

Abstract
Glycine 34 to tryptophan (G34W) substitutions in H3.3 arise in ~90% of giant cell tumour of bone (GCT). Here, we show H3.3G34W is necessary for tumour formation. By profiling the epigenome, transcriptome and secreted proteome of patient samples and tumour-derived cells CRISPR/Cas9-edited for H3.3G34W, we show that H3.3K36me3 loss on mutant H3.3 alters the deposition of the repressive H3K27me3 mark from intergenic to genic regions, beyond areas of H3.3 deposition. This promotes redistribution of other chromatin marks and aberrant transcription, altering cell fate in mesenchymal progenitors and hindering differentiation. Single-cell transcriptomics reveals that H3.3G34W stromal cells recapitulate a neoplastic trajectory from a SPP1+ osteoblast-like progenitor population towards an ACTA2+ myofibroblast-like population, which secretes extracellular matrix ligands predicted to recruit and activate osteoclasts. Our findings suggest that H3.3G34W leads to GCT by sustaining a transformed state in osteoblast-like progenitors which promotes neoplastic growth, pathological recruitment of giant osteoclasts, and bone destruction.
Keywords: H3.3G34W, giant cell tumour of bone, extracellular matrix, osteoprogenitor, osteoclast
INTRODUCTION
Giant cell tumour of bone (GCT) is a locally aggressive yet rarely metastasizing neoplasm that accounts for 20% of benign bone tumours (1). Tumours are composed of three major compartments: multinucleated osteoclast-like giant cells, macrophage-like monocytes, and stromal cells. Although the primary cause of morbidity in GCT is bone resorption by monocyte-derived giant osteoclasts, the actual neoplastic component is formed by cells from the poorly defined mesenchymal stromal compartment, which bears heterozygous somatic mutations in H3F3A, one of two genes encoding histone 3 variant H3.3 (2). These mutations lead to substitution of glycine 34 to tryptophan or, more rarely, to leucine (G34W/L, >92% of cases) (2). Unlike H3.3 glycine 34 to arginine or valine substitutions (H3.3G34R/V) that arise in brain tumours and co-occur with partner TP53 and ATRX mutations (3,4), H3.3G34W/L mutations are the only recurrent molecular alteration in GCT, potentially driving this destructive bone disease (2,5).
While the almost universal occurrence of H3.3G34W/L mutations in GCT (92%) strongly suggests a causal role, how they drive tumourigenesis in the bone remains unknown. Moreover, the cellular processes that result in multinucleated giant cell recruitment have yet to be elucidated. Other H3 mutations, highly recurrent in a wide range of cancers, affect residues that are direct targets of post-translational modifications (PTMs). These lysine to methionine substitutions on K27 (K27M) and K36 (K36M) of the H3 tail impair enzymatic activities of the corresponding methyltransferases (6). This results in genome-wide changes in the deposition and distribution of major antagonistic repressive (H3K27me3) or activating (H3K36me3, H3K36me2, H3K27ac) chromatin marks, and redistribution of the repressive polycomb receptor complex (PRC) 1 and 2 to promote aberrant transcription and tumourigenesis (7-13). In contrast, H3.3G34 mutations are less well understood. They exclusively impact noncanonical H3.3 histones, suggesting an under-investigated H3.3-dependent role in their tumourigenesis. Indeed, deposition of H3.3 is replication-independent and follows a specific temporo-spatial pattern that may be relevant to GCT formation. H3.3 marks active euchromatin at enhancers and gene bodies, but also represents the main H3 variant in silent pericentromeric and telomeric heterochromatin (14). The presence of mutant H3.3G34 could therefore affect chromatin locally in any of these key genomic regions, contributing to tumourigenesis. Although H3.3G34 mutations occur on a residue that does not undergo PTM, they affect the adjacent K36 residue, leading to decreased H3.3K36me3 (6) and potential reciprocal increase of H3.3K27me3 on the mutant H3.3 tail (15). Structural studies suggest that H3.3G34 mutants cause steric hindrance of the catalytic groove of SETD2, an H3K36 trimethyltransferase (16). This hypothesis is further supported by the finding that loss-of-function SETD2 mutations are mutually exclusive with H3.3G34R/V mutations in gliomas, suggesting a convergence of effects (17). H3.3G34R/V have also been suggested to impede H3.3K36 access by KDM2A, an H3K36me2 demethylase (18), or inhibit members of the KDM4 H3K9/K36 demethylase family, resulting in increased H3K36me3 and H3K9me3 at select loci (19).
Altogether, there is no consensus on the mechanism of action of H3.3G34R/V/W/L mutations. The cell-type specific mutation patterns indicate a strong context-dependence that remains poorly understood, and that has so far hampered accurate tumour modeling. GCT models have been mainly restricted to over-expression systems that increase mutant histone expression beyond that observed in tumours (15,20). However, accounting for dosage is key, since oncohistones contribute to less than 20% of the total H3 pool (6). To address these challenges, we generated H3.3G34W (referred to hereafter as G34W) isogenic models using CRISPR/Cas9 in patient-derived GCT stromal cells as well as patient-derived orthotopic xenografts (PDOX). We profiled the epigenome, transcriptome and secreted Golgi proteome of cellular models, and generated transcriptomic data of GCT primary tumours and PDOX at single-cell resolution. Our combined modelling and profiling efforts shed light on the mechanism by which G34W transforms cells to promote tumourigenicity and enables interactions with the tumour microenvironment (TME) of the bone to foster disease.
RESULTS
G34W is required for tumour formation and promotes osteoclast recruitment in GCT.
To determine the role of the H3.3G34W oncohistone on GCT tumourigenesis in pure neoplastic stromal cells, we used CRISPR/Cas9 to target the H3F3A G34W mutation (referred to hereafter as G34W) in three immortalized (Im-GCT) stromal cell lines established from primary tumours (Table S1, Fig. S1A). We generated clones carrying loss-of-function insertions or deletions on the mutant allele (G34W-KO), or clones where the mutation was corrected to wild-type (G34-WT), and confirmed absence of oncohistone expression using a previously validated G34W-specific antibody (21) (Fig. 1A, Fig. S1B). G34W-KO and G34-WT clones showed significantly decreased proliferation (Fig. 1B, Fig. S1C) and colony forming ability in vitro (Fig. 1C, Fig. S1D) in the isogenic cell lines. Since G34W-KO and G34-WT clones behaved similarly in in vitro assays, they are hereafter referred to as edited clones. Orthotopic tibial and subcutaneous tissue implantation of luciferase-tagged G34W and edited Im-GCT-4072 cells showed a weekly increase of bioluminescence and tumour formation only in G34W-mutant cells (Fig. 1D-E, Fig. S1E-H). Notably, no tumours formed when two distinct edited clones were implanted in tibia and subcutaneously, with more than 1-year follow-up post-implantation (Fig. 1E, Fig. S1E).
Figure 1. G34W is necessary for tumourigenesis and promotes aggressive osteolytic bone lesions in an orthotopic xenograft model.
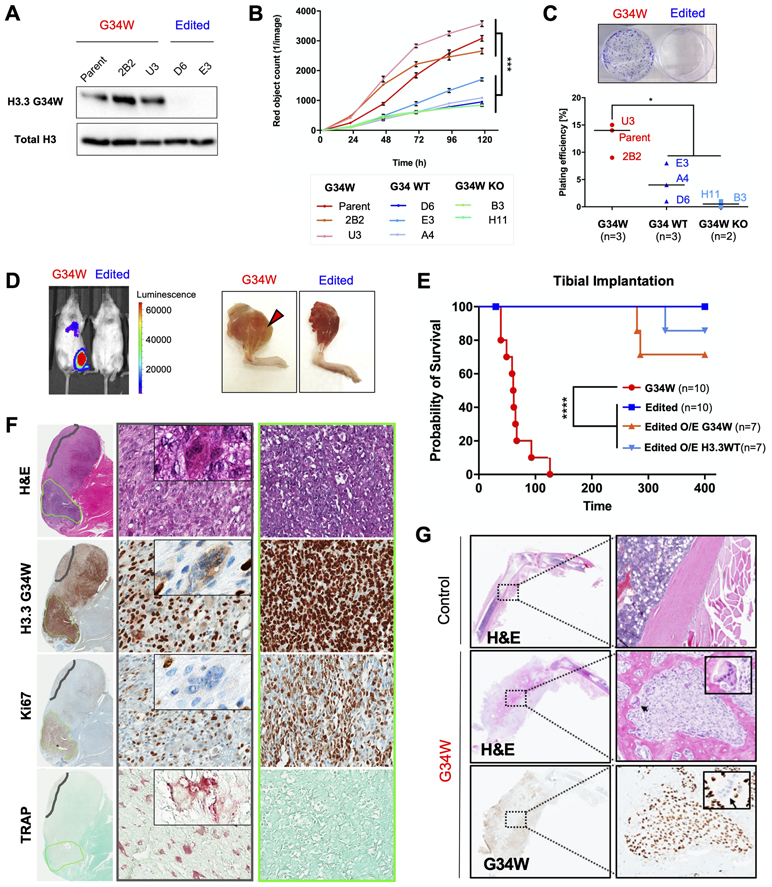
(A) G34W immunoblotting of Im-GCT-4072 G34W (Parent: n=1; Clone: n=2) and edited (Repair to WT: n=2) lines.
(B) G34W lines (Parent: n=1; Clone: n=2) of Im-GCT-4072 proliferate faster than edited lines (Repair to WT: n=3; G34W-KO: n=2), as measured using the IncuCyte live-cell analysis system for 5 consecutive days. Data are presented as mean red object count ± SD from five technical replicates per line. Statistical significance assessed using Student’s t-test based on averaged observations from biological replicates (independent CRISPR clones, labeled).
(C) Representative images of G34W (left) and edited (right) cell colonies in culture plates (top panel). G34W lines (Parent: n=1; Clone: n=2) of Im-GCT-4072 exhibit increased colony formation relative to edited lines (Repair to WT: n=3; G34W KO: n=2), as measured by manual counting of colonies stained with crystal violet after 3 weeks (bottom panel). Two technical replicates were counted per line, and data are presented as an average of biological replicates (independent CRISPR clones, labeled). Statistical significance assessed using Student’s t-test.
(D) Left: Representative bioluminescence Xenogen IVIS 200 imaging of mice implanted in the tibia with Im-GCT-4072 G34W and edited luciferase-tagged lines.
Right: Representative images of mice tibias implanted with G34W and edited cells after skin removal at the time of sacrifice.
(E) Kaplan-Meier survival curve for orthotopic tibial implantation of Im-GCT-4072 G34W (Parent: n=1, Clone: n=2; n=10 mice), edited lines (Repair to WT: n=2, n=10 mice), edited O/E G34W (n=7 mice), edited O/E H3.3WT (n=7 mice) in NRG mice illustrates the dependence of tumour formation on the presence of G34W mutation.
(F) H&E, and H3.3G34W, TRAP (tartrate-resistant acid phosphatase) and Ki67 IHC for a representative tibial xenograft tumour derived from implantation of Im-GCT-4072 G34W parental cells. 20X (70μm) magnified area illustrates a histological compartment (middle panel) with differentiated stromal cells and abundant TRAP+ osteoclasts (an example of giant multinucleated osteoclast is featured in the inset). Right panel illustrates a histological compartment with undifferentiated stromal cells, high Ki67 staining, and absence of TRAP+ osteoclasts.
(G) Representative H&E and G34W IHC of decalcified legs derived from tibial implantation of Im-GCT-6176 G34W parental cells, illustrating the osteolytic effect of G34W stromal cells relative to a control contralateral leg. Inset features reactive G34W-negative osteoclasts observed at the interface between G34W-positive neoplastic stromal cells and normal bone.
To evaluate whether re-introduction of H3.3G34W in edited clones could rescue tumour formation, we overexpressed (O/E) H3.3G34W and H3.3WT constructs in two Im-GCT-4072 G34W-edited clones (Fig. S1I). Edited lines O/E G34W formed tumours, albeit with reduced penetrance and increased latency relative to parental G34W lines (Fig. 1E, Fig. S1J). Histology was similar to parental and unedited lines (Fig. 1F-G, Fig. S1K), reinforcing the oncogenic role of the G34W mutation in GCT.
Our xenograft model was aggressive, prone to disseminate locally and distally (Fig. S1L-M). Orthotopic tibial tumours exhibited osteolytic properties seen in GCTs (Fig. 1F-G). Notably, murine osteoblast and osteoclast cells were recruited to G34W stromal cells adjacent to bone tissue, characteristic of a reactive bone-remodeling process (Fig. 1G). Eponymous multinucleated osteoclasts were evident within the neoplastic stromal cell mass extruding outside bone and were most abundant in regions of xenograft tumours containing differentiated stromal cells (Fig. 1F). Taken together, these results show that G34W is required for tumour formation and promotes recruitment of multinucleated osteoclasts in orthotopic xenograft tumours.
G34W affects the expression of genes involved in muscle function.
We next characterized gene expression changes associated with G34W using RNA-seq (Table S1) and H3K27ac ChIP-seq, a mark associated with transcription and active regulatory elements (22). Removal of G34W led to a global effect on the transcriptome, as shown by the segregation of samples by G34 status in all three isogenic lines using principal component analysis (PCA) (Fig. 2A, Fig. S2A). Despite their different genetic backgrounds, with a large expected individual variability, we identified common dysregulated pathways across the three GCT cell lines. Pathway analysis of upregulated genes in G34W revealed extracellular matrix (ECM) organization genes (e.g. COL6A1/3, EMILIN2, SOX9), a function associated with cartilage and bone-forming chondrocytes and osteoblasts, enriched in two isogenic lines (Fig. 2B-C, Fig. S2B-C, Table S2, Table S3). This transcriptional upregulation of ECM organization genes was associated with H3K27ac deposition at the promoters of these genes, indicative of epigenetic activation (Fig. S2D-E). Notably, H3K27ac profiles displayed distinct genome-wide patterns between G34W and edited lines (Fig. 2D, Fig. S2F). On the other hand, pathways related to actin filament-based processes and muscle contraction were strongly depleted in two out of three G34W cell lines (e.g. ACTA2, CNN1, LMOD1, TAGLN) (Fig. 2B-C, Fig. S2B-C, Table S2, Table S3). Intersection of commonly dysregulated genes across the three isogenic cell lines revealed 27 genes, all of which were downregulated in G34W-mutant cells. These included genes associated with muscle contraction (ADORA1-adenosine A1 receptor, MYL1-myosin light chain 1, PKP2-plakophilin 2, TNNT2-troponin T2) (Fig. 2E-F, S2G-H), suggesting that G34W in neoplastic stromal cells is potentially affecting muscle differentiation, a known lineage for mesenchymal progenitor cells of bone marrow origin (23). To further validate these findings, we obtained expression data from a previously reported dataset of primary G34W cell lines (n=11 G34W vs n=6 WT) (20), which confirmed downregulation in G34W of pathways related to muscle contraction (Table S3). Altogether these results indicate that G34W downregulates genes involved in muscle functions, possibly impairing further differentiation in a mesenchymal progenitor committed to myogenesis.
Figure 2. G34W promotes ECM remodeling and impairs muscle contraction pathways.
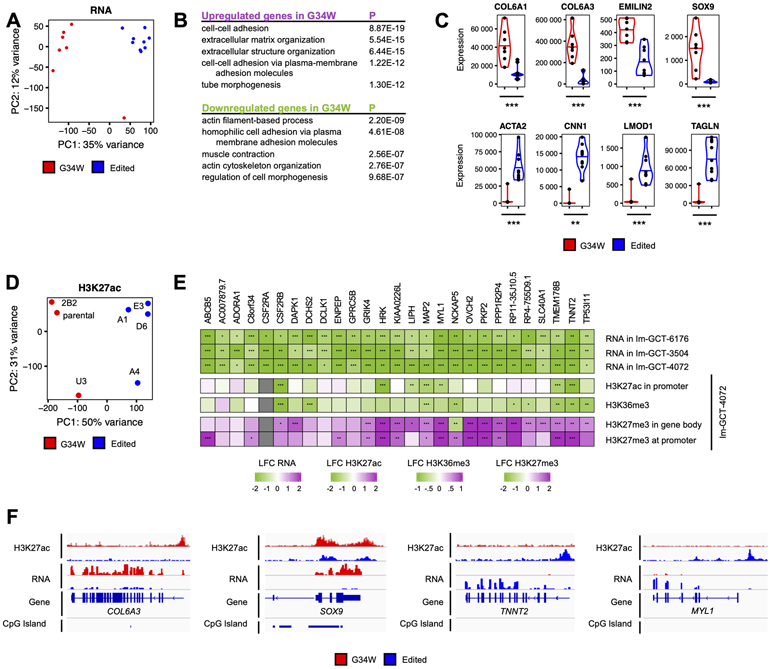
(A) PCA reveals distinct transcriptomic profiles between Im-GCT-4072 G34W (red; n=7) and edited lines (blue; n=9). Read counts were counted over Ensembl genes, normalized using the median-of-ratios procedure and transformed using the variance-stabilizing transformation. The effect of the genotype is captured in PC1 (35% of the variance).
(B) Pathway enrichment analysis of statistically significantly up- (purple) and downregulated (green) genes in G34W compared to edited lines from Im-GCT-4072. Pathway enrichment analysis was performed using g:Profiler. Top 5 statistically significantly enriched terms (GO:BP, term size<1000, P<0.05) are shown. The complete table can be found in Table S3.
(C) Violin plots depicting expression levels of extracellular matrix genes COL6A1, COL6A3, EMILIN2 and SOX9 as well as muscle contraction ACTA2, CNN1, LMOD1 and TAGLN in G34W (red) and edited (blue) lines from Im-GCT-4072. *: P<0.05, **: P<0.01, ***: P<0.001, n.s.: non-significant. Gene expression levels reported in median-of-ratios normalized read counts. Significance was assessed using DESeq2. The complete table can be found in Table S2.
(D) PCA reveals distinct H3K27ac profiles between G34W (red; n=3) and edited lines (blue; n=4) from Im-GCT-4072. Read counts were counted over 10kb genomic bins and normalized to RPKM. The effect of the genotype is captured in PC1 (50% of the variance). The labels correspond to clones. Refer to Table S1 for more details on clones.
(E) Heatmap illustrating transcriptional and epigenetic changes (H3K27ac, H3K36me3, H3K27me3) at consistently transcriptionally deregulated genes across the three isogenic cell models. Differential gene expression and histone mark enrichment are reported in log2 fold-change (LFC) in G34W lines over edited lines of the respective isogenic cell models. Significance was assessed using DESeq2. *: P<0.05, **: P<0.01, ***: P<0.001. The complete table can be found in Table S2.
(F) Genomic tracks highlighting changes in H3K27ac and gene expression at COL6A3, SOX9, TNNT2 and MYL1 loci between G34W (red) and edited (blue) lines from Im-GCT-4072. Signals overlaid by replicates, reported in RPKM, and group auto-scaled by genomic assay.
G34W leads to H3K27me3 redistribution from intergenic to genic regions in GCT.
The G34W phenotypes we observed in driving tumour formation and altering the transcriptome are likely to originate at the epigenome. Thus, we first characterized the in cis effects of G34W on histone marks using histone mass spectrometry (hMS) on four primary G34W GCT cell lines (Table S1), comparing patterns between mutant and WT peptides within each line. Mutant G34W contributed to only 2.6% of the total H3 pool despite accounting for 25.7% of total H3.3 (Fig. 3A). As expected (6,15), we observed significant H3.3K36me3 decrease and concomitant H3.3K27me3 gain on the mutant peptide (Fig. 3B, Fig. S3A-B), reflecting the in cis effects of G34W on the minority of H3.3 mutant histones within the overall H3 pool. Next, we compared isogenic G34W and edited lines and observed a modest, but significant global decrease in H3K27me3 levels on both H3.3 and canonical H3.1/2 in G34W lines (Fig. S3C), while no change was observed in overall H3K36me3 levels (Fig. S3D). This suggests that, even though H3.3G34W accounts for less than 3% of the total H3 pool, this mutant histone exerts an effect in trans, beyond mutant H3.3, that can be detected for the repressive H3K27me3 mark.
Figure 3. G34W is deposited into euchromatin and is associated with redistribution of H3.3 in GCT.
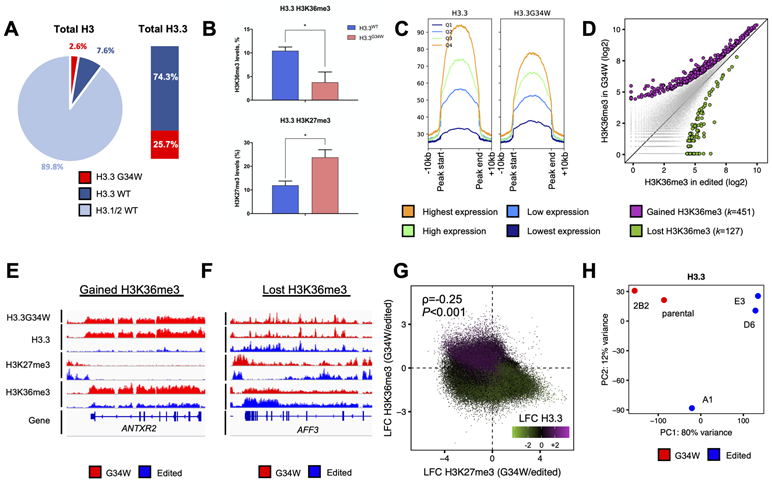
(A) Left: Pie chart illustrating relative abundance of G34W, and wild-type H3.3 and H3.1/2 histones by histone mass spectrometry in the Im-GCT-4072 parental line.
Right: stacked barplot illustrating relative abundance of G34W and wildtype H3.3.
(B) Histone mass spectrometry reveals in cis changes in H3.3K36me3 (top) and H3.3K27me3 (bottom) on G34W (pink) compared to WT (light blue) H3.3 peptides in G34W-mutant GCT cell lines (n=4). *: P<0.05; **: P<0.01, ***: P<0.001, n.s.: non-significant. Significance was assessed using Student’s t-test.
(C) H3.3 (left) and G34W (right) abundances in the ±10kb window around H3.3 peaks. H3.3 peaks are stratified into four quartiles of H3.3 abundance in the parental line Im-GCT-4072.
(D) Scatterplot illustrating changes in H3K36me3 at H3.3 peaks between G34W (y-axis; n=2) and edited (x-axis; n=3) lines from Im-GCT-4072. Statistically significant gains and losses in G34W lines are highlighted in purple and green, respectively. Significance was assessed using DiffBind and read counts reported in log2 scale.
(E-F) Representative tracks at the ANTRX2 locus highlighting gain of H3K36me3 (E) and at the AFF3 locus highlighting loss of H3K36me3 (F) in wild-type- and mutant H3.3-enriched regions in G34W (red; n=2) and edited (blue; n=3) lines from Im-GCT-4072. Signals overlaid by replicates, reported in RPKM, and group auto-scaled by genomic assay.
(G) Scatterplot illustrating changes in H3K36me3 (y-axis), H3K27me3 (x-axis) and H3.3 (color) genome-wide between G34W (n=2) and edited lines (n=3) from Im-GCT-4072. Loci that lose H3K36me3 and gain H3K27me3 concurrently lose H3.3. Reads counted over 10kb bins, averaged per condition, normalized to RPKM and changes were reported as the log2 fold change (LFC) between G34W and edited lines. ρ is the Spearman rank correlation coefficient and significance was calculated from Spearman's ρ statistic.
(H) PCA illustrating genome-wide deposition patterns of H3.3 between G34W (red; n=2) and edited lines (blue; n=3) from Im-GCT-4072. PCA was performed on H3.3 abundance at consensus H3.3 peaks. The effect of the genotype is captured in PC1 (80% of the variance). The labels correspond to clones. Refer to Table S1 for more details on clones.
To further assess the effects of G34W genome wide, we profiled H3.3, G34W, H3K36me3 and H3K27me3 deposition by ChIP-seq in isogenic G34W and edited lines from Im-GCT-4072 (Table S1). G34W histones tracked with H3.3 in the parental line at H3.3 peaks and genome-wide (Fig. 3C, Fig. S3E). Consistent with the expected distribution of H3.3 in euchromatin (14), G34W histones tracked with highly expressed genes with the largest H3.3 abundance at their transcriptional start and end sites (Fig. S3F-G). Next, to identify G34W-enriched loci displaying in cis H3.3K36me3 loss, we compared total H3K36me3 deposited at H3.3 peaks between isogenic lines, as there are no available antibodies specific for H3.3K36me3. Based on the known effects of this mutation on H3.3K36me3, our expectation was to mainly observe areas with decreased H3K36me3 deposition in G34W compared to edited cells. Interestingly, we observed both gains and losses of H3K36me3 at H3.3 peaks when comparing the isogenic pairs (Fig. 3D-F). In G34W-mutant cells, areas which gained H3.3 deposition had concurrent H3K36me3 gain (Fig. 3E, 3G). Notably, areas that gained H3K36me3 also had H3K27me3 loss (Fig. 3E, 3G). Conversely, genomic regions that lost H3.3 deposition had concurrent H3K36me3 loss and H3K27me3 gain (Fig. 3F-G). Together, these results suggest a genome-wide H3.3 redistribution upon G34W removal (Fig. 3H, Fig. S3H), implicating an H3.3G34W-dependent chromatin remodeling process in GCT beyond initial sites of H3.3 deposition. This H3.3 redistribution may result from the spread of H3K27me3 mark upon G34W-induced loss of the antagonistic H3.3K36me3 mark. Spread of H3K27me3 may in turn induce gene silencing and secondary eviction of H3.3 to other genomic areas.
Our investigation of H3K27me3 deposition in the isogenic pairs further identified global changes associated to G34W (Fig. S4A-D). This mark was redistributed genome wide: in G34W-mutant cells, H3K27me3 was lost predominantly in intergenic regions (P<0.001; χ2 test), whereas it was equally gained across genomic compartments, including promoter and intragenic regions (Fig. 4A, Fig. S4E). In addition, G34W re-expression in edited lines also resulted in a modest shift of H3K27me3 deposition from intergenic to promoter and genic regions (Fig. S4F). Redistribution of H3K27me3 was further confirmed by SUZ12 distribution, a core component of the PRC2 complex that catalyzes this repressive mark, as it mirrored H3K27me3 deposition in G34W-mutant cells (Fig. 4B, Fig. S4G). Notably, genes with a chromatin state consistent with an in cis G34W effect (H3.3K36me3 loss/H3K27me3 gain) were enriched in pathways related to actin-myosin contractile functions (Fig. S4H-I, Tables S2, S3), while genes associated with actin filament-based processes showed similar chromatin patterns (Fig. 4C-D, Fig. S4J). These results are in keeping with our transcriptomic data, where downregulated genes between isogenic pairs are mainly involved in muscle function (Fig. 2B, 2E, Fig. S2B, S2G, Table S3).
Figure 4. H3K27me3 is redistributed from intergenic to genic regions during the chromatin remodeling process in G34W GCT cells.
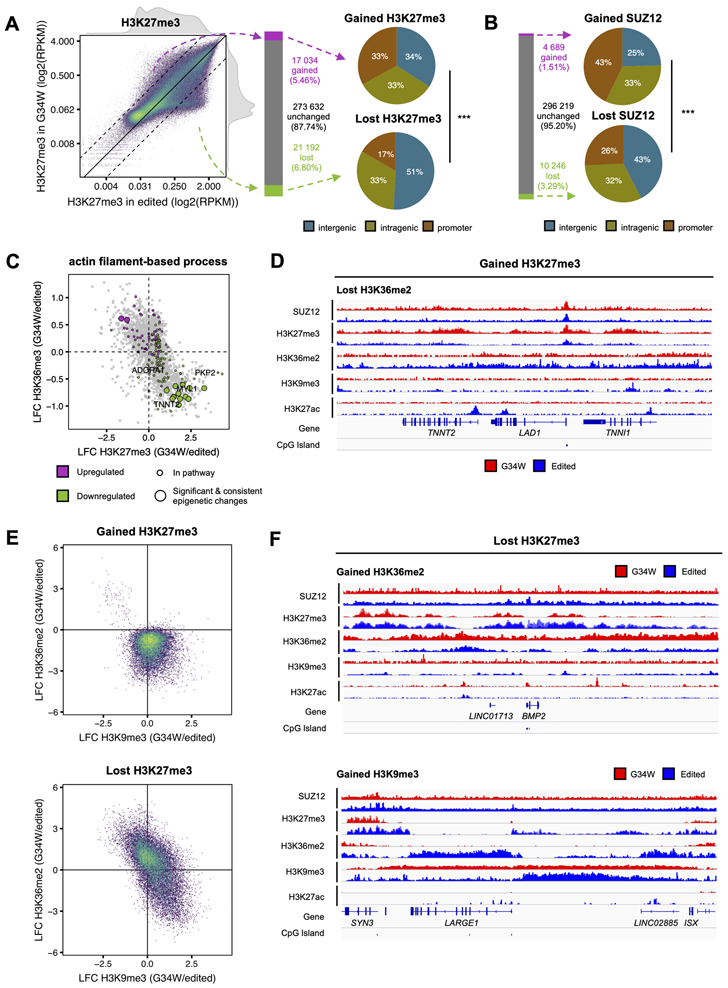
(A) Left: Scatterplot depicting changes in H3K27me3 in G34W (y-axis; n=2) and edited (x-axis; n=3) lines from Im-GCT-4072. Color: point density. Solid line: no change in abundance. Dotted lines: two-fold change in abundance. Reads counted over 10kb bins, averaged per condition, normalized to RPKM and reported in log2 scale.
Middle: Bar plot quantifying the number of 10kb bins with gained (purple), unchanged (grey) and lost (green) H3K27me3 in G34W relative to edited lines. Bins with above-median average H3K27me3 in either condition and with an absolute log2 fold-change (LFC) of H3K27me3 exceeding 1 (2-fold change) were called as gains and losses.
Right: Pie charts illustrating proportion of 10kb bins gaining or losing H3K27me3 that overlap promoters, gene bodies and intergenic regions. ***: P<0.001. Significance was assessed using χ2 test.
(B) Left: Bar plot quantifying the number of 10kb bins with gained (purple), unchanged (grey) and lost (green) SUZ12 in G34W relative to edited lines. Bins with above-median average H3K27me3 in either condition and with an absolute log2 fold-change (LFC) of H3K27me3 exceeding 0.58 (1.5-fold change) were called as gains and losses.
Right: Pie charts illustrating proportion of 10kb bins gaining or losing SUZ12 that overlap promoters, gene bodies and intergenic regions.
(C) Scatterplot illustrating epigenetic changes at significantly deregulated genes in actin filament-based process pathway in G34W lines compared to edited lines from Im-GCT-4072. X-axis: log2 fold-change (LFC) of H3K27me3. Y-axis: LFC of H3K36me3. Grey: significant differentially expressed genes; purple/green: up- and downregulated genes in pathway; big circle: significant changes in genic H3K36me3 and H3K27me3. Replicates: H3K27me3 (G34W n=2; edited n=3), H3K36me3 (G34W n=2; edited n=3), RNA (G34W n=7; edited n=9).
(D) Representative track at the TNNT2, LAD1, and TNNI1 locus illustrating changes in SUZ12, H3K27me3, H3K36me2, H3K9me3, and H3K27ac between G34W (red) and edited lines (blue) from Im-GCT-4072. Signals overlaid by replicates, reported in RPKM, and group auto-scaled by genomic assay.
(E) Top: Scatterplot illustrating that gain of H3K27me3 in G34W lines is associated with a loss of H3K36me2. Color: point density. X-axis: log2 fold-change (LFC) of H3K9me3 between G34W (n=2) and edited (n=2) lines from Im-GCT-4072. Y-axis: LFC of H3K36me2 between G34W (n=2) and edited (n=3) lines.
Bottom: Scatterplot illustrating that loss of H3K27me3 is associated with a gain of either H3K36me2 or H3K9me3.
(F) Representative tracks at the BMP2 (top) and ISX/LARGE1 (bottom) loci illustrating changes in SUZ12, H3K27me3, H3K36me2, H3K9me3, and H3K27ac between G34W (red) and edited lines (blue) from Im-GCT-4072. Signals overlaid by replicates, reported in RPKM, and group auto-scaled by genomic assay.
To assess whether PRC2 redistribution (gain and loss of H3K27me3) in G34W lines is accompanied by secondary chromatin remodelling, we profiled broad intergenic marks with known mutually exclusive deposition patterns relative to H3K27me3, namely H3K36me2 (9) and H3K9me3 (24). An interesting interplay of these marks in intergenic regions was apparent: loci that gained H3K27me3 in G34W lines consistently exhibited loss of H3K36me2 (Fig. 4D-E), while those that lost H3K27me3 gained either H3K9me3 or H3K36me2 (Fig. 4E-F). The TNNI1/TNNT2 locus exemplifies H3K27me3 gain at the expense of H3K36me2 in G34W lines, specifically repressing the TNNI1 and TNNT2 genes encoding subunits of the troponin complex which regulates myofibril contractility (Fig. 4D). In contrast, the locus comprising the negative regulator of muscle differentiation, BMP2, displays H3K36me2 gain at the expense of H3K27me3 (Fig. 4F). The LARGE1 locus, which encodes for a glycosyltransferase important for muscle function (25), shows spread of a heterochromatin H3K9me3 domain into territories occupied by H3K27me3 and H3K36me2 in edited lines, concurrent with repression in G34W (Fig. 4F). Similarly, the spread of H3K9me3 into the MYH cluster likely results in the repression of sarcomeric myosin proteins encoded in the locus (Fig. S4K).
Collectively, these results indicate that deposition of G34W parallels that of wildtype H3.3, and that G34W in cis effects may promote PRC2/H3K27me3 silencing of H3K36me3-depleted nucleosomal substrates, leading to subsequent H3.3 eviction, redistribution, and gene silencing. Recruitment of PRC2 to genic regions may occur at the expense of weaker intergenic substrates where loss of H3K27me3 is replaced by the spread of neighboring antagonistic marks, such as H3K36me2 and H3K9me3. These epigenetic changes are coupled to transcriptional changes affecting mesenchymal cell state, with muscle contractile genes likely constituting early targets of G34W in mesenchymal progenitors at a differentiation stage, yet to be determined, permissive to the generation of neoplastic stromal cells in GCT.
GCT stromal cells resemble specific osteoprogenitors and comprise an ACTA2+ population with features of contractile cells.
GCT stromal cells bearing the G34W mutation are ill-defined and have been alternately described as possessing properties of mesenchymal stem cells, fibroblasts, or pre-osteoblasts based on in vitro differentiation assays and histological studies (26). To better define all cellular components and stromal cell heterogeneity, we performed single-cell RNA-sequencing (scRNA-seq) on four G34W GCT (Table S1). Clustering analysis revealed 17 transcriptionally distinct clusters (Fig. S5A), which we labeled as lymphoid, myeloid, endothelial, and putative stromal cells (Fig. S5B) based on similarity to cell types within the Human Primary Cell Atlas (HPCA) (Fig. S5C) and known markers (Fig. S5D, Table S4). In contrast to other clusters which contained cells from all tumours, putative stromal cell clusters segregated by patient (Fig. S5B), indicating increased variation within this compartment likely reflecting divergent clonal evolution. Putative stromal cell clusters were significantly enriched for the G34W mutation (Fig. S5E) and showed similar pathway enrichment as isogenic G34W stromal cells (e.g. ECM organization) (Fig. S5F, Tables S3, S4). Importantly, stromal clusters across patient tumours displayed the highest enrichment score for the transcriptomic G34W signature derived from our independent isogenic model (Fig. 5A). We therefore conclude that these clusters represent the neoplastic stromal compartment, and that the transcriptional changes induced by G34W in our cellular models are recapitulated in primary tumours.
Figure 5. GCT stromal cells resemble specific osteoprogenitors and a distinct ACTA2+ subset have features of contractile cells.
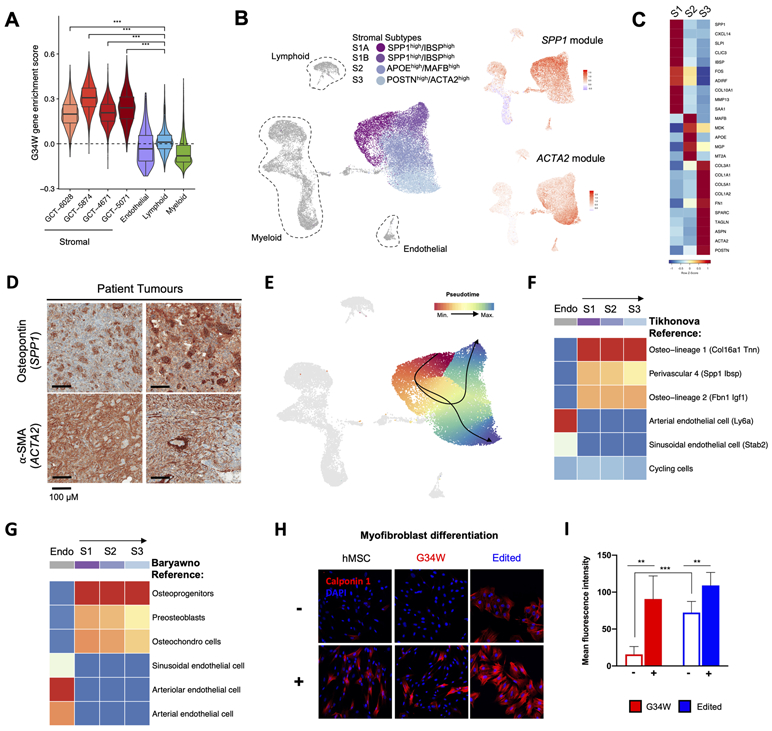
(A) Boxplot displaying higher G34W enrichment scores for single cells in stromal clusters from each patient. The G34W enrichment score is derived from the average expression of differentially expressed genes (LFC>2) between isogenic G34W and edited Im-GCT-4072 lines. ***: P<0.0005, significance was assessed using a Wilcoxon rank sum test.
(B) Left: UMAP plot of Harmony integrated cells from scRNA-seq on n=4 primary GCTs, revealing the 4 stromal subtypes S1A, S1B, S2, and S3.
Right: Average expression of genes highly correlated with S1-specific SPP1 gene (SPP1 module), or with S3-specific ACTA2 gene (ACTA2 module), shown on UMAP plot of Harmony integrated cell clusters. Refer to Table S1 for details.
(C) Row-scaled heatmap showing average expression of differentially expressed genes that characterize each stromal subtype.
(D) Representative IHC for osteopontin (SPP1) and alpha-SMA (ACTA2) in two GCT patient tumours.
(E) Lineage inference by Slingshot showing neoplastic trajectories from S1A to S1B, and S1A to S3. Cells are coloured by pseudotime, with red cells occurring earlier than blue cells in the trajectory.
(F) SingleR classification of each stromal cell subtype (S1, S2, S3) and endothelial control based on Tikhonova et al. reference cell types (28). Stromal cell subtypes most strongly resemble the Osteo-lineage 1 reference cell cluster (labelled as O1 (Col16a1 Tnn) in Tikhonova et al.).
(G) SingleR classification of each stromal cell subtype (S1, S2, S3) and endothelial control based on Baryawno et al. reference cell types (27). Stromal cell subtypes most strongly resemble the osteoprogenitors reference cell cluster (labelled as OLC-2 subtype 8_3 in Baryawno et al.).
(H) Representative immunofluorescence images for the myofibroblast muscle marker calponin 1 in hMSCs and isogenic Im-GCT-4072 cells maintained in non-induced (−) or myofibroblast differentiation media (+) for 2 weeks.
(I) Bar-plot quantifying the mean fluorescence intensity of calponin 1 staining in G34W (n=2 lines; three different fields each) and edited lines (n=2; four different fields each) maintained in non-induced (−) or myofibroblast differentiation media (+).
As expected, the mononuclear stromal component was detected in all samples. This prompted us to use Harmony data integration to reduce the impact of technical and biological inter-tumoural stromal cell heterogeneity, and to query potential neoplastic stromal subpopulations shared by all samples. Clustering analysis identified four clusters within the stromal compartment (S1A, S1B, S2, and S3) (Fig. 5B, S5G-I), with osteopontin (SPP1) and smooth muscle actin (ACTA2) as the most discriminative markers between stromal clusters. Based on differential gene expression analysis and expression modules of genes correlated with SPP1 or ACTA2 (Fig. 5B, Fig. S5H, Table S4), clusters S1A and S1B likely represent a continuum of the same population, leading us to consider three populations: S1 (S1A, S1B), S2 and S3. S1 cells expressed genes associated with osteoblast and chondrocyte functions (e.g. SPP1, IBSP-integrin binding sialoprotein, MMP13-matrix metallopeptidase 13), whereas S3 cells expressed markers of myofibroblasts (e.g. ACTA2-alpha smooth muscle actin, TAGLN-transgelin, POSTN-periostin) and S2, with fewer differentially expressed genes, was considered an intermediate between both states (Fig. 5C, Fig. S5I). We confirmed expression of both osteopontin (SPP1) and alpha smooth muscle actin (ACTA2/α-SMA) in patient tumours (Fig. 5D). In isogenic lines, G34W-mediated effects converge on epigenetic/transcriptional downregulation of muscle contractile genes and upregulation of ECM pathways. The S3 population is defined by similar pathways suggesting that cells within this subgroup are myofibroblast-like and committed to myogenesis, but are restricted by G34W from further differentiation. Indeed, lineage inference analysis identified two possible trajectories across the four populations: from S1A to S1B cells, or from S1A to S3 cells (Fig. 5E, Fig. S5J). This suggests a continuum of progenitor stromal states, with G34W-mutant cells either transitioning between early S1A to S1B states or following a trajectory from an early S1A towards a possible myofibroblast-like progenitor S3 state.
To identify the cell of origin of the neoplastic stromal cells, we next mapped the transcriptional profiles of S1-S3 populations to two recently published scRNA-seq murine bone-marrow stroma datasets (27,28) (Fig. 5F-G). The control endothelial cell cluster mapped, as expected, to reference EC-arterial/arteriolar (27) and V1 arterial (28) cells. Strikingly, S1, S2, and S3 populations strongly mapped to the osteoprogenitor subset in the Baryawno dataset (27). In the Tikhonova dataset (28), they all mapped to the osteo-lineage O1 subset of COL2.3+ osteoblasts, which likely also represents an osteoprogenitor cell type since we show that O1 maps robustly to the osteoprogenitor subset and vice versa (Fig. S5K). S1 and S2 populations mapped more strongly than S3 cells to the less-differentiated perivascular 4 (P4 SPP1/IBSP+) or “preosteoblasts" reference subsets (27,28) (Fig. 5E-F), consistent with their earlier position in the neoplastic trajectory. These findings indicate that GCT neoplastic stromal cells resemble osteoprogenitors or a closely related cell type, with some cells (S3) progressing to display contractile features (Fig. S5L). We further verified that the three stromal subpopulations are also present in four PDOX tumours using single-nuclei (sn) RNA-seq (Fig. S6A). Using the annotated clusters from patient tumours (Fig. 5B), we classified cells from PDOX tumours and found that they were formed predominantly by S3 cells (Fig. S6B-C). Consistent with this result, tibial xenograft tumours displayed high expression of alpha smooth muscle actin (α-SMA/ACTA2), a marker of S3 cells, but low osteopontin expression (marker of S1 cells) (Fig. S6D). Enrichment of the S3 subtype in the xenograft models may explain why they exhibit a more malignant phenotype and pathology compared to relatively benign GCT.
To validate the myofibroblast differentiation potential of GCT stromal cells, we induced differentiation using transforming growth factor beta 1 (TGF-β1), L-ascorbic acid, and platelet derived growth factor (PDGF-AB) in isogenic GCT lines and control human mesenchymal stromal cells (hMSCs). Upon induction, GCT cells expressed high levels of myofibroblast markers including stress fiber-associated protein calponin 1 (CNN1) (Fig. 5H-I) and α-SMA (Fig. S6E). Notably, while both G34W and edited cells were able to differentiate into myofibroblasts, edited cells already exhibited higher expression of calponin 1 at baseline relative to G34W lines (Fig. 5H-I). This is consistent with transcriptomic data showing enrichment of actin-myosin contractile genes following G34W editing (Fig. 2B-C, Fig. S2B-C). Together, these findings reinforce that neoplastic stromal cells transition from progenitor S1 to S3 states, where epigenetic reprogramming by G34W appears to impede differentiation into a terminal myofibroblast-like state.
G34W S3 stromal cells secrete factors promoting ECM remodeling and association with myeloid cells.
In GCT, the formation of giant multinucleated histone WT osteoclasts from monocytic precursors (29) is an essential contributor to the pathogenesis of these tumours and is thought to be mediated by G34W-dependent secretion of factors such as RANKL (TNFSF11) (30). In contrast to previous findings (20), neither RANKL nor its decoy receptor OPG (TNFRSF11B) were differentially expressed between G34W and edited clones (Fig. 6A, S7A), suggesting that RANK/RANKL signalling, while required for osteoclastogenesis, is independent of G34W. We thus investigated G34W-mediated aberrant TME interactions that contribute to the giant cell phenotype. Using additional markers to build upon HPCA-based classification, we first elucidated cell types present in the GCT myeloid compartment (Fig. 6B, S5C-D, S7B, Table S4). We show that the putative osteoclast cluster is enriched for osteoclast differentiation and bone resorption pathways (Fig. S7C). To investigate stromal-cell ligands that promote osteoclastogenesis, we first determined the cell type giving rise to osteoclasts in GCT, using a combination of trajectory inference methods (Fig. 6C, Fig. S7D-G). We identified a bifurcation event where monocytes can either differentiate into macrophages or give rise to osteoclasts through a highly cycling, pre-osteoclast intermediate. These findings confirm the proposed monocytic origin for osteoclasts in GCT (30) and describe a previously unappreciated fate for monocytes towards macrophage differentiation in GCT.
Figure 6. G34W GCT stromal cells secrete factors that promote ECM remodeling and association with myeloid cells.
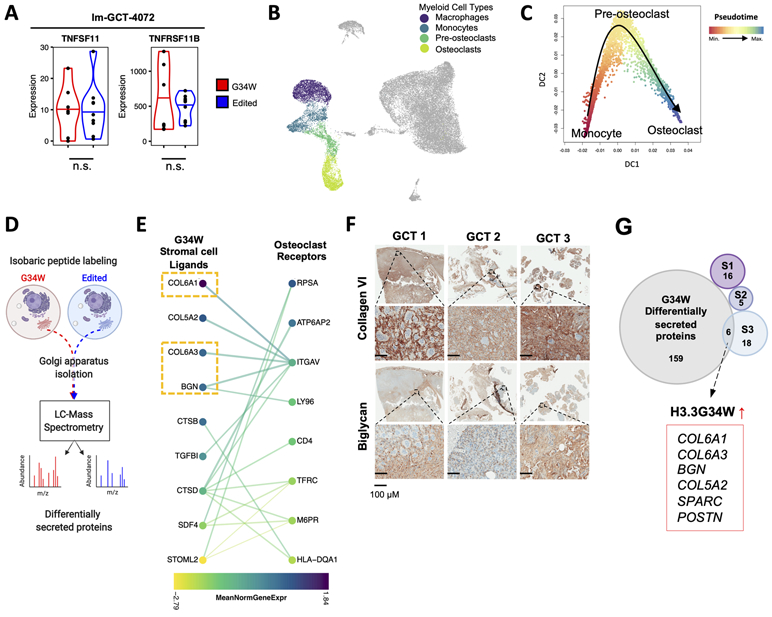
(A) Violin plots depicting expression levels of TNFSF11 (RANKL) and TNFRSF11B (OPG) in G34W (red) and edited (blue) lines from Im-GCT-4072. *: P<0.05, ***: P<0.001, n.s.: non-significant. Gene expression levels reported in median-of-ratios normalized read counts Significance assessed using DESeq2.
(B) UMAP plot highlighting the myeloid compartment of GCT.
(C) Diffusion map showing a trajectory from monocytes (red) to terminally-differentiated osteoclasts (blue) through a pre-osteoclast intermediate along Slingshot-inferred pseudotime.
(D) Schematic of Golgi apparatus isolation and mass spectrometry workflow to identify differentially secreted proteins between isogenic G34W (red) and edited (blue) cells.
(E) CCInx-predicted (31) ligand-receptor interactions between GCT stromal cells (left) and osteoclast cells (right). Colors represent the mean normalized gene expression in each cell type. Only interactions between proteins differentially secreted in G34W cell lines by MS (P<0.05) and expressed by stromal cells are shown on the left and only genes differentially expressed (P<0.05) in osteoclasts (vs. non-myeloid cells) are shown on the right. P values were adjusted for multiple testing using FDR.
(F) Representative IHC for collagen type VI and biglycan in three patient GCTs.
(G) Venn diagram showing overlap of genes with significantly enriched expression in each stromal cell subtype, S1-S3 (Seurat Wilcox test, P<0.05, FDR corrected) and genes with significantly increased protein secretion in G34W cell lines by MS (P<0.05, FDR corrected). The 6 intersecting genes are highlighted.
To identify ligands secreted by G34W stromal cells, we profiled the secreted proteome by isolating Golgi apparatus of isogenic GCT lines and performing mass spectrometry (MS) (Fig. 6D, Table S1), and identified significantly differentially secreted proteins between G34W and edited lines (Fig. S7H, Table S5). Ligand-receptor gene interactions were inferred between stromal cells and the myeloid cell compartment based on curated protein interaction databases (31) (Fig. 6E, Fig. S7I, Table S6). Filtering these predicted ligand-receptor interactions for proteins differentially secreted by G34W lines, we observed stromal cell-specific expression of ECM ligands such as collagens (COL6A1/3, COL5A2) and proteoglycans (BGN) that are predicted to interact with specific integrin receptors on monocyte/macrophage (ITGB2) and osteoclast (ITGAV) cells (Fig. 6E, Fig. S7I). Expression of COL6A1/3 and BGN was further confirmed in PDOX tumours by snRNA-seq (Fig. S7J), and by immunohistochemistry in primary GCT samples as well as PDOX tumours (Fig. 6F, Fig. S7K). Interestingly, when intersecting the Golgi secretome and differentially expressed genes in S1 to S3 populations, the S3 cells were enriched for expression of G34W-specific secreted proteins (Fig. 6G, Fig. S7L), potentially accounting for the over-representation of this subtype in our aggressive PDOX model. Indeed, 6 of the 24 genes specifically expressed by S3 cells are found in the ECM secretome, suggesting that S3 stromal cells are responsible for G34W-driven ECM remodeling, in line with our finding of increased osteoclasts in proximity to the differentiated stromal compartment of a xenograft tumour (Fig. 1F). Taken together, our results indicate that G34W stromal cells resemble osteoblast progenitors stalled in differentiation at a myofibroblast-like progenitor population (S3). These S3 cells secrete ECM remodeling proteins promoting bone destruction by acting locally on myeloid cells in the TME. Therefore, G34W drives both components of GCT pathogenesis: it sustains the neoplastic transformation of the mononuclear stromal cells, as shown by our in vivo orthotopic model, and simultaneously enables the recruitment and formation of pathological giant osteoclasts that largely contribute to the morbidity of this tumour.
DISCUSSION
We comprehensively characterized the epigenetic, transcriptomic (including at the single-cell level) and Golgi proteomics profiles of G34W GCT using primary tumours and isogenic models of tumour-derived cell lines. G34W has been associated with tumourigenesis in previous studies using xenotransplantation of the full tumour (32,33) or transient siRNA knockdown of G34W in GCT-derived cell lines (34). We show that G34W removal is sufficient to prevent tumour formation in vivo, indicating that this mutation is necessary for GCT tumourigenesis. Although G34W occurs on a residue that does not undergo PTM, we show that tumourigenesis is likely the consequence of a global epigenetic remodeling process initiated by the in cis effects of this mutation in the stromal cell-of-origin (Fig. 7A). Indeed, we posit that the earliest event following H3.3K36me3 loss on G34W histones is the deposition of H3K27me3 in active genic regions normally enriched for H3.3 as well as G34W (representing 25% of total H3.3). H3K36me3 loss in genic regions may create improved substrates for the PRC2 complex and result in redistribution of H3K27me3 from lower affinity intergenic regions to promoter and genic regions, in turn promoting gene silencing and eviction of H3.3. Secondary and more widespread changes as a result of H3.3 redistribution may then induce transcriptional changes in the absence of direct epigenetic remodelling.
Figure 7. Schematic illustrating G34W-mediated epigenetic remodeling and ECM remodeling by subpopulations of GCT stromal cells.
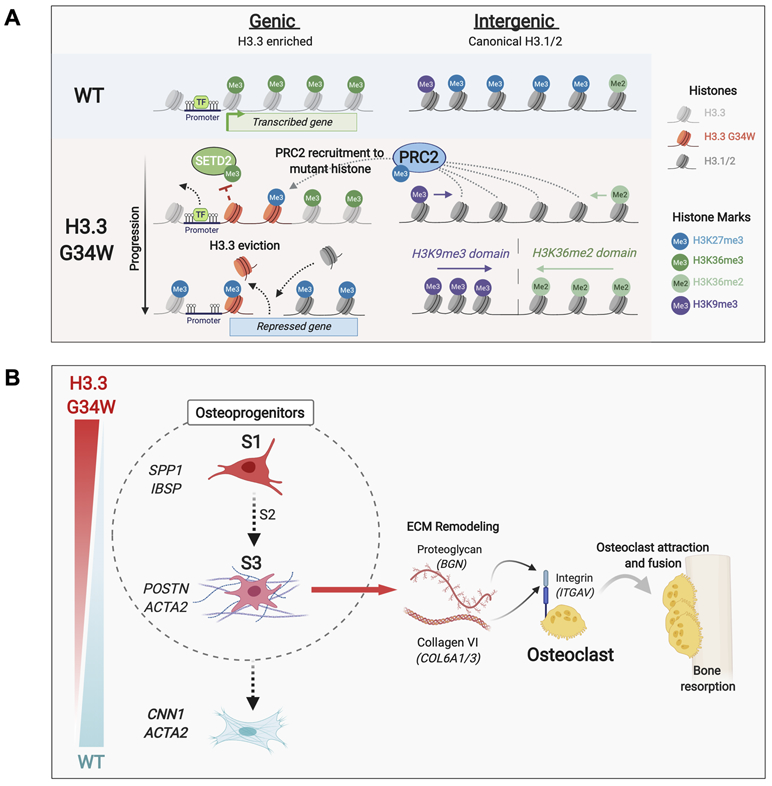
(A) Schematic illustrating G34W-mediated epigenetic remodeling of neoplastic GCT cells.
(B) Schematic illustrating G34W-dependent differentiation trajectory in stromal cells and interactions with osteoclasts in the bone TME.
In intergenic regions, H3K27me3 loss is replaced by H3K36me2 or H3K9me3 in areas vacated by PRC2 (Fig. 7A). These compensatory mechanisms to H3K27me3 loss are likely to have distinct effects. While H3K36me2 enables repressive DNA methylation to be deposited through DNMT3A recruitment (35), H3K36me2-marked regions remain permissive to active regulatory states marked by H3K27ac (e.g. BMP2) (36). In contrast, the constitutive heterochromatin H3K9me3 mark is strongly repressive (37) and its imposition into facultative heterochromatin at a few key loci (e.g. LARGE1, MYH) in G34W-mutant cells may result in undue silencing of genes associated with contractile functions.
The net effect of G34W-mediated epigenetic remodeling affects mesenchymal cell lineage commitment. Indeed, our transcriptomic and epigenetic data converge on a stalled osteoblast-like progenitor population, capable mainly of differentiation along a neoplastic trajectory towards a myofibroblastic cell state. Moreover, at single-cell resolution, G34W GCT stromal cells comprise distinct cell populations related by a neoplastic lineage trajectory with SPP1+ (S1) tumour cells at the origin leading to ACTA2+ myofibroblast progenitor (S3) tumour cells (Fig. 7B). A terminally differentiated myofibroblast cell state is blocked by G34W, as suggested by lower expression of myofibroblast markers such as calponin 1 in G34W-mutant cells. This block can be overcome in vitro using strong differentiation factors (e.g. TGF-β1), an observation which may have implications for GCT treatment, especially in recurring tumours or those where complete surgical resection is not feasible.
Notably, our data suggest that G34W induces the secretion of ECM-remodeling factors in the neoplastic S3 myofibroblast progenitor cells, including collagens and proteoglycans which interact directly with integrin receptors on myeloid cells (Fig. 7B). The presence of neoplastic myofibroblasts has been reported in primary and metastatic GCT using immunohistochemical and ultrastructural studies (38,39). Moreover, scRNA-seq studies of several diseases where stroma plays a major role in pathogenesis have identified ACTA2+ stromal cells expressing ECM ligands (COL6A3, BGN) consistent with the S3 population (40-42). In GCT tumourigenesis, the initiating factor is the oncogenic G34W mutation which leads to a persistent activated progenitor state incapable of further transitioning toward a terminally differentiated myofibroblast state. The presence of myeloid cells in the bone microenvironment niche enables the recruitment and syncytia of osteoclasts through a G34W-mediated ECM remodeling process enriched in S3 ACTA2+ cells. Notably, secreted collagen VI (COL6A1/3) maintains mechanical stiffness within the ECM, a function implicated in the activation of the mechanosensitive Ca2+ channel TRPV4, which is expressed on the plasma membrane of large osteoclasts and regulates terminal osteoclast differentiation (43,44). We previously showed that H3 wild type giant cell lesions of the jaw (GCLJ), a disease that is histologically and radiologically similar to GCT, carry TRPV4 gain-of-function mutations (45), suggesting possible convergence of effects between G34W-mutant GCT and TRPV4-mutant GCLJ on osteoclastogenesis and pathological features.
In conclusion, we show that G34W is necessary to drive the two major pathological features of GCT: the destruction of bone and the maintenance of proliferating osteoprogenitors. G34W in a neoplastic stromal ACTA2+ population mediates the secretion of factors that recruit osteoclasts within the bone TME, resulting in destruction of bone. Few transcriptional changes may be initially needed to maintain this aberrant progenitor state, and these are likely due to G34W’s in cis effects, initiating the global epigenetic remodeling events we observed. This chromatin remodeling, whether a direct effect or indirectly mediated by the mutation, reflects a neoplastic maintenance of a progenitor state, which promotes GCT formation and helps maintain the neoplastic progenitor state. As these changes are potentially reversible, future therapies targeting the epigenome may be of benefit in GCTs.
MATERIALS AND METHODS
Additional Materials and Methods can be found in Supplemental Information.
DATA AND SOFTWARE AVAILABILITY
Raw and processed sequencing data for bulk RNA-seq, scRNA-seq, snRNA-seq, and bulk ChIP-seq data was deposited into the Gene Expression Omnibus (GEO) under accession code GSE149211.
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources/reagents should be directed to, and will be fulfilled by, the Lead Contact Nada Jabado (nada.jabado@mcgill.ca).
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Protocols for this study involving collection of patient samples were approved by the Research Ethics and Review Board of McGill University and affiliated Hospitals Research Institutes. Written informed consent was obtained from all research participants.
Establishing primary cell lines from GCT
Fresh tumour specimens were collected in DMEM medium, washed twice in PBS, minced physically with surgical blades, and dissociated in Collagenase-Dispase (Sigma-Aldrich) with incubation at 37°C in 5% CO2 and agitation for 30 min. The resulting cell pellet was treated with NH4Cl to remove red blood cells, washed twice in DMEM medium (with 4.5 g/L glucose, L-glutamine, phenol red, by Wisent) containing 10% FBS and penicillin/streptomycin, and plated in T-75 flasks in the same medium, and passaged at 75% confluency. DNA from various passages was assessed for H3F3A G34W mutation allele frequency by droplet digital PCR (details available on request). We thank Dr. David Allis for generously sharing the primary GCT-2611 cell line. Three lines were immortalized using hTERT retrovirus (see Supplemental Information for details). All lines were tested monthly for mycoplasma contamination (MycoAlert Mycoplasma Detection kit by Lonza), and STR fingerprinting was regularly performed.
CRISPR/Cas9 gene editing
CRISPR/Cas9 editing (46) was performed using pSpCas9(BB)-2A-Puro (PX459 V2.0) plasmid, a gift from Feng Zhang (Addgene #48139, RRID: Addgene_62988). A single-guide RNA (sgRNA, IDT) targeting H3F3A G34W was cloned into the plasmid, and a single-stranded donor oligonucleotide (ssODN, IDT) template with H3F3A WT sequence (sequences in Supplemental Information) were transfected using lipofectamine 3000 (Thermo Fisher Scientific). Transfected cells were selected using puromycin for 72h and then sorted into 96-well plates. Single-cell clones were expanded and screened for editing events (insertions, deletions, or repair to wild type) by Sanger sequencing, and confirmed through targeted deep sequencing (Illumina MiSeq) and immunoblotting using a H3.3G34W-specific antibody (1:500 dilution, RevMab Biosciences 31-1145-00, RRID:AB_2716434, see Supplemental Information). We refer to H3F3A+/− and H3F3A+/+ clones as “edited”, and not “corrected”, because H3F3A+/− clones are hemizygous and the initial presence of G34W may have caused irreversible transcriptional/epigenetic changes.
Proliferation assay
Cells were seeded as 1,500 cells/well into 96-well plates (five technical replicates per line), and nuclei were stained with NucLight Rapid Red Cell Reagent (1:500 dilution, Essen Bioscience). Cells were imaged using IncuCyte ZOOM System real-time instrumentation (Essen Bioscience) every 2h (10X magnification), and images analyzed after 120h with IncuCyte ZOOMTM 2015A software.
Colony formation assay
Cells were seeded as 250 cells/well in 6-well plates. After 3 weeks, colonies were fixed with 100% methanol for 20 min, and stained with 0.5% crystal violet for 30 min. Colonies (containing >50 cells) were manually counted under an inverted microscope.
Myogenic Differentiation and Immunofluorescence
Cells were seeded as 6000 cells/chamber of 8-well chamber slides in standard medium. At ~70% confluency, differentiation was induced with media containing 5ng/mL TGF-β1 (R&D systems 240-B-002/CF), 30μM L-Ascorbic acid 2-phosphate (Sigma A8960), 5ng/mL PDGF-AB (Peprotech 100-00AB) (47). Fresh differentiation media was added twice weekly for 3 weeks. Immunofluorescence staining was performed using primary antibodies anti-α-smooth muscle actin (1:200, Abcam ab5694, RRID:AB_2223021) and anti-calponin 1 (1:200, Abcam ab46794, RRID:AB_2291941) (see Supplemental Information for details).
Histone post-translational modification quantification with nLC/MS
The complete workflow for histone extraction, LC/MS, and data analysis was described in detail (48,49) (see Supplemental Information for brief description).
Secreted proteome - Golgi Apparatus purification and Mass Spectrometry
Cells were lysed using a 7mL Dounce stainless tissue grinder, and a 2M sucrose solution was added to homogenates to obtain final 1.7M sucrose concentration. Golgi membranes were isolated by isopycnic centrifugation using discontinuous sucrose gradients (50), and processed for MS analyses as described (51). See Supplemental Information for full description of workflow.
ANIMAL MODELS
All mice were housed, bred, and subjected to listed procedures according to the McGill University Health Center Animal Care Committee and in compliance with Canadian Council on Animal Care guidelines. Mice were monitored weekly and euthanized when xenograft tumour volume reached 2,000 mm3 or immediately at clinical endpoint when recommended by veterinary/biological services staff.
Tagging with GFP-Luciferase
pSMAL-GFP-Luc lentiviral vector was a gift from Dr. Kolja Eppert (McGill University). 96h after transduction with virus, GFP-positive cells were sorted using fluorescence-activated cell sorting.
Mouse subcutaneous implantation
1.2-1.5 × 106 cells were prepared as a single-cell suspension in 200 μL PBS-50% Matrigel and implanted subcutaneously into the left flank of 8- to 12-week-old immunodeficient NSG mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ, RRID:IMSR_JAX:005557). Tumour formation was monitored weekly using bioluminescent imaging and palpation starting 4 weeks post-implantation.
Mouse orthotopic intratibial implantation
70% ethanol was used to clean the leg of 8- to 12-week-old immunodeficient NRG mice (NOD.Cg-Rag1tm1Mom Il2rgtm1Wjl/SzJ, RRID:IMSR_JAX:007799). A pre-drilled hole was created in the tibial plateau using rotating movements with a 25G needle. 3 μl of PBS-20% Matrigel containing 1.8-2 × 105 cells were implanted into the tibial diaphysis with a Hamilton syringe (27G needle). Tumour formation was monitored weekly using bioluminescent imaging and tumour volume measured with digital calipers.
Bioluminescence imaging
Bioluminescence imaging was performed as previously described (52) using IVIS 100 system (Caliper). Anesthetized mice were placed in imager 7 min. post intraperitoneal injection of D-luciferin (150 mg/kg).
Immunohistochemistry (IHC)
Xenograft tumours were fixed in 10% buffered formalin for 48h. Bone samples were decalcified using 10% EDTA for 3-4 weeks (solution changed weekly), followed by embedding in paraffin wax and sectioning at a thickness of 5μm. Automated IHC was performed with Ventana Discovery Ultra. Slides were deparaffinized and rehydrated. Antigen retrieval was done using EDTA buffer, and slides were incubated with primary antibodies: H3.3 G34W (1:100, RevMAb Biosciences 31-1145-00, RRID:AB_2716434), Ki67 (1:300, Abcam ab15580, RRID:AB_443209), alpha smooth muscle actin (1:200, Abcam ab5694, RRID:AB_2223021), osteopontin (1:500, Abcam ab8448, RRID:AB_306566), Col VI (1:50, Abcam ab6588, RRID:AB_305585), Biglycan (1:500, Abcam ab49701, RRID:AB_1523212). After washing, secondary antibody (anti-Mouse or Rabbit/Mse HRP) was added and DAB kit chromogen used to detect signal.
Tartrate-resistant acid phosphatase (TRAP) staining
Slides were deparaffinized and rehydrated through graded ethanols to distilled water. Slides were placed in pre-warmed TRAP Staining Solution Mix (4% pararosaniline, 4% sodium Nitrite, 0.1 Acetate buffer pH 11, Naphthol AS-TR Phosphate, Sodium Tartare, pH 5.0), and incubated at 37°C until control was developed, then rinsed in distilled water. Slides were counterstained with 0.1% Fast Green for 1 min, air-dried and mounted. Osteoclasts stain red-violet whereas background stains green using TRAP staining.
NEXT-GENERATION SEQUENCING
Chromatin Immunoprecipitation Library Preparation and Sequencing
ChIP-seq was performed as previously described (8) (see Supplemental Information for details). ChIP reactions for histone modifications were performed on Diagenode SX-8G IP-Star Compact by incubating 2 million cells of sonicated cell lysate with following antibodies: anti-H3K27me3 (1:40, Cell Signaling Technology 9733, RRID:AB_2616029), anti-H3K27ac (1:80, Diagenode C15410196, RRID:AB_2637079), anti-H3K36me3 (1:100, Active Motif 61021, RRID:AB_2614986), anti-H3.3 (1:66, Millipore 09–838, RRID:AB_10845793), anti-H3.3G34W (1:66, RevMAb Biosciences 31-1145-00, RRID:AB_2716434), anti-H3K36me2 (1:50, Cell Signaling Technology 2901), (anti-H3K9me3 (1:66, Abcam ab8898, RRID:AB_306848). SUZ12 ChIPs were performed manually using anti-SUZ12 (1:150, Cell Signaling Technology 3737, RRID:AB_2196850) antibody.
Bulk, single-cell and single-nuclei RNA-seq Library Preparation and Sequencing
RNA-seq was performed as previously described (53) (see Supplemental Information for details).
QUANTIFICATION AND STATISTICAL ANALYSIS
Description of statistical details for each experiment can be found in figure legends. Statistical significance was always adjusted for multiple testing and considered to be attained when P<0.05.
Analysis of ChIP-seq data
The ChIP-seq pipeline from C3G’s GenPipes toolset (RRID: SCR_016376) (54) (v3.1.0) was used. See the Supplemental Information for a complete treatment of sample selection, data processing, data visualization, H3.3 peak enrichment analysis, comparison of H3.3 and H3.3G34W deposition patterns.
Genome-wide chromatin mark enrichment analysis
The abundance of each mark was quantified by counting the number of reads in genomic 10kb bins and normalized to RPKM with the “multiBamSummary bins” functionality of deeptools (RRID:SCR_016366) (55) (v3.1.3). Differential enrichment of each mark at each bin was calculated as the log2 fold-change (LFC) between average abundance in each condition. Thresholds of LFC ±1 (corresponding to a 2-fold-change) and ±0.58 (1.5-fold change) were used to determine large changes in H3K27me3 and SUZ12 in Im-GCT-4072 lines, respectively. For Im-GCT-3504 lines, we used a threshold of LFC ±0.58. For edited lines derived from Im-GCT-4072 overexpressing H3.3G34W or H3.3WT, we used a threshold of LFC ±0.32 (1.25-fold change). To avoid overestimating LFCs, we ignored bins with low counts in both conditions (i.e. below-median average abundance in either condition).
Bins were classified as promoter, intragenic or intergenic regions based on overlap with promoter (i.e. ±1.25kb of a transcription start site), intergenic (i.e. covered in the reference), and intergenic regions (i.e. complement of promoter and intergenic regions) using the UCSC table browser’s (RRID: SCR_00578) “whole” annotation for Ensembl’s ensGene reference (GRCh37; n=60234 genes). The significance of differences in proportion of bins overlapping these regions that also gain or lose a chromatin mark was determined using a chi-square test.
Chromatin mark enrichment analysis at genes
Enrichment of chromatin marks at promoter and gene bodies was obtained by counting reads over promoter and intragenic regions, respectively. Similar to differential gene expression, differential enrichment analysis was computed using DESeq2 (RRID: SCR_015687) (56) (v1.18.1), using unit-normalized library depth as size factors (i.e. total number of uniquely mapped reads). Genes with (1) sufficient coverage (baseMean>25), (2) LFCs exceeding a threshold of ±1 (2-fold) for H3K27me3 and H3K27ac and ±0.58 (1.5-fold) for H3K36me3, and (3) P<0.05, were considered as significantly differentially enriched. For Im-GCT-3504 lines, we used a threshold of ±0.58 (1.5-fold) for changes in H3K27me3. To ensure more accurate estimates of LFCs in the low information setting (i.e. low counts and high dispersion), we enabled shrinkage of LFCs towards zero (56). P values were adjusted for multiple testing using Benjamini-Hochberg.
Analysis of bulk RNA-seq data
Bulk RNA-seq was analyzed as previously described (13). Please refer to Supplemental Information for a complete treatment.
Analysis of single-cell RNA-seq data
Please refer to Supplemental Information for a complete treatment of single-cell RNA-seq data analysis.
Quality control and normalization
Low-quality cells were excluded based on outlier mitochondrial content (indicative of cellular stress or damage) and number of genes expressed using the R package Seurat (RRID: SCR_016341) (57,58) (v3.0.0). The upper threshold on mitochondrial content varied from >15-20% of total genes detected. Since GCT samples contain multinucleated osteoclasts, where a high number of UMI counts is a biological feature of this cell type, we applied a more lenient upper threshold of 3 times the inter-quartile range. Filtered cells were then normalized together using SCTransform (v0.2.0) (59) (with parameter variable.features.n=3000), and mitochondrial content percent was regressed. To reduce the impact of ambient multi-species mRNAs in xenografts, we applied the SoupX (v1.4.5) (60) algorithm.
Identification of cell types
To robustly identify cell types associated with each cell cluster, we first derived markers for each cluster through differential expression (DE) analysis using a Wilcoxon Rank Sum test configured to only test genes expressed in >25% of cells and with log-fold change ≥0.25. We then combined the three following approaches: (1) comparing cluster markers to known cell type markers from the literature (30,61), (2) classifying cell types from reference datasets based on correlation of gene expression profiles, and (3) pathway enrichment analysis of cluster-specific markers. We used SingleR package (v1.0.5) (62) with default parameters to assign cell types to each cluster based on their Spearman correlation to gene expression profiles of known cell types in the Human Primary Cell Atlas (HPCA) reference. Since the stromal and osteoclast cell types were not present in the reference, we performed pathway enrichment analysis on cluster-specific gene markers using G:Profiler (RRID: SCR_006809) with term size set to 1000 and deriving pathways from GO:BP only (63). See Supplemental Information for additional details on stromal and osteoclast cell identification.
Identification of heterogeneity within stromal cells
To examine heterogeneity within stromal cell populations, stromal cells were re-clustered (i.e. in the absence of non-stromal cells), and highly variable genes recalculated. PCA revealed that PC1 was defined by SPP1-correlated genes, and PC2 by ACTA2-correlated genes. To determine if these genes, which represent major sources of variation within the data, defined specific subsets of cells, correlation analysis was performed using the correlateGenes function from R package Scran (RRID: SCR_016944) (v1.12.1) (64). This revealed distinct gene sets differentially enriched in stromal cells, notably “SPP1 module” and “ACTA2 module”.
Technical (i.e. sequencing batch) and biological variance (i.e. divergent tumour evolution and genetic backgrounds) can obscure similarities between neoplastic cells from different tumours. To validate that the gene modules identify distinct stromal cell subsets or states, we used the Harmony data integration method (v0.99.6) (65) (parameters max.iter.harmony=15, theta=2) to correct these sources of variation on the PCA embeddings. Although unsupervised clustering identified distinct S1A and S1B clusters, we collectively defined these stromal subtypes as S1 since they displayed similar enrichment for the “SPP1 module” and modest differential gene expression (Fig S5H). We confirmed that overcorrection did not occur, as the same non-stromal populations were recovered before and after Harmony integration. Both expression of the gene modules derived before correction and cluster-specific markers obtained by differential expression analysis after correction were in agreement.
To confirm the presence of the stromal cell subtypes S1-S3 in xenograft tumours, we created a SingleR reference from Harmony-integrated patient GCT cells using the “aggregateReference” function (with power=0). We then classified cell clusters from xenograft tumours using these references, as described earlier.
Pseudotemporal ordering and trajectory analysis
To determine cell trajectory within each lineage, we employed two complementary trajectory inference methods, Slingshot (RRID: SCR_017012) (v1.3.1) (66) and velocyto.R (RRID: SCR_018167) (https://github.com/velocyto-team/velocyto.R, v0.6) (67). To calibrate parameters for trajectory inference in stromal cells, we first analyzed a well-defined lineage, myeloid cells. To identify lineages, we applied unsupervised Slingshot analysis on a three-dimensional (3D) diffusion map embedding. A 3D diffusion map was constructed using the DiffusionMap function from the R package destiny (v2.14.0) (68), with only high-quality cells included. High quality cells were defined here as cells having < 5% mitochondrial gene content and genes with >10 counts in at least 100 cells. We then used Slingshot (with default parameters and without specifying a start cluster) to identify lineages in the diffusion map embedding (Fig. S7E-F). 3D visualizations were created using R package plotly (v4.9.1) (69). Since Slingshot cannot confidently establish the start of the lineage, we employed velocyto. Briefly, spliced and unspliced reads were counted using the Python implementation of velocyto (v0.17.13). RNA velocity of each gene passing the expression threshold (exonic and intronic reads with a minimum maximum-cluster average of 0.1 and 0.05, respectively, were retained) was estimated using the R package velocyto.R (v0.6) using a gene-relative model with parameters kCells=20 and fit.quantile=0.05 and projected onto UMAP embeddings. Consistent with Slingshot results and literature, velocyto predicted a bifurcated lineage with a mix of monocytes and pre-osteoclasts as a root. Slingshot and velocyto were then applied, using the same parameters, reconstruct trajectories in stromal cells.
Determining stromal cell identity
To better determine a putative cell-of-origin for stromal cells, we classified them using SingleR with custom references derived from two murine bone marrow scRNA-seq datasets Baryawno et al. 2019 (27) and Tikhonova et al. 2019 (28), which better cover the diversity of cells found in in the bone milieu compared to HPCA. Mouse genes were humanized to their human orthologues using Ensembl Biomart (v2.42.0) (70,71). Genes without a human match and irrelevant genes (i.e. ribosomal and housekeeping-related) were removed to decrease the risk of spurious correlation. The humanized expression matrices were then converted into a SingleR reference using the function aggregateReference (with parameter power=0). To ensure that humanization did not remove important genes, we confirmed that there were no errors when classifying clusters from the mouse datasets using their corresponding humanized references. To detect overlapping cell types across the two references, we classified cells from one reference using the other. The fact that neoplastic stromal cells match a cell type that was uniquely matched between the two references further supports the putative identity of the cell of origin (Fig. S5K). Each reference was then used to classify the stromal cells from each of the stromal cell subtype populations.
Cell-to-cell interaction analysis between stromal cells and myeloid cells
Cell-to-cell interaction analysis in the GCT TME was performed as described (31) and described in Supplemental Information.
Analysis of Golgi secretome mass spectrometry data
Data processing, protein enrichment analysis, and identification of secreted factors by stromal cells is described in Supplemental Information.
Supplementary Material
STATEMENT OF SIGNFICANCE.
This study shows that H3.3G34W drives GCT tumourigenesis through aberrant epigenetic remodeling, altering differentiation trajectories in mesenchymal progenitors. H3.3G34W promotes in neoplastic stromal cells an osteoblast-like progenitor state that enables undue interactions with the tumour microenvironment, driving GCT pathogenesis. These epigenetic changes may be amenable to therapeutic targeting in GCT.
Acknowledgments
Financial Support:
This work was supported by funding from: A Large-Scale Applied Research Project grant from Genome Quebec, Genome Canada, the Government of Canada, and the Ministère de l'Économie, de la Science et de l’Innovation du Québec, with the support of the Ontario Institute for Cancer Research through funding provided by the Government of Ontario to NJ, MDT, CLK (13513). Fondation Charles Bruneau to NJ, US National Institutes of Health (NIH grant P01-CA196539 to NJ, PWL, and BAG; R01CA148699 and R01CA159859 to MDT); the Canadian Institutes for Health Research (CIHR grant MOP-286756 and FDN-154307 to NJ and PJT-156086 to CLK); the Canadian Cancer Society (CCSRI grant 705182) and the Fonds de Recherche du Québec en Santé (FRQS) salary award to CLK; NSERC (RGPIN-2016-04911) to CLK; CFI Leaders Opportunity Fund 33902 to CLK, Genome Canada Science Technology Innovation Centre, Compute Canada Resource Allocation Project (WST-164-AB); Data analyses were enabled by compute and storage resources provided by Compute Canada and Calcul Québec. NJ is a member of the Penny Cole Laboratory and the recipient of a Chercheur Boursier, Chaire de Recherche Award from the FRQS. This work was performed within the context of the International CHildhood Astrocytoma INtegrated Genomic and Epigenomic (ICHANGE) consortium, and the Stand Up to Cancer (SU2C) Canada Cancer Stem Cell Dream Team Research Funding (SU2C-AACR-DT-19-15 to MDT, NJ), provided by the Government of Canada through Genome Canada and the Canadian Institute of Health Research, with supplemental support from the Ontario Institute for Cancer Research, through funding provided by the Government of Ontario. Stand Up To Cancer Canada is a Canadian Registered Charity (Reg. # 80550 6730 RR0001). Research Funding is administered by the American Association for Cancer Research International - Canada, the Scientific Partner of SU2C Canada. MDT is supported by The Paediatric Brain Tumour Foundation, The Terry Fox Research Institute, The Canadian Institutes of Health Research, The Cure Search Foundation, b.r.a.i.n.child, Meagan’s Walk, Genome Canada, Genome BC, Genome Quebec, the Ontario Research Fund, Worldwide Cancer Research, V-Foundation for Cancer Research, Canadian Cancer Society Research Institute Impact grant and the Garron Family Chair in Childhood Cancer Research at the Hospital for Sick Children and the University of Toronto. NDJ is a recipient of a fellowship from FRQS and RMGA. We also acknowledge support from the We Love You Connie Foundation (NJ). SCM is supported by Cancer Prevention Research Institute of Texas (CPRIT) Scholar in Cancer Research award (RR170023), and Alex’s Lemonade Stand Foundation (ALSF) A award. LG is supported by the Nicole et François Angers Sarcoma Research Chair from the MGH Foundation, C17 Ewing Sarcoma Grant and the Terry Fox Research Institute Grant (number 1087).
Footnotes
Conflict of Interest Disclosure Statement: The authors declare no potential conflicts of interest.
References
- 1.Board WC of TE. Soft Tissue and Bone Tumours WHO classification of tumours. 2020; 5th edition, 3. [Google Scholar]
- 2.Behjati S, Tarpey PS, Presneau N, Scheipl S, Pillay N, Van Loo P, et al. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat Genet. 2013;45:1479–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fontebasso AM, Papillon-Cavanagh S, Schwartzentruber J, Nikbakht H, Gerges N, Fiset P-O, et al. Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nat Genet. 2014;46:462–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schwartzentruber J, Korshunov A, Liu X-Y, Jones DTW, Pfaff E, Jacob K, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma Nature. Nature Publishing Group; 2012;482:226–31. [DOI] [PubMed] [Google Scholar]
- 5.Presneau N, Baumhoer D, Behjati S, Pillay N, Tarpey P, Campbell PJ, et al. Diagnostic value of H3F3A mutations in giant cell tumour of bone compared to osteoclast-rich mimics. J Pathol Clin Res. 2015;1:113–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lewis PW, Müller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, et al. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science. 2013;340:857–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boileau M, Shirinian M, Gayden T, Harutyunyan AS, Chen CCL, Mikael LG, et al. Mutant H3 histones drive human pre-leukemic hematopoietic stem cell expansion and promote leukemic aggressiveness. Nat Commun. 2019;10:2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harutyunyan AS, Krug B, Chen H, Papillon-Cavanagh S, Zeinieh M, De Jay N, et al. H3K27M induces defective chromatin spread of PRC2-mediated repressive H3K27me2/me3 and is essential for glioma tumorigenesis Nat Commun. Nature Publishing Group; 2019;10:1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu C, Jain SU, Hoelper D, Bechet D, Molden RC, Ran L, et al. Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science. 2016;352:844–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Papillon-Cavanagh S, Lu C, Gayden T, Mikael LG, Bechet D, Karamboulas C, et al. Impaired H3K36 methylation defines a subset of head and neck squamous cell carcinomas. Nat Genet. 2017;49:180–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mohammad F, Weissmann S, Leblanc B, Pandey DP, Højfeldt JW, Comet I, et al. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat Med. 2017;23:483–92. [DOI] [PubMed] [Google Scholar]
- 12.Piunti A, Hashizume R, Morgan MA, Bartom ET, Horbinski CM, Marshall SA, et al. Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas. Nat Med. 2017;23:493–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krug B, De Jay N, Harutyunyan AS, Deshmukh S, Marchione DM, Guilhamon P, et al. Pervasive H3K27 Acetylation Leads to ERV Expression and a Therapeutic Vulnerability in H3K27M Gliomas. Cancer Cell. 2019;35:782–797.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goldberg AD, Banaszynski LA, Noh K-M, Lewis PW, Elsaesser SJ, Stadler S, et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell. 2010;140:678–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shi L, Shi J, Shi X, Li W, Wen H. Histone H3.3 G34 Mutations Alter Histone H3K36 and H3K27 Methylation In Cis. J Mol Biol. 2018;430:1562–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Y, Shan C-M, Wang J, Bao K, Tong L, Jia S. Molecular basis for the role of oncogenic histone mutations in modulating H3K36 methylation. Sci Rep. 2017;7:43906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fontebasso AM, Schwartzentruber J, Khuong-Quang D-A, Liu X-Y, Sturm D, Korshunov A, et al. Mutations in SETD2 and genes affecting histone H3K36 methylation target hemispheric high-grade gliomas. Acta Neuropathol (Berl). 2013;125:659–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheng Z, Cheung P, Kuo AJ, Yukl ET, Wilmot CM, Gozani O, et al. A molecular threading mechanism underlies Jumonji lysine demethylase KDM2A regulation of methylated H3K36. Genes Dev. 2014;28:1758–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Voon HPJ, Udugama M, Lin W, Hii L, Law RHP, Steer DL, et al. Inhibition of a K9/K36 demethylase by an H3.3 point mutation found in paediatric glioblastoma. Nat Commun. 2018;9:3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lim J, Park JH, Baude A, Yoo Y, Lee YK, Schmidt CR, et al. The histone variant H3.3 G34W substitution in giant cell tumor of the bone link chromatin and RNA processing. Sci Rep. 2017;7:13459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lüke J, von Baer A, Schreiber J, Lübbehüsen C, Breining T, Mellert K, et al. H3F3A mutation in giant cell tumour of the bone is detected by immunohistochemistry using a monoclonal antibody against the G34W mutated site of the histone H3.3 variant. Histopathology. 2017;71:125–33. [DOI] [PubMed] [Google Scholar]
- 22.Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A. 2010;107:21931–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Galli D, Vitale M, Vaccarezza M. Bone Marrow-Derived Mesenchymal Cell Differentiation toward Myogenic Lineages: Facts and Perspectives. BioMed Res Int. 2014;2014:7625–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peters AHFM, Kubicek S, Mechtler K, O’Sullivan RJ, Derijck AAHA, Perez-Burgos L, et al. Partitioning and plasticity of repressive histone methylation states in mammalian chromatin. Mol Cell. 2003;12:1577–89. [DOI] [PubMed] [Google Scholar]
- 25.Goddeeris MM, Wu B, Venzke D, Yoshida-Moriguchi T, Saito F, Matsumura K, et al. LARGE glycans on dystroglycan function as a tunable matrix scaffold to prevent dystrophy. Nature. 2013;503:136–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wülling M, Delling G, Kaiser E. The origin of the neoplastic stromal cell in giant cell tumor of bone. Hum Pathol. 2003;34:983–93. [DOI] [PubMed] [Google Scholar]
- 27.Baryawno N, Przybylski D, Kowalczyk MS, Kfoury Y, Severe N, Gustafsson K, et al. A Cellular Taxonomy of the Bone Marrow Stroma in Homeostasis and Leukemia. Cell. 2019;177:1915–1932.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tikhonova AN, Dolgalev I, Hu H, Sivaraj KK, Hoxha E, Cuesta-Domínguez Á, et al. The bone marrow microenvironment at single-cell resolution. Nature. 2019;569:222–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Udagawa N, Takahashi N, Akatsu T, Tanaka H, Sasaki T, Nishihara T, et al. Origin of osteoclasts: mature monocytes and macrophages are capable of differentiating into osteoclasts under a suitable microenvironment prepared by bone marrow-derived stromal cells Proc Natl Acad Sci. National Academy of Sciences; 1990;87:7260–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Noh B-J, Park Y-K. Giant cell tumor of bone: updated molecular pathogenesis and tumor biology. Hum Pathol. 2018;81:1–8. [DOI] [PubMed] [Google Scholar]
- 31.Ximerakis M, Lipnick SL, Innes BT, Simmons SK, Adiconis X, Dionne D, et al. Single-cell transcriptomic profiling of the aging mouse brain. Nat Neurosci. 2019;22:1696–708. [DOI] [PubMed] [Google Scholar]
- 32.Balke M, Neumann A, Szuhai K, Agelopoulos K, August C, Gosheger G, et al. A short-term in vivo model for giant cell tumor of bone. BMC Cancer. 2011;11:241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu L, Wu Z, Zhou Z, Yang X, Xiao J. Intratibial injection of patient-derived tumor cells from giant cell tumor of bone elicits osteolytic reaction in nude mouse. Oncol Lett. 2018;16:4649–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fellenberg J, Sähr H, Mancarella D, Plass C, Lindroth AM, Westhauser F, et al. Knock-down of oncohistone H3F3A-G34W counteracts the neoplastic phenotype of giant cell tumor of bone derived stromal cells. Cancer Lett. 2019;448:61–9. [DOI] [PubMed] [Google Scholar]
- 35.Weinberg DN, Papillon-Cavanagh S, Chen H, Yue Y, Chen X, Rajagopalan KN, et al. The histone mark H3K36me2 recruits DNMT3A and shapes the intergenic DNA methylation landscape. Nature. 2019;573:281–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rao B, Shibata Y, Strahl BD, Lieb JD. Dimethylation of histone H3 at lysine 36 demarcates regulatory and nonregulatory chromatin genome-wide. Mol Cell Biol. 2005;25:9447–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Becker JS, Nicetto D, Zaret KS. H3K9me3-Dependent Heterochromatin: Barrier to Cell Fate Changes. Trends Genet TIG. 2016;32:29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garcia RA, Platica CD, Alba Greco M, Steiner GC. Myofibroblastic differentiation of stromal cells in giant cell tumor of bone: an immunohistochemical and ultrastructural study. Ultrastruct Pathol. 2013;37:183–90. [DOI] [PubMed] [Google Scholar]
- 39.Alberghini M, Kliskey K, Krenacs T, Picci P, Kindblom L, Forsyth R, et al. Morphological and immunophenotypic features of primary and metastatic giant cell tumour of bone. Virchows Arch Int J Pathol. 2010;456:97–103. [DOI] [PubMed] [Google Scholar]
- 40.Farbehi N, Patrick R, Dorison A, Xaymardan M, Janbandhu V, Wystub-Lis K, et al. Single-cell expression profiling reveals dynamic flux of cardiac stromal, vascular and immune cells in health and injury. eLife. 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dominguez CX, Müller S, Keerthivasan S, Koeppen H, Hung J, Gierke S, et al. Single-Cell RNA Sequencing Reveals Stromal Evolution into LRRC15+ Myofibroblasts as a Determinant of Patient Response to Cancer Immunotherapy. Cancer Discov. 2020;10:232–53. [DOI] [PubMed] [Google Scholar]
- 42.Davidson S, Efremova M, Riedel A, Mahata B, Pramanik J, Huuhtanen J, et al. Single-Cell RNA Sequencing Reveals a Dynamic Stromal Niche That Supports Tumor Growth. Cell Rep. 2020;31:107628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zelenski NA, Leddy HA, Sanchez-Adams J, Zhang J, Bonaldo P, Liedtke W, et al. Type VI Collagen Regulates Pericellular Matrix Properties, Chondrocyte Swelling, and Mechanotransduction in Mouse Articular Cartilage Arthritis Rheumatol Hoboken NJ: 2015;67:1286–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Masuyama R, Vriens J, Voets T, Karashima Y, Owsianik G, Vennekens R, et al. TRPV4-mediated calcium influx regulates terminal differentiation of osteoclasts. Cell Metab. 2008;8:257–65. [DOI] [PubMed] [Google Scholar]
- 45.Gomes CC, Gayden T, Bajic A, Harraz OF, Pratt J, Nikbakht H, et al. TRPV4 and KRAS and FGFR1 gain-of-function mutations drive giant cell lesions of the jaw. Nat Commun. 2018;9:4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system Nat Protoc. Nature Publishing Group; 2013;8:2281–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brun J, Lutz KA, Neumayer KMH, Klein G, Seeger T, Uynuk-Ool T, et al. Smooth Muscle-Like Cells Generated from Human Mesenchymal Stromal Cells Display Marker Gene Expression and Electrophysiological Competence Comparable to Bladder Smooth Muscle Cells. PloS One. 2015;10:e0145153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sidoli S, Bhanu NV, Karch KR, Wang X, Garcia BA. Complete Workflow for Analysis of Histone Post-translational Modifications Using Bottom-up Mass Spectrometry: From Histone Extraction to Data Analysis. J Vis Exp JoVE. 2016;111:54112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Karch KR, Sidoli S, Garcia BA. Identification and Quantification of Histone PTMs Using High-Resolution Mass Spectrometry Enzym Epigenetics Part B. Academic Press Inc.; 2016;3–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sircar K, Gaboury L, Ouadi L, Mecteau M, Scarlata E, Saad F, et al. Isolation of human prostatic epithelial plasma membranes for proteomics using mirror image tissue banking of radical prostatectomy specimens. Clin Cancer Res Off J Am Assoc Cancer Res. 2006;12:4178–84. [DOI] [PubMed] [Google Scholar]
- 51.Pfammatter S, Bonneil E, Lanoix J, Vincent K, Hardy M-P, Courcelles M, et al. Extending the Comprehensiveness of Immunopeptidome Analyses Using Isobaric Peptide Labeling. Anal Chem. 2020;92:9194–204. [DOI] [PubMed] [Google Scholar]
- 52.Cosette J, Ben Abdelwahed R, Donnou-Triffault S, Sautès-Fridman C, Flaud P, Fisson S. Bioluminescence-Based Tumor Quantification Method for Monitoring Tumor Progression and Treatment Effects in Mouse Lymphoma Models. J Vis Exp JoVE . 2016;113:536–09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jessa S, Blanchet-Cohen A, Krug B, Vladoiu M, Coutelier M, Faury D, et al. Stalled developmental programs at the root of pediatric brain tumors Nat Genet. Nature Publishing Group; 2019;51:1702–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bourgey M, Dali R, Eveleigh R, Chen KC, Letourneau L, Fillon J, et al. GenPipes: an open-source framework for distributed and scalable genomic analyses. GigaScience. 2019;8:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016;44:W160–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Satija R, Farrell JA, Gennert D, Schier AF, Regev A. Spatial reconstruction of single-cell gene expression data. Nat Biotechnol. 2015;33:495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species Nat Biotechnol. Nature Publishing Group; 2018;36:411–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hafemeister C, Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019;20:296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Young MD, Behjati S. SoupX removes ambient RNA contamination from droplet based single cell RNA sequencing data bioRxiv. Cold Spring Harbor Laboratory; 2018;303727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huang L, Teng XY, Cheng YY, Lee KM, Kumta SM. Expression of preosteoblast markers and Cbfa-1 and Osterix gene transcripts in stromal tumour cells of giant cell tumour of bone. Bone. 2004;34:393–401. [DOI] [PubMed] [Google Scholar]
- 62.Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol. 2019;20:163–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Raudvere U, Kolberg L, Kuzmin I, Arak T, Adler P, Peterson H, et al. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update) Nucleic Acids Res. Oxford Academic; 2019;47:W191–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lun ATL, McCarthy DJ, Marioni JC. A step-by-step workflow for low-level analysis of single-cell RNA-seq data with Bioconductor. F1000Research. 2016;5:2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods. 2019;16:1289–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Street K, Risso D, Fletcher RB, Das D, Ngai J, Yosef N, et al. Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genomics. 2018;19:477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.La Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, et al. RNA velocity of single cells Nature. Nature Publishing Group; 2018;560:494–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Angerer P, Haghverdi L, Büttner M, Theis FJ, Marr C, Buettner F. destiny: diffusion maps for large-scale single-cell data in R. Bioinforma Oxf Engl. 2016;32:1241–3. [DOI] [PubMed] [Google Scholar]
- 69.Sievert C Interactive Web-Based Data Visualization with R, plotly, and shiny. 1 edition. Boca Raton, FL: Chapman and Hall/CRC; 2020. [Google Scholar]
- 70.Durinck S, Moreau Y, Kasprzyk A, Davis S, De Moor B, Brazma A, et al. BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinforma Oxf Engl. 2005;21:3439–40. [DOI] [PubMed] [Google Scholar]
- 71.Durinck S, Spellman PT, Birney E, Huber W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt Nat Protoc. Nature Publishing Group; 2009;4:1184–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw and processed sequencing data for bulk RNA-seq, scRNA-seq, snRNA-seq, and bulk ChIP-seq data was deposited into the Gene Expression Omnibus (GEO) under accession code GSE149211.