

Abstract
Purpose:
Inherited axonopathies (IA) are rare, clinically and genetically heterogeneous diseases that lead to length-dependent degeneration of the long axons in central (hereditary spastic paraplegia (HSP)) and peripheral (Charcot-Marie-Tooth type 2 (CMT2)) nervous systems. Mendelian high-penetrance alleles in over one hundred different genes have been shown to cause IA; yet, about 50% of IA cases do not receive a genetic diagnosis. A more comprehensive spectrum of causative genes and alleles is warranted, including causative and risk alleles, as well as oligogenic multilocus inheritance.
Methods:
Through international collaboration, IA exome studies are beginning to be sufficiently powered to perform a pilot rare variant burden analysis. After extensive quality control, our cohort contained 343 CMT cases, 515 HSP cases, and 935 non-neurological controls. We assessed the cumulative mutational burden across disease genes, explored the evidence for multilocus inheritance, and performed an exome-wide rare variant burden analysis.
Results:
We replicated the previously described mutational burden in a much larger cohort of CMT cases, and observed the same effect in HSP cases. We identified a preliminary risk allele for CMT in the EXOC4 gene (p-value= 6.9×10−6, OR=2.1) and explored the possibility of multi-locus inheritance in IA.
Conclusions:
Our results support the continuing emergence of complex inheritance mechanisms in historically Mendelian disorders.
Keywords: Charcot-Marie-Tooth disease, Hereditary Spastic Paraplegia, inherited axonopathy, mutational burden, oligogenic inheritance
Introduction
Inherited axonopathies (IA) are a group of disorders unified by a common pathological mechanism: length-dependent axonal degeneration. They are traditionally classified into two broad genetic disorders: Hereditary Spastic Paraplegia (HSP) and Charcot-Marie-Tooth disease (CMT) depending on upper or lower motor neuron involvement, respectively. Historically, CMT and HSP are treated as distinct disorders, but their increasingly apparent clinical and genetic overlap challenges this classification. CMT and HSP can be caused by mutations within the same gene (for example, KIF1A, REEP1, and BSCL2), yet the additional factors which determine peripheral or central nerve involvement affected in each IA patient remains unclear. Currently, more than 50% of cases do not receive a genetic diagnosis from next-generation sequencing.1 The high percentage of genetically undiagnosed IA cases may be a result of undiscovered highly penetrant alleles in both known and yet to be associated disease genes, cases without a true genetic etiology, or currently difficult to detect and/or interpret variation such as deep intronic, regulatory, or structural. Additionally, growing evidence suggests that rare mutational mechanisms or modes of inheritances that are likely overlooked in standard WES analysis may also contribute to the overall genetic etiology15. Finally, environmental contributions to phenotypes are very difficult to assess and may have a larger than estimated importance in IA patients.
The phenotypic variability and reduced penetrance observed within IA supports the possibility of multilocus inheritance or genetic modification. These two events are closely related and both result in phenotypic effects that are caused by more than a single Mendelian allele. A distinction between these processes lies in the sufficiency of the primary allele to cause disease.2 If the presence of the primary allele alone manifests the phenotype, then the secondary allele is a genetic modification of the phenotype, such as the severity of progression or the age at onset.2 However, if the presence of an allele in a second gene or multiple genes is required to cause disease, then inheritance is multilocus in nature.2 Non-Mendelian modes of inheritance have been independently demonstrated in both CMT and HSP3,4, but cohort-level sequencing analyses are limited. In this pilot study, we gathered over 800 exomes from IA cases to determine whether multilocus inheritance warrants deeper investigation in classically Mendelian disease groups.
Materials and Methods
Ethics Statement
The cases were collected from the Inherited Neuropathy Consortium, the University of Miami (UM), Children’s Hospital of Philadelphia (CHOP), the University Hospital Tübingen, and McGill University, and all participating individuals gave informed consent prior to initiating this study in agreement with the institutional review boards.
Methods
Families included in the study are affected by IA (either CMT or HSP). CMT cases were diagnosed with CMT (type 1, 2, 4, or intermediate), distal hereditary motor neuropathy, hereditary sensory autonomic neuropathy, or hereditary sensory neuropathy; HSP cases were diagnosed with pure or complicated HSP. WES was performed at UM (CMT and HSP cases), McGill (HSP cases), and at CHOP (controls). Enrolled cases had previously negative testing for key IA genes; however, solved research cases were included in the cohort (8.7% (30/343) CMT cases were solved across 21 disease genes while 5.8% (30/515) HSP cases were solved across 19 disease genes). All samples were sequenced on Illumina HiSeq2000 and joint-genotyped according to the GATK (v.3.3) germline WES best practices. After extensive quality control (including duplication percentage, sex and relatedness, depth and missingness metrics, ancestry), the cohort contained 343 CMT cases, 515 HSP cases, and 935 non-neurological controls of predominantly European ancestry.
To detect risk alleles, a gene-based rare variant association test was performed by the C-alpha test in the PLINK/SEQ suite. Following recommended protocol, tests with an i-statistic greater than 10−3 were removed, and Bonferroni correction was applied.5 To compare the mutational burden across known disease genes (CMT:n=88, HSP:n=95), the number of rare variants (non-synonymous or loss-of-function at ExAC MAF ≤ 0.01 and ≤ 0.001) within disease genes was computed for each sample, and the average counts were compared between case and control using a Mann-Whitney-Wilcoxon test followed by 10,000 iterations of affection permutation for significance. To assess multilocus inheritance, the number of known disease genes carrying at least one qualifying variant was determined per sample. Case and control carrier status was organized into 2×2 contingency tables and assessed by Fisher’s exact test.
CMT disease genes:
AARS,AIFM1,ARHGEF10,ATL3,ATP7A,CHCHD10,CLCF1,COX6A1,CRLF1,DCAF8,DCTN1,DGAT2,DHTKD1,DNAJB2,DNM2,DNMT1,DST,DYNC1H1,EGR2,ERBB3,FAM134B,FBLN5,FBXO38,FGD4,FIG4,GARS,GDAP1,GJB1,GNB4,HARS,HINT1,HK1,HSPB1,HSPB3,HSPB8,IGHMBP2,IKBKAP,INF2,KARS,LITAF,LMNA,LRSAM1,MARS,MED25,MFN2,MME,MORC2,MPZ,MTMR2,NAGLU,NDRG1,NEFH,NEFL,NGF,NTRK1,PDK3,PLEKHG5,PMP2,PMP22,PRPS1,PRX,RAB7A,SBF1,SBF2,SCN10A,SCN11A,SCN9A,SEPTIN,SETX,SH3TC2,SLC5A7,SMN1,SPTLC1,SPTLC2,SYT1,TRIM2,TRPA1,WNK1,YARS
HSP disease genes:
ADD3,AFG3L2,AIMP1,ALDH18A1,AP4B1,AP4E1,AP4M1,AP4S1,AP5Z1,ARSA,ATP13A2,ATXN1,ATXN2,ATXN3,AUH,B4GALNT1,C12ORF65,C19ORF12,CAPN1,CYP27A1,CYP2U1,CYP7B1,DARS2,DDHD1,DDHD2,ELOVL4,ENTPD1,ERLIN1,ERLIN2,FA2H,FAM126A,FBXO7,FLRT1,FXN,GAD1,GALC,GAN,GBA2,GFAP,GJC2,GLRX5,HSPD1,KANK1,KCNA2,KCND3,KIAA0196,KIF1C,KIF5A,KLC2,L1CAM,LMNB1,MAG,MTHFR,MTPAP,NIPA1,NT5C2,OPA1,OPA3,PDYN,PGAP1,PLA2G6,PLP1,PNPLA6,POLR3A,POLR3B,PPP2R2B,PRNP,RTN2,SACS,SLC16A2,SLC33A1,SPAST,SPG20,SPG21,SPG7,STUB1,SYNE1,TBP,TECPR2,TGM6,TUBB4A,VAMP1,VCP,VPS37A,VWA3B,ZFYVE26
CMT and HSP disease genes:
ATL1,BICD2,BSCL2,CCT5,KIF1A,REEP1,SPG11,TFG,TRPV4
Results
Association of EXOC4 with CMT cases
Exome-wide association analysis was performed at 17,637 protein coding loci by the C-alpha test. The PLINK/SEQ suite computes an estimate of the minimal achievable p-value for a locus, the i-statistic. We followed the recommended protocol to filter out loci with an i-statistic greater than 10−3 before Bonferroni correction to remove non-contributing genes.5 Based on the 2,145 remaining loci, the p-value threshold for an experiment-wide significance (alpha=0.05) was 2.3 × 10−5. After filtering results by the PLINK/SEQ i-statistic and applying Bonferroni multiple-testing correction, three genes, KDM5A (p-value= 9.9×10−7, OR=3.6), EXOC4 (p-value= 6.9×10−6, OR=2.6), and CEP78 (p-value= 2.3×10−5, OR=4.4), reached experiment-wide significance (Fig. 1A). KDM5A and EXOC4 both contained a single allele in cases that drove the association: NM_001042603.1(KDM5A):c.11T>G and NM_021807.3(EXOC4):c.1648G>A. Sanger sequencing confirmed the driver allele in EXOC4 (Fig. 1B) and revealed a false positive exome call in KDM5A (indicating a false positive association to KDM5A). We did not follow up with CEP78 since the gene did not contain a single driving allele. At the EXOC4 gene-level, heterozygous carriers were 2.6 (95% CI: 1.28–5.37) times more likely to be affected, while at the driver allele-level, heterozygous carriers were 9.07 times more likely to be affected (95% CI: 2.94–28.01) (Fig. 1C). The variant was a non-synonymous missense change (p.Gly550Arg) predicted to be disease causing by MutationTaster15 with a gnomAD MAF of 0.00411.
Figure 1.
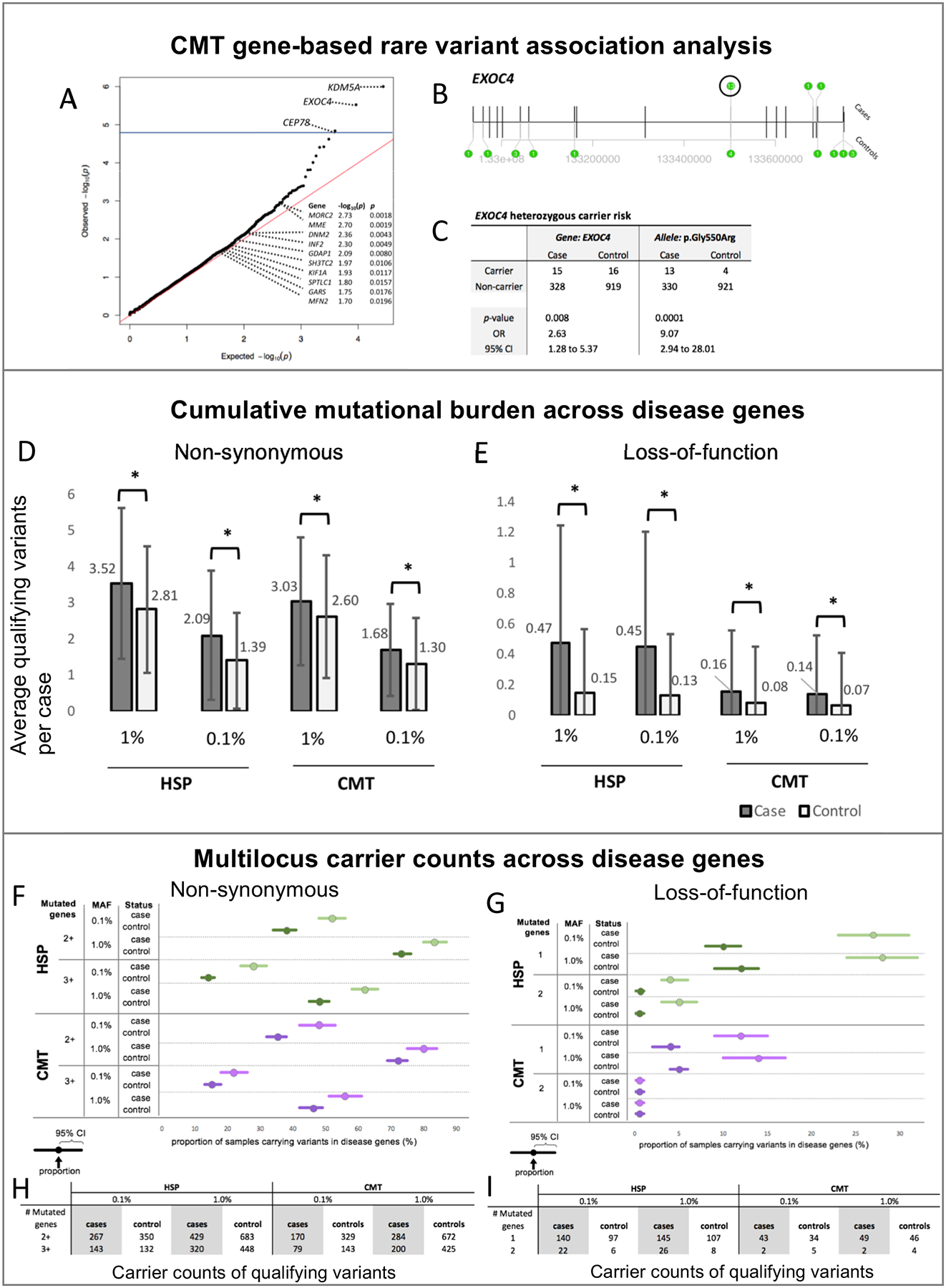
Risk allele and multilocus inheritance in inherited axonopathies. (A-C) CMT gene-based rare variant association analysis. (A) qq plot of the observed p-values from C-alpha gene-based association analysis. Blue line indicates multiple testing correction threshold. Known CMT genes with nominal significance are annotated; (B) Transcript model of EXOC4 annotated with variant positions and counts (green bubble); and (C) Heterozygous carrier risk for CMT at gene and variant level. (D-E) Cumulative mutational burden across disease genes. Distribution of the average count of qualifying variants in known HSP and CMT disease genes per case at 1% and 0.1% ExAC MAF for (D) non-synonymous and (E) loss-of-function variation. Difference in case/control distribution tested with Mann-Whitney U Test (*p-value <= 0.05). (F-I) Multilocus variant counts across disease genes. Proportion (F-G) and absolute counts (H-I) of cases carrying non-synonymous (F,H) and loss-of-function (G,I) variants in the indicated number of mutated disease genes (1, 2, 2+, or 3+) at 0.1% and 1% ExAC MAF.
Increased mutational burden across known disease genes in IA cases
IA cohorts were independently tested for a mutational burden (an excess of rare variants) across known disease genes. In our CMT and HSP cohorts, we identified a significant mutational burden (Mann-Whitney, nominal p-value ≤ 0.05) in each tested variant set (non-synonymous (NS) and loss-of-function (LoF) variation at ExAC MAF ≤ 0.001 or ≤ 0.01; (Fig. 1D–E). As a further test of our observations, we repeated the mutational burden comparison with permutated case/control status over 10,000 iterations. We found that each tested variant set remained statistically significant (empirical p-value ≤ 0.05), thus supporting that the mutational burden found across disease genes is specific to each IA cohort. The number of samples that contained variants above 3 standard deviations upper limit (3SD) are as follows [cohort, n, 3SD]: HSP NS 1%: [cases, 3, 9.8], [controls, 4, 8.1]; HSP NS 0.1%: [cases, 6, 7.5], [controls, 10, 5.4]; HSP LoF 1%: [cases, 9, 2.8], [controls, 19, 1.4]; HSP LoF 0.1%: [cases, 9, 2.7], [controls, 17, 1.3]; CMT NS 1%: [cases, 2, 8.3], [controls, 10, 7.7]; CMT NS 0.1%: [cases, 2, 5.5], [controls, 6, 5.1]; CMT LoF 1%: [cases, 5, 1.4], [controls, 11, 1.2]; CMT LoF 0.1%: [cases, 5, 1.3], [controls, 11, 1.1].
Multilocus inheritance suggested in IA cases
Next, we sought to determine whether the observed mutational burden was more likely to follow a monogenic (single gene), digenic (two genes), or oligogenic (more than two genes) inheritance. Unlike the mutational burden, the significance of each inheritance type was influenced by the minor allele frequency (Fig. 1F–G). HSP cases showed consistent evidence for oligogenic inheritance (≥ 3 genes) of NS variation and monogenic inheritance (1 gene) of LoF variation at both ExAC MAF ≤ 0.01 and ≤ 0.001 (Fisher’s exact, p-value ≤ 0.05). HSP cases also displayed significant di/oligogenic inheritance (≥ 2 genes) of NS variation at the less common ExAC MAF ≤ 0.001 (p-value ≤ 0.05). Furthermore, di/oligogenic inheritance of both NS and LoF variation for HSP cases is suggested at ExAC MAF ≤ 0.01 (p-value = 0.0598 and 0.0572, respectively). Evidence for inheritance types in CMT was not as consistent as in HSP, possibly due to a lower CMT sample size. At ExAC MAF ≤ 0.01, CMT cases demonstrated monogenic inheritance for LoF variation and oligogenic inheritance for NS variation (p-value ≤ 0.05) with potential di/oligogenic inheritance for NS variation (p-value = 0.521). Lastly, at ExAC MAF ≤ 0.001, CMT cases only showed significant evidence for monogenic inheritance of NS variation (p-value ≤ 0.05) with potential evidence for oligogenic NS inheritance and monogenic LoF inheritance (p-value = 0.0641 and 0.0536, respectively). The counts of samples carrying variants is summarized in Fig. 1H–I.
Discussion
As the cost and availability of next-generation sequencing continues to drop, we are now reaching large enough sample sizes to apply statistical approaches to rare diseases. In this study, we sought to assess the mutational burden and multilocus involvement of rare variation in a cohort of inherited axonopathies as well as identify potential risk loci.
To identify genes that could potentially carry non-Mendelian risk alleles, we performed an unbiased exome-wide rare variant burden analysis with the C-alpha test. After filtering results and performing Sanger sequencing, EXOC4 stood out as a candidate CMT gene. EXOC4 is involved in vesicle transport and membrane tethering in polarized cells and is expressed in Schwann cells.6 In a CMT4B1 mouse model, Exoc4 (Sec8) formed a complex with Mtmr2 and Dlg1 to coordinate homeostatic control of myelination.6 Exoc4 is abundantly expressed at the Drosophila neuromuscular junction and required for in vivo regulation of synaptic microtubule formation.7 Furthermore, Exoc4 is suggested to play a central role in oligodendrocyte membrane formation through the regulation of vesicular transport of myelin proteins.8 Though EXOC4 has biologically plausibility, this result should be interpreted with a degree of caution. Stronger genetic evidence for EXOC4 is necessary, including replication of the association or identification of highly penetrant Mendelian variants. Unfortunately, a secondary large CMT exome cohort does not currently exist for follow-up replication analysis.
From the rare variant burden analysis, we were also able to re-identify several established monogenic CMT2 genes, including MME9, MORC16 and MFN210. This is despite a general effort to exclude cases with MFN2 and other common CMT genes from exome analysis. Similarly, known familial ALS genes showed strong associations in a gene-based rare variant burden analysis of sporadic ALS cases.11 These results give us confidence about the utility of association studies in rare disease cohorts, and may indicate the presence of additional risk alleles contributing to the phenotype in these known CMT genes. We interpret these results as additional evidence supporting cohort-level statistical approaches to identify Mendelian and non-Mendelian factors involved in classically monogenic disease.
Additionally, we observed a significant mutational burden across CMT and HSP disease genes in cases compared to non-neurological controls. The aggregation of rare, damaging alleles in disease-associated genes may contribute to risk, severity, and clinical heterogeneity. This inheritance model has been suggested in CMT based on exome sequencing from 37 individuals.12 Gonzaga-Jauregui et al observed an average of 1.8 variants per case across 58 neuropathy genes compared to 1.3 variants per control. They followed up this observation with a second small cohort of 32 cases from Turkish descent, and observed a mutational burden of 2.1 vs 1.6 nonsynonymous rare variants per in cases vs controls. The mutational burden hypothesis was functionally evaluated in vivo in zebrafish experiments, which resulted in increased phenotypic severity when pairs of neuropathy genes were inactivated.11 Our cohort is roughly 10 times larger than the previous cohorts, and is now the third independent CMT cohort to support the mutational burden hypothesis. Furthermore, rare non-synonymous variation was also significantly distributed across 2 or 3 disease genes in our cohort, indicating multilocus inheritance – which remains underexplored in rare diseases because of functional validation challenges. Additional variants in multiple disease genes can have either a combinatorial effect on the same biological pathways or a destabilizing effect on the entire disease module.
The primary goal of this study was to move beyond the ‘one-disease-one-gene’ model to assess an expanded genetic architecture in IA. An appreciation for the extent of allelic and locus heterogeneity, reduced penetrance, and variable expressivity within IA has come from traditional family-based approaches. These insights across Mendelian diseases are driving the genetics community to delineate the more complicated and nuanced patterns of inheritance: 1. A gene-based variant burden test was successfully applied to a cohort of ALS cases and identified a new risk gene.11 Using this approach, we observed an enrichment of qualifying variants (in a candidate gene and in known disease genes) that influence disease risk. We are extremely cautious about overstating any potential involvement of EXOC4 in disease pathogenesis. However, we interpret these results as evidence supporting the hypothesis of ‘risk alleles’ in IA. 2. A mutational burden that can modulate phenotypic severity was observed in two small CMT cohorts, and the increased burden of protein-altering variants was functionally tested in a zebrafish model and demonstrated phenotypic modification.11 We have replicated this finding in a larger cohort of CMT cases and discovered a similar result in HSP cases. Beyond CMT and HSP, a rare variant aggregation has also been show to influence susceptibility to Parkinson disease, and the age-of-onset of ALS.13
These results are subject to several limitations. First, copy number (CNV) and structural variation (SV) was not included in this exome pipeline due to the high false positive rate of CNV from short-read NGS.16 Family-based study designs with genomic regions of interest allow for high confidence filtering approaches. However, with a proband-only design, we were concerned about identifying false associations from false positive calls. Given the importance of SV and CNV in human disease, future studies should consider whole-genome sequencing, long-read technology, or family-based approaches. Another limitation of short-read NGS technology is the challenges in phasing rare variation. As such, we were unable to identify compound heterozygous variation. Finally, this study is limited by the availability of a replication cohort for rare disease. Until the EXOC4 risk allele is replicated or the EXOC4 locus is supported by additional genetic evidence, then this association can only be clarified to a point in functional in vitro or in vivo studies.
Concluding Remarks
Concepts such as risk alleles, mutational burden, and multilocus inheritance within rare Mendelian diseases lie at the intersection of rare and common diseases. Recent discoveries have shed light on the architecture of common disease, including increased risk for a common disease from heterozygous alleles in recessive Mendelian genes.14 However, the impacts of multilocus inheritance on Mendelian disease, including phenotypic severity, oligogenic inheritance, blended phenotypes, and phenotypic expansion, requires further exploration.13 Investigating these non-Mendelian concepts will lead to a unified model of human disease and facilitate precision genetic therapies. Here, we continue pushing these boundaries in IA, suggest potential involvement of EXOC4 in disease pathogenesis of CMT, and provide further evidence supporting a multilocus Mendelian model. Clinicians should be aware of these developments when interpreting negative genetic testing results, and future studies will investigate specific combinations of risk alleles with potential clinical actionability.
Acknowledgements
We thank the patients and their families for their participation. This study was supported by NINDS (U54NS065712-12 to S.Z. as site PI, R01NS105755 to S.Z., R01NS072248 to S.Z.), the Charcot-Marie-Tooth Association, the Hereditary Neuropathy Foundation, and the Muscular Dystrophy Association. This work was done as a part of a Canadian collaboration to study HSP (CanHSP). The CanHSP cohort was funded by CIHR Emerging Team Grant, in collaboration with the Canadian Organization for Rare Disorders (CORD), grant number RN127580 - 260005.
Consortium
Aixa Rodriguez15 (non-author contributor), Alexa Bacha28 (non-author contributor), Ashley Kosikowski16 (non-author contributor), Beth Wood30 (non-author contributor), Brett McCray17 (non-author contributor), Brianna Blume18 (non-author contributor), Carly Siskind19 (non-author contributor), Charlotte Sumner17 (non-author contributor), Daniela Calabrese29 (non-author contributor), David Walk20 (non-author contributor), Dragan Vujovic15 (non-author contributor), Eun Park17 (non-author contributor), Francesco Muntoni27 (non-author contributor), Gabrielle Donlevy21 (non-author contributor), Gyula Acsadi16 (non-author contributor), John Day19 (non-author contributor), Joshua Burns21 (non-author contributor), Jun Li22 (non-author contributor), Karen Krajewski22 (non-author contributor), Kate Eichinger30 (non-author contributor), Kayla Cornett21 (non-author contributor), Krista Mullen20 (non-author contributor), Laura Perez28 (non-author contributor), Laurie Gutmann28 (non-author contributor), Maria Barrett18 (non-author contributor), Mario Saporta22 (non-author contributor), Mariola Skorupinska27 (non-author contributor), Natalie Grant24 (non-author contributor), Paula Bray21 (non-author contributor), Reza Seyedsadjadi24 (non-author contributor), Riccardo Zuccarino28 (non-author contributor), Richard Finkel15 (non-author contributor), Richard Lewis25 (non-author contributor), Sabrina Yum26 (non-author contributor), Sarah Hilbert20 (non-author contributor), Simone Thomas17 (non-author contributor), Steffen Behrens-Spraggins30 (non-author contributor), Tara Jones25 (non-author contributor), Tiffany Grider28 (non-author contributor), Tim Estilow26 (non-author contributor), Vera Fridman18 (non-author contributor), Mary M. Reilly27 (non-author contributor), Michael E. Shy28 (non-author contributor), Chelsea J. Bacon28 (non-author contributor), Shawna M. E. Feely28 (non-author contributor), Alexander M. Rossor27 (non-author contributor), David N. Herrmann30 (non-author contributor)
15Nemours Children's Hospital, Department of Pediatrics, Division of Neurology, Orlando, Florida, USA
16Connecticut Children’s Medical Center, Neurology, Charcot-Marie-Tooth Center of Excellence, Hartford, Connecticut, USA
17Johns Hopkins University, Department of Neurology, Baltimore, Maryland, USA
18University of Colorado Health Neurosciences Center, Anschutz Medical Campus, Aurora, Colorado, USA
19Stanford University, Department of Neurology and Neurological Sciences, Stanford, California, USA
20University of Minnesota, Department of Neurology, Minneapolis, Minnesota, USA
21Paediatric Gait Analysis Service of NSW, The Children’s Hospital at Westmead, Westmead, NSW, Australia
22Department of Neurology, Neuromuscular and Inherited Neuropathies Division, Wayne State University School of Medicine, Detroit, Michigan, USA
24Harvard/Massachusetts General Hospital, Department of Neurology, Boston, Massachusetts, USA
25Cedars-Sinai Medical Center, Department of Neurology, Los Angeles, California, USA
26Children’s Hospital of Philadelphia, Department of Neurology, Philadelphia, Pennsylvania, USA
27Department of Neuromuscular Disease, UCL Queen Square Institute of Neurology and The National Hospital for Neurology, London, UK.
28Department of Neurology, University of Iowa Carver College of Medicine, Iowa City, Iowa, USA.
29Unit of Rare Neurodegenerative and Neurometabolic Diseases, Department of Clinical Neurosciences, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy.
30Department of Neurology, University of Rochester, Rochester, New York, USA.
Footnotes
Disclosure
Nothing to disclose.
Disclosure: The authors declare no conflict of interest.
References
- 1.Cortese A, Wilcox JE, Polke JM, et al. Targeted next-generation sequencing panels in the diagnosis of Charcot-Marie-Tooth disease. Neurology. 2020;94(1):51. doi: 10.1212/WNL.0000000000008672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eldomery MK, Coban-Akdemir Z, Harel T, et al. Lessons learned from additional research analyses of unsolved clinical exome cases. Genome Med. 2017;9(1):26. doi: 10.1186/s13073-017-0412-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bis-Brewer DM, Zuchner S. Genetics modifiers and non-Mendelian aspects of CMT. Brain Research. 2020;1726:146459. doi: 10.1016/j.brainres.2019.146459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bis-Brewer DM, Zuchner S. Perspectives on the Genomics of HSP Beyond Mendelian Inheritance. Front Neurol. 2018;9:25 11. doi: 10.3389/fneur.2018.00958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kiezun A, Garimella K, Do R, et al. Exome sequencing and the genetic basis of complex traits. Nat Genet. 2012;44(6):623 630. doi: 10.1038/ng.2303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bolis A, Coviello S, Visigalli I, et al. Dlg1, Sec8, and Mtmr2 Regulate Membrane Homeostasis in Schwann Cell Myelination. J Neurosci. 2009;29(27):8858 8870. doi: 10.1523/jneurosci.1423-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liebl FL, Chen K, Karr J, Sheng Q, Featherstone DE. Increased synaptic microtubules and altered synapse development in Drosophila sec8 mutants. Bmc Biol. 2005;3(1):27. doi: 10.1186/1741-7007-3-27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anitei M A role for Sec8 in oligodendrocyte morphological differentiation. J Cell Sci. 2006;119(5):807 818. doi: 10.1242/jcs.02785 [DOI] [PubMed] [Google Scholar]
- 9.Auer-Grumbach M, Toegel S, Schabhüttl M, et al. Rare Variants in MME, Encoding Metalloprotease Neprilysin, Are Linked to Late-Onset Autosomal-Dominant Axonal Polyneuropathies. Am J Hum Genetics. 2016;99(3):607 623. doi: 10.1016/j.ajhg.2016.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zuchner S, Mersiyanova I, Muglia M, et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet. 2004;36(5):449 451. doi: 10.1038/ng1341 [DOI] [PubMed] [Google Scholar]
- 11.Cirulli ET, Lasseigne BN, Petrovski S, et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science. 2015;347(6229):1436 1441. doi: 10.1126/science.aaa3650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gonzaga-Jauregui C, Harel T, Gambin T, et al. Exome Sequence Analysis Suggests that Genetic Burden Contributes to Phenotypic Variability and Complex Neuropathy. Cell Reports. 2015;12(7):1169 1183. doi: 10.1016/j.celrep.2015.07.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Posey JE, O’Donnell-Luria AH, Chong JX, et al. Insights into genetics, human biology and disease gleaned from family based genomic studies. Genet Med. 2019;21(4):798–812. doi: 10.1038/s41436-018-0408-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ji W, Foo J, O’Roak BJ, et al. Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat Genet. 2008;40(5):592–599. doi: 10.1038/ng.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frésard L and Montgomery SB. Diagnosing rare diseases after the exome. Cold Spring Harb Mol Case Stud. 2018;4(6):a003392. doi: 10.1101/mcs.a003392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pounraja VJ, Jayakar G, Jensen M, et al. A machine-learning approach for accurate detection of copy number variants from exome sequencing. Genome Res. 2019;29(7):1134–1143. doi: 10.1101/gr.245928.118 [DOI] [PMC free article] [PubMed] [Google Scholar]