Results:
KEAP1/NFE2L2 Mutations are Predictive Biomarkers of LR after RT
We identified 232 consecutive patients who were treated at Stanford University with curative intent with RT or surgery and who underwent tumor genotyping using a clinical hybrid capture-based sequencing assay covering 130-198 genes (20). Our study included three cohorts: 1) 47 patients with locally advanced NSCLC treated with conventionally fractionated RT (CRT cohort), 2) 50 patients with early stage NSCLC treated with high-dose stereotactic ablative radiotherapy (SABR; SABR cohort), and 3) 135 patients with early stage NSCLC treated with surgical resection (Surgery cohort; Fig. 1A). None of these were included in our previously published cohort of RT-treated patients with KEAP1/NFE2L2 tumor genotyping (17). Differences in baseline patient characteristics were reflective of the types of patients routinely selected for each type of treatment (Supplementary Table S1).
Figure 1: KEAP1/NFE2L2 mutations are predictive biomarkers of local recurrence after radiotherapy.
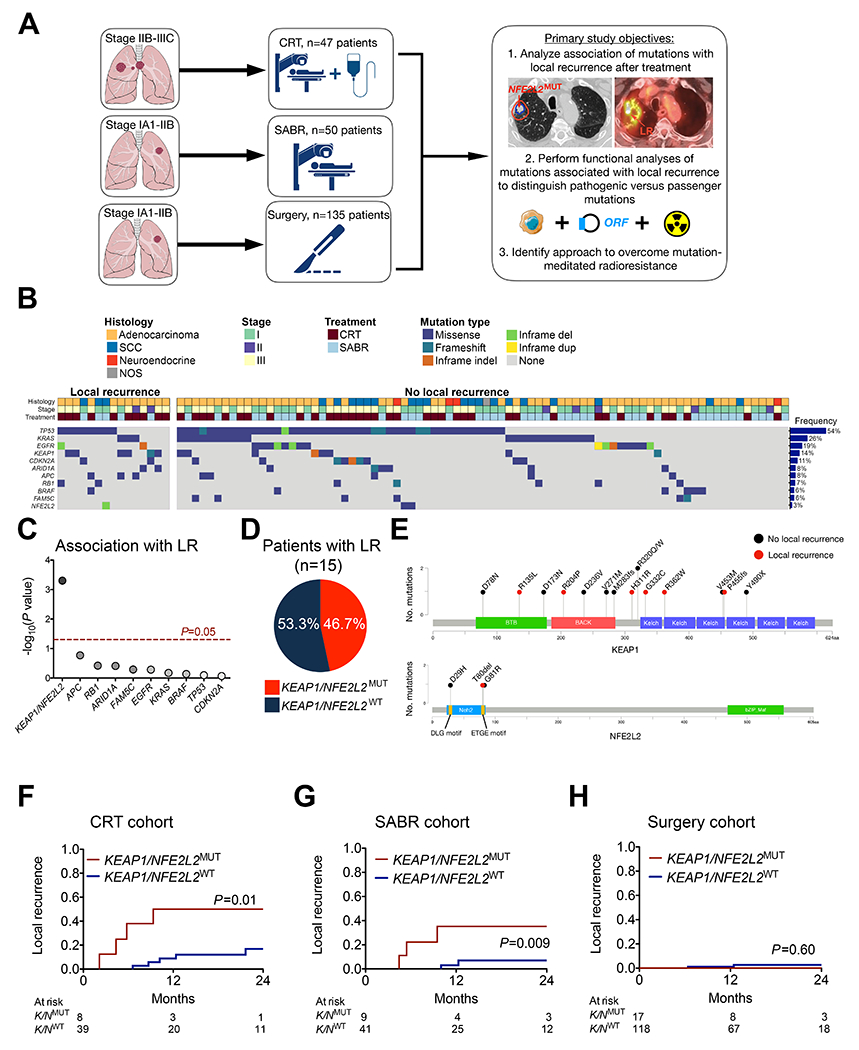
(A) Study design (images were produced and modified from Servier Medical Art, see Acknowledgements). (B) Recurrent mutations in patients with and without local recurrence (LR) after chemoradiotherapy (CRT) or stereotactic ablative radiotherapy (SABR). (C) Association of recurrent mutations with LR. Competing risk comparison performed using Gray’s test with multiple hypothesis testing correction. (D) Pie chart fraction of LR events occurring in tumors with KEAP1 or NFE2L2 mutations. (E) Location of KEAP1 and NFE2L2 mutations from CRT and SABR cohorts. (F) Incidence of local recurrence stratified by KEAP1/NFE2L2 mutation status for patients with stage IIB-IIIC NSCLC receiving CRT. (G) Incidence of LR stratified by KEAP1/NFE2L2 mutation status for patients with stage IA1-IIB NSCLC treated with SABR. (H) Incidence of LR stratified by KEAP1/NFE2L2 mutation status for patients with stage IA1-IIB NSCLC treated with surgical resection.
In order to identify tumor mutations associated with clinical radioresistance, we determined the association between recurrent mutations and LR following in the combined cohort of patients treated with CRT or SABR. LR, defined as tumor re-growth within the radiation field, was chosen as the endpoint of interest since it is most likely to reflect responses of tumor cells exposed to RT. Distant recurrence outside of the prior radiation field could be due to micrometastases that were present at the time of treatment and that were therefore not exposed to RT. Ten mutations met our predetermined recurrence frequency threshold of >5% (Fig. 1B; see Methods for power calculation). Mutations in KEAP1 and NFE2L2 were considered as one group since they result in the same biochemical phenotype (i.e. NFE2L2 overexpression) and display mutual exclusivity (14,17). Strikingly, only KEAP1/NFE2L2 mutations were significantly associated with LR (Fig. 1C; adjusted P=0.005) and these mutations were present in nearly half of tumors that had LR (Fig. 1D). Mutations in KEAP1 were distributed throughout the protein while NFE2L2 mutations were located in the DLG and ETGE hotspots (Fig. 1E). Furthermore, in exploratory analyses we found that KEAP1 mutations were significantly associated with LR on their own (P=0.002) and that neither mutations leading to ERK activation (EGFR/KRAS/BRAF, P=0.49) nor E2F activation (CDKN2A/RB1, P=0.15) were associated with LR when considered together. The frequency of co-mutations in driver genes was similar in KEAP1/NFE2L2 mutant tumors with and without LR (5 out of 7 with LR vs. 4 out of 10 without LR; P=0.33). We also did not observe a significantly different frequency of co-occurring TP53 mutations in KEAP1/NFE2L2 mutant tumors with (3 of 7) versus without LR (8 of 10, P=0.16, Fig. 1B). Thus, among recurrent mutations in NSCLC, KEAP1/NFE2L2 mutations appear to be the dominant cause of clinical radioresistance.
Next we compared LR based on KEAP1/NFE2L2 mutation status in patients with locally advanced NSCLC treated with conventionally fractionated CRT. For the entire CRT cohort, 2-year OS was 65.6% (95%CI=46.9-79.1%) and 2-year LR was 22.8% (95%CI=20.8-24.8%; Supplementary Figure S1A–1B), similar to the rates reported for the standard RT dose arm in RTOG 0617 (21). Patients with KEAP1/NFE2L2 mutant tumors had a significantly increased incidence of LR compared to those without, with 2-year LR of 50.0% (95%CI=36.3-63.7%) vs 16.9% (95%CI=14.6-19.2%), respectively (P=0.01, Fig. 1F). There were no significant differences in clinical characteristics between KEAP1/NFE2L2MUT and wildtype cases (Supplementary Table S2). Metabolic tumor volume (MTV) measured using FDG PET/CT scans and stage did not differ significantly between patients with and without KEAP1/NFE2L2 mutations (P=0.45 and P=0.67, respectively). Furthermore, KEAP1/NFE2L2 mutation status was the only predictor of LR in both univariable analysis (UVA; HR=4.74, 95%CI=1.30-17.20, P=0.02) and multivariable analysis (MVA; HR=5.17, 95%CI=1.30-20.58, P=0.02; Supplementary Table S3). We did not observe a significant association between KEAP1/NFE2L2 mutations and distant recurrence as first recurrence after CRT (P=0.69; Supplementary Table S4 and Supplementary Figure 1C), suggesting that patients with locally advanced NSCLC and KEAP1/NFE2L2 mutations are specifically at high risk of LR.
Patients with stage I-II NSCLC treated with hypofractionated SABR receive significantly higher radiation doses than patients treated with conventionally fractionated CRT. We therefore assessed whether the “ablative” doses delivered during SABR might overcome KEAP1/NFE2L2 mutation-associated radioresistance. Patients in the SABR cohort had 2-year OS of 82.8% (95%CI=68.5%-91.0%) and 2-year LR of 12.4% (95%CI=10.9-13.9%; Supplementary Figure S2A–B). Surprisingly, as in the CRT cohort, we observed a significantly higher incidence of LR after high dose SABR for patients with KEAP1/NFE2L2 mutations versus those without, with 2-year LR of 35.2% (95%CI=23.5-46.9%) vs 7.0% (95%CI=5.6-8.4%), respectively (Fig. 1G, P=0.009). There were no significant differences in clinical characteristics between KEAP1/NFE2L2MUT and wildtype cases (Supplementary Table S2). MTV and stage were not significantly different between patients with and without KEAP1/NFE2L2 mutations (P=0.88 and P=0.71, respectively). On both UVA and MVA, KEAP1/NFE2L2 mutations (HR=8.50, 95%CI=1.56-46.30, P=0.01 and HR=17.92, 95%CI=2.05-156.67, P=0.009, respectively) and MTV (HR=1.42 per 10 cc, 95%CI=1.25-1.62, P=1.2e-7 and HR=1.68, 95%CI=1.27-2.22, P=0.0003, respectively) were significantly associated with LR (Supplementary Table S3). We did not observe a significant association between KEAP1/NFE2L2 mutations and distant recurrence as first recurrence after SABR (P=0.17; Supplementary Table S5 and Supplementary Figure 2C). Thus, KEAP1/NFE2L2 mutations are predictive of LR after SABR.
Lastly, we hypothesized that KEAP1/NFE2L2 mutations are not associated with LR after surgery, since the expression of free radical defense genes induced by these mutations would not be expected to affect tumor resectability. In our 135 patient surgery cohort, LR at the staple line or bronchial stump was a rare occurrence (1- and 2-year rates of 0.9 and 2.3%, respectively) and consistent with our hypothesis did not differ based on KEAP1/NFE2L2 mutation status (Fig. 1H). There were no significant differences in patient age and tumor size between KEAP1/NFE2L2MUT and wildtype cases, but mutant cases were more likely to have non-adenocarcinoma histology and a more significant smoking history (Supplementary Table S2). Our findings are consistent with prior reports showing local recurrence is not a primary pattern of failure after surgery (22). Additionally, we validated there is no difference in prognosis by mutation status within patients with stage I-II lung adenocarcinoma and SCC treated with primary surgery in the TCGA (P=0.71, Supplemental Figure 3). Taken together, our results indicate that KEAP1/NFE2L2 mutations are predictive biomarkers for LR after RT but not surgery.
Functional Evaluation of KEAP1/NFE2L2 Mutations
While our clinical results indicate that KEAP1/NFE2L2 mutations are strongly associated with LR after RT, 58.8% (10 out of 17) of patients with these mutations did not develop tumor regrowth within the radiation field. We therefore hypothesized that a subset of the mutations were passengers that did not affect protein function. To test this hypothesis, we developed isogenic KEAP1 wildtype and knock-out cell lines in which we could express open reading frame (ORFs) constructs containing the mutations we observed in the two RT cohorts and functionally evaluated these for KEAP1/NFE2L2 pathway activity (Fig. 2A). Specifically, we targeted KEAP1 in H1299 human non-small cell lung cancer cells using CRISPR-Cas9-mediated gene editing by directly introducing sgRNA-Cas9 complex (Supplementary Fig. 4A and 4B) (23). KEAP1NULL H1299 cells displayed significant overexpression of NFE2L2 and its targets including NQO1 and SQSTM1 (also known as p62) compared to parental KEAP1WT H1299 cells (Fig. 2B). Additionally, these cells displayed significant resistance to ionizing radiation (Fig. 2C).
Figure 2: Functional analysis of KEAP1/NFE2L2 mutations found in the radiotherapy cohorts identifies pathogenic and passenger mutations.

(A) Strategy for assessing KEAP1/NFE2L2 mutation functional classification using isogenic H1299 knock-out cell lines generated by CRISPR-Cas9 (images were produced and modified from Servier Medical Art, see Acknowledgements). (B) Western blot analysis for KEAP1, NFE2L2, and NFE2L2 target proteins in parental and KEAP1NULL H1299 cells. (C) Clonogenic survival of parental and KEAP1NULL H1299 cells after 10 Gy of ionizing radiation (n=5, **P<0.001). (D) DNA damage assessment by γH2AX foci immunofluoresence analysis 5 min after exposure to 2 Gy of ionizing radiation in parental and KEAP1NULL H1299 cells (n=4, ***P<0.0001). (E) Clonogenic survival of parental and KEAP1NULL H1299 cells in the presence or absence of BSO (100 nM) and/or NAC (100 mM) treated with or without 4 Gy of ionizing radiation (n=4; Student’s t-test, *P<0.01). (F) Expression of the NFE2L2 target gene HMOX1 in KEAP1NULL cells by qRT-PCR after transfection of empty plasmid, plasmid containing wild-type KEAP1, or plasmids containing KEAP1 constructs with mutations observed in the CRT and SABR cohorts (n=4, *P<0.01) (G) Clonogenic survival of KEAP1NULL cells transfected with plasmids containing wild-type or mutant KEAP1 constructs 24 hours before exposure to 5 Gy of ionizing radiation. Experiments were performed in groups, with empty vector and wild-type controls in each group (n=3-4; *P<0.01, ** P<0.001 compared to ‘empty’ by Student’s t-test). (H) Expression of NFE2L2 target genes in NFE2L2NULL or KEAP1NULL cells by qRT-PCR after transfection of plasmids containing wild-type or mutant NFE2L2 (n=4, *P<0.01). (I) Clonogenic survival of NFE2L2NULL cells transfected with plasmids containing wild-type or mutant NFE2L2 constructs 24 hours before exposure to 3 Gy of ionizing radiation (n=3; ** P<0.001 by Student’s t-test). (J) Summary of functional assay results and mutation classification for each KEAP1 and NFE2L2 mutation found in the CRT and SABR cohorts.
To explore the mechanism of radiation resistance induced by KEAP1 loss, we next examined levels of DNA damage induced by ionizing radiation in the isogenic lines. While baseline levels of DNA double strand breaks as measured by γH2AX foci were similar, there were significantly fewer foci in KEAP1NULL compared to KEAP1WT H1299 cells following 2 Gy of ionizing radiation (Fig. 2D). These findings suggest that KEAP1NULL cells are radioresistant due to elevated levels of free radical scavengers and decreased DNA damage production by the indirect effect of ionizing radiation. To test this hypothesis, we examined levels of GSH and found that KEAP1NULL H1299 cells had significantly higher levels of reduces GSH than KEAP1WT cells (Supplementary Fig. 4C). Next, we examined the effects of the free radical scavenger N-acetylcysteine (NAC) and the GSH biosynthesis inhibitor buthionine sulfoximine (BSO) on radiation sensitivity. While treatment with NAC had no effect in unirradiated cells, it significantly enhanced survival of KEAP1WT but not KEAP1NULL H1299 cells after 4 Gy, suggesting that KEAP1NULL H1299 cells are already maximally protected by endogenous free radical scavengers (Fig. 2E). Conversely, BSO treatment had no effect on radiosensitivity of KEAP1WT cells but radiosensitized KEAP1NULL cells to levels similar to those of KEAP1WT cells. These data suggest that elevated levels of GSH are a major driver of the radioresistant phenotype of KEAP1NULL cells.
In order to evaluate the functional effects of the KEAP1 mutations we observed in the RT patients, we individually expressed ORFs carrying the 15 KEAP1 mutations in KEAP1NULL H1299 cells and tested multiple aspects of KEAP1/NFE2L2 pathway activity. We reasoned that pathogenic (i.e. inactivating) mutations should maintain KEAP1/NFE2L2 pathway activity while passenger mutations should act like wild-type KEAP1 and decrease it. We first confirmed that expression levels of both mutant and wild-type KEAP1 constructs were similar (Fig. S5A). Next, we analyzed expression of the NFE2L2 target gene HMOX1 by qRT-PCR and observed that all 6 constructs containing KEAP1 mutations from LR cases did not alter expression, consistent with a loss-of-function phenotype (Fig. 2F). Conversely, 6 of 9 (67%) constructs containing mutations from non-LR cases decreased expression similarly to wild-type KEAP1, suggesting that these mutations do not alter KEAP1 function. Similar results were observed when examining expression of HMOX1 and SQSTM1 at the protein level (Supplementary Fig. S5B–F). Furthermore, we performed in vitro clonogenic survival assays using 5 Gy of ionizing radiation and observed that all 6 constructs containing KEAP1 mutations from LR cases did not re-sensitize KEAP1NULL H1299 cells while 6 of 9 (67%) constructs containing mutations from non-LR cases did (Fig. 2G). Finally, we observed the same pattern when examining resistance to hydrogen peroxide (Supplementary Fig. S5G).
In an analogous fashion, we developed isogenic NFE2L2 wildtype and knock-out cell lines. NFE2L2NULL H1299 cells displayed loss of NFE2L2 protein, reduced target gene expression, and greater sensitivity to ionizing radiation compared to parental NFE2L2WT H1299 cells (Supplementary Fig. S6A and S6B). We expressed ORFs carrying each of the three NFE2L2 mutations from our RT cohorts in NFE2L2NULL H1299 cells (Supplementary Fig. S6C and D). To remove any native NFE2L2, we generated a KEAP1 overexpression cell model using the NFE2L2NULL H1299 cells transfected with multiple copies of wild-type KEAP1 (Supplementary Fig. 6D). While wild-type NFE2L2 did not significantly increase NFE2L2 target gene expression, each of the three mutant constructs did, suggesting KEAP1-mediated degradation of wild-type but not mutant NFE2L2 (Fig. 2G). Analysis of protein expression of NFE2L2 and targets HMOX1 and GCLM confirmed stabilization of the mutant but not wild-type NFE2L2 constructs (Supplementary Fig. S6D). Finally, all 3 mutant but not wild-type NFE2L2 constructs induced increased protein expression of HMOX1 (Fig. 2H) and resistance to ionizing radiation and hydrogen peroxide in NFE2L2NULL H1299 cells (Fig. 2I; Supplementary Fig. S6E). Fig. 2J provides a summary of functional assay results and mutation classification for each KEAP1 and NFE2L2 mutation found in the CRT and SABR cohorts.
Functional classification of KEAP1/NFE2L2 mutations improves the association with LR
Based on these molecular analyses, we classified the KEAP1 and NFE2L2 mutations observed in our RT cohorts as either pathogenic (i.e. loss-of-function for KEAP1 or gain-of-function for NFE2L2) or passenger (i.e. neutral). For KEAP1, 9 of 15 (60%) mutations were pathogenic while 6 (40%) were passengers and for NFE2L2 all 3 mutations were pathogenic (Fig. 2I). Notably, we only observed LR in patients with pathogenic KEAP1 or NFE2L2 mutations and in none of the patients with passenger mutations (Fig. 3A). Additionally, we found that patients with pathogenic mutations who did not develop LR had significantly smaller tumors (median 2.7 cc) than those who did develop LR (median 64.1 cc; P=0.03, Fig. 3B). Among patients with pathogenic mutations, 28.6% (2 of 7) with tumors <20.4 cc experienced a LR while 100% (5 of 5) with tumors ≥20.4 cc experienced a LR. These findings suggest that doses of RT delivered during CRT or SABR can control some tumors carrying pathogenic mutations if tumors are small and fewer clonogens are present.
Figure 3: Pathogenic KEAP1/NFE2L2 mutations but not passenger mutations or expression analysis predict local recurrence after radiotherapy.
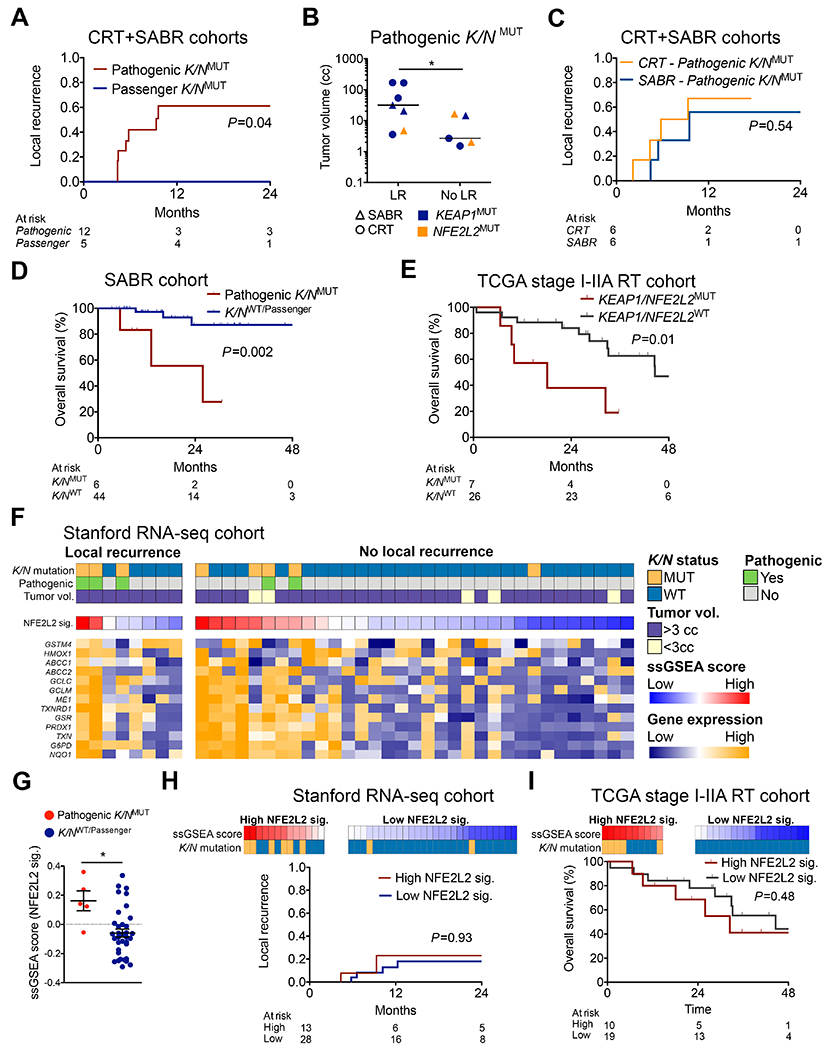
(A) Incidence of local recurrence after CRT or SABR in patients with KEAP1/NFE2L2 mutations stratified by functional classification. (B) Tumor volumes for patients with pathogenic KEAP1/NFE2L2 mutations (K/NMUT). For patients who did not develop local recurrence (LR) the volume of the largest lesion is shown (*P=0.03). (C) Incidence of local recurrence in patients with pathogenic KEAP1/NFE2L2 mutations stratified by radiotherapy type. (D) Overall survival of patients in the SABR cohort stratified by presence or absence of pathogenic KEAP1/NFE2L2 mutations (WT, wild-type). (E) Overall survival of stage I-II patients from the TCGA lung adenocarcinoma and squamous cell cohorts who were treated with RT and not surgery, stratified by presence or absence of KEAP1/NFE2L2 mutations. (F) RNA-seq analysis of tumor cells from FFPE tumor biopsies of patients in the CRT and SABR cohorts (n=41). CIBERSORTx was used to deconvolve tumor cell expression (26). The heatmap depicts single sample gene set enrichment analysis (ssGSEA) scores of a previously defined NFE2L2 target gene expression signature (NFE2L2 sig.) and expression of the individual signature genes (27). (G) NFE2L2 target gene ssGSEA scores in tumor biopsies from patients in the CRT and SABR cohorts stratified by the presence or absence of pathogenic KEAP1/NFE2L2 mutations. (H) Incidence of local recurrence after CRT or SABR stratified by ssGSEA scores for NFE2L2 signature. Legend is same as in F. Stratification threshold was obtained by choosing the highest significance value by log-rank for LR based on 1,000 re-sampling iterations (I) Overall survival of patients from the TCGA lung adenocarcinoma and squamous cell cohorts who were treated with RT and not surgery, stratified by ssGSEA scores for NFE2L2 signature. The optimal cutpoint identified in H was used.
Re-analyzing LR rates in CRT- and SABR-treated patients based on the presence of pathogenic mutations increased separation between mutant and wild-type patients (Supplementary Fig. 7A, B). Strikingly, rates of LR did not differ significantly between patients treated with CRT or SABR when mutation functional classification was considered (Fig. 3C, P=0.54). Patients with pathogenic KEAP1/NFE2L2 mutations who were treated with SABR had a significantly worse OS than non-mutant patients (Fig. 3D, P=0.002). Similarly, the presence of KEAP1/NFE2L2 mutations was associated with inferior survival in stage I-IIA NSCLC patients from TCGA who were treated with radiation and not surgery (Fig. 3E, P=0.01) (24). Thus, functional classification of KEAP1/NFE2L2 mutations improves their association with LR and the lack of local control associated with pathogenic mutations appears to lead to higher rates of death.
NFE2L2 Target Gene Expression does not predict LR after RT
Since prior studies have suggested an association of the expression of NFE2L2 target genes and resistance to radiation (25), we next explored whether NFE2L2 target gene expression is also associated with LR after RT. To do so we performed RNA-seq on formalin fixed paraffin embedded biopsy samples from 41 (42.3%) patients in the SABR and CRT cohorts who had sufficient tissue remaining after DNA sequencing. In order to focus on gene expression of tumor cells, we used CIBERSORTx digital cytometry to infer tumor cell-specific gene expression profiles and scored samples for the presence of a previously defined NFE2L2 target gene signature (Fig. 3F) (26,27). We observed significantly higher expression of NFE2L2 target genes in tumors harboring pathogenic KEAP1/NFE2L2 versus wild-type/passenger mutations (P=0.01; Fig. 3G). Expression of NFE2L2 targets was not significantly associated with LR by Cox regression (HR=2.33 95%CI=0.06-84.30, P=0.64) and did not stratify risk of LR in competing risk analysis (P=0.93; Fig. 3H). Similarly, there was no difference in OS in the TCGA radiation cohort (Fig. 3I). Taken together, these results suggest that although KEAP1/NFE2L2 mutation status is predictive of clinical response to radiation, expression of NFE2L2 target genes is not.
Glutaminase Inhibition Selectively Radiosensitizes KEAP1/NFE2L2 Mutant Cells
Finally, we wished to identify a potential approach for radiosensitizing KEAP1/NFE2L2 mutant tumors. Recent work has demonstrated that KEAP1 loss promotes dependence on glutamine metabolism and sensitivity to glutaminase inhibition (28,29). Since glutamine metabolism includes production of glutathione (GSH), a critical antioxidant that has been linked to resistance to ionizing radiation, we hypothesized that glutaminase inhibition can preferentially radiosensitize KEAP1/NFE2L2 mutant cells (Fig. 4A). To test this hypothesis, we first examined expression of the glutamine transporter alanine-serine-cysteine transporter 2 (ASCT2) in our isogenic cell lines and observed increased expression in KEAP1NULL and decreased expression in NFE2L2NULL H1299 cells (Fig. 4B and Supplementary Fig. S8A). Similarly, siRNA-mediated knock-down of NFE2L2 in H1975 NSCLC cells (KEAP1/NFE2L2 wildtype) resulted in decreased ASCT2 expression. Furthermore, RNA-seq analysis of both KEAP1NULL compared to KEAP1WT H1299 cells and tumor biopsy specimens from patients with pathogenic KEAP1/NFE2L2 mutations (n=5) compared to patients without (n=41) revealed significant overexpression of genes involved in glutamine metabolism (Fig. 4C and D). Thus, both KEAP1/NFE2L2 mutant human tumors and our KEAP1NULL cell lines display upregulation of genes involved in glutamine metabolism.
Figure 4: Glutaminase inhibition preferentially radiosensitizes KEAP1 mutant lung cancer cells.

(A) Schematic depicting potential interaction between glutaminase inhibition and ionizing radiation in KEAP1 mutant cells (images were produced and modified from Servier Medical Art, see Acknowledgements). (B) Western blot analysis for ASCT2, NFE2L2, and HMOX1. Left: parental, KEAP1NULL, and NFE2L2NULL H1299 cells. Right: transient siRNA knock-down of NFE2L2 in H1975 NSCLC cells (wild-type for KEAP1 and NFE2L2). (C) Gene set enrichment analysis (GSEA) of a previously defined glutamine metabolism signature using RNA-seq data from parental and KEAP1NULL H1299 cells. (D) As in C but using RNA-seq data from the tumor biopsies of patients in the CRT and SABR cohorts, comparing samples with and without pathogenic KEAP1 mutations. (E) Oncoprint of genes recurrently mutated in NSCLC (24) in the three cell line used for the experiments with CB-839. (F) Clonogenic survival of parental and KEAP1NULL H1299 cells in the presence or absence of CB-839 (100 nM; 24 hour pre-treatment) and 4 Gy of ionizing radiation (n=4; *P<0.01, **P<0.001). Results were normalized against untreated cells. (G) Clonogenic survival of KEAP1NULL and parental H1299 cells (KEAP1 wildtype) in the presence or absence of CB-839 (100 nM; 24 hour pre-treatment) and 4 Gy of ionizing radiation (n=4; *P<0.01). (H) Clonogenic survival of H1437 cells (KEAP1 wildtype) with and without siRNA knock-down of KEAP1 in the presence or absence of 2 Gy and 500 nM CB-839 (n=4; *P<0.01). (I) Clonogenic survival of parental and KEAP1 transfected A549 cells (KEAP1 mutant) in the presence or absence of CB-839 (0.1 nM; 24 hour pre-treatment) and 2 Gy of ionizing radiation (n=4; *P<0.01, **P<0.001). Results for panel F through I were normalized to untreated cells. (J) Intracellular reactive oxygen species (ROS) levels measured by DCFDA intensity via FACS in parental and KEAP1NULL H1299 cells in the presence or absence of CB-839 (100 nM; 24 hour pre-treatment) and 2 Gy of ionizing radiation (n=4; *P<0.01). (K) Glutathione (GSH) to glutathione disulfide (GSSG) ratio in parental and KEAP1NULL H1299 cells in the presence or absence of CB-839 (1 μM; 24 hour pre-treatment; n=4; *P< 0.05, **P< 0.01). (L) Clonogenic survival of KEAP1NULL H1299 cells treated with or without 2 Gy of ionizing radiation and/or 100 nM CB-839 in the presence or absence of NAC (1 mM) (n=4; *P<0.01). (M) Western blot analysis for γH2AX in parental and KEAP1NULL H1299 cells in the presence or absence of CB-839 (100 nM) treated with or without 2 Gy IR.
To explore if targeting of glutamine metabolism can preferentially radiosensitize KEAP1/NFE2L2 mutant cells, we tested the combination of RT and the small-molecule glutaminase inhibitor CB-839, which is currently in Phase 1/2 clinical trials, in NSCLC cell lines of differing genetic backgrounds (Fig. 4E). Although KEAP1NULL and KEAP1WT H1299, H1437, and A549 cells displayed minimal sensitivity to CB-839 doses in the range of 10-100 nM, the combination of ionizing radiation and CB-839 preferentially killed KEAP1NULL cells (Fig. 4F–I). The sensitization effect was substantial, with a dose modifying factor for CB-839 in H1299 KEAP1NULL cells of 3.4 (at 63% survival, Fig. 4F). Importantly, we observed this effect starting with parental cell lines that were either KEAP1 wildtype (H1299 and H1437) or mutant (A549). For the latter, treating A549 cells with 100 nM CB-839 did not have a dramatic effect on cell survival but sensitized cells to ionizing radiation (Fig. 4I and Supplementary Fig. S8B–C). However, expression of wildtype KEAP1 in A549 cells increased radiation sensitivity and abolished the sensitizing effect of CB-839. Thus, CB-839 preferentially radiosensitizes KEAP1NULL cells.
Lastly, we investigated the mechanism by which CB-839 preferentially radiosensitizes KEAP1NULL cells and hypothesized that glutaminase inhibition counteracts the enhanced free radical defenses caused by loss of KEAP1. We first examined the effects of CB-839 and ionizing radiation on intracellular ROS levels. KEAP1NULL H1299 cells displayed significantly lower baseline levels of ROS than their wildtype counterparts. However, unlike in KEAP1WT H1299 cells, the combination of CB-839 and radiation significantly increased ROS levels in KEAP1NULL H1299 cells compared to radiation alone (Fig. 4J). Additionally, CB-839 treatment decreased GSH to GSSG ratios significantly more in KEAP1NULL than KEAP1WT H1299 cells (Fig. 4K). This suggests that CB-839 leads to decreased free radical scavenging capacity in KEAP1NULL cells. We next tested if exogenous addition of a free radical scavenger can rescue KEAP1NULL cells from CB-839-mediated radiosensitization. Treatment of H1299 KEAP1NULL cells with the ROS scavenger NAC did not significantly affect cell survival by itself or when combined with either 2 Gy or CB-839 alone. However, NAC significantly rescued the enhanced cell killing caused by the combination of 2 Gy and CB-839 (Fig. 4L), suggesting that CB-839 radiosensitization is mediated by depletion of free radical scavengers. Finally, we reasoned that if this mechanism is correct, CB-839 should increase the amount of DNA damage induced by ionizing radiation. To test this, we measured DNA double strand breaks via γH2AX before and after ionizing radiation in the presence of absence of CB-839. Irradiation with 2 Gy induced approximately 2-fold higher levels of γH2AX in KEAP1WT compared to KEAP1NULL cells (Fig. 4M). Strikingly, twenty-four hour pretreatment with CB-839 resulted in elevation of γ-H2AX in KEAP1NULL but not KEAP1WT cells. These data indicate that CB-839 preferentially radiosensitizes KEAP1NULL cells by decreasing free radical scavenging capacity and therefore increasing the amount of DNA damage caused by the indirect effect of ionizing radiation.
Discussion:
In summary, in this study we demonstrate that KEAP1/NFE2L2 mutations are a common cause of LR in patients with localized NSCLC treated with RT but not surgery. These mutations are therefore clinically relevant predictive biomarkers of radioresistance. Using functional analyses including radiation clonogenic assays, we found that 60% of KEAP1 mutations and all NFE2L2 mutations in our cohort were pathogenic, and that pathogenic mutations were associated with a ~60% rate of LR after RT but not surgery. Additionally, we show that the glutaminase inhibitor CB-839 preferentially sensitizes KEAP1/NFE2L2 mutant NSCLC cells to RT.
Of the recurrent mutations found in our cohort, only KEAP1/NFE2L2 mutations were statistically significantly associated with LR. This finding both validates and extends our prior analysis of a smaller, independent cohort in which we only examined KEAP1/NFE2L2 mutations and found that these were associated with LR after RT (17). Overall, nearly half of all LRs occurred in tumors with these mutations, suggesting that KEAP1/NFE2L2 mutations are a dominant biological driver of clinical radioresistance in localized NSCLC. Co-mutations in other frequently mutated genes such as lung cancer drivers were not associated with LR. Additionally, unlike other prior studies, we did not observe an association of KRAS and/or TP53 mutations with LR (12,13). One potential explanation for this discrepancy is that KEAP1 mutations often co-occur with both KRAS and TP53 mutations and KEAP1/NFE2L2 genotyping was not performed in the prior studies. In an exploratory analysis we also examined if STK11 mutations were associated with LR after RT since these have been implicated in treatment resistance to other therapies (28,30,31). However, only one of five STK11 mutant tumors in our RT cohorts developed LR and this was not statistically significantly different than wildtype tumors (Supplementary Fig. 9; P=ns). Of note, this tumor also carried a pathogenic KEAP1 mutation, suggesting that it was the likely cause of LR. However, we cannot rule out that there may be additional mutations beyond KEAP1/NFE2L2 that are associated with radioresistance but that our study was underpowered to detect. That said, our findings suggest that discovering such mutations will require very large cohorts and that other mutations are unlikely to match KEAP1/NFE2L2 mutations in recurrence frequency and/or effect size.
We were surprised to find high rates of LR rates in patients with KEAP1/NFE2L2 mutations who were treated with SABR, which delivers significantly higher doses of ionizing radiation to tumors than conventionally fractionated RT. We found no significant difference in the 2-year LR rate after CRT (67%) and SABR (56%), suggesting dose escalation alone is not sufficient to significantly improve local control in these patients. However, it is possible that our study was underpowered to detect a modest benefit of dose escalation. Indeed, the fact that smaller tumors with KEAP1/NFE2L2 mutations had better local control suggests dose escalation may be useful in some settings. Notably, our results and prior studies have shown that increasing rates of LR in patients with stage I-II NSCLC are associated with worse OS, highlighting the need for achieving better local tumor control in this setting (2,32).
By expressing mutant alleles in knock-out cell lines, we found that ~30% of KEAP1 mutations in our RT cohorts have neutral effects on KEAP1 function and were therefore likely passenger mutations. This is in line with a prior study in which 4/18 (22%) KEAP1 mutations found in a lung squamous cell carcinoma cohort were determined to have neutral effects on KEAP1 activity (33). We compared four functional assays for KEAP1/NFE2L2 pathway activity including NFE2L2 target mRNA expression, NFE2L2 target protein expression, resistance to hydrogen peroxide, and resistance to ionizing radiation. The majority of variants behaved either as loss of function (LOF) or neutral alleles in all four assays while one mutation (R320Q) displayed a hypomorphic pattern. Notably, two of the mutations (R204P and R320Q) found in our SABR cohort had also been included in prior studies and were classified as hypomorphic variants (33,34). One of these (R204P) behaved like a loss-of-function allele in our assays and was found in a patient who developed LR, while the other (R320Q) behaved like a passenger mutation and was found in a patient who did not have LR. Thus, functional classification of KEAP1 variants may be context dependent. However, the knock-out cell line system we established appears to faithfully recapitulate radiation resistance phenotypes observed clinically.
Since pathogenic KEAP1/NFE2L2 mutations result in over-expression of NFE2L2 target genes, we tested whether analysis of gene expression could also identify tumors at highest risk of LR. Interestingly, we observed that while tumors with KEAP1/NFE2L2 mutations did tend to overexpress NFE2L2 target genes, a significant subset of wildtype tumors also displayed overexpression. Unlike genotyping for KEAP1/NFE2L2 mutations, high expression of NFE2L2 target genes did not stratify LR or OS. One potential explanation for this observation is that stress induced by ischemia and/or tissue handling might affect expression of NFE2L2 target genes, as has been demonstrated for NQO1 (35). Separately, NFE2L1 (also called NRF1) shares many downstream target genes with NFE2L2 and thus it is possible that KEAP1/NFE2L2 wildtype tumors with high expression of NFE2L2 target genes may have enhanced NFE2L1 activity (36). Although more work is needed in this area, our results suggest that tumor genotyping for KEAP1/NFE2L2 mutations is the most reliable method for identifying radioresistant NSCLCs.
A key implication of our findings is the need to develop strategies for overcoming KEAP1/NFE2L2-mediated radioresistance. Our analysis of DNA double strand breaks after ionizing radiation suggests that KEAP1 loss leads to radioprotection by decreasing DNA damage via enhanced free radical scavenging. Consistent with this hypothesis, KEAP1NULL cells display lower ROS levels and higher levels of glutathione, a key intracellular antioxidant that has previously been implicated in contributing to radioresistance (29,37). Furthermore, depletion of GSH via BSO radiosensitized KEAP1NULL cells to levels similar to those in KEAP1WT cells, suggesting the mechanisms of radioresistance is largely mediated by increased GSH production. We therefore tested the hypothesis that glutaminase inhibition, which decreases GSH production, preferentially radiosensitizes KEAP1 mutant cells. Our interest in glutaminase was in part based on prior observations that KEAP1 mutant NSCLC is dependent on glutamine metabolism (28,29) and that inhibition of glutamine metabolism may be of therapeutic benefit in NSCLC (38–42). Strikingly, we found that low doses of the glutaminase inhibitor CB-839 (1-500 nM), which is currently in clinical trials in other contexts, can preferentially radiosensitize KEAP1 mutant but not isogenic wildtype cells in three different NSCLC cell line models. CB-839 sensitized KEAP1NULL cells to similar levels as KEAP1WT cells, suggesting that this approach could improve local control of KEAP1 mutant tumors to similar levels as that of wildtype tumors. Additionally, we expect that the ~2-fold increase in cell killing we observed in KEAP1NULL cells treated with CB-839 after a single dose of RT would be compounded over a multi-fraction course of RT, leading to potentially dramatic increases in total cell killing.
Mechanistic analyses demonstrated that CB-839 pre-treatment leads to depletion of free radical scavengers and increased DNA damage via the indirect effect of radiation. While our analyses were focused on in vitro experiments, a recent study by another group demonstrated that CB-839 can radiosensitize KEAP1 mutant cells in vivo. Specifically, Boysen et al. treated H460 NSCLC xenografts grown in nude mice with CB-839, 12-18 Gy of radiation, or both (43). The combination of CB-839 plus radiation significantly delayed tumor growth compared to radiation alone. Although not mentioned by Boysen et al., H460 cells carry the pathogenic KEAP1 D236H mutation (44). Thus, CB-839 radiosensitization appears to be a promising approach for overcoming KEAP1/NFE2L2 mutation-mediated radioresistance.
We have now validated the association of KEAP1/NFE2L2 mutations with inferior outcomes after RT in four independent cohorts of NSCLC patients (one from our prior report and three in the current report) (17). Our findings therefore have potential implications for the clinical management of patients with KEAP1/NFE2L2 mutant NSCLC. For medically operable patients with node negative disease who are candidates for surgery or SABR, surgery may be preferable since it does not appear to be associated with increased rates of LR. For medically operable patients with lymph node-positive NSCLC, the presence of KEAP1/NFE2L2 mutations is more complicated since these patients usually receive RT and/or surgery plus chemotherapy and these mutations have also been linked to chemoresistance (45,46). Such patients may therefore be ideal candidates for trimodality therapy consisting of surgery, RT, and chemotherapy, since surgical debulking should increase the chance that RT and chemotherapy can eliminate any clonogens remaining after surgery. Our finding that local control of KEAP1/NFE2L2 mutant tumors appears to be better in smaller tumors with fewer clonogens supports this idea. For medically inoperable early stage patients a radiosensitization strategy such as treatment with CB-839 might be beneficial. Lastly, based on several randomized phase II trials demonstrating survival benefits, use of SABR in NSCLC patients with oligometastatic disease appears to be a promising approach for improving survival (47–49). Our findings suggest that oligometastatic NSCLC patients with KEAP1/NFE2L2 mutant tumors will likely have inferior local control if treated with SABR alone and therefore might benefit from radiosensitization with CB-839. Since RT is frequently delivered with systemic therapies, future studies will also need to ensure that CB-839 does not increase toxicity in combination therapy settings.
Limitations of our study include its retrospective nature and that all patients were treated at a single institution. Thus, although we have validated the association of KEAP1/NFE2L2 mutations with LR after RT in multiple cohorts, it would be useful to repeat these analyses in cohorts from other institutions in the future. Additionally, LR was relatively uncommon and thus forced us to limit our analyses to mutations with sufficient recurrence frequency. Furthermore, since our cohort was genotyped using a targeted assay it is possible that mutations in genomic regions that were not covered might also be associated with LR. Lastly, there were significant differences in patient characteristics between the SABR and surgery cohorts that reflect the types of patients who receive each therapy and that could have confounded the comparison of the two groups. However, the fact that tumor size/stage, which to our knowledge is the only consistently reported parameter associated with LR after either treatment, did not differ between the two cohorts decreases this risk (9,50).
In conclusion, in a large cohort of patients with localized NSCLC treated with definitive radiation or surgery, we have demonstrated that KEAP1/NFE2L2 mutations are strongly predictive of LR after RT but not surgery. Furthermore, functional classification of KEAP1/NFE2L2 mutations revealed that only pathogenic mutations that lead to radioresistance in vitro were associated with LR clinically. Finally, glutaminase inhibition may offer a strategy for a precision radiation oncology approach for radiosensitizing KEAP1/NFE2L2 mutant tumors.
METHODS:
Study design and patient selection
Using prospective registries of patients with NSCLC treated with radiotherapy (2009-2018) or surgery (2015-2018) at Stanford University School of Medicine, we performed a retrospective study with institutional review board approval of all consecutive patients with AJCC 8th edition stage IA1-IIIC NSCLC who had clinical next generation sequencing (NGS) performed on tumor tissue with baseline characteristics shown in Supplementary Table S1. We excluded patients that were included in our previously published cohort of RT-treated patients with KEAP1/NFE2L2 tumor genotyping in order to ensure analysis of a completely independent cohort (17).
Our primary goal was to evaluate the association between somatic tumor mutations and LR in patients with localized NSCLC receiving CRT or SABR. Given that these cohorts totaled 97 patients, we calculated that in order to achieve 80% power to detect an absolute difference in local recurrence rate of ≥15% at two years (alpha = 0.05) a gene would need to be mutated in ≥6 patients. We therefore only considered genes above this recurrence threshold.
Patient cohorts
The CRT cohort (Supplementary Table S4) consisted of patients with stage IIB-IIIC NSCLC and the SABR cohort (Supplementary Table S5) consisted of patients with stage IA1-IIB NSCLC. We have previously reported our RT treatment protocols (10,51). Follow-up post-RT consisted of CT and/or PET-CT imaging at 3-month intervals during the first 2 years, 6-month intervals during the next 2 years, and yearly thereafter.
Patients in the surgery cohort had stage IA1-IIB NSCLC and underwent surgery without neoadjuvant or adjuvant chemotherapy. Patients underwent wedge resection, segmentectomy, or lobectomy with or without selective ipsilateral hilar and mediastinal nodal dissections. Follow-up visits occurred at 6-month intervals with CT performed at each visit.
LR was defined as tumor regrowth within the RT planning target volume (PTV) for the RT cohorts and recurrence at the staple line or bronchial stump for the surgery cohort. LR was scored using pathologic confirmation or by radiologic changes consistent with tumor re-growth. Radiologic criteria for LR were 1) interval increase in size of a mass-like lesion on CT and/or 2) interval increase in FDG uptake in a focal pattern on PET. Whenever possible, we confirmed radiologic findings on serial imaging in the context of overall disease progression. Radiologic scoring of LR was done by a single investigator (M.S.B.) blinded to the tumor genotyping results.
Metabolic tumor volume analysis
We used the ‘PET Edge’ tool in MIMvista software (Cleveland, OH), a gradient based algorithm, to delineate the metabolic tumor volume (MTV) of individually distinct lesions on attenuation corrected PET scans as previously described (10,52). For lesion-specific MTV analyses in CRT patients, the MTV of the lesion that recurred was used in patients with LR and the MTV of the largest targeted lesion was used in patients who did not develop LR.
Tumor genotyping
Tumor genotyping was performed in the Stanford Molecular Pathology CLIA laboratory on formalin fixed paraffin embedded tumor tissue via a laboratory developed test NGS assay called the ‘STanford Actionable Mutation Panel’ (STAMP), as previously described (20). Two versions of the assay were used during the years of the study covering 130 or 198 genes (both including all exons of KEAP1, NFE2L2, TP53, and KRAS). Mutation calls were extracted from clinical genotyping reports. Lung cancer driver genes were defined as EGFR, KRAS, NRAS, PIK3CA, HER2, BRAF, MET, ALK, ROS1, RET, and NTRK1/2/3. Copy number alteration analysis was performed as previously described considering both on-target and off-target reads (53). Activating mutations in KRAS (codons 12 and 13) and EGFR (exons 18-21 (54)) were considered pathogenic . All other mutations with Combined Annotation Dependent Deletion (‘CADD’) PHRED scores ≥20 and with a population frequency <1% in the genome aggregation database (Broad institute, v3,(55)) were considered pathogenic. Mutations in NFE2L2 were considered pathogenic if they were located in the Neh2 domain (amino acids 16-86) that interacts with KEAP1 and had CADD PHRED scores ≥20. Final tumor genotyping used for analysis is summarized in Supplementary Table S6.
RNA-sequencing
Isolation of RNA from formalin fixed paraffin embedded tumor tissue was performed for cases with sufficient tissue remaining using the RNAstorm kits (CELLDATA, Fremont, CA) with DNAse digestion (Qiagen, Hilden, Germany) and subsequent purification with RNA Clean & Concentrator kits (Zymo Research, Irvine CA). Sequencing libraries were prepared using the SMARTer Stranded total RNA-Seq Kit v2 (Takara Bio USA, Inc., Mountain View, CA) and sequenced on the Illumina HiSeq4000 platform with paired-end 150bp reads and 14-16 samples per lane. Gene expression levels were quantified using Salmon (v0.8.2) under quasi-mapping mode to Gencode version 27 (56). Transcript per million (TPM, Table S7) values were used as input for CIBERSORTx digital cytometry, allowing for inference of tumor cell-specific gene expression profiles using a reference transcriptome of flow-sorted malignant, endothelial, immune, and fibroblast cell expression profiles (26,57). Differential gene expression analysis and normalization was conducted with the package ‘DESeq2’. Single and bulk sample gene set enrichment and gene set variation analyses were performed with ‘GSVA’.
Cell culture, plasmids and nucleic acid delivery
H1299 and A549 cells were purchased from the American Type Culture Collection (ATCC). The H1299 cell line was cultured in RPMI 1640 supplemented with 10% fetal bovine serum (FBS) and A549 cells were maintained in DMEM with 10% FBS. All cell lines were tested for mycoplasma and were negative using PlasmoTest (Invivogen). KEAP1 or NFE2L2 plasmids were purchased from Addgene (#87545 for KEAP1 and #21555 FOR NFE2L2) and the KEAP1 or NFE2L2 cDNA was subcloned into the pcDNA4-V5-His vector (Invitrogene, #V861-20). Site-directed mutagenesis was performed to generate KEAP1 and NFE2L2 alleles bearing mutations identified in patients from the RT cohorts using QuickChange II site-Directed Mutagenesis Kit (Agilent, #200524) and confirmed with sanger sequencing. Transient expression of these constructs as well as siRNAs was performed using Lipofectamine 3000 as per the manufacturer’s protocol (Invitrogen). Cell viability was measured using CellTiter-Glo® Luminescent Cell Viability Assay kit (Promega, #G7572) or clonogenic assays. For the latter, cells were cultured 1-2 weeks and colonies greater than 50 densely packed cells were counted using crystal violet staining. Quantitation was performed using Image J software as previously described (58). The Survival fraction was calculated based on the plating efficiency, number of colonies, and number of seeded cells [SF= Colonies counted / (Cells seeded x Plating efficiency)]. For experiments involving mutant alleles, cells were transfected with plasmids ≥72 hours prior to treatment with ionizing radiation and/or CB-839.
Antibodies
Anti-ASCT2 (#8057), anti-GAPDH (#5174), anti-HMOX1 (#5061), anti-KEAP1 (#8047, used to detect endogenous KEAP1 protein), anti-NQO1 (#3187), anti-P62 (#88588), anti-phospho-Histone H2AX (#9718), anti-ubiquitin (#3936) and anti-Vinculin (#13901) antibodies were purchased from Cell Signaling Technology and used at 1:1000 dilution for Western blot analysis and 1:100 dilution for immunoprecipitation and immunofluorescence analysis. Polyclonal anti-KEAP1 antibody (#10503), used to detect KEAP1 mutant proteins since some lacked binding site for the monoclonal anti-KEAP1 antibody, was purchased from Protein-Tech and used at 1:1000 dilution for Western blot analysis. Anti-V5 antibody (#MA5-15253) was purchased from Invitrogen and used at 1:5000 dilution for Western blot analysis. Anti-flag M2 antibody (F3165) was purchased from Sigma Aldrich and used at 1:100 dilution for immunoprecipitation analysis. All antibodies were monoclonal except anti-HMOX1 and anti-KEAP1 (#10503).
Isogenic cell line generation
The sgRNAs for KEAP1 or NFE2L2 gene were designed using the Genetic Perturbation Portal (Broad Institute). To generate knock-out cell lines, sgRNAs and Cas9 protein (Integrated DNA Technologies, Inc., # 1081058) complexes were nucleofected into H1299 cell. After 48 hrs, single cells were sorted into 96 well plates. After 2-3 weeks, colonies were harvested and KEAP1 or NFE2L2 knock out was confirmed by qRT-PCR, Western blot, and gDNA sequencing analyses. In order to prevent exogenous overexpression of wild-type NFE2L2 from overcoming the capacity of endogenous KEAP1 to degrade it, we co-transfected a wild-type KEAP1 containing plasmid with plasmids containing NFE2L2 constructs into NFE2L2NULL H1299 cells (Fig. 2I and J; Supplementary Fig. 6C–E). To generate A549-KEAP1 cells, KEAP1 cDNA was subcloned into the pCDH lentiviral vector (System Bioscience). A549 cell were then transduced with the virus, followed by selection using puromycin (1 ug/ml) for 7 days. Stable cell line generation was confirmed by qRT-PCR and Western blot analyses. The sequences of all sgRNAs and siRNAs are listed in Supplementary Fig. S4.
Classification of KEAP1 and NFE2L2 mutations
KEAP1 mutations were scored as being neutral (i.e. passengers) in the various assays if they displayed: 1) decreased HMOX1 mRNA expression compared to KEAP1NULL cells transfected with empty vector (P<0.05); 2) increased H2O2 sensitivity in CellTiter Glo assays compared to KEAP1NULL H1299 cells transfected with empty vector (P<0.05); 3) decreased NFE2L2 target gene protein expression compared to KEAP1WT H1299 cells by visual scoring; and 4) increased radiation sensitivity in clonogenic assays compared to KEAP1NULL H1299 cells transfected with empty vector (P<0.05). KEAP1 mutations not meeting these criteria were scored as loss-of-function (i.e. pathogenic). NFE2L2 mutations were scored as being gain-of-function (i.e. pathogenic) in the various assays if they displayed: 1) increased HMOX1 mRNA expression compared to NFE2L2NULL cells transfected with NFE2L2WT vector (P<0.05); 2) increased NFE2L2 target gene protein expression compared to NFE2L2NULL cells transfected with NFE2L2WT vector by visual scoring; and 3) increased radiation resistance in clonogenic assays compared to NFE2L2NULL cells transfected with NFE2L2WT vector (P<0.05).
Cell cytotoxicity assay
A549, H1299 and their isogenic cell lines (1.0*10^3 cells) were subcultured into 96 well plate. After 24 hrs, the cells were treated with CB-839 for 72 hrs or hydrogen peroxide for 24 hrs as indicated concentration in the figure legend, the cells are subsequently subject to CellTiter-Glo® Luminescent Cell Viability Assay (Promega, #G7572) to determine cell viability as followed by manufacturer’s protocol.
Measurement for intracellular reactive oxygen species and glutathione
Cells were pre-incubated with vehicle, CB-839, or N-acetylcystein (Abcam, #ab143032) for 24 hours and subsequently exposed to ionizing radiation. After an additional 24 hours, cells were stained with 25 μM DCFDA for 45 min (Abcam) and evaluated via FACS analysis. GSH:GSSG ratio was measured using GSH/GSSG Ratio Detection Assay Kit (Abcam, # ab138881) according to the manufacturer’s protocol and quantified using a microplate reader (Ex/Em= 490/520nm).
Statistical Analysis
Follow up was measured using the reverse Kaplan-Meier method from completion of treatment until last thoracic imaging. OS was estimated using the Kaplan-Meier method from the time of completion of RT. All other statistical analyses were conducted with adjustment for the competing risk of death. The cumulative incidence of LR and out-of-field recurrence (recurrence outside of the RT PTV) were measured using the R package ‘cmprsk’. Univariable and multivariable competing risk regressions were conducted with the R package ‘crrSC’. Variables with P<0.1 on univariable analysis were included in MVA. Adjusted Benjamini & Hochberg P-values were calculated with the R package ‘stats’. Sample size calculation was conducted with the R package ‘samplesize’. For comparison of experimental data the Mann-Whitney-U test was used to calculate P-values unless otherwise specified. All statistical analyses were conducted with R version 3.6 (Vienna, Austria) and PRISM version 6 (San Diego, CA). Unless otherwise specified, all error bars represent standard deviation. All P-values were two-sided and considered significant at P<0.05.
Supplementary Material
Statement of significance:
This study shows that mutations in KEAP1 and NFE2L2 predict for local recurrence after radiotherapy but not surgery in patients with NSCLC. Approximately half of all local recurrences are associated with these mutations and glutaminase inhibition may allow personalized radiosensitization of KEAP1/NFE2L2 mutant tumors.
ACKNOWLEDGEMENTS:
The schematic for Fig. 1A, Fig. 2A, and Fig. 4A were produced using Servier Medical Art (https://smart.servier.com). Servier Medical Art by Servier is licensed under a Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/).
Funding sources:
This work was supported by grants from the National Cancer Institute (M.D., A.A.A., R01CA188298), the US National Institutes of Health Director’s New Innovator Award Program (M.D., 1-DP2-CA186569), the Ludwig Institute for Cancer Research (M.D., A.A.A.), the CRK Faculty Scholar Fund (M.D.), and in part by PA-14-015, Grant Number T32 CA 121940, awarded by Ruth L. Kirschstein National Research Service Award (NRSA), and the National Research Foundation of Korea (NRF) grant funded by the Korea government (Y.J., NRF-2020R1F1A1071579).
Disclosure of potential conflicts of interest:
SKP reports grant support from EpicentRx, Boehringer Ingelheim, Bayer and serves as a consultant for AstraZeneca, AbbVie, G1 Therapeutics, and Pfizer. M. Das reports grant support from Verily, United Therapeutics, Celgene, and Abbvie and serves as a consultant for Bristol Myers Squibb. JWN reports grant support from Genentech/Roche, Merck, Novartis, Boehringer Ingelheim, Exelixis, ARIAD/Takeda, and Nektar and serves as a consultant for ARIAD/Takeda, AstraZeneca, Genentech/Roche, Lilly, Exelixis, Loxo Oncology and Jounce Therapeutics. HAW reports advisory board participation (compensated) from Janssen, Mirati, Daiichi Sankyo, Helsinn and advisory board (without compensation) from Merck and Genentech/Roche. HAW also reports trial support paid to institution: ACEA Biosciences, Arrys Therapeutics, AstraZeneca/Medimmune, BMS, Celgene, Clovis Oncology, Exelixis, Genentech/Roche, Gilead, Lilly, Merck, Novartis, Pfizer, Pharmacyclics, Xcovery. AAA holds ownership interest in CiberMed and Foresight Diagnostics, and is a consultant/advisory board member for Roche. BWL reports research support from Varian Medical Systems and serves as a board member of TibaRay. M. Diehn receives research support from Varian Medical Systems and Illumina, holds ownership interest in CiberMed and Foresight Diagnostics, and is a consultant/advisory board member for Roche, AstraZeneca, Illumina, BioNTech, RefleXion, Gritstone Oncology, and Novartis.
REFERENCES:
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7–30. [DOI] [PubMed] [Google Scholar]
- 2.Machtay M, Paulus R, Moughan J, Komaki R, Bradley JE, Choy H, et al. Defining local-regional control and its importance in locally advanced non-small cell lung carcinoma. J Thorac Oncol. 2012;7:716–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bradley JD, Paulus R, Komaki R, Masters G, Blumenschein G, Schild S, et al. Standard-dose versus high-dose conformal radiotherapy with concurrent and consolidation carboplatin plus paclitaxel with or without cetuximab for patients with stage IIIA or IIIB non-small-cell lung cancer (RTOG 0617): a randomised, two-by-two factorial phase 3 study. Lancet Oncol. 2015;16:187–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Senthi S, Lagerwaard FJ, Haasbeek CJ, Slotman BJ, Senan S. Patterns of disease recurrence after stereotactic ablative radiotherapy for early stage non-small-cell lung cancer: a retrospective analysis. Lancet Oncol. 2012;13:802–9. [DOI] [PubMed] [Google Scholar]
- 5.Ball D, Mai GT, Vinod S, Babington S, Ruben J, Kron T, et al. Stereotactic ablative radiotherapy versus standard radiotherapy in stage 1 non-small-cell lung cancer (TROG 09.02 CHISEL): a phase 3, open-label, randomised controlled trial. Lancet Oncol. 2019;20:494–503. [DOI] [PubMed] [Google Scholar]
- 6.Ohri N, Bodner WR, Kabarriti R, Shankar V, Cheng H, Abraham T, et al. Positron Emission Tomography-Adjusted Intensity Modulated Radiation Therapy for Locally Advanced Non-Small Cell Lung Cancer. Int J Radiat Oncol Biol Phys. 2018;102:709–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Diessen JN, Chen C, van den Heuvel MM, Belderbos JS, Sonke JJ. Differential analysis of local and regional failure in locally advanced non-small cell lung cancer patients treated with concurrent chemoradiotherapy. Radiother Oncol. 2016;118:447–52. [DOI] [PubMed] [Google Scholar]
- 8.Werner-Wasik M, Swann RS, Bradley J, Graham M, Emami B, Purdy J, et al. Increasing tumor volume is predictive of poor overall and progression-free survival: secondary analysis of the Radiation Therapy Oncology Group 93-11 phase I-II radiation dose-escalation study in patients with inoperable non-small-cell lung cancer. Int J Radiat Oncol Biol Phys. 2008;70:385–90. [DOI] [PubMed] [Google Scholar]
- 9.Zhao L, Zhou S, Balter P, Shen C, Gomez DR, Welsh JD, et al. Planning Target Volume D95 and Mean Dose Should Be Considered for Optimal Local Control for Stereotactic Ablative Radiation Therapy. Int J Radiat Oncol Biol Phys. 2016;95:1226–35. [DOI] [PubMed] [Google Scholar]
- 10.Gensheimer MF, Hong JC, Chang-Halpenny C, Zhu H, Eclov NCW, To J, et al. Mid-radiotherapy PET/CT for prognostication and detection of early progression in patients with stage III non-small cell lung cancer. Radiother Oncol. 2017;125:338–43. [DOI] [PubMed] [Google Scholar]
- 11.Lockney NA, Yang TJ, Barron D, Gelb E, Gelblum DY, Yorke E, et al. PIK3CA mutation is associated with increased local failure in lung stereotactic body radiation therapy (SBRT). Clin Transl Radiat Oncol. 2017;7:91–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mak RH, Hermann G, Lewis JH, Aerts HJ, Baldini EH, Chen AB, et al. Outcomes by tumor histology and KRAS mutation status after lung stereotactic body radiation therapy for early-stage non-small-cell lung cancer. Clin Lung Cancer. 2015;16:24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cassidy RJ, Zhang X, Patel PR, Shelton JW, Escott CE, Sica GL, et al. Next-generation sequencing and clinical outcomes of patients with lung adenocarcinoma treated with stereotactic body radiotherapy. Cancer. 2017;123:3681–90. [DOI] [PubMed] [Google Scholar]
- 14.Best SA, Sutherland KD. “Keaping” a lid on lung cancer: the Keap1-Nrf2 pathway. Cell Cycle. 2018;17:1696–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cancer Genome Atlas Research N. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cancer Genome Atlas Research N. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jeong Y, Hoang NT, Lovejoy A, Stehr H, Newman AM, Gentles AJ, et al. Role of KEAP1/NRF2 and TP53 Mutations in Lung Squamous Cell Carcinoma Development and Radiation Resistance. Cancer Discov. 2017;7:86–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singh A, Bodas M, Wakabayashi N, Bunz F, Biswal S. Gain of Nrf2 function in non-small-cell lung cancer cells confers radioresistance. Antioxid Redox Signal. 2010;13:1627–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McDonald JT, Kim K, Norris AJ, Vlashi E, Phillips TM, Lagadec C, et al. Ionizing radiation activates the Nrf2 antioxidant response. Cancer Res. 2010;70:8886–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chuang JC, Stehr H, Liang Y, Das M, Huang J, Diehn M, et al. ERBB2-Mutated Metastatic Non-Small Cell Lung Cancer: Response and Resistance to Targeted Therapies. J Thorac Oncol. 2017;12:833–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bradley JD, Hu C, Komaki RR, Masters GA, Blumenschein GR, Schild SE, et al. Long-Term Results of NRG Oncology RTOG 0617: Standard- Versus High-Dose Chemoradiotherapy With or Without Cetuximab for Unresectable Stage III Non–Small-Cell Lung Cancer. J Clin Oncol. 2019;JCO.19.01162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taylor MD, Nagji AS, Bhamidipati CM, Theodosakis N, Kozower BD, Lau CL, et al. Tumor Recurrence After Complete Resection for Non-Small Cell Lung Cancer. Ann Thorac Surg. 2012;93:1813–21. [DOI] [PubMed] [Google Scholar]
- 23.Seki A, Rutz S. Optimized RNP transfection for highly efficient CRISPR/Cas9-mediated gene knockout in primary T cells. J Exp Med. 2018;215:985–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell. 2018;173:400–416. e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abazeed ME, Adams DJ, Hurov KE, Tamayo P, Creighton CJ, Sonkin D, et al. Integrative radiogenomic profiling of squamous cell lung cancer. Cancer Res. 2013;73(20):6289–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Newman AM, Steen CB, Liu CL, Gentles AJ, Chaudhuri AA, Scherer F, et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol. 2019;37:773–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim JW, Botvinnik OB, Abudayyeh O, Birger C, Rosenbluh J, Shrestha Y, et al. Characterizing genomic alterations in cancer by complementary functional associations. Nat Biotechnol. 2016;34:539–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Galan-Cobo A, Sitthideatphaiboon P, Qu X, Poteete A, Pisegna MA, Tong P, et al. LKB1 and KEAP1/NRF2 Pathways Cooperatively Promote Metabolic Reprogramming with Enhanced Glutamine Dependence in KRAS-Mutant Lung Adenocarcinoma. Cancer Res. 2019;79:3251–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Romero R, Sayin VI, Davidson SM, Bauer MR, Singh SX, LeBoeuf SE, et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat Med. 2017;23:1362–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kitajima S, Ivanova E, Guo S, Yoshida R, Campisi M, Sundararaman SK, et al. Suppression of STING Associated with LKB1 Loss in KRAS-Driven Lung Cancer. Cancer Discov. 2019;9:34–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Skoulidis F, Goldberg ME, Greenawalt DM, Hellmann MD, Awad MM, Gainor JF, et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018;8:822–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hamaji M, Chen F, Matsuo Y, Ueki N, Hiraoka M, Date H. Treatment and Prognosis of Isolated Local Relapse after Stereotactic Body Radiotherapy for Clinical Stage I Non-Small-Cell Lung Cancer: Importance of Salvage Surgery. J Thorac Oncol. 2015;10:1616–24. [DOI] [PubMed] [Google Scholar]
- 33.Hast BE, Cloer EW, Goldfarb D, Li H, Siesser PF, Yan F, et al. Cancer-derived mutations in KEAP1 impair NRF2 degradation but not ubiquitination. Cancer Res. 2014;74:808–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berger AH, Brooks AN, Wu X, Shrestha Y, Chouinard C, Piccioni F, et al. High-throughput Phenotyping of Lung Cancer Somatic Mutations. Cancer Cell. 2016;30:214–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Caboux E, Paciencia M, Durand G, Robinot N, Wozniak MB, Galateau-Salle F, et al. Impact of Delay to Cryopreservation on RNA Integrity and Genome-Wide Expression Profiles in Resected Tumor Samples. PLOS ONE. 2013;8:e79826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Y, Xiang Y. Molecular and cellular basis for the unique functioning of Nrf1, an indispensable transcription factor for maintaining cell homoeostasis and organ integrity. Biochem J. 2016;473:961–1000. [DOI] [PubMed] [Google Scholar]
- 37.Estrela JM, Ortega A, Obrador E. Glutathione in cancer biology and therapy. Crit Rev Clin Lab Sci. 2006;43:143–81. [DOI] [PubMed] [Google Scholar]
- 38.LeBoeuf SE, Wu WL, Karakousi TR, Karadal B, Jackson SR, Davidson SM, et al. Activation of Oxidative Stress Response in Cancer Generates a Druggable Dependency on Exogenous Non-essential Amino Acids. Cell Metab. 2020;31:339–350. e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meijer TWH, Peeters WJM, Dubois LJ, van Gisbergen MW, Biemans R, Venhuizen J-H, et al. Targeting glucose and glutamine metabolism combined with radiation therapy in non-small cell lung cancer. Lung Cancer Amst Neth. 2018;126:32–40. [DOI] [PubMed] [Google Scholar]
- 40.Hassanein M, Hoeksema MD, Shiota M, Qian J, Harris BK, Chen H, et al. SLC1A5 mediates glutamine transport required for lung cancer cell growth and survival. Clin Cancer Res Off J Am Assoc Cancer Res. 2013;19:560–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vanhove K, Derveaux E, Graulus G-J, Mesotten L, Thomeer M, Noben J-P, et al. Glutamine Addiction and Therapeutic Strategies in Lung Cancer. Int J Mol Sci. 2019;20(2):252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Momcilovic M, Bailey ST, Lee JT, Fishbein MC, Braas D, Go J, et al. The GSK3 Signaling Axis Regulates Adaptive Glutamine Metabolism in Lung Squamous Cell Carcinoma. Cancer Cell. 2018;33:905–921. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boysen G, Jamshidi-Parsian A, Davis MA, Siegel ER, Simecka CM, Kore RA, et al. Glutaminase inhibitor CB-839 increases radiation sensitivity of lung tumor cells and human lung tumor xenografts in mice. Int J Radiat Biol. 2019;95:436–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Singh A, Misra V, Thimmulappa RK, Lee H, Ames S, Hoque MO, et al. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med. 2006;3:e420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jeong Y, Hellyer JA, Stehr H, Hoang NT, Niu X, Das M, et al. Role of KEAP1/NFE2L2 mutations in the chemotherapeutic response of non-small cell lung cancer patients. Clin Cancer Res. 2020;26:274–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goeman F, Nicola FD, Scalera S, Sperati F, Gallo E, Ciuffreda L, et al. Mutations in the KEAP1-NFE2L2 Pathway Define a Molecular Subset of Rapidly Progressing Lung Adenocarcinoma. J Thorac Oncol. 2019;14(11):1924–1934. [DOI] [PubMed] [Google Scholar]
- 47.Iyengar P, Wardak Z, Gerber DE, Tumati V, Ahn C, Hughes RS, et al. Consolidative Radiotherapy for Limited Metastatic Non-Small-Cell Lung Cancer: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2018;4:e173501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gomez DR, Tang C, Zhang J, Blumenschein GR, Hernandez M, Lee JJ, et al. Local Consolidative Therapy Vs. Maintenance Therapy or Observation for Patients With Oligometastatic Non-Small-Cell Lung Cancer: Long-Term Results of a Multi-Institutional, Phase II, Randomized Study. J Clin Oncol Off J Am Soc Clin Oncol. 2019;37:1558–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Palma DA, Olson R, Harrow S, Gaede S, Louie AV, Haasbeek C, et al. Stereotactic ablative radiotherapy versus standard of care palliative treatment in patients with oligometastatic cancers (SABR-COMET): a randomised, phase 2, open-label trial. The Lancet. Elsevier; 2019;393:2051–8. [DOI] [PubMed] [Google Scholar]
- 50.Dziedzic DA, Rudzinski P, Langfort R, Orlowski T, Polish Lung Cancer Study G. Risk Factors for Local and Distant Recurrence After Surgical Treatment in Patients With Non-Small-Cell Lung Cancer. Clin Lung Cancer. 2016;17:e157–67. [DOI] [PubMed] [Google Scholar]
- 51.Trakul N, Chang CN, Harris J, Chapman C, Rao A, Shen J, et al. Tumor volume-adapted dosing in stereotactic ablative radiotherapy of lung tumors. Int J Radiat Oncol Biol Phys. 2012;84:231–7. [DOI] [PubMed] [Google Scholar]
- 52.Ohri N, Bodner WR, Halmos B, Cheng H, Perez-Soler R, Keller SM, et al. (18)F-Fluorodeoxyglucose/Positron Emission Tomography Predicts Patterns of Failure After Definitive Chemoradiation Therapy for Locally Advanced Non-Small Cell Lung Cancer. Int J Radiat Oncol Biol Phys. 2017;97:372–80. [DOI] [PubMed] [Google Scholar]
- 53.Przybyl J, Chabon JJ, Spans L, Ganjoo KN, Vennam S, Newman AM, et al. Combination Approach for Detecting Different Types of Alterations in Circulating Tumor DNA in Leiomyosarcoma. Clin Cancer Res. 2018;24:2688–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gazdar A. Activating and resistance mutations of EGFR in non-small-cell lung cancer: role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene. 2009;28:S24–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv. 2019;531210. [Google Scholar]
- 56.Srivastava A, Sarkar H, Gupta N, Patro R. RapMap: a rapid, sensitive and accurate tool for mapping RNA-seq reads to transcriptomes. Bioinformatics. 2016;32:i192–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gentles AJ, Hui AB-Y, Feng W, Azizi A, Nair RV, Knowles DA, et al. Clinically-relevant cell type cross-talk identified from a human lung tumor microenvironment interactome. bioRxiv. 2019;637306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guzmán C, Bagga M, Kaur A, Westermarck J, Abankwa D. ColonyArea: An ImageJ Plugin to Automatically Quantify Colony Formation in Clonogenic Assays. PLOS ONE. 2014;9:e92444. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.