

SUMMARY
Metabolic reprogramming is a hallmark of T cell activation and function. As our understanding of T cell metabolism increases, so does our appreciation of its inherent complexity. The metabolic heterogeneity of T cells that reside in different locations, such as lymphoid and non-lymphoid tissues, presents a challenge to developing therapies that exploit metabolic vulnerabilities. The roots of metabolic heterogeneity are only beginning to be understood. Here, we propose four factors that contribute to the adaptation of T cells to their dynamic tissue environment: (1) functional status of T cells, (2) local factors unique to the tissue niche, (3) type of inflammation, and (4) time spent in a specific tissue. We review emerging concepts about tissue-specific metabolic reprogramming in T cells with particular attention to explain how such metabolic properties are used as an adaptation mechanism.
Adaptation of immune cells to the local microenvironment is critical for their persistence and function. Here, Varanasi et al. review the role and types of metabolic adaptation acquired by T cells in tissues and how these adaptations might differ between tissue type, disease state, and functionality of a T cell.
INTRODUCTION
Activation of naive T cells within lymphoid tissues leads to metabolic reprograming from oxidative catabolic metabolism in naive T cells to anabolic metabolism in activated T cells. This metabolic reprograming is necessary to support T cell proliferation and differentiation into different T cell subsets. Metabolic reprogramming in effector T cells subsets (Th1, Th2, Th17, and CD8+ effector T cells) mainly involves increased glycolysis coupled with lactate production (referred to as aerobic glycolysis), glutaminolysis-driven mitochondrial respiration, serine, and one carbon metabolism. These features of metabolism are less evident in a subset of T cells with regulatory functions (referred to as Tregs), which depend mainly on mitochondrial respiration for their survival and immunosuppressive functions (Gerriets et al., 2015; Klysz et al., 2015; Ma et al., 2017; Macintyre et al., 2014; Makowski et al., 2020). However, this dependence has mainly been shown with murine Tregs where their canonical transcription factor Foxp3 is known to inhibit expression of Glut-1 needed for glucose uptake as well as some enzymes involved in glycolysis (Angelin et al., 2017; Gerriets et al., 2016). Curiously, human Treg cells might engage in glycolytic activity although identification of human Treg cells is less precise and the effects of gene silencing on suppressive functions still need to be perfected. Nevertheless, inhibition of metabolic reprogramming during effector T cell differentiation, as occurs during nutrient deprivation, either leads to insufficient activation/differentiation/proliferation or in some cases results in the generation of Tregs (Gerriets et al., 2015; Ho et al., 2015; Klysz et al., 2015; Macintyre et al., 2014; Michalek et al., 2011).
The rules for T cell subset metabolic requirements were largely developed by using in vitro culture conditions rich in nutrients or from cells isolated from lymphoid tissues (mainly spleen). Upon differentiation in vivo, T cell subsets egress from lymphoid tissues and enter non-lymphoid tissues where their function is mainly focused. We know little about the metabolic programs that various T cell subsets might need to adopt in order to reside and function within different non-lymphoid tissue locations. In such environments, nutrient availability might be significantly different from lymphoid tissues or in vitro culture conditions. We expand upon this notion and suggest that the key to survival and function within non-lymphoid tissues involves adaptation to the metabolic climate unique to different tissues. Should this fail, the consequences could include tissue-specific inflammatory diseases. Here, we describe current understanding of how various T cell subsets adjust their metabolic needs to the local tissue environment during homeostasis and different tissue pathologies that were described mostly by using murine models. We do not discuss events involved in initiating tissue location by T cell subsets (i.e., T cell homing), as this topic has been extensively covered in other reviews (Rosato et al., 2020; Szabo et al., 2019).
ADAPTATION TO LOW-GLUCOSE AND HIGH-LACTATE ENVIRONMENTS
The idea of tissue-specific metabolic reprograming and adaptation was first implied from studies with Treg cells (Angelin et al., 2017). In conditions where glucose and oxygen are abundant, Treg cells maintain their characteristic high levels of oxidative phosphorylation (OXPHOS) by diverting glucose-driven pyruvate to mitochondrial respiration that generates ATP and metabolites that support their survival and function (Angelin et al., 2017; Gerriets et al., 2016; Howie et al., 2017). In contrast, when glucose is limiting, along with an abundance of lactate (such as in some tissues with underlying disease), Treg cells rely on an alternative energy source. The tissues with high lactate include tumors and inflammatory sites such as occurs in arthritic joints, atherosclerotic plaques, adipose tissue in obese individuals or during ischemia, where lactate levels rise to 10–40 mM as opposed to physiological levels of 1.5–3 mM (Amorini et al., 2014; Pucino et al., 2017, 2019). Lactate abundance in the tissues or plasma is maintained by plasma bicarbonate buffering of lactic acid. Lactate and lactic acid can have different effects on T cell subsets. For example, in circumstances where lactate is abundant, Treg cells use lactate to power mitochondrial respiration and are able to convert lactate to pyruvate. This mechanism was mainly restricted to Treg cells and not T effectors, given that Treg cells maintain high levels of NAD+ generated from mitochondrial respiration, which is required for lactate-dehydrogenase-dependent oxidation of lactate. However, in effector T cells NAD+ consumption, due to high glycolysis, leads to a low NAD+/NADH ratio that is insufficient to support the utilization of lactate (Angelin et al., 2017). Within inflammatory tissues during autoimmune conditions, where high lactate levels accumulate, effector T cells display an adaptation mechanism that enables them to oxidize lactate. For example, CD4+ effector T cells within arthritic joints during rheumatoid arthritis express a lactate transporter, SLC5A12, that feeds lactate to mitochondrial respiration that in turn supports their survival and differentiation into Th1 and Th17 cells. However, Treg cells within those inflamed environments expressed lower levels of SLC5A12 and were less influenced by lactate exposure (Haas et al., 2015; Pucino et al., 2019). The utilization of lactate over glucose in inflammatory tissues is also supported by the evidence that T cells within rheumatoid arthritis lesions downregulate their expression of Phosphofructokinase, an enzyme primarily involved in glycolysis thereby decreasing their glucose consumption and glycolytic flux (Yang et al., 2013). This suggests that T cells in synovial joints during rheumatoid arthritis are poised to utilize lactate over glucose as an alternate source of fuel. Although, lactate exposure can inhibit effector T cell proliferation both in vitro and in vivo (Angelin et al., 2017; Rundqvist et al., 2019), evidence also suggests that lactate could significantly increase CD8+ T effector functions, mainly the expression of Gzmb and IFN-γ (Rundqvist et al., 2019). Of interest, injection of lactate into animals reduced tumor growth in a CD8+ T cell-dependent manner (Rundqvist et al., 2019). This suggests that expression of the lactate transporter (SLC5A12) and the abundance of lactate within inflamed tissues could positively influence effector functions of T cells. In contrast to the inflamed tissues, tumors could predominantly accumulate lactic acid over lactate, which would impede cytotoxic and effector T cell activity but would still support Treg survival. This represents an obstacle to anti-cancer immunity (Brand et al., 2016; Rundqvist et al., 2019; Wang et al., 2020a). Thus, exposure of effector T cells to lactic acid, but not to lactate, inhibits the function and survival of both CD4+ and CD8+ effector T cells (Brand et al., 2016; Haas et al., 2015; Rundqvist et al., 2019). Consequently, rebalancing lactate and lactic acid within tissues to reshape effector and Treg cell ratios could represent an effective therapeutic strategy to counteract immune mediated diseases. For instance, diminishing lactic acid levels in tumors by identifying approaches that can promote the generation of lactate over lactic acid, could enhance effector T cell responses and tumor control. Interestingly, lactate and lactic acid were also shown to impair CD4+ and CD8+ T cell migratory abilities (in a subset specific manner), so promoting their retention within tissue sites in vivo (Haas et al., 2015), a mechanism that also could be attributed to inhibition of T cell glycolysis.
The differential effects of lactate and lactic acid could be attributed to their utilization of different transporters such as SLC5A12 or SLC16A1 and fates within T cells. Thus, lactic acid leads to intracellular acidification and inactivation of NFAT signaling, but lactate feeding into metabolism mainly favors mitochondrial respiration (Angelin et al., 2017; Brand et al., 2016; Pucino et al., 2019; Rundqvist et al., 2019). Interestingly, although inhibition of NFAT signaling inhibited effective T cell responses (Brand et al., 2016; Vaeth et al., 2012), NFAT signaling had minimal effects on thymus-derived naturally occurring Treg cells (Vaeth et al., 2012). This implies that some Treg cells might possess the ability to survive in both lactic-acid- and lactate-rich environments.
Taken together, the above observations indicate that within different tissues or during different disease conditions, T cell subsets could gain the ability to utilize and adapt to either lactate or lactic-acid-rich environments. Thus, identifying factors that drive the distinct adaptation profiles of T cell subsets could lead to useful therapies to change the pattern of T cell responsiveness.
MITOCHONDRIAL METABOLISM IS KEY FOR T CELL SURVIVAL WITHIN TISSUES
In addition to aerobic glycolysis being an important determinant for effector T cell differentiation, active mitochondrial respiration might be critical for survival and function of T cells especially within tissue environments. Interestingly, a recent study showed that mitochondrial respiration occurs at higher rates in physiologically primed effector T cells compared with cells primed in vitro (Ma et al., 2019). The latter study used in vivo infusion of isotope-labeled glucose and flux analysis and showed that CD8+ T cells primed in vivo after an acute infection had their glucose diverted to mitochondrial respiration, whereas T cells that were activated in vitro used mainly glycolysis (Ma et al., 2019). This in vivo preference for mitochondrial respiration over glycolysis might also explain the defective ability of in-vitro-activated cells to utilize/oxidize lactate as an alternative energy source.
It is also becoming evident that mitochondrial respiration and more specifically individual components of the mitochondrial electron transport chain are differentially required to sustain both effector and Treg functions. For example, under differentiating conditions in vitro, expression of IFN-γ by Th1 cells requires active complex II and III, whereas, IL-17 expression by Th17 cells is dependent on complex I and II (Bailis et al., 2019). Additionally, T cells lacking complex III of the electron transport chain failed to generate mitochondrial-derived reactive oxygen species (ROS) and displayed defective effector T cell responses (in both CD4+ and CD8+) during acute infection (Sena et al., 2013). In addition, effector T cell responses were increased in mice that lack MCJ/DnaJC15, an endogenous break for mitochondrial respiration in cells, because of increased OXPHOS (Champagne et al., 2016). Additionally, mitochondrial metabolism (biogenesis, morphology, and function) is critical for memory T cell responses although this has been mostly studied in circulating memory T cells (Buck et al., 2016; Dumauthioz et al., 2020; O’Sullivan et al., 2014). Similar to effectors and memory CD8+ T cells, Treg cells also display differential dependence on discrete components of the mitochondrial electron transport chain. For example, mitochondrial complex III, but not I, is specifically required to maintain Treg suppressive capacity, but not their proliferation and survival (Weinberg et al., 2019). However, it is still unclear which exact mechanism within mitochondrial metabolism supports regulatory functions of Treg cells such as IL-10 production.
Mitochondrial metabolism within lymphocytes varies depending on the type of tissue where they reside. For instance, intraepithelial lymphocytes (IELs) located at the intestinal barrier display a distinct mitochondrial metabolic state to that of effector or memory T cells from the spleen (Konjar et al., 2018). Despite, harboring high numbers of mitochondria per cell, IELs displayed reduced spare respiratory capacity that could indicate low availability of fuel for mitochondrial metabolism, at least during the quiescent state. However, IELs also have a distinct mitochondrial membrane cardiolipin composition that is altered to support their rapid proliferation and effector function upon re-stimulation (Konjar et al., 2018). Similarly, inducing mitochondrial biogenesis in T cells by using PGC-1a overexpression (which led to increased mitochondrial number and function) did not influence the abundance of memory T cells in the liver after listeria infection, despite showing an increased number of circulating memory T cells (Dumauthioz et al., 2020). It is yet to be ascertained if long term residency of T cells in the liver is altered in cells that maintain high mitochondrial metabolism. However, this might not be the case for lung-resident memory T cells as they seem to be dependent on mitochondrial metabolism. Thus, lung-resident T cells expressed a transcription factor Bhlhe40 that predominantly regulated genes involved in mitochondrial metabolism (Li et al., 2019). Subsequently, T cells lacking Bhlhe40 showed diminished expression of genes involved in mitochondrial metabolism and/or OXPHOS along with their defective persistence in the lungs as tissue-resident memory T cells (TRM). However, their early infiltration into the tissue as well as their persistence as splenic memory T cells remained unchanged (Li et al., 2019). Besides mitochondrial metabolic genes, Bhlhe40 also regulated expression of other immune and non-immune genes, but the contribution of mitochondrial metabolism to the Bhlhe40-knockout T cell phenotype still needs to be established. Overall, however, it appears that mitochondrial metabolism could be regulated in a tissue-specific manner, although this issue merits further investigation. Our current understanding of mitochondrial metabolism of TRM and Treg cells in different tissues is summarized in Figure 1.
Figure 1. Tissue-Specific Metabolic Adaptations of T Cells.
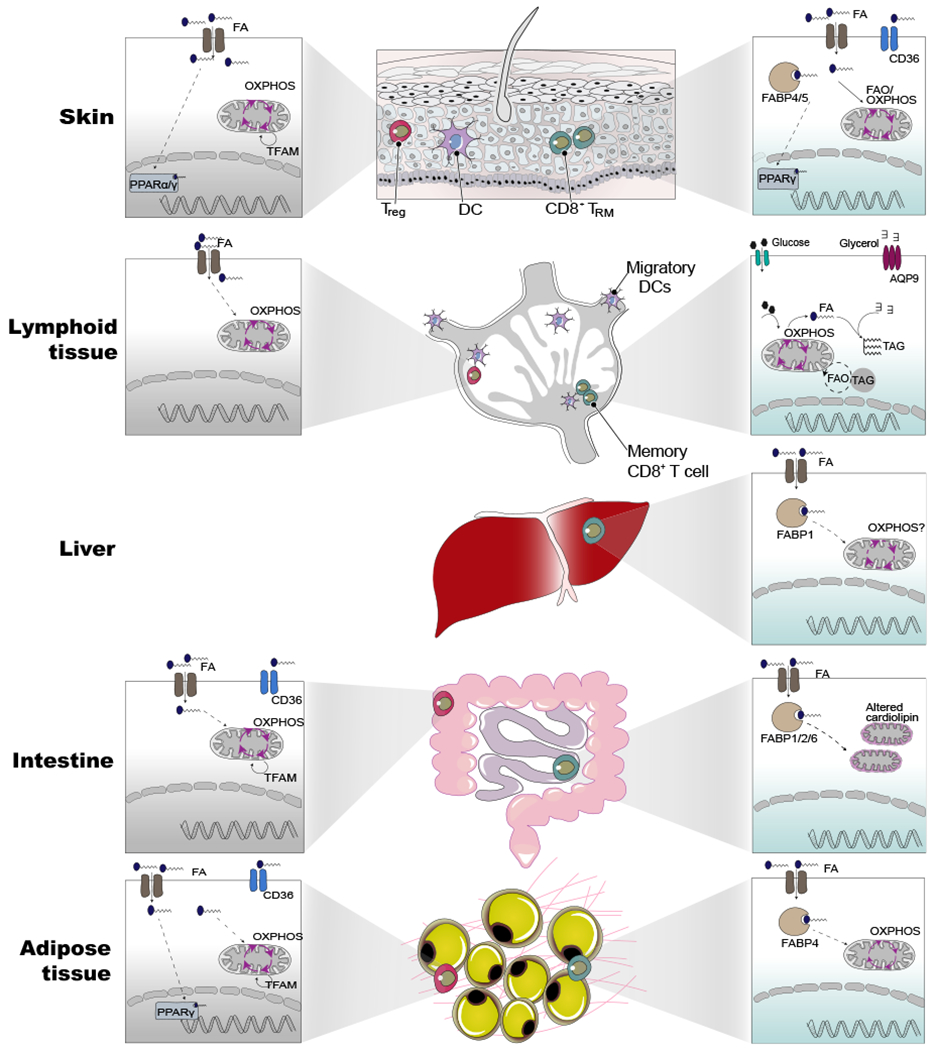
Tissue-resident memory and Tregs from different tissues share a core metabolic program involving in fatty acid metabolism and mitochondrial metabolism compared with their counterparts in lymphoid tissues. Beyond this core metabolic program, each population possesses additional tissue-specific programs shaped by the local environment. For instance, skin TRM show the expression of PPARγ and express fatty-acid-binding protein isoform, FABP4/5, whereas liver TRM and adipose tissue express the isoform FABP1 and FABP4, respectively. Another notable difference is the altered cardiolipin content of mitochondrial membranes in the intestinal interepithelial lymphocytes. Interestingly, skin Treg cells express either PPARα/γ isoforms whereas, adipose-tissue-specific Tregs express only PPARγ. However, Treg cells in non-lymphoid tissues, but not in the lymphoid tissues, depended on mitochondrial DNA replication and transcription orchestrated by Tfam. For each tissue, we have shown Treg (gray box) and CD8+ TRM (green box) depicting known metabolic pathways or genes related to metabolism. The dotted lines indicate mechanism/pathway that are unclear.
The aspect of regulation of mitochondrial metabolism within tissues was well illustrated by studies on the maintenance of the mitochondrial genome by mitochondrial transcription factor A (Tfam). This is a nuclear-encoded mitochondrial protein that regulates replication, transcription as well as the stability of mitochondrial DNA. Cell-type-specific deletion of Tfam in T cells by using the CD4-Cre approach causes severe mitochondrial respiration defects, lysosomal storage disorders, and enhanced IFN-γ production by CD4+ T cells (Baixauli et al., 2015), resulting in severe autoimmunity (Desdín-Micó et al., 2020). Tfam depletion in Treg cell alone led to similar mtDNA depletion and mitochondrial respiratory deficiencies but had no obvious effect on Treg maintenance in the thymus and spleen. However, the number and function of Treg cells in non-lymphoid tissues were markedly impaired in Tfam-deficient Treg cells resulting in severe autoimmunity (Chapman et al., 2018; Fu et al., 2019). One potential explanation for Tfam deletion causing an increased effector response in T cells despite showing defective mitochondrial respiration, could be because of reduced NAD levels. Thus, restoring NAD levels by supplementation with the nicotinamide precursor NAM partially rescued the Tfam deficiency and ameliorated the impaired Th1 responses (Desdín-Micó et al., 2020). Another potential explanation for Tfam-deletion-induced effector T cell responses could be attributed to defective packaging of the mitochondrial DNA upon Tfam deletion. This would result in mitochondrial DNA release into the cytoplasm and activation of cytosolic cGAS-stimulator of the interferon gene (STING) DNA-sensing pathway and inflammatory cytokine expression (Chung et al., 2019; West et al., 2015). In addition, it remains unclear how the reduced ratio of oxidized NAD+ to its reduced form NADH (leading to reductive stress), regulates the effector functions of T cells. It is widely recognized that reactive oxygen and nitrogen species are required for T cell activation, and one potential way to address the role of reductive stress in modulating T cell responses is to employ a genetic tool wherein a bacterial NADH oxidase (LbNOX) is overexpressed in a compartment-specific manner (mitochondrial versus cytosol) to increase the NAD+/NADH ratio (Goodman et al., 2020; Patgiri et al., 2020). It is also reasonable to assume that mitochondrial metabolism of Treg cells might possess a tissue homeostatic advantage over effector T cells in the same tissues.
With regard to mitochondrial metabolism within tumors, mounting evidence suggests that tumor-specific CD8+ T cells when within tumors display impaired mitochondrial function and increased accumulation of mitochondrial ROS as compared with their circulating counterparts (Siska et al., 2017; Zhang et al., 2017). This impaired mitochondrial function of T cells within tumors was associated with their diminished effector function. However, the effector T cell function could be rescued by overexpression of PGC-1α, specifically within the CD8+ T cells, and this led to an increase in both mitochondrial number and function (Bengsch et al., 2016; Dumauthioz et al., 2020; Scharping et al., 2016). Although the exact mechanism by which the functional restoration of mitochondria led to increased effector functions of T cells is unclear, changes in T cell differentiation state could be one potential explanation. Thus, increased mitochondrial respiration imposed a memory-like state in the effector T cells and enhanced their antitumor responses (Buck et al., 2016; Dumauthioz et al., 2020). It will be interesting to see if PGC-1a-restored cells resemble the recently described stem-like memory cells that were shown to be highly relevant in immunity to tumors and some chronic infections (Brummelman et al., 2018; Miller et al., 2019; Siddiqui et al., 2019; Utzschneider et al., 2016).
In contrast to effector T cells within tumors, Treg cells remain highly functional and show intact and higher rates of mitochondrial activity (Field et al., 2020; Maj et al., 2017). This is in contrast to autoimmune lesions where Treg cells display dysfunctional mitochondrial metabolism. Treg cells from multiple sclerosis patients and in its mouse model experimental autoimmune encephalitis (EAE) displayed signatures of depolarized mitochondria and the apoptotic signaling pathway. This status was also associated with reduced mitochondrial potential, increased production of mitochondrial ROS, and increased apoptotic cell death in Treg cells from EAE. Additionally, treating EAE mice with MitoTEMPO, a mitochondria-specific superoxide scavenger increased Treg cell accumulation in the spinal cord and delayed the disease onset (Alissafi et al., 2020). This suggests that mitochondrial metabolism plays a central role in conferring T cell adaptation within tissues during both disease and homeostatic states. And that within the same microenvironment, some T cell subsets can adapt and stay functional whereas others cannot. Taken together, regulating T cell function and adaptation by mitochondrial metabolism goes beyond the classical function of mitochondria, ATP production, but includes other activities such as mitochondrial DNA signaling, TCA intermediates-dependent signaling, mitochondrial ROS signaling, and levels of NAD+/NADH. Our current understanding of mitochondrial metabolism of T cells and Treg cells in different disease contexts is summarized in Figure 2.
Figure 2. Different T Cell Subsets Adapt Differently within Tissues of Different Disease States.
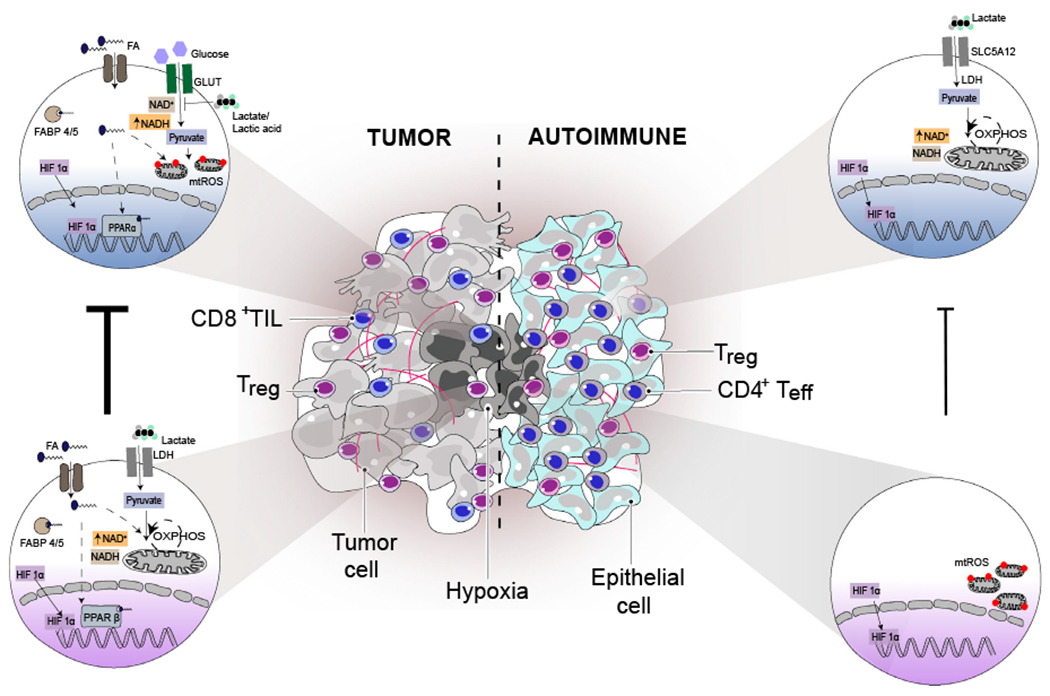
Tumors and autoimmune tissues consist of metabolically distinct environments where both Treg and T effector display distinct metabolic features. Treg cells in tumors display high suppressor functions that is correlated with intact mitochondrial metabolism that is powered by PPARβ signaling and/or lactate utilization (due to high NAD+/NADH ratio). Although both Treg and CD8 T cells in tumor express FABP4/5 and take up fatty acids, PPARα-driven signaling in tumor-infiltrating CD8+ T cells is insufficient to sustain their effector functions as CD8 T cells in tumors also display dysfunctional mitochondrial metabolism with increased mitochondrial ROS production that contributes to CD8+ T cell dysfunction. CD8+ T cells in tumors also lack the ability to utilize either lactate or lactic acid (due to low NAD+/NADH ratio), which later inhibits T cell glycolysis and T effector functions. In contrast to Treg cells in tumors, Treg cells in autoimmune lesions display dysfunctional mitochondrial metabolism with increased mitochondrial ROS production (similar to CD8+ T cells in tumors), which is associated with their inadequate suppressor functions. Whereas, effector T cells (mainly CD4) in autoimmune tissue display heightened ability to utilize lactate (due to high NAD+/NADH ratio) to power their mitochondrial metabolism and sustain high effector functions. Thus, intact mitochondrial metabolism is essential for sustaining T cell function (both Treg and T effector) within tissues during different disease states. Although there are some distinct metabolic features between Treg cells and effector T cells within tumors and autoimmune tissue, there are some common adaptation mechanisms such as expression of HIF1α in the infiltrating T cell subsets (both Treg and T effector) that is critical for their infiltration and survival within these tissues. Thus, 4 factors contribute to the adaptation of T cells to their dynamic tissue environment. These are (1) functional status of T cells (Treg/effector or highly functional/dysfunctional), (2) local factors unique to a niche of the tissue (lactate/lactic acid or type of fatty acids), (3) type of inflammation that might be occurring in the tissue (cancer/autoimmune), and (4) time spent in a specific tissue (early infiltrators/long term resident T cells). The dotted lines indicate mechanism/pathway that are unclear, red dots on mitochondria represent mitochondrial ROS, and CD8+ TIL represent CD8+ tumor-infiltrating lymphocytes.
ADAPTATION TO HYPOXIC ENVIRONMENT IS KEY FOR EFFECTIVE T CELL RESPONSES
It is now known that tissues with an ongoing inflammation or tumors are associated with increased oxygen consumption resulting in localized tissue hypoxia. A key signaling molecule that regulates cellular adaptation to hypoxic tissue microenvironments, especially to regulate both glycolysis and mitochondrial respiration, is HIF1α. Under hypoxia, HIF1α in Treg cells directed glucose away from mitochondrial respiration and toward glycolysis, which is critical for Treg migration. Deficiency of HIF1α in Treg cells led to impaired glycolysis and augmented mitochondrial OXPHOS. Despite displaying high suppressive capacity, HIF1α-deficient Treg cells had impaired migratory and survival abilities within the colon and brain, but not in lymphoid tissues. This defective Treg cell migration and survival led to increased protective effector T cell responses within the colon during T cell-mediated colitis (Clambey et al., 2012) and in the brain during glioma (Miska et al., 2019). Additionally, hypoxia-induced HIF1α also plays similar role in effector T cells. For instance, the hypoxic environment within kidneys during murine lupus nephritis induces the expression of HIF1α effector T cells (CD4+ and CD8+) contributing to T-cell-dependent injury. In fact, deletion of HIF1α CD4+ T cells reduced T cell numbers specifically within kidneys and protected mice from kidney damage. Whereas, T cell responses in the spleen remained intact (Chen et al., 2020b). Similarly, CD8+ T cells within tumors, but not in lymphoid tissues, express HIF1α, and the CD8+ T cells that lacked HIF1α displayed reduced infiltration and survival within tumors resulting in increased tumor burden. Nevertheless, CD8+ T cells persisted in the spleen and lymph nodes (Chapman et al., 2018; Palazon et al., 2017). Given that the loss of HIF1α in Treg and effector T cells resulted in reduced tissue infiltration (Chapman et al., 2018; Chen et al., 2020b; Clambey et al., 2012; Miska et al., 2019; Palazon et al., 2017). Additionally inhibition of glycolysis with lactate or lactic acid inhibited the migration of both CD4+ and CD8+ T cells (Haas et al., 2015), indicating a critical role for glycolysis in T cell migration. Subsequently, glucokinase, an enzyme involved in glycolysis, promotes cytoskeletal rearrangements by associating with actin. Of interest, Treg cells lacking glucokinase failed to migrate to skin allografts and graft rejection was augmented (Kishore et al., 2017). Thus, even though glycolysis could be less critical for Treg cell generation and function, it could be an important factor for Treg cell migration. Taken together, these observations support the idea that within tissues where oxygen is limiting, T cells adapt by expressing HIF1α that supports their metabolism and function within such hypoxic microenvironments (Figure 2).
WITHOUT ADEQUATE FATTY ACIDS IN TISSUES T CELLS MALFUNCTION
An important metabolic constituent of tissues and tumor environments is the abundance of fatty acids. Many studies reported that tissue-infiltrating T cells, compared with circulating T cells, express gene signatures involved in fatty acid metabolism, particularly those associated with fatty acid uptake, binding, or oxidation. For instance, TRM isolated from tissues such as skin, liver, gut, adipose tissue, and bone marrow (Collins et al., 2019; Frizzell et al., 2020; Han et al., 2017; Pan et al., 2017) expressed disparate isoforms of fatty-acid-binding protein (FABP), which are critical for their survival and persistence within those tissues. For instance, CD8+ TRM in the skin after vaccinia virus infection took up fatty acids and expressed FABP4/5, which, if knocked down in CD8+ T cells, impaired TRM skin accumulation (Pan et al., 2017). However, circulating T cell responses remained intact. Similarly, TRM in adipose tissue after Yersinia infection expressed higher levels of FABP4 (Han et al., 2017), and liver derived TRM expressed high FABP1 levels (Frizzell et al., 2020). Moreover, T cells depleted of FABP1 no longer accumulated in the liver, but their splenic distribution pattern was unchanged. Curiously, expression of various FABP isoforms is not permanently imprinted within a particular species of TRM. Instead, these genetic programs can apparently be rewired in tissue-specific manner. For instance, transfer of liver TRM into naive mice resulted in TRM accessing the intestines and skin. However, they lost the liver-specific FABP1 isoform but expressed the isoform characteristic of the respective tissue (Frizzell et al., 2020). This is in contrast to circulating and in-vitro-generated memory T cells that utilize lipolysis of intracellularly stored lipids for their generation and persistence (Cui et al., 2015; O’Sullivan et al., 2014). The differences in fatty acid metabolism in different TRM and Treg cells are summarized in Figure 1. Similar to skin TRM, CD8+ T cells infiltrating skin tumors also take up and catabolize fatty acids and displayed enhanced expression of FABP4/5. This fatty acid signaling and catabolism was mainly observed in CD8+ T cells from advanced tumors (30 days) compared with early tumors (14 days), or T cells from the spleen (Zhang et al., 2017). This also correlates well with the changes in gene expression observed in the skin infiltrating CD8+ T cells after acute infection where gene signatures involved in fatty acid metabolism appears mainly from day 25 post infection, and this was maintained until day 90 (Pan et al., 2017). This indicates that the time spent by the CD8+ T cells within a given tissue during either an acute infection or in tumors could influence their dependence on fatty acid metabolism.
Interestingly, a key difference in fatty acid metabolism between skin TRM and skin tumor-infiltrating CD8+ T cells was the expression of peroxisome proliferator-activated receptor (PPAR), a nuclear receptor involved in fatty acid signaling. Whereas skin TRM expressed the gamma isoform of PPAR (PPARγ) (Pan et al., 2017), CD8+ T cells in skin tumors expressed the alpha isoform (PPARα) (Zhang et al., 2017). It still needs to be shown if these individual isoforms have different substrate specificities and if they can differently regulate T cell functions. However, deletion of PPARα in CD8+ T cells impaired their persistence and function within tumors and led to increased tumor burden. In contrast, promoting PPARα signaling using the PPARα-specific agonist-fenofibrate improved CD8+ T cell function and reduced the tumor burden (Zhang et al., 2017). Similar to skin tumors, CD8+ T cells from human gastric adenocarcinomas with a TRM phenotype (CD69+CD103+) also displayed enhanced fatty acid uptake and expressed FABP4/5. This allowed them to remain functional compared with the CD8+ T cells with the non-TRM phenotype within tumors, which in consequence became dysfunctional (Lin et al., 2020). Together, these observations indicate the CD8+ T cells within different tissues and during different disease contexts display elevated signatures of fatty acid metabolism that are critical for their persistence in those tissues.
As with CD8+ T cells, Treg cells also display different fatty acid metabolic signatures within non-lymphoid tissues compared with lymphoid tissues. For instance, single-cell RNA sequencing analysis of Treg cells from lymphoid and different non-lymphoid tissues revealed that Treg cells in lymphoid tissues expressed genes involved in glycolysis. In contrast, Treg cells in the colon and skin expressed genes involved in fatty acid metabolism (Miragaia et al., 2019). The skin Treg cells specifically expressed PPARγ and DGAT2 that are involved in fatty acid signaling and storage, respectively, compared with Treg cells in lymphoid tissues (Delacher et al., 2020; Miragaia et al., 2019). Although the role of PPARγ in skin Treg is not yet explored, it is possible that skin Treg cells also express other isoforms of PPAR such as PPARα. In support of this possibility, Treg cells from PPARα-deficient mice display diminished suppressor functions and reduced abundance in skin upon skin sensitization with an allergen (Dubrac et al., 2011). In addition, Treg cells in visceral adipose tissues (VATs) take up fatty acids and expressed the lipid transporter CD36 as well as a nuclear receptor PPARγ compared with their counterparts in the spleen. Deletion of PPARγ in Treg cells impaired their survival within VAT tissue, but not in the spleen. Additionally, promoting PPARγ signaling, by using the PPARγ-specific agonist (pioglitazone), increased Treg cell numbers specifically in VATs and reduced high-fat-diet-induced insulin resistance in a Treg-specific-PPARγ-dependent manner (Cipolletta et al., 2012). The expression of CD36 in both CD4+ and CD8+ T cells from VATs was also observed in humans, indicating that some features of metabolism are conserved between humans and mice (Couturier et al., 2019). Similarly, Treg cells in colonic lamina propria intestines express receptors for CD36 compared with splenic Treg cells. This might allow intestinal Treg cells to take up and supply lipids to fuel mitochondrial respiration, but this still needs to be described (Kabat et al., 2016). Tregs in mouse tumor models might also take up lipids and express lipid transporters, such as CD36 (Pacella et al., 2018; Wang et al., 2020a) and FABP4/5 (Field et al., 2020). In addition, the expression of CD36 along with lipid uptake was also observed in Treg cells from patients with non-small-cell lung carcinoma and breast cancer (Wang et al., 2020a). In fact, CD36-mediated lipid uptake promoted mitochondrial respiration in Treg cells via PPARβ signaling. This mitochondrial fitness expressed by Tregs is likely critical for their survival in lactic-acid-rich tumor microenvironments. Thus, ablation of CD36 or PPARβ specifically in Treg cells reduced their persistence and suppressive activity in tumors but fortunately left Treg cells in lymphoid tissues intact (Wang et al., 2020a). We can state that both Treg cells and CD8+ T cells express some features of fatty acid metabolism within tissues and also express different isoforms of nuclear receptors during various disease contexts (Figure 2). It still remains to be known the role of fatty acid metabolism in both effector T cells and Treg cells within autoimmune tissue. Understanding if the reliance of Treg cells on fatty acid metabolism holds true within additional tissues, especially barrier tissues such as the lungs and the eye where Treg cells were shown to attain tissue repair functions (Arpaia et al., 2015; Varanasi et al., 2018), will be an important area for future investigation.
We can conclude that fatty acid metabolism might play a central role in the adaptation of T cells (both effector and regulatory) within the majority of tissues that display lipid-rich compositions. Because each tissue has a different composition of fatty acid and lipid species, the fatty acid or lipid preferences of TRM or Treg cells to bind and activate different FABP/PPAR isoforms in a tissue-specific manner needs further evaluation. It is also not known whether fatty acid uptake and binding to fatty acids stimulates fatty acid β-oxidation in lysosomes. Accordingly, CPT1a was shown to be dispensable for the generation and maintenance of peripheral T cell responses (CD8+ effector, memory, and Treg cells) (Raud et al., 2018), although it is still unclear whether CPT1a- or CPT2-dependent fatty acid β-oxidation might be crucial for maintaining T cells within non-lymphoid tissues or tumors.
TISSUE-SPECIFIC ADAPTATIONS GOES BEYOND CELLULAR METABOLISM
Along with metabolites that can be utilized for energy provision, T cells also encounter other metabolites that are enriched in a tissue-specific manner. These metabolites might not only regulate T cell metabolism but could also be important for regulating immune functions occurring as a consequence of downstream signaling within T cells. An appreciation of how these mechanisms might operate within different tissues is only beginning to emerge, and this information comes mainly from studies of T cells in the intestinal mucosa (Figure 3). Intestinal sites are abundant in fats, fat-soluble vitamins, bile acids, and microbial-derived metabolites. Moreover, intestinal segments descending from proximal to distal regions are responsible for absorption of different nutrients and these can regulate immune responses in a specific manner. Although the small intestine is responsible for the majority of nutrient absorption, the abundant microbial community of the large intestine is essential for the fermentation of indigestible material such as fibers and starch (Flint et al., 2012; Hillman et al., 2017). This compartmentation appears to have a discrete impact on immune responses generally and on lymphocyte function more specifically. Germ-free mice lacking microbiota at barrier surfaces, especially within the gut, displayed reduced Treg cell numbers within the colon (Lathrop et al., 2011; Sefik et al., 2015). However, Treg cells in other segments of the intestines and lymphoid tissues were largely unaffected. One mechanism by which the gut microbiota regulates Treg cell abundance, especially in the colon, is by producing short-chain fatty acids and secondary bile acids. In addition, SCFA derived from bacterial fermentation of dietary fiber regulate Treg cell (RORγ+) abundance via free fatty acid receptor (FFAR2/GPR43)-dependent inhibition of histone deacetylase (HDAC) activity (Arpaia et al., 2013; Furusawa et al., 2013; Smith et al., 2013). In contrast, secondary bile acids regulate the abundance of RORγ+ Treg in a vitamin D receptor (VDR)-dependent manner (Song et al., 2020). Subsequently, Treg cells within the colon, but not in the small intestine and spleen, expressed FFAR2/GPR43 or VDR. Additionally, mice lacking FFAR2 or VDR specifically in Treg cells displayed reduced numbers and function of colonic RORγ+ Treg. However, Treg abundance within other locations was intact. Consequently, feeding mice with short-chain fatty acids, or various secondary bile acid metabolites, increased colonic RORγ+ Treg and ameliorated intestinal inflammation (Arpaia et al., 2013; Furusawa et al., 2013; Sefik et al., 2015; Smith et al., 2013; Song et al., 2020). Interestingly, the gut-microbiota-derived colonic Treg cells were mostly restricted to a subset of Treg cells that express the transcription factor RORγ (these constitute to more than 50% of colonic Treg cells). Thus, mice depleted of gut microbiota, or germ-free mice, were deficient in RORγ+ Treg in part dependent on secondary bile acids and short-chain fatty acid stimulation (Sefik et al., 2015; Song et al., 2020). The metabolic consequences of dysbiosis observed during different disease conditions such as inflammatory bowel disease or cancer, particularly the impact on SCFA and bile acid profiles, could provide new mechanistic insight into how dysbiosis alters T cell adaptation and function during disease pathogenesis.
Figure 3. Tissue-Specific Adaptations of T Cells Goes beyond Cellular Metabolism.
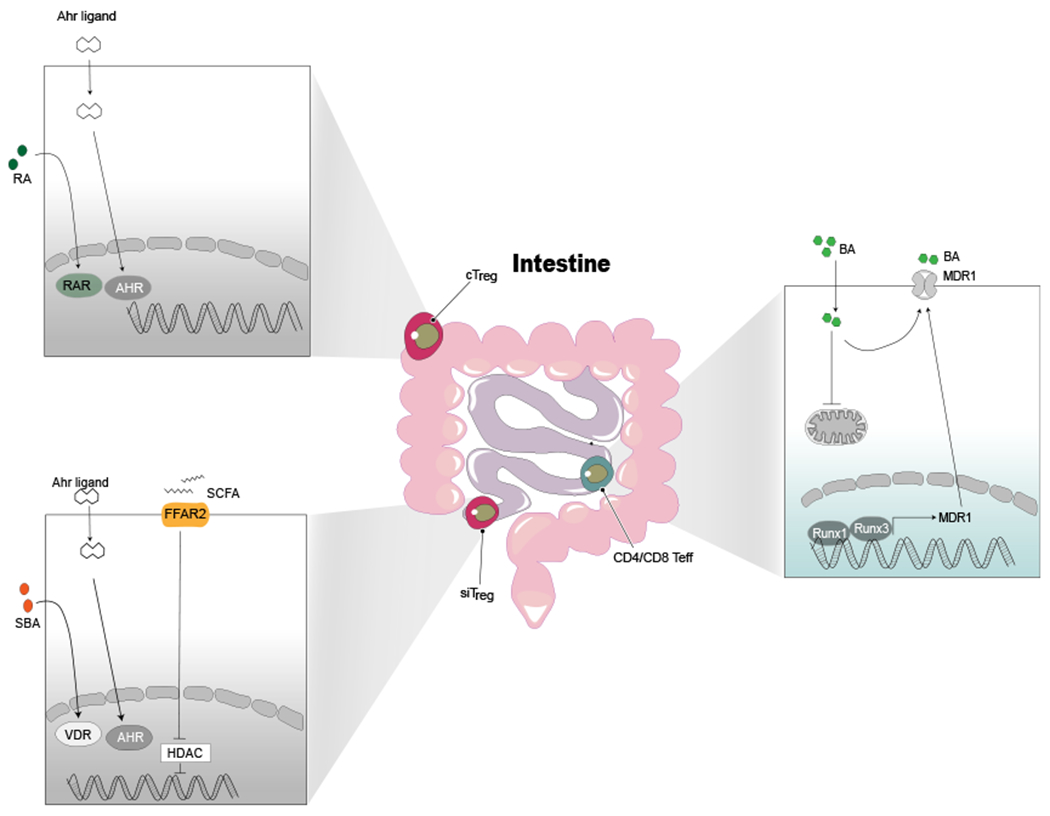
To survive within different tissues that harbor a variety of metabolites, Treg cells and effector T cells evolve new ways to adapt. cTregs (colonic Treg cells) and siTregs (small intestine Treg cells) sense and signal the AHR. Ligands that activates AHR induced expression of different target genes are key to survival within these tissues. Additionally, cTreg cells also sense colon-specific metabolites such as retinoic acid via the retinoid acid receptor (RAR). siTreg cells sense and respond to SCFAs via FFAR2-mediated HDAC signaling and SBAs via VDR signaling, both of which are critical for generation and survival of siTreg cells. CD4/CD8 Teff cells adapt to bile acids in the small intestine by efflux of bile acid (BA) via MDR1.
Whereas microbial-derived metabolites drove Treg cell differentiation and persistence within the colon, Treg cells within the small intestine are primarily driven by dietary antigens or dietary supplements. In fact, germ-free mice devoid of dietary antigens derived from solid foods had fewer Treg cells (Kim et al., 2016). Another dietary factor that shapes the small intestinal immune response is vitamin A, a fat-soluble vitamin that is absorbed in the proximal parts of the small intestine where it is metabolized to its bioactive form, retinoic acid. Together with TGF-β, retinoic acid drives the generation of Treg cells mainly within the small intestine and its draining lymph nodes (Sun et al., 2007). In support of this, feeding mice that are deficient in vitamin A prevented the development of Treg cells and promoted Th17 cells (Ohnmacht et al., 2015). Together with TGF-β, retinoic acid drives the generation of RORγ+ Treg within the small intestine and its draining lymph nodes (Sun et al., 2007). Feeding mice that are deficient in vitamin A prevented the development of RORγ+ Tregs in the small intestine (Ohnmacht et al., 2015). Interestingly, within different regions of the small intestine, zonation occurs with metabolites and immune subsets differing in abundance. While the proximal small intestine and its draining LNs (deodenum/jejunum) preferentially accumulate tolerogenic T cell responses, distal LNs (cecum) favors pro-inflammatory responses, mainly mediated by Th17 cells (Esterházy et al., 2019). However, the link between metabolite abundance within different zones of the small intestine and the spectrum of immune cell types present requires further study.
Of interest, other metabolites generated by host cells, diet, and microbiota (e.g., amino acid tryptophan metabolites) that trigger the transcription factor aryl hydrocarbon receptor (AHR) in both small and large intestines also are involved in Treg maintenance. Thus, Treg cells in the colon and small intestine express higher levels of AHR than Treg cells in other locations express. In consequence, mice with Treg-specific AHR deficiency display a reduction in the number of Treg cells in both intestinal compartments (Ye et al., 2017). Additionally, tissue-infiltrating T cells need to manage some of the toxic metabolites within the intestines to avoid toxic stress and to remain functionally effective. For instance, CD4+ and CD8+ T cells that are within the small intestine lamina propria, but not in lungs, express a xenobiotic transporter-MDR1 (multidrug resistance protein) that is critical for efflux of bile acids that were engulfed (Cao et al., 2017; Chen et al., 2020a). CD4+ T cells lacking MDR1 failed to persist within intestinal inflammatory sites (Cao et al., 2017), and as a consequence, bile acids induced mitochondrial toxicity (Cao et al., 2017; Chen et al., 2020a). Interestingly, the expression of MDR1 in T cells is regulated in part by a group of Runt-related transcription factors (Runx). Thus, knockdown of Runx1 or Runx3 inhibited the expression of MDR1 (Chen et al., 2020a). We still need to understand how and when the various adaptation signals are acquired by tissue-infiltrating T cells and if the adaptations change over time in the case of cells that are short lived compared with cells that establish long term residence.
A possible mechanism by which tissue-infiltrating T cells acquire adaptation signals is by interacting with dendritic cells that drain the tissue to the lymph nodes. For instance, migratory dendritic cells that express both the transcription factor Batf3 and αV integrins are critical for the generation of skin-resident memory cells, especially cells that reside in the epidermis, but not the dermis or circulating memory T cells (Iborra et al., 2016; Mani et al., 2019). Although it remains to be shown if priming by these dendritic cells imprints any metabolic features onto skin-resident memory cells, dendritic cells draining other tissues can imprint some metabolic signatures. For example, the expression of MDR1 within intestinal T cells could be induced by migratory dendritic cells isolated from the gut (Cao et al., 2017). Similarly, Treg cells generated within the small intestine depend on dendritic-cell-mediated conversion of vitamin A to retinoic acid via oxidation by retinal dehydrogenases (Coombes et al., 2007; Sun et al., 2007). However, within the colon, secondary bile acids (SBAs) such as isoDCA-induced colonic Treg cells required activation via the bile acid nuclear receptor-FXR on dendritic cells (Campbell et al., 2020). We summarize these adaptations in Figure 3. Another potential explanation for adaptation is that the metabolites or compounds within the tissues could directly regulate the expression of various tissue-specific metabolic proteins. For example, culturing in-vitro-activated T cells with tissue lysates from the skin or liver induced the expression of tissue-specific FABP isoforms (Fabp4 when cultured with skin extracts and the expression of Fabp1 when cultured with liver extracts) (Frizzell et al., 2020). However, the exact factors present in these tissues and the mechanisms that drive the expression of tissue-specific isoforms of FABP are yet to be discovered. Taken together, this indicates that different zones within tissues (especially the intestinal tract) present some unique and different metabolic microenvironments. These effects could influence T cell responses, and this presents a need for them to differentially adapt to stay functional.
CONCLUDING REMARKS
Research on immunometabolism has transformed our understanding and control of diseases that include cancers, autoimmunity, cardiovascular diseases, and infections. Different T cell subsets and other cell types involved in those diseases rely on different metabolic pathways to support their functions, and these pathways can potentially be manipulated therapeutically to change the disease outcome. We have discussed evidence showing how cells with different functions take cues from their tissue location and adjust their metabolism to carry out their activities at that tissue site. Many, but not all, of the cues are metabolic precursors or products, with different immune cells responding differentially to the cues to perform their functions. For example, upon entering into the various tissues, T cells can sense nutrient levels present at the site and can then rewire their metabolic activities to a pattern that allows them to survive and function optimally at that site. We refer to these metabolic adjustments made by T cells at tissue-reactive sites as T cell adaptations as was also recently reviewed for Tregs (Wang et al., 2017; Wang et al., 2020b). We propose that 4 factors contribute to the adaptation of T cells to their dynamic tissue environment. These are (1) functional status of T cells, (2) local factors unique to a niche of the tissue, (3) type of inflammation that might be occurring in the tissue, and (4) time spent in a specific tissue. These adaptations differ between different tissue locations and disease events, and this can be either beneficial or detrimental for disease control. Identifying the mechanisms of T cell adaptation could result in developing therapeutic approaches to change such events when untoward happenings occur. We have described the observations pertinent to this topic and highlight some of the open questions that remain to be answered at a mechanistic level. Thus, although it is possible that some adaptations are tissue specific, others might be global and common to all tissues. So far, our understanding of T cell adaptations within different tissues is restricted to few genes or pathways and is also focused on one specific tissue per study. Future studies should resolve how adaptations are controlled at a global scale and should compare multiple tissues simultaneously. Moreover, these adaptations need to be comparatively studied within both functional and dysfunctional T cells.
Other issues needing evaluation that could affect adaptations include differences between humans and mouse models and the influence of sex, diet, and age. With recent advancements in measuring changes in T cell metabolism at the single-cell level (Hartmann et al., 2020), and in the near future spatial level, we could correlate differences in metabolism to the T cell function in space and time and to be able to develop more focused therapeutic approaches. The ultimate payoff will be to identify extracellular and intracellular factors that regulate tissue-specific adaptations and then be able to modulate them specifically at a tissue level with currently available or developing drugs. This approach could overcome the problem that most currently used immune or metabolic modulating drugs are designed on the basis of changes observed in lymphoid tissues or blood as opposed to tissues and act at a whole-body level that can result in low efficacy or unwanted complications and toxicity. As a rapidly maturing field, immunometabolism is now poised to move forward to address.
ACKNOWLEDGMENTS
We thank Mark Sundrud for insightful edits and comments on the manuscript. B.T.R. is supported by R01-EY005093-36 and R21-AI 142862-02 from National Eye Institute (NEI) and National Institute of Allergy and Infectious Diseases (NIAID), respectively. V.S.K. is supported by Cancer Research Institute Postdoctoral fellowship (ID: 60810).
REFERENCES
- Alissafi T, Kalafati L, Lazari M, Filia A, Kloukina I, Manifava M, Lim JH, Alexaki VI, Ktistakis NT, Doskas T, et al. (2020). Mitochondrial oxidative damage underlies regulatory T cell defects in autoimmunity. Cell Metab. 32, 591–604.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amorini AM, Nociti V, Petzold A, Gasperini C, Quartuccio E, Lazzarino G, Di Pietro V, Belli A, Signoretti S, Vagnozzi R, et al. (2014). Serum lactate as a novel potential biomarker in multiple sclerosis. Biochim Biophys Acta 1842, 1137–1143. [DOI] [PubMed] [Google Scholar]
- Angelin A, Gil-de-Gómez L, Dahiya S, Jiao J, Guo L, Levine MH, Wang Z, Quinn WJ III, Kopinski PK, Wang L, et al. (2017). Foxp3 reprograms T cell metabolism to function in low-glucose, high-lactate environments. Cell Metab. 25, 1282–1293.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, Deroos P, Liu H, Cross JR, Pfeffer K, Coffer PJ, and Rudensky AY (2013). Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 504, 451–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arpaia N, Green JA, Moltedo B, Arvey A, Hemmers S, Yuan S, Treuting PM, and Rudensky AY (2015). A distinct function of regulatory T cells in tissue protection. Cell 162, 1078–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailis W, Shyer JA, Zhao J, Canaveras JCG, Al Khazal FJ, Qu R, Steach HR, Bielecki P, Khan O, Jackson R, et al. (2019). Distinct modes of mitochondrial metabolism uncouple T cell differentiation and function. Nature 571, 403–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baixauli F, Acín-Pérez R, Villarroya-Beltrí C, Mazzeo C, Nuñez-Andrade N, Gabandé-Rodriguez E, Ledesma MD, Blázquez A, Martin MA, Falcón-Pérez JM, et al. (2015). Mitochondrial respiration controls lysosomal function during inflammatory T cell responses. Cell Metab. 22, 485–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengsch B, Johnson AL, Kurachi M, Odorizzi PM, Pauken KE, Attanasio J, Stelekati E, McLane LM, Paley MA, Delgoffe GM, and Wherry EJ (2016). Bioenergetic insufficiencies due to metabolic alterations regulated by the inhibitory receptor PD-1 are an early driver of CD8(+) T cell exhaustion. Immunity 45, 358–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, Matos C, Bruss C, Klobuch S, Peter K, et al. (2016). LDHA-associated lactic acid production blunts tumor immunosurveillance by T and NK cells. Cell Metab. 24, 657–671. [DOI] [PubMed] [Google Scholar]
- Brummelman J, Mazza EMC, Alvisi G, Colombo FS, Grilli A, Mikulak J, Mavilio D, Alloisio M, Ferrari F, Lopci E, et al. (2018). High-dimensional single cell analysis identifies stem-like cytotoxic CD8+ T cells infiltrating human tumors. J. Exp. Med 215, 2520–2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck MD, O’Sullivan D, Klein Geltink RIK, Curtis JD, Chang CH, Sanin DE, Qiu J, Kretz O, Braas D, van der Windt GJ, et al. (2016). Mitochondrial dynamics controls T cell fate through metabolic programming. Cell 166, 63–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell C, McKenney PT, Konstantinovsky D, Isaeva OI, Schizas M, Verter J, Mai C, Jin W-B, Guo CJ, Violante S, et al. (2020). Bacterial metabolism of bile acids promotes generation of peripheral regulatory T cells. Nature 581, 475–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao W, Kayama H, Chen ML, Delmas A, Sun A, Kim SY, Rangarajan ES, McKevitt K, Beck AP, Jackson CB, et al. (2017). The xenobiotic transporter Mdr1 enforces T cell homeostasis in the presence of intestinal bile acids. Immunity 47, 1182–1196.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champagne DP, Hatle KM, Fortner KA, D’Alessandro A, Thornton TM, Yang R, Torralba D, Tomá s-Cortá zar J, Jun YW, Ahn KH, et al. (2016). Fine-tuning of CD8(+) T cell mitochondrial metabolism by the respiratory chain repressor MCJ dictates protection to influenza virus. Immunity 44, 1299–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman NM, Zeng H, Nguyen T-LM, Wang Y, Vogel P, Dhungana Y, Liu X, Neale G, Locasale JW, and Chi H (2018). mTOR coordinates transcriptional programs and mitochondrial metabolism of activated T reg subsets to protect tissue homeostasis. Nat. Commun 9, 2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ML, Sun A, Cao W, Eliason A, Mendez KM, Getzler AJ, Tsuda S, Diao H, Mukori C, Bruno NE, et al. (2020a). Physiological expression and function of the MDR1 transporter in cytotoxic T lymphocytes. J. Exp. Med 217, e20191388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P-M, Wilson PC, Shyer JA, Veselits M, Steach HR, Cui C, Moeckel G, Clark MR, and Craft J (2020b). Kidney tissue hypoxia dictates T cell–mediated injury in murine lupus nephritis. Sci. Transl. Med 12, eaay1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung KW, Dhillon P, Huang S, Sheng X, Shrestha R, Qiu C, Kaufman BA, Park J, Pei L, Baur J, et al. (2019). Mitochondrial damage and activation of the STING pathway lead to renal inflammation and fibrosis. Cell Metab. 30, 784–799.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipolletta D, Feuerer M, Li A, Kamei N, Lee J, Shoelson SE, Benoist C, and Mathis D (2012). PPAR-γ is a major driver of the accumulation and phenotype of adipose tissue T reg cells. Nature 486, 549–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clambey ET, McNamee EN, Westrich JA, Glover LE, Campbell EL, Jedlicka P, de Zoeten EF, Cambier JC, Stenmark KR, Colgan SP, and Eltzschig HK (2012). Hypoxia-inducible factor-1 alpha–dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc. Natl. Acad. Sci. USA 109, E2784–E2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins N, Han SJ, Enamorado M, Link VM, Huang B, Moseman EA, Kishton RJ, Shannon JP, Dixit D, Schwab SR, et al. (2019). The bone marrow protects and optimizes immunological memory during dietary restriction. Cell 178, 1088–1101.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coombes JL, Siddiqui KR, Arancibia-Cá rcamo CV, Hall J, Sun CM, Belkaid Y, and Powrie F (2007). A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta– and retinoic acid–dependent mechanism. J. Exp. Med 204, 1757–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couturier J, Nuotio-Antar AM, Agarwal N, Wilkerson GK, Saha P, Kulkarni V, Lakhashe SK, Esquivel J, Nehete PN, Ruprecht RM, et al. (2019). Lymphocytes upregulate CD36 in adipose tissue and liver. Adipocyte 8, 154–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui G, Staron MM, Gray SM, Ho PC, Amezquita RA, Wu J, and Kaech SM (2015). IL-7-induced glycerol transport and TAG synthesis promotes memory CD8+ T cell longevity. Cell 161, 750–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delacher M, Imbusch CD, Hotz-Wagenblatt A, Mallm JP, Bauer K, Simon M, Riegel D, Rendeiro AF, Bittner S, Sanderink L, et al. (2020). Precursors for nonlymphoid-tissue Treg cells reside in secondary lymphoid organs and are programmed by the transcription factor BATF. Immunity 52, 295–312.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desdín-Micó G, Soto-Heredero G, Aranda JF, Oller J, Carrasco E, Gabandé-Rodríguez E, Blanco EM, Alfranca A, Cussó L, Desco M, et al. (2020). T cells with dysfunctional mitochondria induce multimorbidity and premature senescence. Science 368, 1371–1376. [DOI] [PubMed] [Google Scholar]
- Dubrac S, Elentner A, Schoonjans K, Auwerx J, and Schmuth M (2011). Lack of IL-2 in PPAR-α-deficient mice triggers allergic contact dermatitis by affecting regulatory T cells. Eur. J. Immunol 41, 1980–1991. [DOI] [PubMed] [Google Scholar]
- Dumauthioz N, Tschumi B, Wenes M, Marti B, Wang H, Franco F, Li W, Lopez-Mejia IC, Fajas L, Ho P-C, et al. (2020). Enforced PGC-1α expression promotes CD8 T cell fitness, memory formation and antitumor immunity. Cell. Mol. Immunol 10.1038/s41423-020-0365-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esterházy D, Canesso MCC, Mesin L, Muller PA, de Castro TBR, Lockhart A, ElJalby M, Faria AMC, and Mucida D (2019). Compartmentalized gut lymph node drainage dictates adaptive immune responses. Nature 569, 126–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field CS, Baixauli F, Kyle RL, Puleston DJ, Cameron AM, Sanin DE, Hippen KL, Loschi M, Thangavelu G, Corrado M, et al. (2020). Mitochondrial integrity regulated by lipid metabolism is a cell-intrinsic checkpoint for Treg suppressive function. Cell Metab. 31, 422–437.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint HJ, Scott KP, Duncan SH, Louis P, and Forano E (2012). Microbial degradation of complex carbohydrates in the gut. Gut Microbes 3, 289–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frizzell H, Fonseca R, Christo SN, Evrard M, Cruz-Gomez S, Zanluqui NG, von Scheidt B, Freestone D, Park SL, McWilliam HEG, et al. (2020). Organ-specific isoform selection of fatty acid–binding proteins in tissue-resident lymphocytes. Sci. Immunol 5, eaay9283. [DOI] [PubMed] [Google Scholar]
- Fu Z, Ye J, Dean JW, Bostick JW, Weinberg SE, Xiong L, Oliff KN, Chen ZE, Avram D, Chandel NS, and Zhou L (2019). Requirement of mitochondrial transcription factor A in tissue-resident regulatory T cell maintenance and function. Cell Rep. 28, 159–171.e4. e154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, Nakanishi Y, Uetake C, Kato K, Kato T, et al. (2013). Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504, 446–450. [DOI] [PubMed] [Google Scholar]
- Gerriets VA, Kishton RJ, Johnson MO, Cohen S, Siska PJ, Nichols AG, Warmoes MO, de Cubas AA, MacIver NJ, Locasale JW, et al. (2016). Foxp3 and toll-like receptor signaling balance T reg cell anabolic metabolism for suppression. Nat. Immunol 17, 1459–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerriets VA, Kishton RJ, Nichols AG, Macintyre AN, Inoue M, Ilkayeva O, Winter PS, Liu X, Priyadharshini B, Slawinska ME, et al. (2015). Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J. Clin. Invest 125, 194–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman RP, Markhard AL, Shah H, Sharma R, Skinner OS, Clish CB, Deik A, Patgiri A, Hsu Y-HH, Masia R, et al. (2020). Hepatic NADH reductive stress underlies common variation in metabolic traits. Nature 583, 122–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas R, Smith J, Rocher-Ros V, Nadkarni S, Montero-Melendez T, D’Acquisto F, Bland EJ, Bombardieri M, Pitzalis C, Perretti M, et al. (2015). Lactate regulates metabolic and pro-inflammatory circuits in control of T cell migration and effector functions. PLoS Biol. 13, e1002202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han SJ, Glatman Zaretsky A, Andrade-Oliveira V, Collins N, Dzutsev A, Shaik J, Morais da Fonseca D, Harrison OJ, Tamoutounour S, Byrd AL, et al. (2017). White adipose tissue is a reservoir for memory T cells and promotes protective memory responses to infection. Immunity 47, 1154–1168.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann FJ, Mrdjen D, McCaffrey E, Glass DR, Greenwald NF, Bharadwaj A, Khair Z, Verberk SGS, Baranski A, Baskar R, et al. (2020). Single-cell metabolic profiling of human cytotoxic T cells. Nat. Biotechnol 10.1038/s41587-020-0651-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillman ET, Lu H, Yao T, and Nakatsu CH (2017). Microbial ecology along the gastrointestinal tract. Microbes Environ. 32, 300–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho P-C, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, Tsui Y-C, Cui G, Micevic G, Perales JC, et al. (2015). Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell 162, 1217–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howie D, Cobbold SP, Adams E, Ten Bokum A, Necula AS, Zhang W, Huang H, Roberts DJ, Thomas B, Hester SS, et al. (2017). Foxp3 drives oxidative phosphorylation and protection from lipotoxicity. JCI Insight 2, e89160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iborra S, Martínez-López M, Khouili SC, Enamorado M, Cueto FJ, Conde-Garrosa R, del Fresno C, and Sancho D (2016). Optimal generation of tissue-resident but not circulating memory T cells during viral infection requires crosspriming by DNGR-1+ dendritic cells. Immunity 45, 847–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabat AM, Harrison OJ, Riffelmacher T, Moghaddam AE, Pearson CF, Laing A, Abeler-Dörner L, Forman SP, Grencis RK, Sattentau Q, et al. (2016). The autophagy gene Atg16l1 differentially regulates Treg and TH2 cells to control intestinal inflammation. eLife 5, e12444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KS, Hong S-W, Han D, Yi J, Jung J, Yang B-G, Lee JY, Lee M, and Surh CD (2016). Dietary antigens limit mucosal immunity by inducing regulatory T cells in the small intestine. Science 351, 858–863. [DOI] [PubMed] [Google Scholar]
- Kishore M, Cheung KCP, Fu H, Bonacina F, Wang G, Coe D, Ward EJ, Colamatteo A, Jangani M, Baragetti A, et al. (2017). Regulatory T cell migration is dependent on glucokinase-mediated glycolysis. Immunity 47, 875–889.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klysz D, Tai X, Robert PA, Craveiro M, Cretenet G, Oburoglu L, Mongellaz C, Floess S, Fritz V, Matias MI, et al. (2015). Glutamine-dependent α-ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation. Sci. Signal 8, ra97. [DOI] [PubMed] [Google Scholar]
- Konjar Š, Frising UC, Ferreira C, Hinterleitner R, Mayassi T, Zhang Q, Blankenhaus B, Haberman N, Loo Y, Guedes J, et al. (2018). Mitochondria maintain controlled activation state of epithelial-resident T lymphocytes. Sci. Immunol 3, eaan2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lathrop SK, Bloom SM, Rao SM, Nutsch K, Lio CW, Santacruz N, Peterson DA, Stappenbeck TS, and Hsieh CS (2011). Peripheral education of the immune system by colonic commensal microbiota. Nature 478, 250–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Zhu B, Son YM, Wang Z, Jiang L, Xiang M, Ye Z, Beckermann KE, Wu Y, Jenkins JW, et al. (2019). The transcription factor Bhlhe40 programs mitochondrial regulation of resident CD8+ T cell fitness and functionality. Immunity 51, 491–507.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin R, Zhang H, Yuan Y, He Q, Zhou J, Li S, Sun Y, Li DY, Qiu H-B, Wang W, et al. (2020). Fatty acid oxidation controls CD8+ tissue-resident memory T-cell survival in gastric adenocarcinoma. Cancer Immunol. Res 8, 479–492. [DOI] [PubMed] [Google Scholar]
- Ma EH, Bantug G, Griss T, Condotta S, Johnson RM, Samborska B, Mainolfi N, Suri V, Guak H, Balmer ML, et al. (2017). Serine is an essential metabolite for effector T cell expansion. Cell Metab. 25, 345–357. [DOI] [PubMed] [Google Scholar]
- Ma EH, Verway MJ, Johnson RM, Roy DG, Steadman M, Hayes S, Williams KS, Sheldon RD, Samborska B, Kosinski PA, et al. (2019). Metabolic profiling using stable isotope tracing reveals distinct patterns of glucose utilization by physiologically activated CD8+ T cells. Immunity 51, 856–870.e5. [DOI] [PubMed] [Google Scholar]
- Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, Anderson SM, Abel ED, Chen BJ, Hale LP, and Rathmell JC (2014). The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. 20, 61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maj T, Wang W, Crespo J, Zhang H, Wang W, Wei S, Zhao L, Vatan L, Shao I, Szeliga W, et al. (2017). Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat. Immunol 18, 1332–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makowski L, Chaib M, and Rathmell JC (2020). Immunometabolism: from basic mechanisms to translation. Immunol. Rev 295, 5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani V, Bromley SK, T. Äijö, Mora-Buch R, Carrizosa E, Warner RD, Hamze M, Sen DR, Chasse AY, Lorant A, et al. (2019). Migratory DCs activate TGF-β to precondition naïve CD8+ T cells for tissue-resident memory fate. Science 366, eaav5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, Sullivan SA, Nichols AG, and Rathmell JC (2011). Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol 186, 3299–3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, Yates KB, Lako A, Felt K, Naik GS, et al. (2019). Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat. Immunol 20, 326–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miragaia RJ, Gomes T, Chomka A, Jardine L, Riedel A, Hegazy AN, Whibley N, Tucci A, Chen X, Lindeman I, et al. (2019). Single-cell transcriptomics of regulatory T cells reveals trajectories of tissue adaptation. Immunity 50, 493–504.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miska J, Lee-Chang C, Rashidi A, Muroski ME, Chang AL, Lopez-Rosas A, Zhang P, Panek WK, Cordero A, Han Y, et al. (2019). HIF-1α is a metabolic switch between glycolytic-driven migration and oxidative phosphorylation-driven immunosuppression of Tregs in glioblastoma. Cell Rep. 27, 226–237.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan D, van der Windt GW, Huang SC-C, Curtis JD, Chang C-H, Buck MD, Qiu J, Smith AM, Lam WY, DiPlato LM, et al. (2014). Memory CD8(+) T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity 41, 75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnmacht C, Park JH, Cording S, Wing JB, Atarashi K, Obata Y, Gaboriau-Routhiau V, Marques R, Dulauroy S, Fedoseeva M, et al. (2015). MUCOSAL IMMUNOLOGY. The microbiota regulates type 2 immunity through RORγt+ T cells. Science 349, 989–993. [DOI] [PubMed] [Google Scholar]
- Pacella I, Procaccini C, Focaccetti C, Miacci S, Timperi E, Faicchia D, Severa M, Rizzo F, Coccia EM, Bonacina F, et al. (2018). Fatty acid metabolism complements glycolysis in the selective regulatory T cell expansion during tumor growth. Proc. Natl. Acad. Sci. USA 115, E6546–E6555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palazon A, Tyrakis PA, Macias D, Veliç a P, Rundqvist H, Fitzpatrick S, Vojnovic N, Phan AT, Loman N, Hedenfalk I, et al. (2017). An HIF-1α/VEGF-A axis in cytotoxic T cells regulates tumor progression. Cancer Cell 32, 669–683.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y, Tian T, Park CO, Lofftus SY, Mei S, Liu X, Luo C, O’Malley JT, Gehad A, Teague JE, et al. (2017). Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature 543, 252–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patgiri A, Skinner OS, Miyazaki Y, Schleifer G, Marutani E, Shah H, Sharma R, Goodman RP, To TL, Robert Bao XR, et al. (2020). An engineered enzyme that targets circulating lactate to alleviate intracellular NADH: NAD+ imbalance. Nat. Biotechnol 38, 309–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pucino V, Bombardieri M, Pitzalis C, and Mauro C (2017). Lactate at the crossroads of metabolism, inflammation, and autoimmunity. Eur. J. Immunol 47, 14–21. [DOI] [PubMed] [Google Scholar]
- Pucino V, Certo M, Bulusu V, Cucchi D, Goldmann K, Pontarini E, Haas R, Smith J, Headland SE, Blighe K, et al. (2019). Lactate buildup at the site of chronic inflammation promotes disease by inducing CD4+ T cell metabolic rewiring. Cell Metab. 30, 1055–1074.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raud B, Roy DG, Divakaruni AS, Tarasenko TN, Franke R, Ma EH, Samborska B, Hsieh WY, Wong AH, Stüve P, et al. (2018). Etomoxir actions on regulatory and memory T cells are independent of Cpt1a-mediated fatty acid oxidation. Cell Metab. 28, 504–515.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosato PC, Wijeyesinghe S, Stolley JM, and Masopust D (2020). Integrating resident memory into T cell differentiation models. Curr. Opin. Immunol 63, 35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rundqvist H, Veliça P, Barbieri L, Gameiro P, Cunha PP, Gojkovic M, Mijwel S, Ahlstedt E, Foskolou IP, Ruiz-Pérez MV, et al. (2019). Lactate potentiates differentiation and expansion of cytotoxic T cells. bioRxiv. 10.1101/571745v1. [DOI] [Google Scholar]
- Scharping NE, Menk AV, Moreci RS, Whetstone RD, Dadey RE, Watkins SC, Ferris RL, and Delgoffe GM (2016). The tumor microenvironment represses T cell mitochondrial biogenesis to drive intratumoral T cell metabolic insufficiency and dysfunction. Immunity 45, 374–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sefik E, Geva-Zatorsky N, Oh S, Konnikova L, Zemmour D, McGuire AM, Burzyn D, Ortiz-Lopez A, Lobera M, Yang J, et al. (2015). MUCOSAL IMMUNOLOGY. Individual intestinal symbionts induce a distinct population of RORγ+ regulatory T cells. Science 349, 993–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, Wang CR, Schumacker PT, Licht JD, Perlman H, et al. (2013). Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 38, 225–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui I, Schaeuble K, Chennupati V, Fuertes Marraco SA, Calderon-Copete S, Pais Ferreira D, Carmona SJ, Scarpellino L, Gfeller D, Pradervand S, et al. (2019). Intratumoral Tcf1+ PD-1+ CD8+ T cells with stemlike properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity 50, 195–211.e10. [DOI] [PubMed] [Google Scholar]
- Siska PJ, Beckermann KE, Mason FM, Andrejeva G, Greenplate AR, Sendor AB, Chiang Y-CJ, Corona AL, Gemta LF, Vincent BG, et al. (2017). Mitochondrial dysregulation and glycolytic insufficiency functionally impair CD8 T cells infiltrating human renal cell carcinoma. JCI Insight 2, e93411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly-Y M, Glickman JN, and Garrett WS (2013). The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 341, 569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Sun X, Oh SF, Wu M, Zhang Y, Zheng W, Geva-Zatorsky N, Jupp R, Mathis D, Benoist C, and Kasper DL (2020). Microbial bile acid metabolites modulate gut RORγ+ regulatory T cell homeostasis. Nature 577, 410–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun CM, Hall JA, Blank RB, Bouladoux N, Oukka M, Mora JR, and Belkaid Y (2007). Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J. Exp. Med 204, 1775–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo PA, Miron M, and Farber DL (2019). Location, location, location: tissue resident memory T cells in mice and humans. Sci. Immunol 4, eaas9673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utzschneider DT, Charmoy M, Chennupati V, Pousse L, Ferreira DP, Calderon-Copete S, Danilo M, Alfei F, Hofmann M, Wieland D, et al. (2016). T cell factor 1-expressing memory-like CD8(+) T cells sustain the immune response to chronic viral infections. Immunity 45, 415–427. [DOI] [PubMed] [Google Scholar]
- Vaeth M, Schliesser U,Müller G, Reissig S, Satoh K, Tuettenberg A, Jonuleit H, Waisman A, Müller MR, Serfling E, et al. (2012). Dependence on nuclear factor of activated T-cells (NFAT) levels discriminates conventional T cells from Foxp3+ regulatory T cells. Proc. Natl. Acad. Sci. USA 109, 16258–16263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varanasi SK, Rajasagi NK, Jaggi U, and Rouse BT (2018). Role of IL-18 induced amphiregulin expression on virus induced ocular lesions. Mucosal Immunol. 11, 1705–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Franco F, and Ho PC (2017). Metabolic regulation of Tregs in cancer: opportunities for immunotherapy. Trends Cancer 3, 583–592. [DOI] [PubMed] [Google Scholar]
- Wang H, Franco F, Tsui YC, Xie X, Trefny MP, Zappasodi R, Mohmood SR, Ferná ndez-Garcıá J, Tsai C-H, Schulze I, et al. (2020a). CD36-mediated metabolic adaptation supports regulatory T cell survival and function in tumors. Nat. Immunol 21, 298–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Lu CH, and Ho P-C (2020b). Metabolic adaptation orchestrates tissue context-dependent behavior in regulatory T cells. Immunol. Rev 295, 126–139. [DOI] [PubMed] [Google Scholar]
- Weinberg SE, Singer BD, Steinert EM, Martinez CA, Mehta MM, Martı ńez-Reyes I, Gao P, Helmin KA, Abdala-Valencia H, Sena LA, et al. (2019). Mitochondrial complex III is essential for suppressive function of regulatory T cells. Nature 565, 495–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, Bestwick M, Duguay BA, Raimundo N, MacDuff DA, et al. (2015). Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520, 553–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Fujii H, Mohan SV, Goronzy JJ, and Weyand CM (2013). Phosphofructokinase deficiency impairs ATP generation, autophagy, and redox balance in rheumatoid arthritis T cells. J. Exp. Med 210, 2119–2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Qiu J, Bostick JW, Ueda A, Schjerven H, Li S, Jobin C, Chen ZE, and Zhou L (2017). The aryl hydrocarbon receptor preferentially marks and promotes gut regulatory T cells. Cell Rep. 21, 2277–2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Kurupati R, Liu L, Zhou XY, Zhang G, Hudaihed A, Filisio F, Giles-Davis W, Xu X, Karakousis GC, et al. (2017). Enhancing CD8+ T cell fatty acid catabolism within a metabolically challenging tumor microenvironment increases the efficacy of melanoma immunotherapy. Cancer Cell 32, 377–391.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]