

Abstract
Background and Aims:
Chromosomal instability is a hallmark of cancer that results in broad and focal copy number alterations (CNAs), two events associated with distinct molecular, immunological and clinical features. In hepatocellular carcinoma (HCC), the role of CNAs has not been thoroughly assessed. Thus, we dissected the impact of CNA burdens on HCC molecular and immune features.
Methods:
We analyzed SNP-array data from 452 paired tumor/adjacent resected HCCs and 25 dysplastic nodules. For each sample, broad and focal CNA burdens were quantified using CNApp, and the resulting broad scores (BS) and focal scores (FS) were correlated with transcriptomic, mutational and methylation profiles, tumor immune composition, and with clinico-pathological data.
Results:
HCCs with low broad CNA burdens (defined as BS≤4; 17%) presented high inflammation, active infiltrate signalling, high cytolytic activity, and enrichment of the ‘HCC-Immune-Class’ and gene signatures related to antigen presentation. Conversely, tumors with chromosomal instability (high broad CNA loads, BS≥11; 40%), displayed immune excluded traits and were linked to proliferation, TP53 dysfunction, and DNA repair. Candidate determinants of the low cytotoxicity and immune exclusion features of high-BS tumors included alterations in antigen presenting machinery (i.e. HLA), widespread hypomethylation and decreased rates of observed/expected neoantigenic mutations. High focal scores were independent of tumor immune features, but were related to proliferation, TP53 dysfunction and progenitor cell traits.
Conclusions:
HCCs with high chromosomal instability exhibit features of immune exclusion, whereas tumors displaying low burdens of broad CNAs present an immune active profile. These CNA scores can be tested to predict response to immunotherapies.
Keywords: Broad scores, Focal scores, HCC immune class, inflamed tumors, immune exclusion, cold tumors, immunotherapy, biomarker, methylation
Introduction
Liver cancer is the fourth leading cause of cancer-related death and a major health problem globally[1]. Among liver cancers, hepatocellular carcinoma (HCC) is the most common form (∼90% of primary liver cancers), with over 850,000 new cases per year worldwide, and is a type of tumor that occurs more commonly among men[1]. Despite recent major advances in the understanding of the molecular pathogenesis of HCC, current therapeutic options remain limited, with curative approaches only available for a minority of patients diagnosed at early stages[2,3]. Of note, around 40% of HCC patients are diagnosed at advanced stages, where the kinase inhibitors sorafenib and lenvatinib are first-line standard systemic therapies, while regorafenib, cabozantinib and ramucirumab are administered in second line[2]. Only recently, the first-line combination therapy with the immune checkpoint inhibitor (ICI) atezolizumab and the VEGF inhibitor bevacizumab (monoclonal antibodies against PD-L1 and VEGF-A, respectively) showed significantly improved survival rates in patients with unresectable HCC compared to the standard of care sorafenib in the IMbrave 150 phase III clinical trial[4]. Response rates in this trial and in phase-1/2 trials testing nivolumab and pembrolizumab (anti-PD-1) or combinations of nivolumab and ipilimumab (anti- CTLA-4)[5-7] ranged between 15-30% of patients, highlighting the need for biomarkers of response/resistance in HCC and an overall better comprehension of the tumor immune responses[8].
In this regard, we have recently characterized the immune landscape of HCC tumors and identified the HCC Immune Class, a subgroup of approximately one third of HCCs whose immune profile is characterized by high TILs, high expression levels of PD-L1 and PD1 and markers of cytolytic activity[9]. Additionally, we have described the HCC Exclusion Class, with exclusion of TILs and other ‘cold’ tumors traits, and prevalent in ~30% of HCCs[10].
Copy number changes of chromosomal segments, also known as aneuploidy, are a common feature of human cancers and have been proposed as a driving force of tumorigenesis[11]. Aneuploidy encompasses broad somatic copy number alterations (CNAs), involving large portions of chromosomes, and smaller focal CNAs. Recently, it has been uncovered that the loads of broad and focal CNAs differentially correlate with gene expression markers related to hallmarks of cancer, cell proliferation and immune evasion[12,13], suggesting that they are involved in the carcinogenic process through distinct mechanisms. While high levels of focal events correlate with proliferation markers[12] and can have increased prognostic value compared to broad CNAs[14], high levels of broad events were strongly associated with markers of immune evasion (in several tumor types) and with a reduced response to immunotherapy (e.g. in NSCLC and melanoma)[12,13,15]. Thus, the interaction between the cancer genome and the immune system might be regulated through a general gene dosage imbalance governing over specific gene alterations. Nevertheless, none of the above mentioned studies –or any other published- has specifically deepened on such analysis in HCC. In addition, recent reports have shown that the intensity and direction of the association between CNAs and tumor immunity may not be universal in all cancer types[15-18], underlining the need to investigate the impact of aneuploidy considering the tumor tissue-specific context.
In a previous work, we assessed whether the number of statistically-significant recurrent amplifications or deletions in HCC correlated with the immune classes[9], but a thorough characterization of the CNA burden was unaddressed. Thus, we analyzed the effect of focal and broad CNA loads using a newly-described scoring strategy[19] and correlated the scores with molecular and immune features. Our findings suggest that HCCs with high burden of broad CNAs correlate with immune exclusion profiles and proliferation signatures; on the other end, tumors with lower burdens of broad CNAs were enriched in the HCC Immune Class[9] and in pro-inflammatory pathways, presented higher cytolytic activity and exhibited absence of CTNNB1 mutations. Consequently, we propose that the ‘hot’ tumor traits of HCCs with low CNA burdens may make them potential candidates to a sustained favorable response to immune checkpoint inhibitors. In contrast, the ‘non-inflamed’ traits and reduced cytotoxic immunophenotype of HCCs with higher levels of broad CNAs are consistent with the notion that broad events may be implicated in immune escape mechanisms and could be indicators of resistance to immunotherapies, as observed in other cancer types[12].
Materials and methods
Study cohorts
A total of 452 samples were analyzed (Supplementary Figure 1A), including a discovery cohort (n=107)[20] and a validation cohort (n=345; TCGA)[21]. The main clinico-pathological features of these two cohorts are summarized in Supplementary Table 1. The Heptromic cohort is characterized by mostly caucasian patients, median age 66 years old, males (78%), with HCV-related HCC (46%), at early stages of the disease (86% BCLC 0-A) undergoing resection for single tumors (77%) with AFP <400ng/ml (85%) in patients with well-preserved liver function (Child-Pugh A: 98%, platelet count >100,000: 82%). At pathological examination, 36% of tumors presented microvascular invasion and 24%, satellites. Patients from the TCGA cohort were characterized by a median age of 61 years old, 68% were males, with 12% of cases related to HCV infection, at early stages of the disease (75% AJCC-T1 and AJCC-T2), and the majority of them presented a well-preserved liver function (Child-Pugh A: 91%, platelet count > 100,000: 94%). At pathological examination, 35% of tumors presented microvascular invasion. In addition to this two cohorts, previously published pre-malignant samples (n=25) and very early HCCs (veHCC, n=18) were included in the study[20,22]. Finally, a publicly available transcriptome dataset corresponding to 65 pre-treatment samples of cancer patients treated with anti-PD-1[23] was used to run the subclass mapping. More details on the cohorts have been included in Supplementary Data File.
CNA data processing and determination of the CNA level
The HEPTROMIC and TCGA-LIHC SNP array data, and the CNApp web tool[19], were used to quantify the individual CNA burdens of each sample, and to generate the broad and focal CNA scores (BS and FS, respectively). Details are provided in the Supplementary Data.
Results
Profiling broad and focal copy number alterations in human HCC
To determine the impact of the genomic imbalances in the molecular and immune features of HCC, we analyzed 452 samples profiled by SNP array: 107 HCCs samples (HEPTROMIC, discovery cohort) and 345 HCC samples (TCGA-LIHC, validation cohort; Supplementary Figure 1). Using the validated CNApp re-segmentation approach (see Materials and methods), we first generated each sample’s genome-wide CNA landscape, and then categorized the detected chromosomal amplifications and deletions into broad and focal events[12]. HCC tumors displayed a median of 6.8 and 8.3 broad CNAs per sample in the HEPTROMIC and TCGA-LIHC cohorts, respectively; and a median of 21.0 and 30.3 focal CNAs per sample. In terms of lengths, broad chromosomal fragments had, respectively, a median size of 69.7Mb and 62.4Mb (HEPTROMIC: range of 9.5Mb–243Mb; LIHC-TCGA: range of 8.8Mb-245Mb). Focal CNAs had a median length of 1.7 Mb in HEPTROMIC and 0.6 Mb in LIHC-TCGA, and 84% of all focal CNAs were below 12 Mb[24] (81% in HEPTROMIC (n=107); 85% in LIHC-TCGA (n=345); Supplementary Figure 1). Overall, no major differences were identified between the CNA landscapes of the two cohorts (Supplementary Figure 1), and the profiled landscapes of the most frequent events were consistent with previously published analysis applying the GISTIC algorithm on other HCC cohorts[24-26]. In short, the most recurrent broad gains in the two cohorts (n=452) were in 1q (52%) and 8q (47%), and the most frequent broad losses were in 8p (52%) and 17p (45%). We also detected slightly less-prevalent broad losses in 4q (30%), 6q (24%) and 16q (28%). CNApp also identified frequent high focal gains characteristic of HCC such as the ones located in 5p15.33 (in 63/452 HCCs (14%); involving TERT), in 11q13.3 (>6%; affecting CCND1 and FGF19), or in 6p21.1 (10%; including VEGFA), among others. Despite the similar genomic landscapes, the two analyzed cohorts presented some differences at the clinico-pathological level (Supplementary Table 1). TCGA-LIHC patients were at more advanced stage and presented poorer liver function. Median age was also slightly different (in HEPTROMIC was 66 vs 61 in the TCGA (p<2.6E-5)) and etiology distribution differed in the two cohorts, HCV-related HCC in HEPTROMIC accounted for 45% of cases and alcohol-related HCC in TCGA-LIHC accounted for 32% of cases. These differences however, did not impact the CNA profiles as no significant associations were found between CNAs and the analyzed clinico-pathological variables. Next, we quantified the load of broad and focal CNAs in each sample using CNApp. The power of CNApp at capturing the actual fraction of altered genome through scores has been previously demonstrated using the TCGA pan-cancer dataset of >10,000 samples[19]. Summarized, for each tumor CNApp provided a broad and a focal score (BS and FS, respectively) accounting for the number, amplitude and length of broad and focal segments.
In order to rank the aneuploidy levels of HCC among the cancer spectrum, we conducted a pan-cancer analysis using the CNApp-derived broad and focal scores of 10,635 TCGA samples representing 33 tumor types[19] (Supplementary Table 2). This analysis revealed that in HCC, the BS and FS distributions were intermediate when compared with the other 32 tumor types (Figure 1A and 1C).
Figure 1. Diagrams representing CNApp broad and focal score distributions and their correlation with the ESTIMATE immunity score in a pan-cancer cohort.
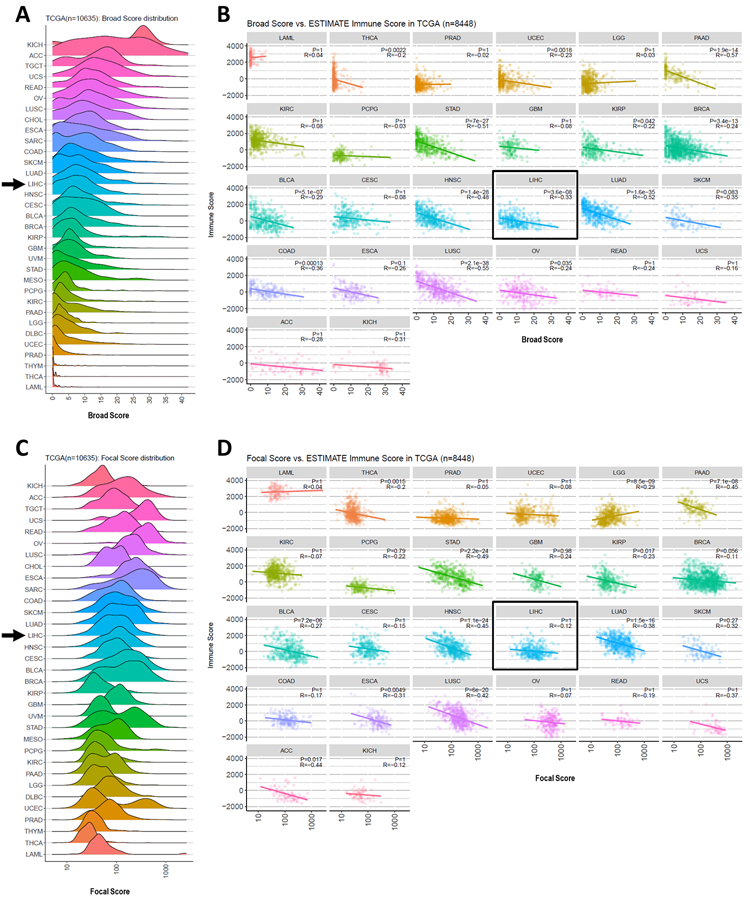
(A) Broad score distribution across 33 cancer types from the TCGA. (B) Dot-plots displaying the correlation between broad score and ESTIMATE-derived immune score per cancer type. (C) Focal score distribution across 33 cancer types from the TCGA. (D) Dot-plot with focal score and immune score correlations per cancer type. P: spearman correlation p-value; R: Spearman regression coefficient. Arrow in (A) and (C) and box in (B) and (D) indicate HCC. TCGA cancer project abbreviations: LAML, Acute Myeloid Leukemia; THCA, Thyroid Carcinoma; PRAD, Prostate Adenocarcinoma; UCEC, Uterine Corpus Endometrial Carcinoma; LGG, Brain Lower Grade Glioma; PAAD, Pancreatic Adenocarcinoma; KIRC, Kidney Renal Clear Cell Carcinoma; PCPG, Pheochromocytoma and Paraganglioma; STAD, Stomach Adenocarcinoma; GBM, Glioblastoma Multiforme; KIRP, Kidney Penal Papillary Cell Carcinoma; BRCA, Breast Invasive Carcinoma; BLCA, Bladder Urothelial Carcinoma; CESC, Cervical Squamous Cell Carcinoma and Endocervical Adenocarcinoma; HNSC, Head and Neck Squamous Cell Carcinoma; LIHC, Liver Hepatocellular Carcinoma; LUAD, Lung Adenocarcinoma; SKCM, Skin Cutaneous Melanoma; COAD, Colon Adenocarcinoma; ESCA, Esophageal Carcinoma; LUSC, Lung Squamous Cell Carcinoma; OV, Ovarian Serous Cystadenocarcinoma; READ, Rectum Adenocarcinoma; UCS, Uterine Carcinosarcoma; ACC, Adrenocortical Carcinoma; KICH, Kidney Chromophobe Cell Carcinoma.
To explore the impact of the CNA profiles on the molecular and immune characteristics of the tumors, we categorized each sample into high/low-BS and high/low-FS according to the BS and FS quartiles. The threshold for high-BS tumors was set at ≥11 (the upper quartile of both the TCGA pan-cancer cohort and the discovery cohort), and for low-BS at ≤4 (coinciding with the lower quartile in the discovery cohort). In terms of FS, low-FS samples were defined as those with FS ≤ 13.5, while high-FS was defined as those with FS ≥ 47, considering the quartile values in the discovery cohort. Overall, 17% of the analyzed tumors presented low broad CNA burdens (75/452) and 40%, high loads of broad CNAs (183/452). In terms of focal events, 20% of cases were low-FS (92/452) and 43%, high-FS (196/452).
Burdens of broad and focal copy number alterations are associated with distinct molecular features
Stratifying HCC tumors according to their BS and assessing the correlation with molecular and immune features revealed that HCCs with fewer broad genomic imbalances (those with low-BS) were characterized by high inflammation (increase of hallmarks of inflammatory response, TNF-alpha, IL2-JAK-STAT signalling) and interferon signalling (Figure 2A). Interestingly, these tumors presented enrichment of the HCC Immune class and Active Immune subclass[9] when compared to tumors with higher BS (p=0.02 and p=0.04, respectively, Figure 2A). In addition, low-BS tumors were deficient in S2[27] proliferative traits (p=0.006), presented significant activation of different immune-related pathways and had LXR/FXR/RXR signaling as one of the top canonical pathways (p<0.001; Supplementary Table 3).
Figure 2: HCC tumors with a low burden of broad copy number chromosomal alterations display higher immune infiltration when compared to HCCs with higher loads of broad events.
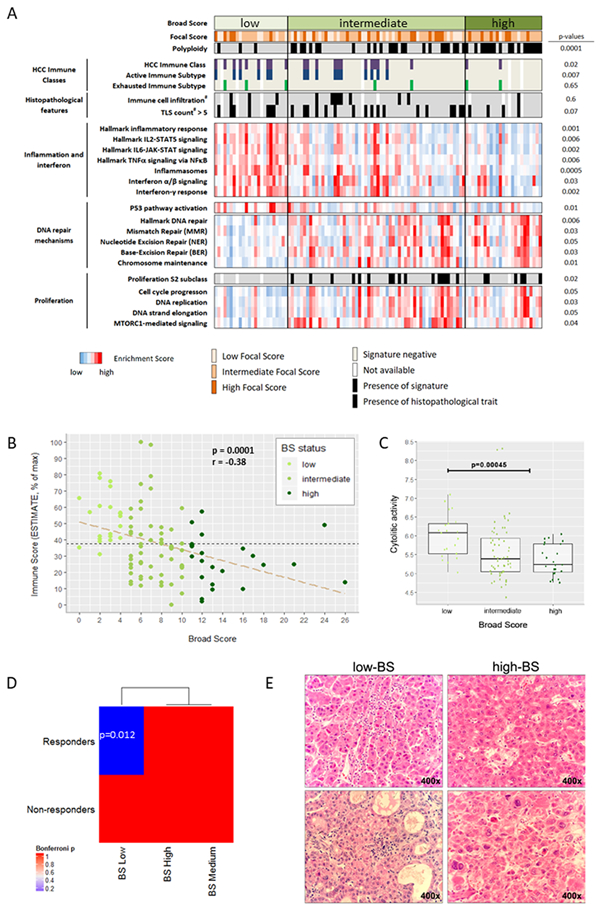
(A) Scores to quantify the burden of broad and focal chromosomal alterations (BS and FS) were computed using CNApp. Tumors with low burdens of broad chromosomal alterations (low BS) were significantly associated with the HCC Immune Class, and were mainly diploid and had functional p53. In contrast, tumors with high BS were markedly polyploid, and were enriched in cell proliferation features and DNA repair hallmarks including TP53 mutations/losses. #: pathologically assessed. Displayed p values were calculated comparing low-BS tumors with high-BS tumors. (B) Transcriptome-based estimation of immune cell infiltration in HCC negatively correlates with broad CNA burdens (n=102, discovery cohort). Data are presented normalized to the maximum Immune Score. r: Spearman’s rank correlation coefficient. (C) BS-low HCC tumors exhibited significantly higher cytolytic activity compared to the rest (n=102, discovery cohort). (D) Molecular similarity between low-BS HCC patients and tumors responsive to PD-1 therapy. Submap analysis was applied considering 2 groups in the HCC cohort (low-BS vs rest) and 2 groups (anti-PD1 responders and non-responders) in the anti-PD1 treated dataset. Low-BS HCC tumors were significantly similar to tumors responding to anti-PD1 (p=0.01). (E) Representative images of immune cell infiltration in low-BS and high-BS HCC tumors from the HEPTROMIC cohort.
In terms of ploidy, we found 40% of HCCs being polyploid (3.9 ± 0.7 sets of chromosomes on average), and the remaining 60% being diploid (2 ± 0.2 sets of chromosomes). Of note, polyploid tumors were significantly enriched in high-BS HCCs (17/25 (68%) in the discovery cohort, p<0.001; 69/158 (44%) in the validation cohort; p<0.0001; Supplementary Table 5 and 6). Low-BS HCCs were mainly diploids (p=0.002; Supplementary Tables 5 and 6). At the clinico-pathological level, none of the variables assessed (including etiology and tumor stage) were linked to low-BS (Supplementary Table 4).
On the other hand, HCC tumors with high burden of broad CNAs presented a completely different molecular profile. High-BS tumors were characterized by hallmarks of mismatch repair and proliferation and presented low immune infiltration -inferred from the tumor transcriptomic profile[28]- (Figure 2A, Supplementary Table 5). Consistently they were significantly excluded from the HCC Immune class (p=0.02). Pathway analysis further supported these findings, providing cell cycle control of chromosomal replication and DNA damage as top canonical pathways (Supplementary Table 3). In addition, Ingenuity Pathway Analysis (IPA) revealed a role for the arginase pathway in these tumors. The parallel evaluation of the TCGA cohort confirmed these observations (Supplementary Figure 2, Supplementary Table 6).
When classifying the tumors according to their FS, we observed that those with high levels of focal events were characterized by classical proliferative features and were associated with poor survival signatures both in the discovery and in the validation cohorts (Figure 3A; Supplementary Figure 3A; Supplementary Tables 7 and 8). Consistently, transcriptome-based pathway analysis revealed cell cycle-related pathways as the top canonical pathways for high-FS tumors. In addition, these tumors were enriched in the HCC proliferation subclass[24] (p<0.004) and were excluded from the CTNNB1-subclass[24] (p=0.032). This molecular profile was aligned with CTNNB1 mutations being significantly excluded from high-FS tumors (p<0.001), while TP53 mutations were enriched among them (p<0.001, Supplementary Figure 3A and Supplementary Table 8). In addition, high-FS tumors were positive for the signatures of hepatic progenitors[29] (p=0.004), CK19[30,31] (p=0.01), vascular invasion[32] (p=0.02), MET[33] (p=0.029) and late TGFβ signaling[34] (p=0.005). At the clinico-pathological level, these tumors were associated with poor cell differentiation (p<0.001)(Supplementary Tables 7 and 8). Interestingly, we did not find a significant association between FS and tumor immune profiles (Figure 3A; Supplementary Figure 3; Supplementary Tables 7 and 8). Despite displaying traits of poor prognosis, FS status did not correlate with patient’s overall survival. Levels of broad CNAs were also not associated with patient’s outcome.
Figure 3: HCC tumors with a high burden of focal copy number chromosomal alterations exhibit proliferation and poor prognosis traits but no association with tumor immunity.
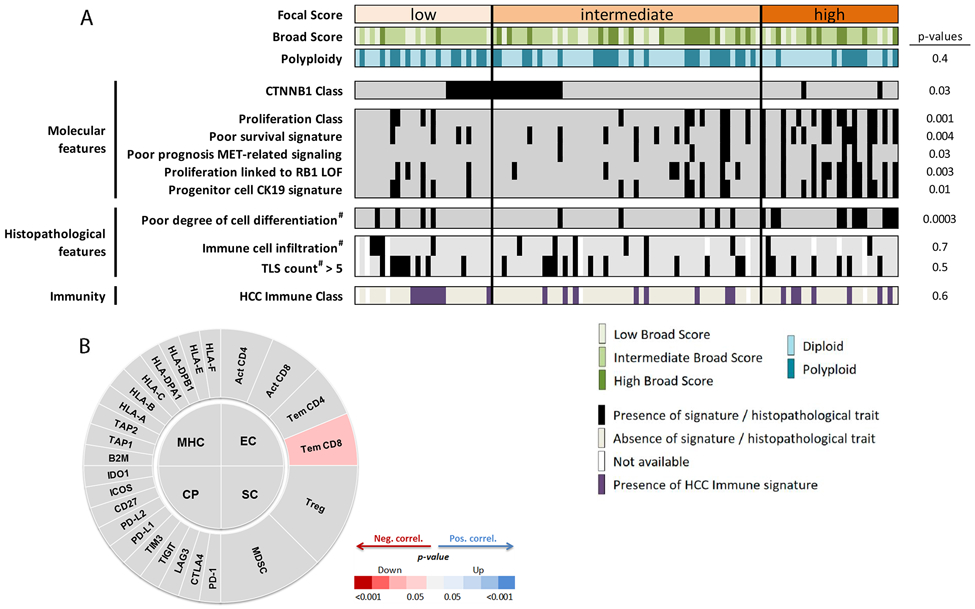
(A) Tumors with a high level of focal events (high Focal Score (high-FS)) were enriched in poor prognosis and proliferation signatures, and presented a higher degree of cell differentiation. HCCs with low or intermediate levels of focal copy number alterations (low-FS and intermediate-FS) were enriched in WNT-CTNNB1 signalling. Focal Scores were not associated with immune signalling. #: pathologically assessed. (B) Immunophenoscore diagram correlating FS with expression features from effector cells (EC), immune checkpoints (CP), major histocompatibility complex-related components (MHC) and suppressor cells (SP). Focal scores did not correlate with any of the immunophenoscore groups of genes.
Overall, and as in other cancer types, burdens of broad CNAs, but not focal, impact on the HCC immune profile, and this also supports a differential role for focal and broad events as determinants of HCC anti-tumor immunity.
Low broad CNAs loads correlate with the HCC immune class, and high burdens of broad CNAs correlate with immune exclusion
As above mentioned, low BS tumors presented hallmarks of inflammatory and interferon signalling and were characterized by immune traits as seen by an enrichment of the HCC Immune Class[9]. Chemokine and cytokine signalling as well as signatures of innate (cytotoxicity mediated by NK cells, monocytes, dendritic cells) and adaptive immunity (cytotoxic T cells, T helper lymphocytes, B and T cell receptor signalling) were also enriched in low-BS tumors (Supplementary Figure 4A). Interestingly, expression levels of immune checkpoints such as PD1 and CTLA4 were higher in these tumors compared to HCCs with intermediate or high-BS (Supplementary Figure 4A). The presence of higher immune infiltrate in low-BS HCCs was confirmed at the pathological level and using transcriptome-based independent algorithms (Figure 2)[28,35,36]. Firstly, pathological assessment of tumor immune infiltrates in our cohort revealed a trend towards higher immune cell infiltration (p=0.054) and higher count of Tertiary Lymphoid Structures (TLS; p=0.06; Figure 2A and 2E, Supplementary Table 5) in low-BS tumors. TLS density has been reported to correlate with CD8+ and CD4+ T cell density in tumors and its presence is associated with favorable prognosis[37,38]. Secondly, levels of immune cell infiltrate were inferred from the tumor whole genome expression profiles[28], and were found significantly increased among HCCs with low CNA burden (low-BS; p=0.0001; Figure 2B). Thirdly, we adapted the Immunophenoscore[35] (Supplementary Table 9) to further dissect the transcriptome of our discovery and validation cohorts and confirmed the above mentioned immune traits of low-BS HCCs (Supplementary Figures 2C and 4B). We also inferred the cytolytic activity of each tumor[36] and observed that it was higher in tumors with reduced broad CNA burdens (p<0.001, Figure 2C). In addition, we performed a submap analysis comparing the molecular profile of our HCC tumors grouped according to BS, and a publicly-available molecular print of treated tumors[23], classified according to their response to anti-PD1 treatment. Remarkably, we only observed a significant association between low-BS tumors and tumors responding to anti-PD1 therapy (p=0.01, Figure 2D). Further, we identified significantly-elevated expression of HLA-A molecules in BS low tumors when compared to the rest (p<0.001, Supplementary Figure 4). Finally, pathway analysis revealed several interleukins and IFNγ among the top upstream regulators in low-BS HCC tumors (Supplementary Table 3), as well as a consistent association with different immune-related pathways [(antigen presentation (p<0.001); communication between innate and adaptive immune cells (p<0.001); differential regulation of cytokines (p<0.001); IL-17-mediated production of immune cells (p<0.001); and B cell development (p<0.001)].
Regarding HCC tumors with a high burden of broad CNAs (high-BS), in addition to low immune infiltration, they were significantly excluded from the HCC Immune Class (n=2/25; p=0.02) and the HCC Active Immune subclass (n=0/25; p=0.01; Supplementary Table 5). This was mirrored in the TCGA cohort, where high-BS tumors had significantly less HCC Immune-Class patients compared to low-BS tumors (17% vs 37%, p=0.006). In addition, in both cohorts high-BS HCCs were not related to immunosuppressive features (Supplementary Figures 2C and 4B). This is consistent with the notion that aneuploidy may play a role as a mechanism of tumor immune evasion[12].
In this regard, a previous study analysing the aneuploidy profile of 12 cancer types (not including HCC) proposed that high levels of CNAs were drivers of immune exclusion[12]. Thus we assessed whether our scores were associated to immunity in a large spectrum of cancer types. To this end, we correlated the BS and FS scores from 10,635 TCGA samples representing 27 tumor types, with a transcriptomic surrogate of the immune infiltrate level: the Immune score from the ESTIMATE tool[28]. Strikingly, in terms of broad alterations, we observed a significant negative correlation between Immune score and BS in 14 out of the 27 tumor types analyzed, including HCC (Figure 1B and 1D). In terms of focal events, our FS correlated significantly with the Immune score in 12 out of 27 tumor types, but HCC was not among them (Figure 1B and 1D). This places HCC in the subset of tumors were broad aneuploidy events adversely affect immune cell action against tumors[12], but also corroborates that the relation between immunity and CNA alterations is not universal across cancer types, highlighting the importance of considering the tumor specific context[15-18].
Pre-neoplastic lesions and very early HCC tumors with high loads of broad CNAs also present reduced immune features
Following the identification of immune features associated to low-BS HCCs, we sought to explore whether the burden of broad copy-number changes (BS) could also influence the modulation of the immune system at the early stages of hepatocarcinogenesis. To this end, we analyzed a total of 25 pre-malignant dysplastic nodules (DN) and 18 veHCCs[20,22]. Since the pre-neoplastic lesions presented very few broad chromosomal rearrangements, we categorized them according to the presence or absence of broad CNAs, rather than to compute their BS. Overall, we found that the presence of broad alterations in 4/25 (16%) of the analyzed DNs (‘presence’ being equivalent to ‘BS-high’) and it was linked to reduced molecular features of active antitumor immunity (Supplementary Figure 5). More precisely, BS-high DNs displayed trends towards reduced cytolytic activity values (Supplementary Figure 5A) and towards reduced immune cell infiltration (Supplementary Figure 5B). On the other end, lesions without broad CNAs presented enrichment of effector cell features, immune checkpoints, MHC-related components and suppressor cells (Supplementary Figures 5C and 5D). Similarly, analysis of veHCCs also revealed a significant negative correlation between BS and cytolytic activity, as well as the expression-derived Immune Score reflecting immune infiltration (Supplementary Figure 6).
Overall, our results suggest that there is a continued association between BS and immunity throughout the hepatocarcinogenic process, from DNs to veHCCS to more advanced tumors.
Potential mechanisms determining the distinct molecular and immune landscapes of tumors with different types of copy number profiles
a). Tumor Mutational Burden (TMB), neoantigens and TP53 status
The fact that differences in broad CNA burdens were associated with distinct immune profiles suggests that mechanisms related to overall gene imbalance may contribute to determining the activation of molecular and immune pathways in HCC. Here, we assessed the potential contribution of the mutational load (both silent and non-silent mutations), the TP53 status, the total predicted neoantigens and the ratio of observed/expected neoantigens in determining the different HCC immune characteristics of specific CNA profiles. For this purpose, we used the genomic profiles of 179 patients available from the TCGA HCC cohort[36], that presented an average of reported non-silent mutations per Megabase of 2.5 (TMB), with only 2% of the samples (n=6/302) with TMB>10 mutations/Mb[39]. Our analysis on the total number of mutations per sample revealed that this was significantly lower among low-BS tumors as compared to the rest (p=0.023, Figure 4A). Consistently, pan-cancer studies have described increased mutational burdens among tumors with increased aneuploidy[15]. Since the ability to trigger a tumor immune response does not only depend on the number of mutations but also on their antigenicity, we also calculated the ratio of observed/expected neoantigens. This ratio measures how much the number of observed neo-epitopes (defined based on the mutation’s potential to bind HLA with high affinity, and taking into accound the gene expression) deviates from the number of expected neoantigens (inferred using a pan-cancer empirical model) in each sample[36]. Interestingly, we observed that low-BS tumors displayed higher ratios of observed/expected neoantigens when compared with high-BS tumors (p=0.015) and that BS negatively correlated with this ratio (p=0.028, Figure 4B). In terms of mutations, low-BS tumors presented a positive correlation between total number of mutations and ratio of observed/expected neoantigens (p=0.028), whereas such correlation was not observed in the rest of the cohort (Figure 4C). Overall, this suggests that, despite presenting lower loads of mutations, low-BS tumors rendered more neoantigens than expected. In contrast, the higher mutational burdens together with the lower ratios of observed/expected neoantigens among high-BS tumors point towards a negative selection of neoantigens that could be rendering higher rates of less immunogenic mutations among tumors accumulating broad CNAs (Figure 4A to 4D).
Figure 4: Impact of the mutational landscape in HCC CNA profiles.
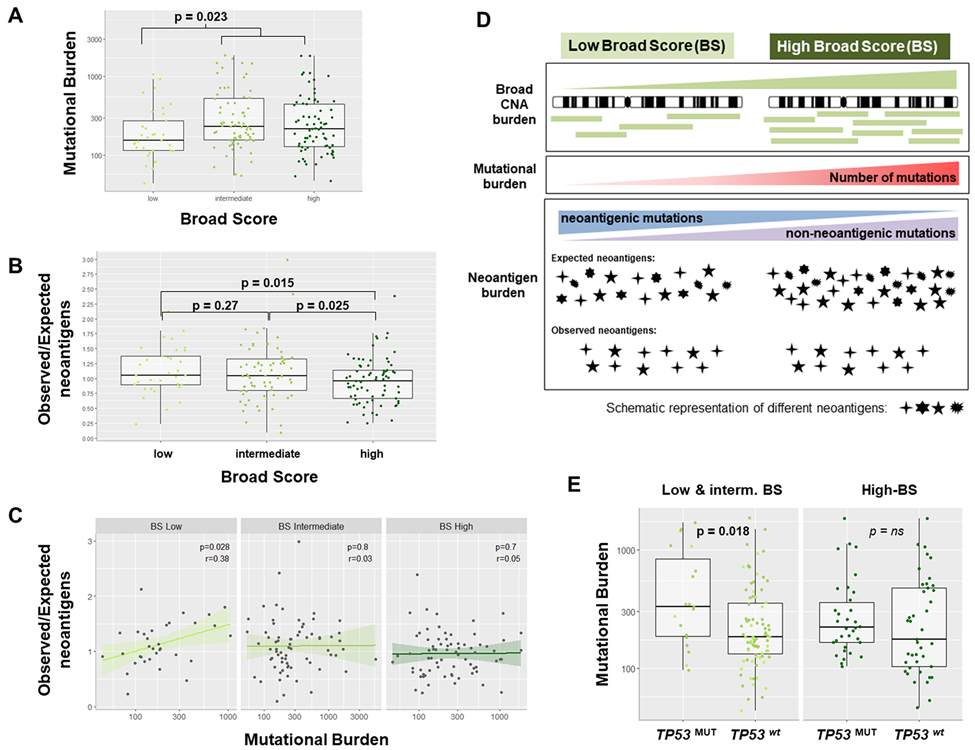
(A) HCC tumors with a low burden of broad copy number alterations (low-BS) exhibited a significantly lower mutational load (silent + non-silent mutations) when compared to the rest (n=179 LIHC-TCGA; p=0.023). (B) BS-high tumors were associated with lower ratios of observed/expected neoantigens, suggesting a negative selection of low immunogenic mutations in these tumors (n=179). (C) Mutational burden in HCC significantly correlated with the ratio of observed/expected neoantigens only in BS-low tumors. (D) Diagram summarizing the relationship between broad CNA loads, mutational burden and observed/expected neoantigens in HCC. (E) Total number of mutations in HCC tumors characterized by high burdens of broad CNAs (high-BS) was not related to TP53 mutational status. In contrast, low-BS HCCs (and intermediate) tumors with non-functional TP53 (TP53MUT) displayed a significantly higher number of mutations compared to those with wild type TP53 (TP53wt). r: Spearman’s rank correlation coefficient.
We next assessed whether our observed association between CNA burden and immune infiltration further depended on the mutational status of TP53, given its role in preventing genome mutations. We observed that the total number of mutations (silent and non-silent) was independent from the TP53 mutational status among high-BS tumors, whereas for the rest of tumors, the number of mutations was higher when TP53 was non-functional (p=0.018, Figure 4E). A similar observation was made regarding neoantigen burdens: predicted neoantigens were independent from the TP53 status only in high-BS tumors. In the rest of tumors, those with mutated TP53 displayed significantly higher numbers of predicted neoantigens as compared with those with wild-type TP53 (p=0.0082, Supplementary Figure 7A). Thus, our analysis suggests that TP53 could have a role in shaping the immunogenicity of tumors that do not accumulate high loads of broad genomic imbalances.
b). T cell-related and antigen presenting machinery-related mechanisms
Additionally, we assessed whether amplification or deletion of specific immune-related genes could contribute to explain the link between immunity and BS score levels in HCC. Screening the CNApp segmentation output, we identified copy number losses significantly enriched in high-BS tumors from the discovery and validation cohorts, potentially contributing to the observed immune phenotype. These included losses in genes from the antigen-presentation machinery, T cell receptor components and immune checkpoints (Supplementary Figure 7). Remarkably, few of these alterations were detected in low-BS tumors. Among others, we found significant losses of HLA-DQB1, coding for the type MHC class II heterodimer, in high-BS tumors [65/158 high-BS HCCs (35%) vs 6/51 low-BS (12%); p<0.0001]. This was accompanied by a significant reduction of HLA-DQB1 expression in high-BS tumors compared to low-BS tumors in the discovery cohort (p=0.002) and a trend in the validation cohort. Of note, the loss of HLA-DQB1 could mediate a deficit in cytotoxic cell responses in high-BS HCCs. In this regard, in lung cancer, HLA-DQB1 expression on tumor cells has been positively correlated with CD4- and CD8-positive lymphocyte infiltration[40].
c). Epigenetic features
In addition, considering recent evidence revealing the correlation between global demethylation and CNAs across different tumor types[41], and given that this may promote immune evasion in tumors with high copy number load, we analyzed the methylation data available for our cohorts. In the discovery cohort, we observed that among the 17,090 probes differentially methylated between low-BS tumors and the rest, 13,252 (77%) presented higher methylation levels (Supplementary Figure 8). Similarly, in the validation cohort, 65,361 (88%) out of 74,195 differentially methylated probes displayed higher methylation levels among low-BS samples as compared to the rest (Supplementary Figure 8). Therefore, our data align with the fact that the accumulation of broad CNAs leads to a widespread hypomethylation[41], and suggests an impact of epigenetic regulation in HCC immune profiles. The fact that low-BS tumors are characterized by high immune infiltration and higher methylation levels aligns with our previous observation that tumors within the HCC immune class present higher overall levels of methylation[9].
d). Specific contribution of CNA gains and losses
Here, we questioned whether CNA gains and losses contributed equaly in determining the tumor immune profile. This was motivated by a report in melanoma suggesting that the total number of genes with copy number losses was a marker of resistance to immune checkpoint-related treatments[42]. Similar to melanoma, we observed a significant negative correlation between broad losses and cytolytic activity in HCC, both in the discovery and validation cohorts (p=0.0004 in HEPTROMIC; p=0.01 in LICH-TCGA; Supplementary Figure 9). Thus, the more broad losses the tumors have, the less cytolytic activity they present; and therefore less chances to respond to immune checkpoint inhibitors. This was also consistent with our finding that high-BS HCCs are depleted from tumor immune features and suggest that CNA losses may contribute more to this phenotype. No consistent correlations were found in terms of gains (neither broad nor focal) and in terms of focal losses.
Discussion
Copy number alterations are considered a hallmark of cancer. Recently, pan-cancer studies have described broad and focal CNA burdens as genomic features able to determine tumor immune infiltration and immune exclusion in several cancer types[12,15]. Notably, the strength and direction of this immunogenomic correlations may not be common in all cancer types[16-18], mainly due to the different context of each tumor[43,44]. The impact of CNAs in the setting of HCC has not been comprehensively addressed. Here, we used data from a high-resolution whole genome SNP-array to understand the immune and molecular impact of genomic CNA burdens in HCC, taking into account each tumor’s landscape determined using CNApp[19]. Similar to other tumor types, in HCC we observed that different broad copy number landscapes presented distinct immune profiles, while focal CNAs did not impact tumor immunity. Specifically, we observed that HCC tumors with a high burden of broad chromosomal alterations were linked to DNA-repair and proliferation signatures and presented immune exclusion. On the other hand, tumors with low levels of broad CNAs displayed features of immunologically ‘hot’ HCCs: higher immune infiltration and cytolytic activity, as well as up-regulation of pro-inflammatory cues. Thus, like it has been described in melanoma[12], we hypothesized that HCCs with low-BS might represent potential responders to immunotherapies, whereas high-BS HCCs, potential non-responders. Our hypothesis is sustained by the fact that i) tumors responding to anti-PD1 treatment have a molecular profile similar to low-BS HCCs and ii) patients with tumors responding to anti-CTLA-4 or anti-PD-L1 correlate with the expression of IFNγ and GZMB, both overexpressed among low-BS tumors[45-47]. With regard to focal events, HCCs with a high burden of focal CNAs were characterized by pro-proliferative and more aggressive features, and exhibited no CTNNB1 mutations. Overall, in our study we propose a strategy to classify HCCs (which usually do not present high TMB) into immune/inflamed and excluded/non-inflamed tumors based on CNAs, regardless of the mutational load. This classification expands on our proposed HCC immune classification[9,10]. In particular, we propose a broad score ≤4 (low-BS) as a putative marker to identify HCC patients with inflamed tumors, and broad scores ≥11 (high-BS) for HCCs lacking immune-related features (Figure 5). The fact that immune traits may be predicted by broad, but not focal CNA loads is consistent with the above mentioned pan-cancer study[12]. According to this study, tumors with fewer broad copy number events may sustain higher infiltration of cytotoxic immune cells, and the genetic variability induced in high-BS tumors may provide tumor cells with an advantage to avoid immunological recognition. However, since no molecular data of HCC patients treated with immunotherapies are publicly available to date, the predictive value of the scores warrants validation.
Figure 5: Immune classification of HCC.
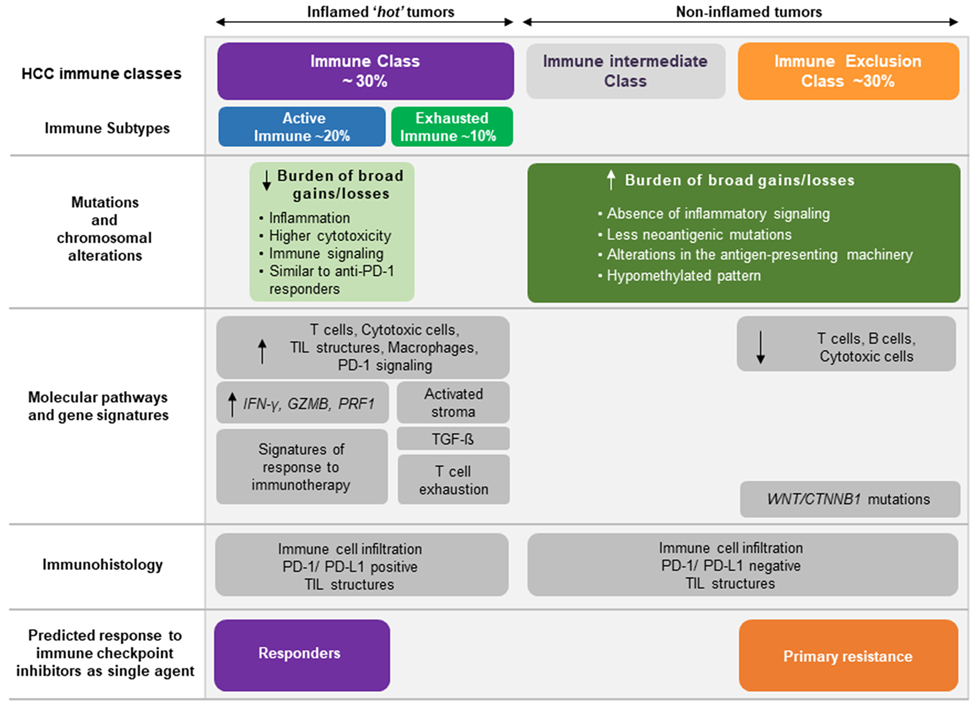
Our results complement the previously proposed immune classification of HCC[9,10]. Tumors with low burden of broad copy number alterations (those with low-BS) presented inflammatory features and were enriched in the HCC immune class. The HCC immune class has been previously reported to display high levels of immune cell infiltration, expression of programmed cell death 1 (PD-1) and/or programmed cell death 1 ligand 1 (PD-L1), activation of IFNγ signaling, and markers of cytolytic activity (such as granzyme B and perforin 1). On the other hand, HCCs with high loads of broad CNAs lacked immune features, paralleling the HCC exclusion class, enriched in CTNNB1 mutations, and are proposed to represent those tumors with innate resistance to anti–PD-1/PD-L1 inhibitors. TIL: tumor-infiltrating lymphocyte.
We also analyzed the potential mechanisms by which an increase in broad CNA burdens might be associated with exclusion of immune infiltration. It has been described that neo-epitopes drive cytolytic activity in tumors[36], and therefore high mutational loads could render higher neoantigen burdens and greater immune infiltration. In the present study we observed that low-BS HCCs displayed lower burdens of mutations in comparison with high-BS tumors. However, the ratio of observed/expected neo-epitopes was lower among high-BS tumors, suggesting that the accumulation of broad CNAs would be linked to an enhanced pruning of neoantigenic mutations, regardless of overall mutation burden. Since the anti-tumor immune responses are conditioned by the functional presentation of tumor antigens[48], such pruning could contribute to the descreased anti-tumor immunity features of high-BS HCCs. Of note, an additional reason could be the reduced antigenicity of neoantigenic mutations among high-BS tumors. In other cancer types, it has been reported that not only the number of neo-antigens was a determinant of the tumor immune status, but also the ability of neo-antigens to induce an immune response[49,50].
In addition, specific CNAs involving immune-related genes could impact the tumor immune features. In particular, gene losses involved in antigen presentation, found enriched among high-BS HCCs (i.e. HLA-DQB1), could drive the failure to attract effector T cells in these tumors. This, and the fact that high-BS HCCs were enriched in arginine degradation pathways, suggests that these tumors may evade immune destruction by impairing the MHC-mediated antigen presentation, as in lung cancer[48], rather than developing an immunosuppressive microenvironment. Overall, our findings are in concordance with recent studies on NSCLC (TRACERx[51]). Tumors sparsely infiltrated exhibited decreased neoantigen editing indicative of copy-number losses of clonal neoantigens, and dysfunction in the neoantigen presenting machinery, among the mechanisms of immune evasion[48,51].
In parallel, the demethylation observed in high-BS tumors could also promote lack of immune infiltration[41]. Even though our data suggests potential explanations, further mechanistic studies will be required to determine the causality between tumor CNA burden and anti-tumor immunity.
On the other hand, the observation that HCCs with high levels of broad CNAs were markedly polyploid, with proliferative features and associated to poor prognosis signatures, is consistent with a recent publication[52] that proposes ploidy as a prognosis marker.
With regard to focal copy number events, the fact that high-FS HCCs were more proliferative is consistent with data from other tumor types[12], and has even been demonstrated experimentally through the depletion of CDKN2A in human cell lines[53].
Lastly, we observed that our findings in HCC were extensible to pre-neoplastic lesions and very early HCCs. Although further confirmatory studies using larger cohorts would be required, these data aligns with the idea that CNAs may contribute to the escape from anti-tumor immune pressure[12,54].
Overall, our study revealed differential associations between broad and focal genomic CNA burdens and several molecular features of HCC. Most importantly, our data reinforce the evidence supporting a differential role for broad CNAs in determining anti-tumor immune profiles in HCC and provide evidence in favour of chromosomal stability as a hallmark of tumor immunogenicity, and consequently, patient response to immunotherapies.
Supplementary Material
Translational Relevance.
In HCC, immune checkpoint inhibitors achieve responses in 15-20% of patients, however, there is no established biomarker predictive of response. Recently, several pan-cancer studies have uncovered that chromosomal alterations strongly associated with specific immune traits recapitulating known ‘immune/inflamed’ and ‘excluded/non-inflamed’ tumor subtypes. These subtypes have been linked to potential response or primary resistance to checkpoint inhibitors, respectively. Since the intensity and direction of the interaction between CNAs and tumor immunity is not uniform in all cancer types, and different tissue-specific features may affect the immune response of tumors, we analyzed the particularities of HCC. Here, we propose a strategy based on the CNA profile of each tumor that allows its classification into active/excluded immune profiles. Our study revealed that while HCCs with high loads of broad CNAs (high-BS) present immune excluded features and proliferation signatures; tumors with low levels of broad CNAs (low-BS) displayed immune active-hot features. We hypothesize that the immune profile of low-BS HCCs may indicate a favorable response to immunotherapies.
Acknowledgements
We would like to thank Dr Carla Montironi for her contribution in the manuscript. This study has been developed in part in the Centre Esther Koplowitz from IDIBAPS / CERCA Programme / Generalitat de Catalunya. The results shown here are in part based on data generated by the TCGA Research Network: http://cancergenome.nih.gov/.
Footnotes
Conflict of interest
J.M.L. receives research support from Bayer HealthCare Pharmaceuticals, Eisai Inc, Bristol-Myers Squibb, Boehringer-Ingelheim and Ipsen; and consulting fees from Bayer HealthCare Pharmaceuticals, Merck, Eisai Inc, Bristol-Myers Squibb, Celsion Corporation, Exelixis, Eli Lilly, Roche, Genentech, Glycotest, Nucleix, Can-Fite Biopharma and AstraZeneca. The rest of authors have nothing to disclose.
Financial disclosures
J.M.L. is supported by grants from the European Commission (EC) Horizon 2020 Program (HEPCAR, proposal number 667273-2), the US Department of Defense (CA150272P3), the National Cancer Institute (P30 CA196521), the Samuel Waxman Cancer Research Foundation, the Spanish National Health Institute (MICINN, SAF-2016-76390 and PID2019-105378RB-I00), through a partnership between Cancer Research UK, Fondazione AIRC and Fundación Científica de la Asociacion Española Contra el Cáncer (HUNTER, Ref. C9380/A26813), and by the Generalitat de Catalunya (AGAUR, SGR-1358). L.B., R.E.F. and R.M. were supported respectively by a Beatriu de Pinos Grant (AGAUR, 2016BP00161), a MICINN fellowship (BES-2017-081286) and a FSEOM-Boehringer Ingelheim Grant. D.S. is supported by the Klion Young Scientist Award and the PhD Scientist Innovative Research Award. C.E.W. is supported by a Sara Borrell fellowship (CD19/00109) from the ISCIII and the European Social Fund. J.C. is supported by grants from the Instituto de Salud Carlos III and co-funded by the European Regional Development Fund (ERDF) (CPII18/00026, PI17/01304), the CIBEREHD program, and the Agència de Gestió d’Ajuts Universitaris i de Recerca, Generalitat de Catalunya (2017 SGR 1035). S.F-E. was supported by a CIBEREHD contract.
References
- 1.Ferlay J, Colombet M, Soerjomataram I, Mathers C, Parkin DM, Piñeros M, et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J cancer 2019;144:1941–1953. [DOI] [PubMed] [Google Scholar]
- 2.Llovet JM, Montal R, Sia D, Finn RS. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat Rev Clin Oncol 2018;15:599–616. [DOI] [PubMed] [Google Scholar]
- 3.Llovet JM, Zucman-Rossi J, Pikarsky E, Sangro B, Schwartz M, Sherman M, et al. Hepatocellular carcinoma. Nat Rev Dis Prim 2016;2:16018. [DOI] [PubMed] [Google Scholar]
- 4.Galle PR, Finn RS, Qin S, Ikeda M, Zhu AX, Kim T-Y, et al. Patient-reported outcomes (PROs) from the Phase III IMbrave150 trial of atezolizumab (atezo) + bevacizumab (bev) vs sorafenib (sor) as first-line treatment (tx) for patients (pts) with unresectable hepatocellular carcinoma (HCC). J Clin Oncol 2020;38:476. [Google Scholar]
- 5.El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017;389:p2492–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhu AX, Finn RS, Edeline J, Cattan S, Ogasawara S, Palmer D, et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): a non-randomised, open-label phase 2 trial. Lancet Oncol 2018;19:940–952. [DOI] [PubMed] [Google Scholar]
- 7.Yau T, Kang Y-K, Kim T-Y, El-Khoueiry AB, Santoro A, Sangro B, et al. Nivolumab (NIVO) + ipilimumab (IPI) combination therapy in patients (pts) with advanced hepatocellular carcinoma (aHCC): Results from CheckMate 040. J Clin Oncol 2019;37:4012. [Google Scholar]
- 8.Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017; 168:707–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sia D, Jiao Y, Martinez-Quetglas I, Kuchuk O, Villacorta-Martin C, Castro de Moura M, et al. Identification of an Immune-specific Class of Hepatocellular Carcinoma, Based on Molecular Features. Gastroenterology 2017;153:812–826. [DOI] [PubMed] [Google Scholar]
- 10.Pinyol R, Sia D, Llovet JM. Immune exclusion-Wnt/CTNNB1 class predicts resistance to immunotherapies in HCC. Clin Cancer Res 2019; 25:2021–2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sansregret L, Vanhaesebroeck B, Swanton C. Determinants and clinical implications of chromosomal instability in cancer. Nat. Rev. Clin. Oncol 2018; 15:139–150. [DOI] [PubMed] [Google Scholar]
- 12.Davoli T, Uno H, Wooten EC, Elledge SJ. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science 2017;355:eaaf8399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thorsson VV, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH, et al. The Immune Landscape of Cancer. Immunity 2018;48:812–830.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith JC, Sheltzer JM. Systematic identification of mutations and copy number alterations associated with cancer patient prognosis. Elife 2018;7:e39217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taylor AM, Shih J, Ha G, Gao GF, Zhang X, Berger AC, et al. Genomic and Functional Approaches to Understanding Cancer Aneuploidy. Cancer Cell 2018;33:676–689.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Budczies J, Seidel A, Christopoulos P, Endris V, Kloor M, Győrffy B, et al. Integrated analysis of the immunological and genetic status in and across cancer types: impact of mutational signatures beyond tumor mutational burden. Oncoimmunology 2018;7, e1526613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiménez-Sánchez A, Cybulska P, Mager KLV, Koplev S, Cast O, Couturier DL, et al. Unraveling tumor–immune heterogeneity in advanced ovarian cancer uncovers immunogenic effect of chemotherapy. Nat Genet 2020;52, 582–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Braun DA, Hou Y, Bakouny Z, Ficial M, Sant’ Angelo M, Forman J, et al. Interplay of somatic alterations and immune infiltration modulates response to PD-1 blockade in advanced clear cell renal cell carcinoma. Nat Med 2020;26, 909–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Franch-Expósito S, Bassaganyas L, Vila-Casadesús M, Hernández-Illán E, Esteban-Fabró R, Díaz-Gay M, et al. CNApp, a tool for the quantification of copy number alterations and integrative analysis revealing clinical implications. Elife 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Villanueva A, Portela A, Sayols S, Battiston C, Hoshida Y, Méndez-González J, et al. DNA methylation-based prognosis and epidrivers in hepatocellular carcinoma. Hepatology 2015;61:1945–1956. [DOI] [PubMed] [Google Scholar]
- 21.Ally A, Balasundaram M, Carlsen R, Chuah E, Clarke A, Dhalla N, et al. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017;169:1327–1341.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Torrecilla S, Sia D, Harrington AN, Zhang Z, Cabellos L, Cornella H, et al. Trunk mutational events present minimal intra- and inter-tumoral heterogeneity in hepatocellular carcinoma. J Hepatol 2017;67:1222–1231. [DOI] [PubMed] [Google Scholar]
- 23.Prat A, Navarro A, Paré L, Reguart N, Galván P, Pascual T, et al. Immune-related gene expression profiling after PD-1 blockade in non–small cell lung carcinoma, head and neck squamous cell carcinoma, and melanoma. Cancer Res 2017;77:3540–3550. [DOI] [PubMed] [Google Scholar]
- 24.Chiang DY, Villanueva A, Hoshida Y, Peix J, Newell P, Minguez B, et al. Focal gains of VEGFA and molecular classification of hepatocellular carcinoma. Cancer Res 2008;68:6779–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guichard C, Amaddeo G, Imbeaud S, Ladeiro Y, Pelletier L, Maad I Ben, et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet 2012;44:694–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schulze K, Imbeaud S, Letouzé E, Alexandrov LB, Shinde J, Soysouvanh F, et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet 2015;47:505–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoshida Y, Nijman SMBS, Kobayashi M, Chan J a, Brunet J-P, Chiang DY, et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res 2009;69:7385–7392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoshihara K, Shahmoradgoli M, Martínez E, Vegesna R, Kim H, Torres-Garcia W, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun 2013;4:2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee J, Heo J, Libbrecht L, Chu I, Kaposi-Novak P, Calvisi DF, et al. A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nat Med 2006;12:410–6. [DOI] [PubMed] [Google Scholar]
- 30.Andersen JB, Loi R, Perra A, Factor VM, Ledda-Columbano GM, Columbano A, et al. Progenitor-derived hepatocellular carcinoma model in the rat. Hepatology 2010;51:1401–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Villanueva A, Hoshida Y, Battiston C, Tovar V, Sia D, Mazzaferro V, et al. Combining clinical, pathology, and gene expression data to predict recurrence of hepatocellular carcinoma. Gastroenterology 2011;140:1501–1512.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mínguez B, Hoshida Y, Villanueva A, Toffanin S, Cabellos L, Thung S, et al. Gene-expression signature of vascular invasion in hepatocellular carcinoma. J Hepatol 2011;55:1325–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaposi-Novak P, Lee J-S, Gòmez-Quiroz L, Coulouarn C, Factor VM, Thorgeirsson SS. Met-regulated expression signature defines a subset of human hepatocellular carcinomas with poor prognosis and aggressive phenotype. J Clin Invest 2006;116:1582–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Coulouarn C, Factor VM, Thorgeirsson SS. Transforming growth factor-beta gene expression signature in mouse hepatocytes predicts clinical outcome in human cancer. Hepatology 2008;47:2059–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Charoentong P, Finotello F, Angelova M, Mayer C, Efremova M, Rieder D, et al. Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep 2017;18:248–262. [DOI] [PubMed] [Google Scholar]
- 36.Rooney MSS, Shukla SAA, Wu CJJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015;160:48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sautès-Fridman C, Petitprez F, Calderaro J, Fridman WH. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat. Rev. Cancer 2019; 19:307–325. [DOI] [PubMed] [Google Scholar]
- 38.Calderaro J, Petitprez F, Becht E, Laurent A, Hirsch TZ, Rousseau B, et al. Intra-tumoral tertiary lymphoid structures are associated with a low risk of early recurrence of hepatocellular carcinoma. J Hepatol 2019, 70, 58–65. [DOI] [PubMed] [Google Scholar]
- 39.Hoadley KA, Yau C, Hinoue T, Wolf DM, Lazar AJ, Drill E, et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018; 173: p291–304.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang L, Li M, Deng B, Dai N, Feng Y, Shan J, et al. HLA-DQB1 expression on tumor cells is a novel favorable prognostic factor for relapse in early-stage lung adenocarcinoma. Cancer Manag Res 2019;11:2605–2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jung H, Kim HS, Kim JY, Sun JM, Ahn JS, Ahn MJ, et al. DNA methylation loss promotes immune evasion of tumours with high mutation and copy number load. Nat Commun 2019;10:4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roh W, Chen PL, Reuben A, Spencer CN, Prieto PA, Miller JP, et al. Integrated molecular analysis of tumor biopsies on sequential CTLA-4 and PD-1 blockade reveals markers of response and resistance. Sci Transl Med 2017;9:eaah3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bianchi JJ, Zhao X, Mays JC, Davoli T. Not all cancers are created equal: Tissue specificity in cancer genes and pathways. Curr. Opin. Cell Biol 2020;63:135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ben-David U, Amon A. Context is everything: aneuploidy in cancer. Nat. Rev. Genet 2020;21:44–62. [DOI] [PubMed] [Google Scholar]
- 45.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014;515:568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science, 2015;348:124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chan TA, Wolchok JD, Snyder A. Genetic Basis for Clinical Response to CTLA-4 Blockade in Melanoma. N Engl J Med 2015;373:1984–1984. [DOI] [PubMed] [Google Scholar]
- 48.Rosenthal R, Cadieux EL, Salgado R, Bakir M Al, Moore DA, Hiley CT, et al. Neoantigen-directed immune escape in lung cancer evolution. Nature 2019; 567:479–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McGranahan N, Furness AJSS, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science, 2016;351:1463–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Turajlic S, Litchfield K, Xu H, Rosenthal R, McGranahan N, Reading JL, et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: a pan-cancer analysis. Lancet Oncol 2017;18:1009–1021. [DOI] [PubMed] [Google Scholar]
- 51.Jamal-Hanjani M, Wilson GA, McGranahan N, Birkbak NJ, Watkins TBK, Veeriah S, et al. Tracking the Evolution of Non–Small-Cell Lung Cancer. N Engl J Med 2017; 376:2109–2121. [DOI] [PubMed] [Google Scholar]
- 52.Hélias-Rodzewicz Z, Lourenco N, Bakari M, Capron C, Emile J-FF. CDKN2A Depletion Causes Aneuploidy and Enhances Cell Proliferation in Non-Immortalized Normal Human Cells. Cancer Invest 2018;36:338–348. [DOI] [PubMed] [Google Scholar]
- 53.Bou-nader M, Caruso S, Donne R, Celton-Morizur S, Calderaro J, Gentric G, et al. Polyploidy spectrum: a new marker in HCC classification. Gut 2019;gutjnl-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Meylan M, Petitprez F, Lacroix L, Di Tommaso L, Roncalli M, Bougoüin A, et al. Early hepatic lesions display immature tertiary lymphoid structures and show elevated expression of immune inhibitory and immunosuppressive molecules. Clin Cancer Res 2020; clincanres.2929.2019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.