

Abstract
Purpose:
Pediatric high-grade glioma (pHGG) diagnosis portends poor prognosis and therapeutic monitoring remains difficult. Tumors release cell-free tumor DNA (cf-tDNA) into cerebrospinal fluid (CSF), allowing for potential detection of tumor-associated mutations by CSF sampling. We hypothesized that direct, electronic analysis of cf-tDNA with a handheld platform (Oxford Nanopore MinION) could quantify patient-specific CSF cf-tDNA variant allele fraction (VAF) with improved speed and limit of detection compared to established methods.
Patients and Methods:
We performed ultra-short fragment (100–200 base pair) PCR amplification of cf-tDNA for clinically actionable alterations in CSF and tumor samples from patients with pHGG (n = 12) alongside non-tumor CSF (n = 6). PCR products underwent rapid amplicon-based sequencing by Oxford Nanopore Technology (Nanopore) with quantification of VAF. Additional comparison to next-generation sequencing (NGS) and droplet digital PCR (ddPCR) was performed.
Results:
Nanopore demonstrated 85% sensitivity and 100% specificity in CSF samples (n = 127 replicates) with 0.1 femtomol DNA limit of detection and 12-hour results, all of which compared favorably to NGS. Multiplexed analysis provided concurrent analysis of H3F3A and HIST1H3B mutations in a non-biopsied patient and results were confirmed by ddPCR. Serial CSF cf-tDNA sequencing by Nanopore demonstrated correlation of radiological response on a clinical trial, with one patient showing dramatic multi-gene molecular response that predicted long-term clinical response.
Conclusions:
Nanopore sequencing of ultra-short pHGG CSF cf-tDNA fragments is feasible, efficient, and sensitive with low-input samples, thus overcoming many of the barriers restricting wider use of CSF cf-tDNA diagnosis and monitoring in this patient population.
Keywords: pediatric brain tumors, high-grade glioma, molecular response, molecular diagnosis, cerebrospinal fluid analysis
Introduction:
Pediatric high-grade glioma (pHGG), including diffuse intrinsic pontine glioma (DIPG), is a group of aggressive brain cancers with < 10% surviving at two years from diagnosis (1). Overall median survival for pHGG is 13.5 months (21.4% two-year survival) in midline gliomas and 11 months (5.2% two-year survival) for DIPG specifically (2,3). Unlike in adult HGG, pHGG is most commonly midline or infratentorial in location (1). Midline HGGs are not usually surgically resectable and standard treatment relies on palliative radiation and chemotherapy. Improved, targeted treatment with rapid time to diagnosis is desperately needed for pHGG.
The World Health Organization reclassified most gliomas to include molecular features, including the H3K27M mutational status of diffuse midline gliomas. This mutation occurs in > 50% of pHGGs and in > 80% of DIPGs and portends a universally poor prognosis (4,5). Pediatric HGGs also frequently have mutations in tumor-driving genes such as ATRX, TP53, ACVR1, BRAF, EGFR, and PIK3CA.
Historically, diagnosis has been made based on clinical and imaging findings. Prior to the early 2000s, surgical biopsy for DIPG and other midline tumors was considered unsafe, especially when therapeutic options were lacking (6). While MRI has been important for diagnosis and assessing response to treatment of brain tumors, accurate assessment may be confounded by radiation-related swelling (pseudo-progression). Molecular diagnosis of pHGG through biopsy carries major drawbacks (7–9). Liquid biopsy with cerebrospinal fluid (CSF) sampling offers a safer and less expensive alternative to determine molecular diagnosis than surgical biopsy. Brain tumors shed their DNA into the CSF (hereafter referred to as cell-free tumor DNA, cf-tDNA), and this cf-tDNA can be detected via CSF sampling (10–12).
Prior work has demonstrated the feasibility of detecting the H3K27M mutation in CSF using droplet digital PCR (ddPCR) with high sensitivity and specificity (11,13,14). However, ddPCR requires expensive and time-consuming assay design, and is best used for recurring point mutations such as H3K27M. For genes that have clinically significant mutations throughout multiple exons (e.g. TP53, PIK3CA), ddPCR has significant limitations. Additionally, technical complexity limits use of ddPCR for characterization of a larger quantity of alterations.
As liquid biopsy technology advances, alternative methods to detect and monitor cf-tDNA from CSF are being explored, including sequencing. Next-generation sequencing (NGS) has been successfully employed to isolate patient-specific alterations in CSF cf-tDNA (12,15). These studies have solely examined adult tumors (primary brain tumors or metastases) and required considerable amounts of CSF (3.5 mL to 5 mL) to obtain sequencing results. For the pediatric population, especially with brainstem or midline tumors, this volume can be risky or impossible to obtain. Additionally, NGS methods remain relatively expensive with sophisticated and time-intensive informatics; this has limited its widespread use for CSF cf-tDNA diagnostics (16). Additionally, few studies have explored the serial monitoring of CSF cf-tDNA for HGG (in children or adults) with comparison to clinical and radiographic outcomes. Confirmed feasibility of a clinical trial design with safe serial lumbar puncture and rapid cf-tDNA analysis at time of MRI would represent an important step for future clinical trials in HGG.
Here we explore the use of an alternative platform for sequencing with the goal of tackling some of the issues that have limited the use of CSF cf-tDNA in HGG serial monitoring. Oxford Nanopore Technology (Nanopore) is an inexpensive, handheld device that sequences through direct, electronic analysis of nucleic acids fed through >1,000 biological pores that sequence DNA fragments simultaneously. Previous work has confirmed its feasibility and efficiency (same-day) in molecular characterization of adult glioma tumor samples (17). We hypothesized that the low DNA input required for Nanopore sequencing would be optimal for CSF cf-tDNA, which is often in the nanogram range for an entire sample; this is especially important for pediatric patients, where larger volume samples are difficult to obtain safely. In order to improve clinical utility, we were interested to see if the sequencing efficiency and fairly straightforward informatics previously associated with Nanopore could be applied to liquid samples.
Here we show that Nanopore sequencing of ultra-short pHGG CSF cf-tDNA fragments is feasible, efficient, and sensitive with very low-input samples. Additionally, we describe the feasibility of a clinical trial design with serial correlate lumbar puncture in pHGG, which allowed us to track multi-gene molecular response in two patients with H3K27M-mutant HGG that correlated with long-term radiological response.
Materials and Methods:
Patient samples
Pediatric patients with high-grade glioma who were seen at the University of Michigan and registered into the IRB-approved studies Brain Tumor CSF Registry (HUM0013228) or ONC201 phase 1 clinical trial in pediatric patients with H3K27M-mutant glioma (HUM00144364) were included in the study. Patients with tissue were also enrolled in the IRB-approved Pediatric MiOncoseq study (HUM00056496) for next-generation DNA (1,700 gene panel) and RNA sequencing (Illumina) of tumor and germline samples using previously established methods (18).
Patients enrolled in the ONC201 trial underwent sedated MRI with and without contrast plus CSF draw by LP at the time of study initiation, at two months into treatment, and six months into treatment. For patients who underwent clinical CSF collection (Brain Tumor CSF Registry or ONC201 trial), the amount of CSF collected ranged from 1 mL to 5 mL at each draw.
Normal control CSF was obtained from patients with normal pressure hydrocephalus through the IRB-approved Negative Controls for CSF Studies protocol (HUM00128299).
The study was conducted in accordance with the Declaration of Helsinki and International Ethical Guidelines for Biomedical Research Involving Human Subjects (CIOMS). All patients provided written consent and the study protocols were IRB-approved.
Sample processing and DNA extraction
Collected CSF was immediately processed by centrifuging at 4 °C for 10 minutes at 2,000 rpm, then aliquoted as 0.5 mL to 1 mL into separate vials and stored at −80 °C. Control CSF samples were processed and stored in 2 mL to 10 mL aliquots at −80 °C.
For samples obtained through research autopsy, CSF was collected by lumbar puncture, cisterna magna puncture, and/or lateral ventricle puncture. The brain (including tumor) was then extracted, placed on ice, and immediately transported for processing. Fresh brain and tumor were sectioned into 2 mL cryovials, flash-frozen with liquid nitrogen, then placed into −80 °C. CSF was immediately processed as described above.
DNA was extracted from ~20 mg of tissue (either normal brain or tumor specimens depending on the experiment) using DNeasy Blood and Tissue Kit (Qiagen) according to manufacturer’s protocol. DNA was extracted from 1 mL CSF using the QIAamp Circulating Nucleic Acid Extraction Kit (Qiagen) following the manufacturer’s protocol.
Evaluation of DNA fragment size
For DNA fragment size analysis, 1 mL each of normal CSF and tumor CSF was used. Following extraction as described above for CSF, the DNA was sent to the University of Michigan Advanced Genomic Core for High Sensitivity DNA ScreenTape (TapeStation) analysis.
Primer design and PCR
Using information obtained by TapeStation analysis, primers were designed to amplify a ~100–200 bp sequence around mutations of interest (see Supplemental Table S1 for gene panel and primer sequences) and ordered from IDT with attached Nanopore barcoding tags. Primers were reconstituted to 100 uM stock with Buffer AE (Qiagen) and stored at −20 °C, while working solutions were diluted to 10 uM and stored at 4 °C.
Extracted DNA from patient samples underwent PCR followed by gel electrophoresis to confirm amplification of target genes. All amplicons (by patient) were then pooled, underwent 1X bead clean-up with AMPure XP Beads (Beckmann-Coulter), and eluted in molecular biology grade nuclease-free water (Corning). The concentration of pooled samples was again checked via Qubit.
Targeted DNA sequencing by Oxford Nanopore Technology MinION device
Patient samples were run in triplicate and underwent library preparation according to Oxford Nanopore Technology. For experiments requiring multiple replicates or multiple patients in one sequencing run, all replicates underwent library preparation, barcoding, and adapter ligation using Native Barcoding Amplicons (with EXP-NBD104, EXP-NBD114, and SQK-LSK109) protocol (Nanopore, https://dx.doi.org/10.17504/protocols.io.bgzxjx7n). For experiments not requiring barcoding, samples underwent library preparation and adapter ligation according to Amplicons by Ligation (SQK-LSK109) protocol (Nanopore, https://dx.doi.org/10.17504/protocols.io.bgzxjx7n). For all experiments, after adapter ligation, the concentration of the sample was checked and the DNA library prepared by adding 37.5 uL Sequencing Buffer (SQB) and 25.5 uL Loading Beads (Nanopore) to the DNA library. The DNA library was then run on a FLO-MIN106 (r9.4.1) flow cell using the Nanopore MinION handheld sequencing device. Sequencing runs were allowed to progress until 100,000 reads per sample was achieved, assuming equal distribution of samples across the flow cell. In general, ~80–90% of reads were deemed mappable (e.g. 86.4% for patient UMPED57 tumor, 80.3% for patient UMPED22B CSF), and only mapped reads were included for VAF calculation. For detailed materials and methods, see Oxford Nanopore Technology Library Preparation section in supplemental materials and methods.
Nanopore bioinformatics analysis
Once the sequencing was complete on the MinION flow cell, Nanopore MinKNOW sequencing software output FAST5 files were basecalled to FASTQ files and separated by barcode using Oxford Nanopore’s Guppy basecaller (version 2.3.5 or other where noted). The reads in the barcoded FASTQ files were aligned to human genome 19 using minimap2 version 2.17 (19) and resulting BAM files were sorted using SAMtools version 1.9 (20). Variant allele fraction (VAF) for each sample and gene of interest was then determined using Integrated Genome Viewer (IGV, version 2.61). VAF was calculated as mutant reads divided by the sum of mutant and wild type reads at that chromosomal location (see Supplemental Table S1 for chromosomal locations for SNPs of interest). When possible, up to three replicates were performed for sequencing. Reported VAF represents the mean of replicate values. Positive values are those with mean replicate values greater than our lower limit VAF (see Results).
Next-generation sequencing (Illumina)
Cf-tDNA was subjected to PCR using an amplicon library designed for patient-specific SNPs and optimized for Illumina sequencing following manufacturer’s protocol (Swift Biosciences). Final library quality was assessed using the TapeStation 4200 (Agilent) and libraries were quantified by Kapa qPCR (Roche). Pooled libraries were subjected to 150 bp paired-end sequencing according to the manufacturer’s protocol (Illumina MiSeq, University of Michigan Sequence Core Facility). Bcl2fastq2 Conversion Software (Illumina) was used to generate de-multiplexed FASTQ files.
Limit of detection analysis
Both normal control and tumor cf-DNA were amplified with H3F3A primers and underwent gel electrophoresis to verify amplification. Bead clean-up and library preparation (including barcoding) proceeded as described in supplemental materials and methods. Prior to sequencing, the concentration was determined for both conditions. Ten separate samples with 75 fmol of normal DNA plus ever-decreasing amount of tumor DNA (Supplemental Table S2) were run independently on the flow cell and sequencing progressed to about 200,000 reads. The data for each sample was then analyzed using the computational pipeline described above to determine H3F3A K27M VAF.
Error and depth rate analysis
PCR was performed for both samples using H3F3A primers with the same PCR settings as described in supplemental materials and methods, followed by gel electrophoresis and bead clean-up. Library preparation proceeded with the Oxford Nanopore SQK-LSK109 kit without barcoding. In the Nanopore MinKNOW software, automatic basecalling was turned off. The sequencing runs progressed until > 100,000 reads were obtained per sample, as estimated by the MinKNOW sequencer software. The resulting signal data from each sample was basecalled using the GPU accelerated version of Oxford Nanopore’s Guppy basecaller (version 3.2.1 and 3.6.0 for supplemental figure 2). Reads were then aligned to Gh37/hg19 human assembly using minimap2 version 2.17 (19), and sorted by read timestamp added by the basecaller. We then generated bins of 250 consecutive reads, considering only reads which mapped to the target region (e.g. chr1:226252135 for H3F3A K27M). Each time-sorted bin was then re-aligned to hg19 and a pileup was generated using samtools pileup command (20). A custom python script was used to parse the pileup and extract VAF corresponding to the locus for the target mutation. VAF was computed as described earlier.
Multiplexed Nanopore PCR
Multiplexed PCR was performed to identify the presence of HIST1H3B or H3F3A K27M mutation in CSF for sample with known H3K27M status. Control DNA included DNA from a patient with known H3F3A K27M mutation and from a patient with known HIST1H3B K27M mutation (by Illumina sequencing upon biopsy). Each of the three patient samples were individually amplified using H3F3A and HIST1H3B primers and following the Q5 High-Fidelity DNA Polymerase Protocol (NEB). The PCR tubes were prepared by combining 5X Q5 Reaction Buffer (NEB), 10 uM dNTPS (NEB), Q5 DNA Polymerase (NEB), GC Enhancer (NEB), H3F3A and HIST1H3B primers (for final concentration 0.15 uM), and DNA. The samples then underwent PCR amplification, gel electrophoresis, bead clean-up, DNA end repair, barcoding, and adapter ligation as described, then were sequenced on the Nanopore MinION device.
Statistical Analysis
Statistical significance in all experiments was defined as a two-sided P ≤ 0.05, with comparisons between groups made using Welch’s t-test. All analyses were conducted with GraphPad Prism software (version 7.00).
Results:
CSF cf-tDNA fragment size analysis
In order to improve limit of detection for low-input samples such as pediatric CSF samples, we utilized Nanopore sequencing due to its low input requirements. Prior to sequencing, we characterized the cell-free DNA fragment distribution in the CSF of both pHGG and control patients.
TapeStation analysis of cell-free DNA extracted from normal control CSF demonstrated two noticeable peaks of cf-DNA at 139 bp and 636 bp, while in tumor CSF there were three peaks at 184 bp, 338 bp, and 551 bp, with 184 bp being the largest peak (Figure 1). Previous work has demonstrated that, early in apoptosis, DNA is cleaved into very small fragments of about 180–200 bp (21). Based on the notable ~180 bp distribution that was enriched in CSF from patients with tumors, we designed primers to amplify DNA fragments of 100–200 bp in length in order to enrich for analysis of apoptotic tumor DNA from CSF samples. We have previously designed and tested primers that amplify larger sequences (900–3,000 bp) with notably low amplification efficiency in CSF samples despite excellent amplification efficiency in tissue samples (data not shown).
Figure 1. CSF cf-tDNA fragment analysis.
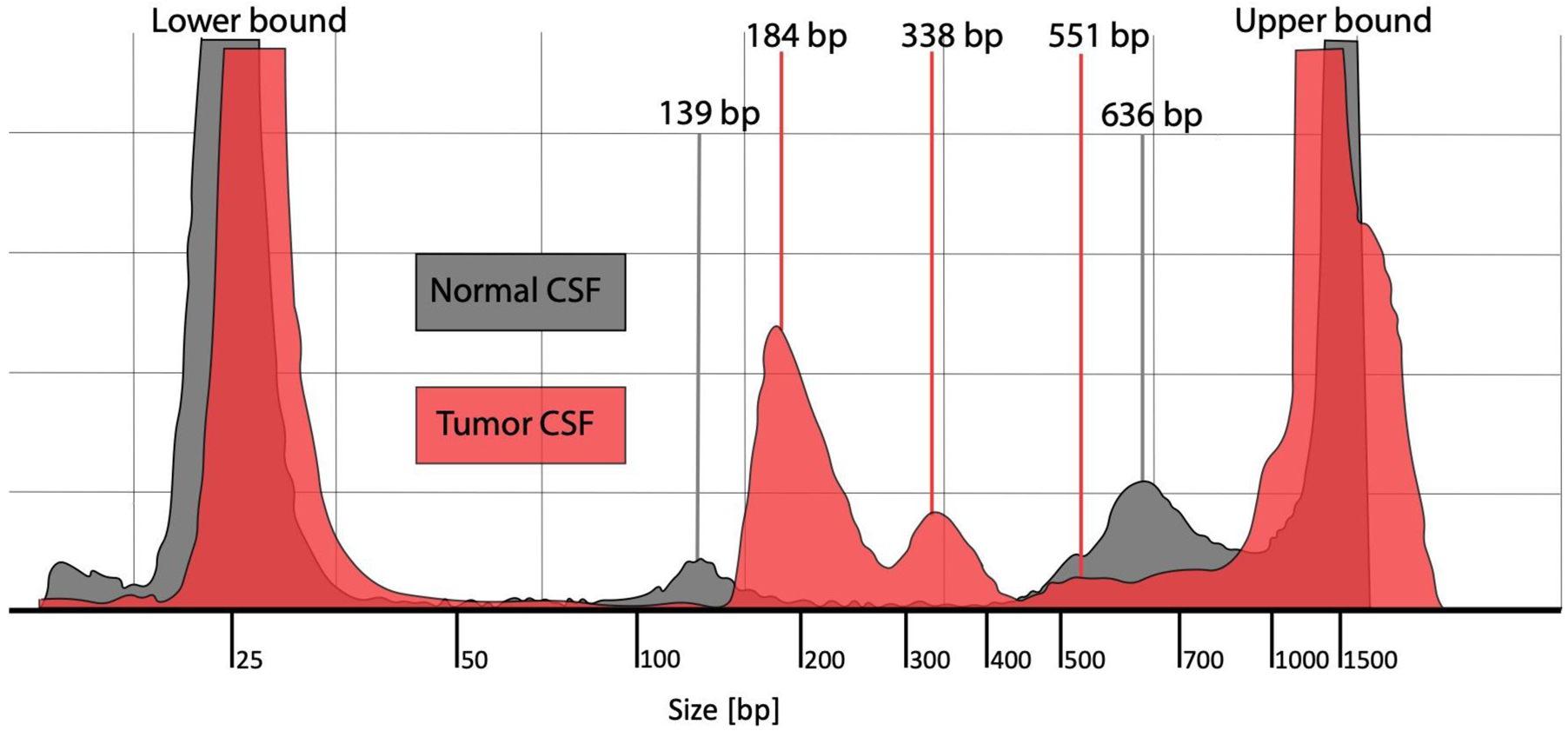
Fragment analysis of cell-free DNA from normal CSF (gray color) and from tumor CSF from a patient with DIPG (red color). The figure shows DNA fragment sizing and quantification using TapeStation High Sensitivity DNA ScreenTape Bioanalyzer for each of the two sample types.
Abbreviations: CSF = cerebrospinal fluid, DIPG = diffuse intrinsic pontine glioma, bp = base pairs
CSF cf-tDNA Nanopore sequencing limit of detection analysis
We compiled CSF samples from pHGG patients through multiple IRB-approved clinical protocols and research autopsies. Samples from 12 patients were included, with average age of 10 years (range 3 years to 17 years) at time of first sample donation (CSF or tissue) (Table 1). Eleven of our patients had biopsy-confirmed diffuse midline glioma (brainstem = seven patients, thalamic = three patients, spinal cord = one patient); nine of these were H3K27M positive and one patient had radiographic DIPG that did not undergo biopsy but did undergo CSF collection at autopsy. All patients had undergone standard treatment prior to or during enrollment, including radiation and chemotherapy. We collected and stored specimens using identical methods regardless of protocol and sample time point. We then designed 100–200 bp primer sets for a panel of recurrent and actionable gene mutations found across our pHGG samples (Supplemental Table S1).
Table 1.
Patient Demographics
| Sample ID (UMPED #) | Age (years) | Gender | Clinical/Radiographic Diagnosis | Histopathological Diagnosis | Tumor Genomics | Therapies | Time(s) of Tissue Samples | Time(s) of CSF Samples | Location of CSF Samples | Overall Survival (From Diagnosis) |
|---|---|---|---|---|---|---|---|---|---|---|
| UMPED18 | 8 | Male | DIPG | Diffuse midline glioma, H3K27M- mutant | H3F3A K27M, PIK3CA E545K | Radiation + AZD1775, everolimus + panobinostat | 1 month | At autopsy | Lumbar Puncture | 9 months |
| UMPED22 | 4 | Male | DIPG | Diffuse midline glioma, H3K27M-mutant | ACVR1 G328E, HIST1H3B K27M, TP53 R213* | Radiation, bevacizumab | 2 weeks | At autopsy | Cisterna Magna | 12 months |
| UMPED24 | 3 | Female | DIPG | Diffuse midline glioma, H3K27M-mutant | H3F3A K27M, PTPN11 T468P | Radiation, bevacizumab, trametinib | 11 days | At diagnosis | Lumbar Puncture | 14 months |
| UMPED25 | 14 | Male | DIPG | Diffuse midline glioma, H3K27M-mutant | FGFR3 K650E, H3F3A K27M | Radiation, ponatinib, re-irradiation, bevacizumab , panobinostat, pazopanib + everolimus | At diagnosis | At diagnosis | Ventricular | 20 months |
| UMPED37 | 13 | Male | Thalamic HGG | Diffuse midline glioma, H3K27M-wildtype | Diagnosis: EGFR V292L, EGFR T483_G485del; Autopsy: TP53 H214R, EGFR V292L, EGFR T483_G485del | Chemotherapy (TPCV), radiation, osimertinib | At diagnosis; at autopsy | At autopsy | Ventricular | 17 months |
| UMPED39 | 9 | Male | DIPG (secondary to medulloblastoma) | Diffuse midline glioma, H3K27M-wildtype | PIK3CA G106V, PIK3CA R88Q, PIK3CA H1065L | Radiation, everolimus, LY3023414 | At diagnosis | At autopsy | Cisterna Magna | 11 months |
| UMPED55 | 8 | Female | DIPG | Not biopsied at diagnosis | Radiation, bevacizumab, ONC201 | At time of death | Lumbar Puncture | 9 months | ||
| UMPED57 | 9 | Female | DIPG | Diffuse midline glioma, H3K27M-mutant | H3F3A K27M, PPM1D E525* | Radiation, ONC201 | At diagnosis | At autopsy | Lumbar Puncture | 8 months |
| UMPED58 | 9 | Male | DIPG | Diffuse midline glioma, H3K27M-mutant | ATRX Q119*, H3F3A K27M | Radiation, bevacizumab, intra-arterial therapy (multiple compounds, unknown regimen) | 1 week | At autopsy | Lumbar Puncture | 21 months |
| UMPED60 | 17 | Female | Thalamic HGG | Diffuse midline glioma, H3K27M-mutant | H3F3A K27M, PIK3CA E545G, TP53 R158G, TP53 R248Q | Radiation + temozolomide, bevacizumab , ONC201 | At diagnosis | 0, 2, and 6 months on ONC201 | Lumbar Puncture | 22 months and ongoing |
| UMPED62 | 13 | Male | Spinal cord HGG | Diffuse midline glioma, H3K27M-mutant | ATRX I2248fs, FGFR1 N546K, H3F3A K27M, H3F3A G35W, PPM1D W427*, PTPN11 E69K | Temozolomide + irinotecan; subtotal resection, radiation, bevacizumab, ONC201 | At progression | At progression | Lumbar Puncture | 19 months from progression |
| UMPED77 | 13 | Male | Thalamic HGG | Diffuse midline glioma, H3K27M-mutant | H3F3A K27M by IHC | Radiation + temozolomide, bevacizumab , ONC201 | At diagnosis | 0, 2, and 6 months on ONC201 | Lumbar Puncture | 14 months and ongoing |
The table gives patient demographics for NGS-sequenced tumors including tumor type and H3K27M status, all tumor-driving or clinically actionable mutations determined at diagnosis and explored via Nanopore analysis, therapies each patient received prior to or during the study, and timing and location of sample collection. Two patients did not have surgical biopsy but provided CSF for analysis at time of death.
Abbreviations: DIPG = diffuse intrinsic pontine glioma,
= stop codon mutation, del = deletion mutation, fs = frameshift mutation, HGG = high-grade glioma, TPCV= temozolomide, procarbazine, CCNU, vincristine
We determined the minimum amount of cf-tDNA that was required to calculate reliable VAF. In our pediatric patient population, the amount of CSF safely obtained by lumbar puncture ranged from 1 mL to 5 mL per CSF draw. When receiving only 1 mL of CSF, the amount of cf-tDNA is limited to about 15 ng per sample on average. When amplifying the DNA for sub-200 bp fragments, 15 ng of input DNA usually results in picomolar amounts of amplified cf-tDNA (see Methods section for PCR settings).
Given that Nanopore MinION flow cells are able to withstand a maximum of 200 fmol of input DNA, we hypothesized that we could obtain reliable results with 1 mL of CSF from pediatric patients with HGG. We were able to detect the H3F3A K27M mutation at a non-significantly different VAF in the tumor CSF down to 0.1 fmol of input cf-tDNA (Figure 2A). This represented a depth of only 139 reads (Figure 2B) for a sample with VAF of 24%.
Figure 2. Limit of Detection and Error Rate of Nanopore CSF cf-tDNA Sequencing.
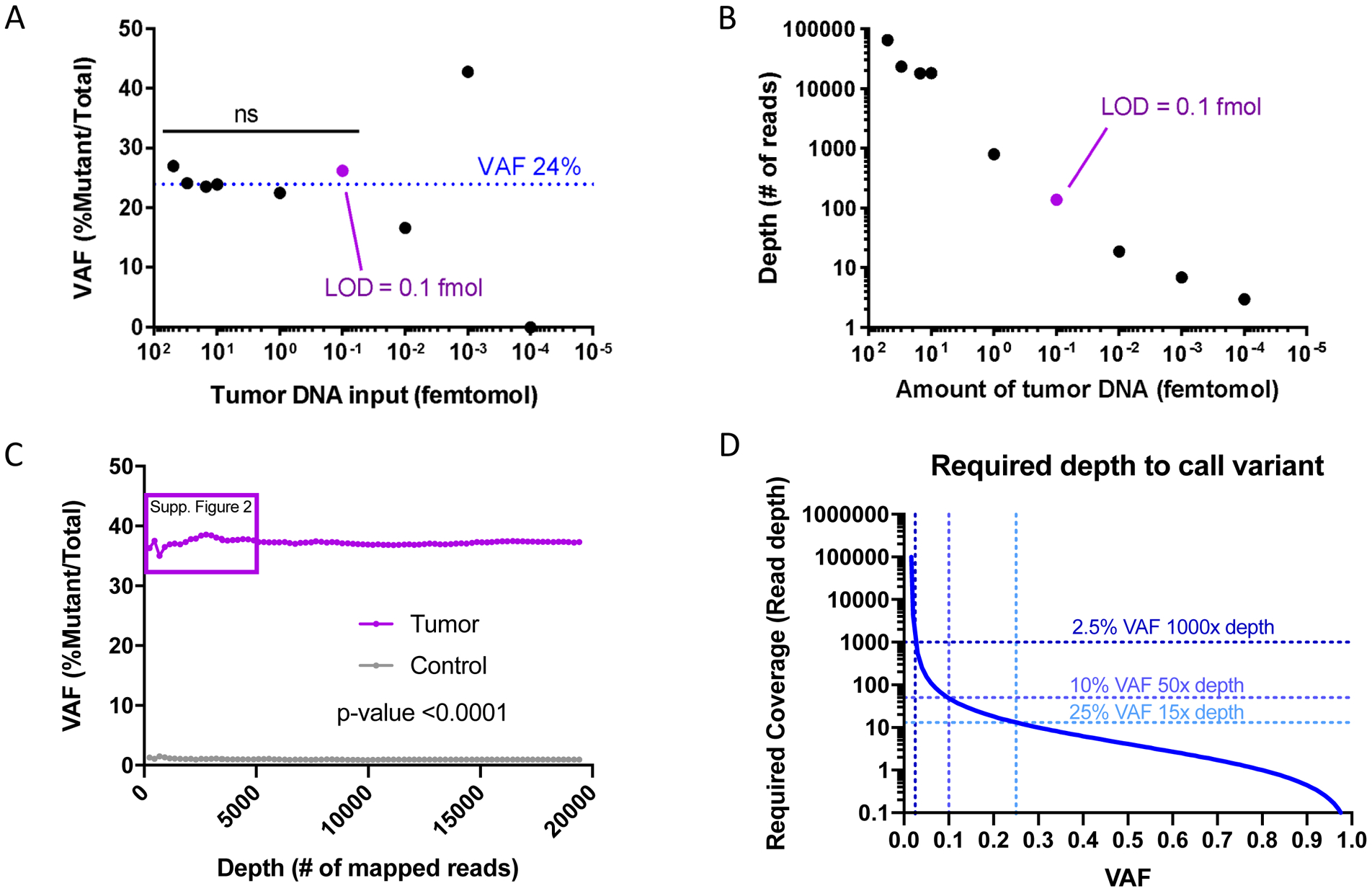
A) Limit of detection by fmol of tumor DNA input onto the Nanopore flow cell needed to detect stable reference VAF of 24% H3F3A K27M obtained by Illumina sequencing at diagnosis. The purple dot shows the limit of detection. B) The graph shows read depth versus input DNA, demonstrating the depth required to call VAF of 24% at each amount of input DNA. The purple dot is the limit of detection. C) The graph shows the error rate of Nanopore in the negative control sample (gray line) as well as the VAF for H3F3A K27M for the tumor sample (purple line). D) Cochran’s equation was used to estimate the number of samples (reads) required to call various VAF’s where the lower bound of the 95% confidence interval is equal to the maximum observed false positive rate.
Abbreviations: VAF = Variant Allele Fraction, fmol = femtomol, Supp. = supplemental
We then determined the error rate of Nanopore CSF cf-tDNA sequencing. We compared CSF cf-DNA from UMPED57, a 9-year-old female with established (solid) tumor H3F3A (H3.3) K27M mutation VAF (47%), with CSF cf-DNA from a patient without tumor (normal). We examined the H3.3 K27M VAF in bins of 250 reads each (plotted in bins of 1,000X for ease of visualization) and found that at even low depth of < 1,000X, the normal CSF cf-DNA maintained a false positive error rate of about 1% VAF for H3F3A K27M mutation; this is consistent with established false positive rates for recent high-accuracy basecallers targeting Oxford Nanopore sequencers (Figure 2C) (17,22–24). Using the same data and separating by 1,000X depth bins, only the smallest bin (0–1,000X depth) was significantly different for our 100,000X data VAF (Supplementary Figure S2A), which is consistent with the depth range from our limit of detection analysis (Figure 2B). For normal CSF, the mean VAF was 0.98% (std dev 0.44%, range 0.17% to 1.58%) when depth was >1000X, across all point mutations listed in Table 1.
In order to explore depth requirements for lower VAFs, we used a statistical analysis to estimate the coverage required to call a mutation with a desired confidence interval (95%) in relation to our established false positive rate of ~1% (Figure 2D). We used Cochran’s equation to estimate a sample size (total wildtype and mutant reads) required to call a VAF within a 95% confidence interval (CI). For each VAF, we set the error margin so that the lower bound of the tumor sample CI is equal to the upper-bound of our non-tumor false positive samples. The required read depth increases with proximity of underlying VAF to the false positive rate (Figure 2D). Our analysis indicates that at 1,000X (wildtype + mutant) depth with our error rate, Nanopore sequencing can call mutations with a VAF as low as 2.5% with 95% CI. Based on these data, we considered our lower limit VAF 2.5% for all samples with at least 1,000X depth. Sequencing of an additional sample (UMPED22, H3.1 K27M) demonstrated a definitive VAF of 8%, with statistical significance from negative control even at lowest bin (first 250 mapped reads) (Supplementary Figure S2B).
CSF cf-tDNA Nanopore sequencing sensitivity, specificity, and efficiency analysis
We performed Nanopore sequencing of liquid and solid samples from our pHGG cohort (n = 188 total replicates, 127 CSF replicates). All samples were run with normal control (either normal brain tissue or normal CSF). Compared to diagnostic NGS-based tumor results, Nanopore sequencing detected clinically relevant genetic mutations in pHGG with 83% sensitivity and 100% specificity (Supplemental Table S3A and S3B). In CSF alone, the sensitivity was 85% and the specificity 100% (Supplemental Table S3A and S3B). Patient status did not influence sensitivity (living – 83%, autopsy – 82% (Supplemental Table S3A and S3B). For autopsy samples, ventricular CSF sample location showed the highest cf-DNA yield (7.42 ng/uL, n=1), compared to lumbar puncture (mean 1.41 ng/uL, n=3), and cisterna magna (mean 0.47 ng/uL, n=2). Lumbar puncture samples obtained from living patients yielded lowest average yield (mean 0.14 ng/mL). However, 1 mL of CSF was sufficient to generate >200 fmol in all samples. Within the limits of our sample size, sample type did not influence sensitivity for Nanopore sequencing.
In order to determine the efficiency of Nanopore sequencing for CSF cf-tDNA using ultra-short amplicons, we compiled the time to results for samples analyzed immediately after sample collection. We sequenced using Nanopore immediately after sample collection for a sample from patient UMPED60. We performed Nanopore library preparation using Nanopore SQK-LSK109 and EXP-NBD104 kits and sequenced on Nanopore MinION device using an R9.4.1 flow cell in under 1.5 hours to total > 300,000 DNA fragment reads, which was consistent with other Nanopore sequencing runs. Using our software pipeline (see Methods), we calculated VAF and detected the desired gene mutations with statistical significance (Figure 3). From time of CSF draw to results, provided as VAF of the gene mutations, the total time required was around 12 hours (Figure 3). We furthermore found that time to 1,000X depth (minimum depth suggested to achieve VAF with statistical certainty) was less than 10 minutes for multiple gene mutations of interest (Figure 3), consistent with other comparable studies (17). These results suggested that Nanopore sequencing was both rapid and reliable in detecting mutations with oncogenic significance in CSF of patients with HGG.
Figure 3. Nanopore sequencing efficiency analysis.
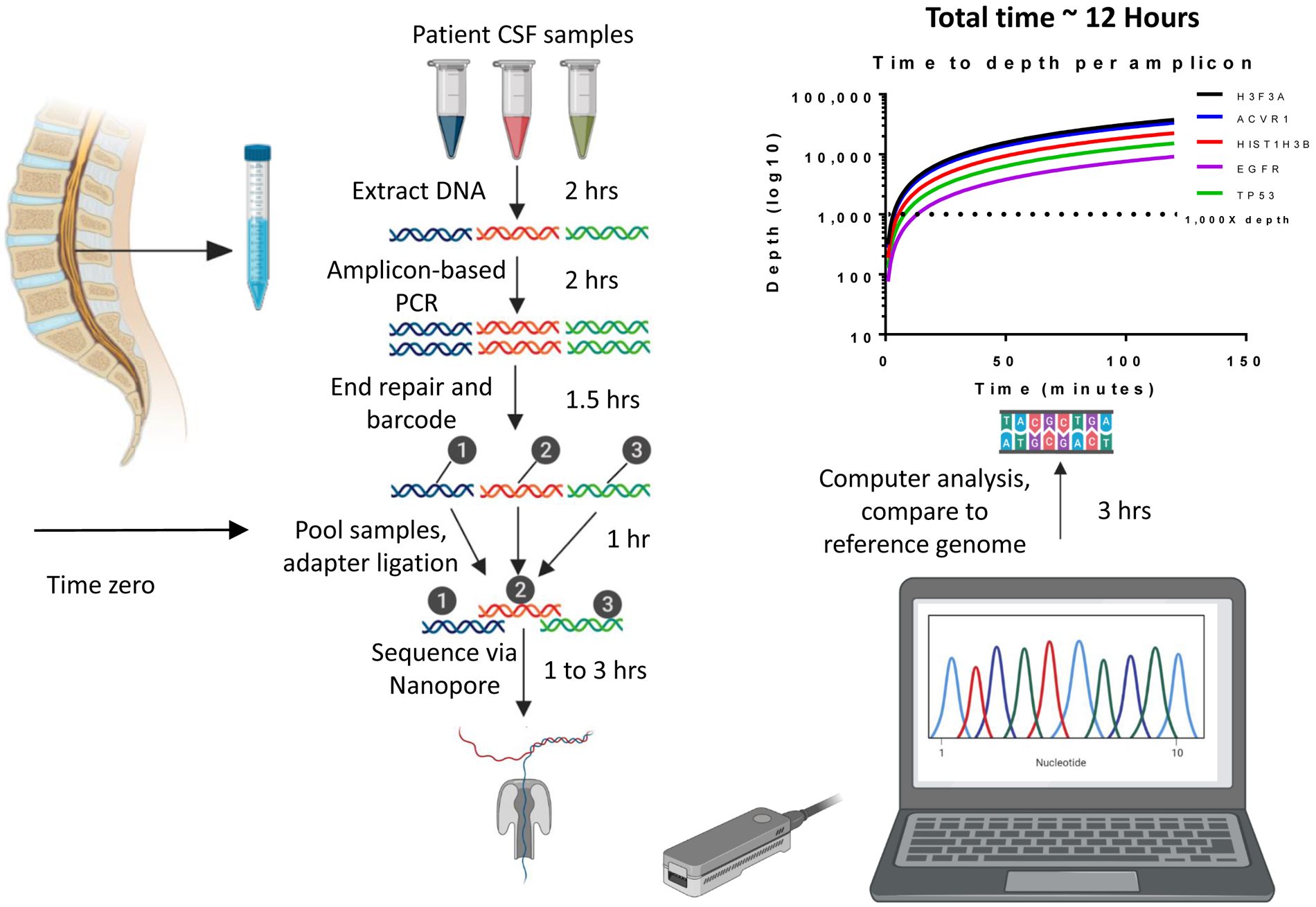
CSF was drawn via lumbar puncture at time zero of processing. After DNA extraction, the sample(s) underwent amplification with pediatric HGG-specific genes, followed by sequencing on the MinION device. Raw sequencing output was basecalled using Guppy, then reads were aligned using Minimap2 to the human reference genome hg19. Integrated Genome Viewer was used to calculate variant allele fraction of aligned amplicons. Time required to sequence to a depth of 1000X ranged from 4 minutes to 14 minutes, with several genes shown as examples. The total time to results (variant allele fraction of SNPs) was less than 12 hours. Samples were run in triplicate.
Comparison of NGS and Nanopore sequencing of pHGG tumor and CSF cf-tDNA samples
To further establish the feasibility and reliability of Nanopore sequencing, we performed comparison NGS (Illumina) sequencing of CSF and tumor samples for patients with matched samples at diagnosis and autopsy (n = 6). Patient-specific driver mutations were determined through NGS sequencing, and amplicon-based Nanopore and NGS sequencing were performed on CSF and tumor samples (Figure 4)
Figure 4. Comparison of Nanopore and NGS sequencing of pHGG tumor and CSF cf-tDNA.
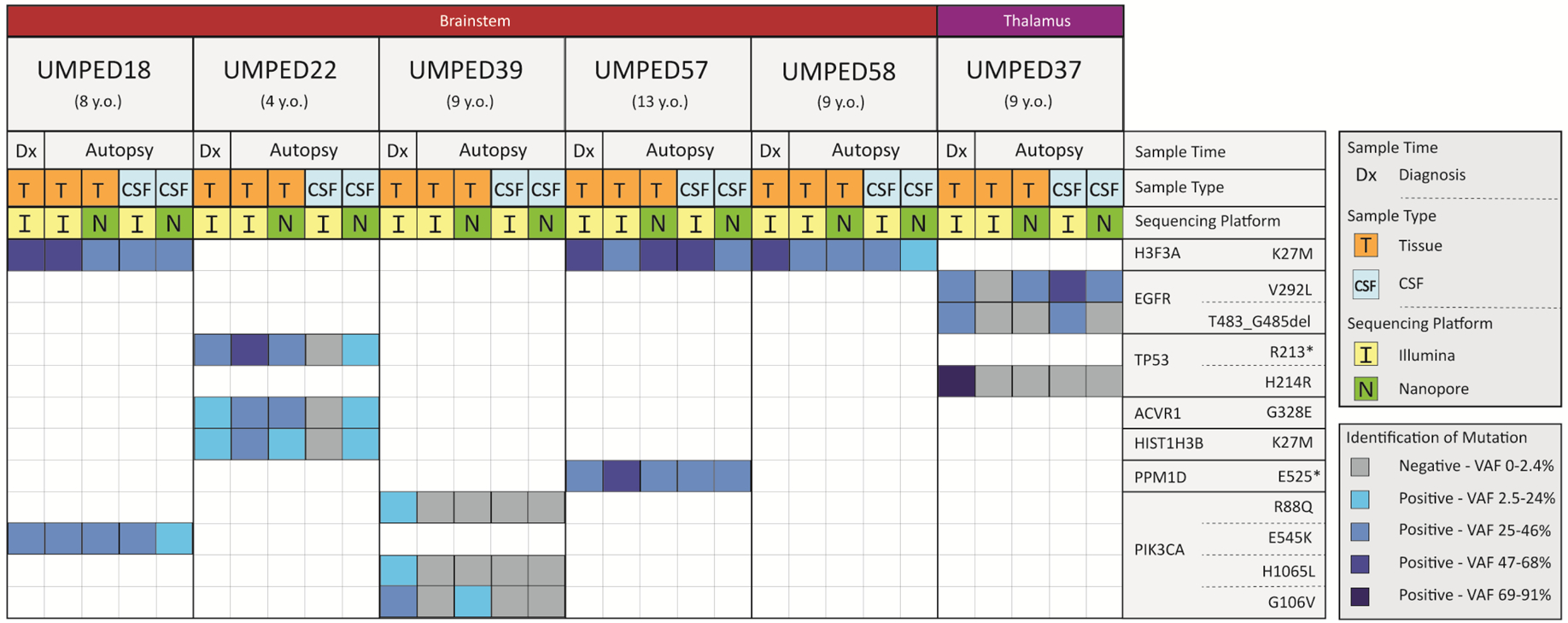
Each UMPED number is a pediatric patient who had Illumina-based tissue sequencing at time of diagnosis and who donated tissue and CSF at time of death (age at time of death shown beneath the UMPED number). Anatomical location of the patient’s HGG is shown across the top while the tumor-driving mutations found at time of diagnosis are shown on the right. Sample type (tissue or CSF) and whether it was from diagnosis or autopsy is indicated beneath the age at death. Each sample is marked as being sequenced by Illumina or Nanopore and its corresponding column indicates if it was positive or negative for the mutation given for that row. For samples where mutation was found, the VAF was determined. Darker shades of blue indicate higher VAF quartiles. Samples were run in triplicate for Nanopore sequencing.
Abbreviations: del = deletion, * = stop codon, VAF = variant allele fraction
Sample volumes were scaled to optimal machine input requirements. For Nanopore sequencing, 1 mL of CSF was used for each patient. To achieve the required nanogram input required for NGS (Illumina NextSeq), 2 mL to 3 mL was required. Nanopore sequencing detected patient-specific mutations in 67.9% (19/28) of the samples and was concordant with Illumina in 70.0% (14/20) of those. Nanopore sequencing detected the patient-specific mutation in five samples that Illumina did not detect, while Illumina detected a patient-specific mutation in one sample that Nanopore did not detect. In CSF, Nanopore found 64.3% (9/14) of the correct mutations, while Illumina detected 50% (7/14) of the correct mutations (Figure 4 and Supplemental Table S4). For three of the mutations, neither Illumina nor Nanopore were able to detect the mutation at autopsy, raising question of tumor evolution from diagnosis.
CSF cf-tDNA multiplex H3F3A and HIST1H3B analysis for patient with unknown H3K27M status
While the majority of our patient samples have Illumina-based molecular analysis results available by which to compare and confirm our results, we had one pediatric patient (UMPED55) who was radiographically diagnosed with DIPG and who donated CSF by LP at time of death. H3K27M mutation can denote a K27M mutation affecting either the H3.1 (HIST1H3B) or H3.3 (H3F3A) protein. We performed multiplexed PCR on CSF from our patient with unknown H3 status alongside positive control CSF samples from patients with H3F3A K27M mutation and a patient with known HIST1H3B K27M mutation, amplifying with both H3F3A and HIST1H3B primers in all three samples. All samples were run together and in triplicate on the Nanopore MinION device. We found that the patient with unknown H3 status had about 20% VAF for H3F3A K27M and about 1% VAF for HIST1H3B (background error rate of Nanopore) (Supplemental Figure 1). This result was later confirmed on ddPCR analysis of H3F3A (data not shown) using previously established methods (18).
Multi-time point CSF cf-tDNA Nanopore sequencing of pHGG samples
We next explored whether Nanopore sequencing could be employed for serial monitoring of CSF cf-tDNA for pHGG patients and compared to clinical and radiographic outcomes. We established a clinical trial design with serial lumbar puncture and rapid cf-tDNA analysis at time of MRI, and this was employed as Arm D of the multi-site ONC201 phase 1 trial for children with H3K27M-mutant glioma (NCT03416530). ONC201 is a dopamine receptor DRD2 antagonist and allosteric ClpP agonist, which has early reports of efficacy in patients with H3 K27M-mutant DMG (25–27). Patients undergo LP at time of trial initiation (baseline), two months into treatment, and six months into treatment (Figure 5A). We performed Nanopore sequencing on serial samples from two patients with pHGG.
Figure 5. Clinical trial schema for serial CSF cf-tDNA molecular response assessment.

A) The ONC201 clinical trial timeline. B) Axial brain T1 with contrast MRI images at each time point on the ONC201 trial for patient UMPED77 (dotted line denotes estimate tumor border). Total tumor area is noted (in mm2, shown in purple). C) Nanopore sequencing results for H3F3A K27M VAF (orange) and tumor area (blue) for each CSF collection time point for the patient in 5B. D) Axial brain T1 with contrast MRI images at each time point on the ONC201 trial for patient UMPED60. Total tumor area is noted (in mm2, shown in purple). E) Nanopore sequencing results (shown as VAF) for four key tumor-driving mutations (identified from surgical biopsy) for the patient in 5D, as well as tumor area for each of the clinical trial time points. All samples were run in triplicate for Nanopore sequencing.
Patient UMPED77 is a 13-year-old male with a large cortical and thalamic H3K27M-mutant diffuse midline glioma (Figure 5B). He was enrolled on Arm D of the ONC201 trial shortly after radiation therapy. Unfortunately, tumor fixation did not allow for baseline tumor DNA library preparation/sequencing, but immunohistochemistry (IHC) analysis confirmed H3K27M mutation in tumor. The patient has remained on treatment for six months with clinical and radiographic stability. This was mirrored on CSF cf-tDNA Nanopore analysis, which showed overall stability of H3K27M VAF in CSF obtained from lumbar puncture, with slight trend towards increase at six-month time point (Figure 5C).
Patient UMPED60 is a 17-year-old female with a unilateral thalamic H3K27M-mutant diffuse midline glioma. She was enrolled on Arm D of ONC201 trial shortly after radiation therapy after initial concerns for early progression. NGS sequencing at time of biopsy (prior to radiation) demonstrated four clinically relevant mutations, including TP53 R158G, TP53 R248Q, PIK3CA E545G, and H3F3A K27M (Supplemental Table S5). The patient demonstrated clinical and radiographic stability at two months, with sustained left-sided mild hemiparesis and tremor. At six months on the trial she had symptomatic improvement as well as 40% decrease in tumor size on MRI compared to baseline, suggesting that the tumor was responding to ONC201 (Figure 5D). CSF cf-tDNA Nanopore sequencing demonstrated stable-to-increased VAF of her four patient-specific alterations at two months, but reduction to 10% or less VAF for all at the six-month time point CSF sample (Figure 5E). This molecular response predicted a sustained clinical and radiographic response, which has continued for nearly two years on therapy at the time of this report. These results suggest that Nanopore sequencing can be used to track the multi-gene molecular response of a tumor to ONC201.
Discussion:
Here we present a novel use of Oxford Nanopore Technology (Nanopore) sequencing as a way to detect molecular alterations in CSF cf-tDNA of pHGG patients for both diagnosis and monitoring treatment response. Our data demonstrate that Nanopore sequencing of ultra-short pHGG CSF cf-tDNA fragments is feasible, efficient, and sensitive with low-input samples, thus overcoming many of the barriers restricting wider use of CSF cf-tDNA diagnosis and monitoring in this patient population. We also present a unique clinical trial design with standardized, serial CSF collections by lumbar puncture and establish feasibility for serial treatment monitoring via Nanopore sequencing of CSF in patients with HGG.
Nanopore MinION sequencing has recently been established as an alternative option to complement or improve some of the constraints of NGS, including in adult brain tumor samples (17). Euskirchen et al. found that detection of mutations by Nanopore was reliable compared to standard methods, including NGS or Sanger sequencing, in agreement with our results. We built upon this to highlight some of the features of Nanopore that are particularly well-suited for CSF cf-tDNA analysis, such as low tumor DNA input. Our work also establishes a rational framework for amplicon design for cf-tDNA using ultra-short (100–200 bp) sequences.
Although published research on detecting glioma cf-tDNA in CSF is limited, there are three studies in particular that have shown the feasibility of detecting HGG cf-tDNA in CSF (12,15,23). Unlike our studies, Miller et al. and Pentsova et al. examined adult patients. Pentsova et al. found 63% cf-tDNA positivity in solid tumor metastasis and 50% positivity for primary brain tumors using 5 mL CSF per patient and NGS-based assay (15). Miller et al. was able to detect cf-tDNA in 49.4% of adults with gliomas and in 58.7% of patients with grade IV glioma also using NGS-based assay (12). Another study by Huang et al. had sensitivity of 87.5% for HGG in CSF from pediatric patients using Sanger sequencing with sub-2 mL sample inputs (23). Our data compares favorably to these alternate platforms. Even when utilizing low-volume samples (1 mL or less), Nanopore sequencing demonstrated 83% sensitivity and 100% specificity for pediatric patient-specific alterations.
While difficult to compare Nanopore and NGS sequencing directly, our data highlights some potential benefits to Nanopore for cf-tDNA analysis. Nanopore demonstrated sensitivity at an extremely low limit of detection at 0.1 fmol DNA input. For comparison, Illumina devices generally require picomolar to nanomolar amounts of DNA input for reliable results, which equates to 2 mL to 3 mL of CSF at a minimum (28). Even for our comparison samples that used optimal amounts of input DNA for each platform, we found Nanopore to provide a slightly higher rate of detection of patient-specific mutations than matched NGS analysis. Additionally, we have shown that Nanopore sequencing can provide results in as little as 12 hours from the time of CSF draw by LP, with less than 30 minutes of sequencing time for suitable depth. It is important to note that NGS can offer significant advantages, including well-established whole exome or whole genome sequencing methodology and a much lower error rate. While amplicon-based Nanopore sequencing is suitable for calling recurrent hotspot mutations or regions, NGS is less error-prone and more suitable for isolating novel mutations or loss-of-functions mutations in large genes such as ATRX. In our work, we examined error only at specific codons (such as the H3F3A K27M mutation) rather than the entire read region, which may affect error rate. Future studies may employ both platforms for liquid and solid tumor diagnostics to take advantage of complementary features. Additional future work may examine error rate of short read sequencing versus long read sequencing.
Importantly, we established and implemented serial spinal fluid collection within a glioma clinical trial. While serial LP is not typically standard of care for the management of glioma, our study demonstrates that timed CSF collection by LP has potential clinical use across multiple high-grade brain tumors. We showed that Nanopore sequencing allows for serial monitoring of patient-specific mutations and that this information correlates with radiographic and clinical response to treatment. Our patients tolerated the procedure well, with procedure being performed at time of clinically indicated anesthesia. Multi-time point CSF assessment in conjunction with MRI has the potential to clarify molecular changes over time and to track clinical response and pseudo-response. We are now adopting this trial platform for multiple early-phase studies for high-grade glioma. With further validation, real-time results of serial CSF analysis via Nanopore/NGS could be used to generate an integrated radiographic/molecular response assessment in future clinical trials.
Limitations of the study include the anecdotal nature of our serial and multi-focal CSF assessments. Additionally, our sensitivity may have been influenced favorably by the number of autopsy samples, which may have been enriched for apoptotic cf-tDNA. Future studies will build on our initial results in prospective clinical trials.
In summary, our data establish Nanopore sequencing of ultra-short pHGG CSF cf-tDNA fragments as an efficient and sensitive alternative to NGS. Additionally, we describe the feasibility of a clinical trial design with serial correlate lumbar puncture in pHGG, which allowed us to track multi-gene molecular response. We believe this platform will push the field towards improved diagnostic and therapeutic assessments for patients with HGG.
Supplementary Material
Translational Relevance:
We describe one of the few primary studies demonstrating feasibility of detecting cell-free tumor DNA (cf-tDNA) in cerebrospinal fluid (CSF) of patients with brain cancer. The utility of detecting cf-tDNA in CSF to avoid potentially hazardous brain biopsies of eloquent tissue has broad implications in patients of all ages, but especially in pediatric patients. We demonstrate the ability of electronic sequencing (Nanopore) to rapidly and reliably detect key tumor-driving mutations based on a panel of recurrently mutated genes in pediatric high-grade glioma (pHGG). Nanopore sequencing of ultra-short CSF cf-tDNA fragments is feasible, efficient, and sensitive with low-input samples, thus overcoming many barriers restricting wider use of CSF cf-tDNA diagnosis and monitoring in this patient population. Furthermore, we describe the feasibility of a clinical trial design with serial correlate lumbar puncture in pHGG, which allowed us to track multi-gene molecular response that predicted long-term clinical response.
Acknowledgments:
The authors thank the patients and their families for participation in this study. CK is supported by NIH/NINDS K08-NS099427-01, the University of Michigan Chad Carr Pediatric Brain Tumor Center, the Evans family, the Chad Tough Foundation, Hyundai Hope on Wheels, Catching up With Jack, the Prayers from Maria Foundation, the U CAN-CER VIVE Foundation, the Morgan Behen Golf Classic, and the DIPG Collaborative.
Footnotes
Disclosure of Potential Conflicts of Interest:
None of the authors have potential conflicts of interest.
References:
- 1.Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DT, Konermann C, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 2012;22(4):425–37 doi 10.1016/j.ccr.2012.08.024. [DOI] [PubMed] [Google Scholar]
- 2.Mackay A, Burford A, Carvalho D, Izquierdo E, Fazal-Salom J, Taylor KR, et al. Integrated Molecular Meta-Analysis of 1,000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma. Cancer Cell 2017;32(4):520–37 e5 doi 10.1016/j.ccell.2017.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoffman LM, Veldhuijzen van Zanten SEM, Colditz N, Baugh J, Chaney B, Hoffmann M, et al. Clinical, Radiologic, Pathologic, and Molecular Characteristics of Long-Term Survivors of Diffuse Intrinsic Pontine Glioma (DIPG): A Collaborative Report From the International and European Society for Pediatric Oncology DIPG Registries. J Clin Oncol 2018;36(19):1963–72 doi 10.1200/JCO.2017.75.9308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lulla RR, Saratsis AM, Hashizume R. Mutations in chromatin machinery and pediatric high-grade glioma. Sci Adv 2016;2(3):e1501354 doi 10.1126/sciadv.1501354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lu VM, Alvi MA, McDonald KL, Daniels DJ. Impact of the H3K27M mutation on survival in pediatric high-grade glioma: a systematic review and meta-analysis. J Neurosurg Pediatr 2018;23(3):308–16 doi 10.3171/2018.9.PEDS18419. [DOI] [PubMed] [Google Scholar]
- 6.Hargrave D, Chuang N, Bouffet E. Conventional MRI cannot predict survival in childhood diffuse intrinsic pontine glioma. J Neurooncol 2008;86(3):313–9 doi 10.1007/s11060-007-9473-5. [DOI] [PubMed] [Google Scholar]
- 7.Pincus DW, Richter EO, Yachnis AT, Bennett J, Bhatti MT, Smith A. Brainstem stereotactic biopsy sampling in children. J Neurosurg 2006;104(2 Suppl):108–14 doi 10.3171/ped.2006.104.2.108. [DOI] [PubMed] [Google Scholar]
- 8.Roujeau T, Machado G, Garnett MR, Miquel C, Puget S, Geoerger B, et al. Stereotactic biopsy of diffuse pontine lesions in children. J Neurosurg 2007;107(1 Suppl):1–4 doi 10.3171/PED-07/07/001. [DOI] [PubMed] [Google Scholar]
- 9.Hamisch C, Kickingereder P, Fischer M, Simon T, Ruge MI. Update on the diagnostic value and safety of stereotactic biopsy for pediatric brainstem tumors: a systematic review and meta-analysis of 735 cases. J Neurosurg Pediatr 2017;20(3):261–8 doi 10.3171/2017.2.PEDS1665. [DOI] [PubMed] [Google Scholar]
- 10.Wang Y, Springer S, Zhang M, McMahon KW, Kinde I, Dobbyn L, et al. Detection of tumor-derived DNA in cerebrospinal fluid of patients with primary tumors of the brain and spinal cord. Proc Natl Acad Sci U S A 2015;112(31):9704–9 doi 10.1073/pnas.1511694112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bonner ER, Bornhorst M, Packer RJ, Nazarian J. Liquid biopsy for pediatric central nervous system tumors. NPJ Precis Oncol 2018;2:29 doi 10.1038/s41698-018-0072-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miller AM, Shah RH, Pentsova EI, Pourmaleki M, Briggs S, Distefano N, et al. Tracking tumour evolution in glioma through liquid biopsies of cerebrospinal fluid. Nature 2019;565(7741):654–8 doi 10.1038/s41586-019-0882-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stallard S, Savelieff MG, Wierzbicki K, Mullan B, Miklja Z, Bruzek A, et al. CSF H3F3A K27M circulating tumor DNA copy number quantifies tumor growth and in vitro treatment response. Acta Neuropathol Commun 2018;6(1):80 doi 10.1186/s40478-018-0580-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Panditharatna E, Kilburn LB, Aboian MS, Kambhampati M, Gordish-Dressman H, Magge SN, et al. Clinically Relevant and Minimally Invasive Tumor Surveillance of Pediatric Diffuse Midline Gliomas Using Patient-Derived Liquid Biopsy. Clin Cancer Res 2018;24(23):5850–9 doi 10.1158/1078-0432.CCR-18-1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pentsova EI, Shah RH, Tang J, Boire A, You D, Briggs S, et al. Evaluating Cancer of the Central Nervous System Through Next-Generation Sequencing of Cerebrospinal Fluid. J Clin Oncol 2016;34(20):2404–15 doi 10.1200/JCO.2016.66.6487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McEwen AE, Leary SES, Lockwood CM. Beyond the Blood: CSF-Derived cfDNA for Diagnosis and Characterization of CNS Tumors. Front Cell Dev Biol 2020;8:45 doi 10.3389/fcell.2020.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Euskirchen P, Bielle F, Labreche K, Kloosterman WP, Rosenberg S, Daniau M, et al. Same-day genomic and epigenomic diagnosis of brain tumors using real-time nanopore sequencing. Acta Neuropathol 2017;134(5):691–703 doi 10.1007/s00401-017-1743-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koschmann C, Wu Y-M, Kumar-Sinha C, Lonigro R, Vats P, Kasaian K, et al. Clinically Integrated Sequencing Alters Therapy in Children and Young Adults With High-Risk Glial Brain Tumors. JCO Precision Oncology 2018(2):1–34 doi 10.1200/po.17.00133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li H Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 2018;34(18):3094–100 doi 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009;25(16):2078–9 doi 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Di Filippo M, Bernardi G. The early apoptotic DNA fragmentation targets a small number of specific open chromatin regions. PLoS One 2009;4(4):e5010 doi 10.1371/journal.pone.0005010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leija-Salazar M, Sedlazeck FJ, Toffoli M, Mullin S, Mokretar K, Athanasopoulou M, et al. Evaluation of the detection of GBA missense mutations and other variants using the Oxford Nanopore MinION. Mol Genet Genomic Med 2019;7(3):e564 doi 10.1002/mgg3.564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang TY, Piunti A, Lulla RR, Qi J, Horbinski CM, Tomita T, et al. Detection of Histone H3 mutations in cerebrospinal fluid-derived tumor DNA from children with diffuse midline glioma. Acta Neuropathol Commun 2017;5(1):28 doi 10.1186/s40478-017-0436-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Orsini P, Minervini CF, Cumbo C, Anelli L, Zagaria A, Minervini A, et al. Design and MinION testing of a nanopore targeted gene sequencing panel for chronic lymphocytic leukemia. Sci Rep 2018;8(1):11798 doi 10.1038/s41598-018-30330-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arrillaga-Romany I, Odia Y, Prabhu VV, Tarapore RS, Merdinger K, Stogniew M, et al. Biological activity of weekly ONC201 in adult recurrent glioblastoma patients. Neuro Oncol 2020;22(1):94–102 doi 10.1093/neuonc/noz164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hall MD, Odia Y, Allen JE, Tarapore R, Khatib Z, Niazi TN, et al. First clinical experience with DRD2/3 antagonist ONC201 in H3 K27M-mutant pediatric diffuse intrinsic pontine glioma: a case report. J Neurosurg Pediatr 2019:1–7 doi 10.3171/2019.2.PEDS18480. [DOI] [PubMed] [Google Scholar]
- 27.Chi AS, Tarapore RS, Hall MD, Shonka N, Gardner S, Umemura Y, et al. Pediatric and adult H3 K27M-mutant diffuse midline glioma treated with the selective DRD2 antagonist ONC201. J Neurooncol 2019;145(1):97–105 doi 10.1007/s11060-019-03271-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quail MA, Smith M, Coupland P, Otto TD, Harris SR, Connor TR, et al. A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genomics 2012;13:341 doi 10.1186/1471-2164-13-341. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.