

Abstract
Despite widespread utilization of immunotherapy, treating immune-cold tumors has proven to be a challenge. Here, we report that expression of the immune checkpoint molecule B7-H4 is prevalent among immune-cold triple negative breast cancers, where its expression inversely correlates with that of PD-L1. Glycosylation of B7-H4 interferes with its interaction/ubiquitination by AMFR, resulting in B7-H4 stabilization. B7-H4 expression inhibits doxorubicin-induced cell death through the suppression of eIF2α phosphorylation required for calreticulin exposure vis-à-vis the cancer cells. NGI-1, which inhibits B7-H4 glycosylation causing its ubiquitination and subsequent degradation, improves the immunogenic properties of cancer cells treated with doxorubicin, enhancing their phagocytosis by dendritic cells and their capacity to elicit CD8+ interferon-γ-producing T cell responses. In preclinical models of TNBC, a triple combination of NGI-1, camsirubicin (a non-cardiotoxic doxorubicin analog) and PD-L1 blockade was effective in reducing tumor growth. Collectively, our findings uncover a novel strategy for targeting the immunosuppressive molecule B7-H4.
Keywords: B7-H4, AMFR, STT3A, ubiquitination, glycosylation, tumor immunogenicity, immunogenic cell death
INTRODUCTION
Despite the widespread use of immunotherapy, the poor clinical response by immune-cold tumors is a current challenge. Triple negative breast cancer (TNBC) is the most aggressive mammary carcinoma subtype. Although a PD-L1 inhibitor has been FDA approved for the treatment of metastatic TNBC, the majority of TNBC patients show limited responses, particularly when their tumors are “cold” or “non-inflamed” (1). In contrast with “hot” tumors, these immune-cold tumors are characterized by a scarcity of T lymphocyte infiltration, commensurate with their failure to elicit anticancer immunity (2). Our endeavor to identify suitable targets for improving anticancer immune responses against TNBC therapy has drawn our attention to B7-H4.
B7-H4, also known as V-set domain containing T cell activation inhibitor 1 (VTCN1/B7x/B7 homolog4/B7S1) is a type I transmembrane protein and belongs to the co-inhibitory B7 family ligands (3–5). It regulates T cells, neutrophils as well as macrophages, and its local expression in the tumor has been linked to poor prognosis for ovarian, uterus, gastric, lung, kidney, and breast cancers (6). Although B7-H4 levels are known to be regulated at the posttranscriptional level, a detailed exploration of its posttranslational modifications and regulation has been elusive, further spurring our interest in this gene product.
Cancer cells are known to evade the immune system through ‘don’t eat me’ signals including beta-2 microglobulin, CD24, CD47 and perhaps even PD-L1 (7–12). These signals allow malignant cells to escape recognition and engulfment by dendritic cells (DCs), thus resulting in a failure to generate anti-tumor immune responses. Some chemotherapeutic drugs such as anthracyclines and microtubule-destabilizing agents possess an immune-stimulatory function by triggering immunogenic cell death (13). ICD is usually characterized by the cell membrane exposure of damage-associated molecular patterns (DAMPs) such as calreticulin (CALR) and heat shock proteins (HSP)70 and HSP90, which serve as ‘eat me’ signals to facilitate the recognition of stressed cancer cells by DCs (14). Extensive biochemical analyses have revealed the translocation of CALR from the endoplasmic reticulum (ER) to the cell surface relies on a particular ER stress signal, the phosphorylation of eukaryotic initiation factor 2a (eIF2α), which is the pathognomonic hallmark of ICD (7,15). Nevertheless, how CALR exposure may be suppressed to favor immune evasion remains elusive.
In the current study, we unravel a novel regulatory mechanism by which B7-H4 protein turnover is governed by asparagine (N)-linked glycosylation. N-linked glycosylation is a co- and post-translational modification important for protein stability, folding, trafficking, and physiological function (16,17). Glycosylation of B7-H4 catalyzed by specific glycosyltransferases (STT3A and UGGG1) antagonizes the B7-H4 ubiquitination by E3 ligase autocrine motility factor receptor (AMFR), thereby preventing the degradation of B7-H4 and hence stabilizing the protein. Abundant B7-H4 can inhibit the phosphorylation of eIF2α, CALR exposure, and cancer cell immunogenicity. We designed a strategy for inhibiting the oligosaccharyltransferase (OST) complex that stabilizes B7-H4, thus reducing its expression and enhancing the immunogenicity TNBC cells in the context of ICD-eliciting chemotherapy and PD-L1-targeting immunotherapy.
RESULTS
Accumulation of B7-H4 in immune-cold breast cancers inversely correlates with PD-L1 expression.
To identify potential therapeutic targets for immune-cold breast cancer, we performed a bioinformatic analysis of proteomic data concerning 100 immune-relevant proteins from 81 breast cancer patients in The Cancer Genome Atlas (TCGA) (18). Elevated expression of three proteins, including PROM1, B7-H4 (VTCN1) and MMP7, was detected in the basal (TNBC) subtype of breast cancer compared with other subtypes (Fig. 1A).
Figure 1. Accumulation of B7-H4 is associated with immune-cold breast cancer and reduced PD-L1 expression.
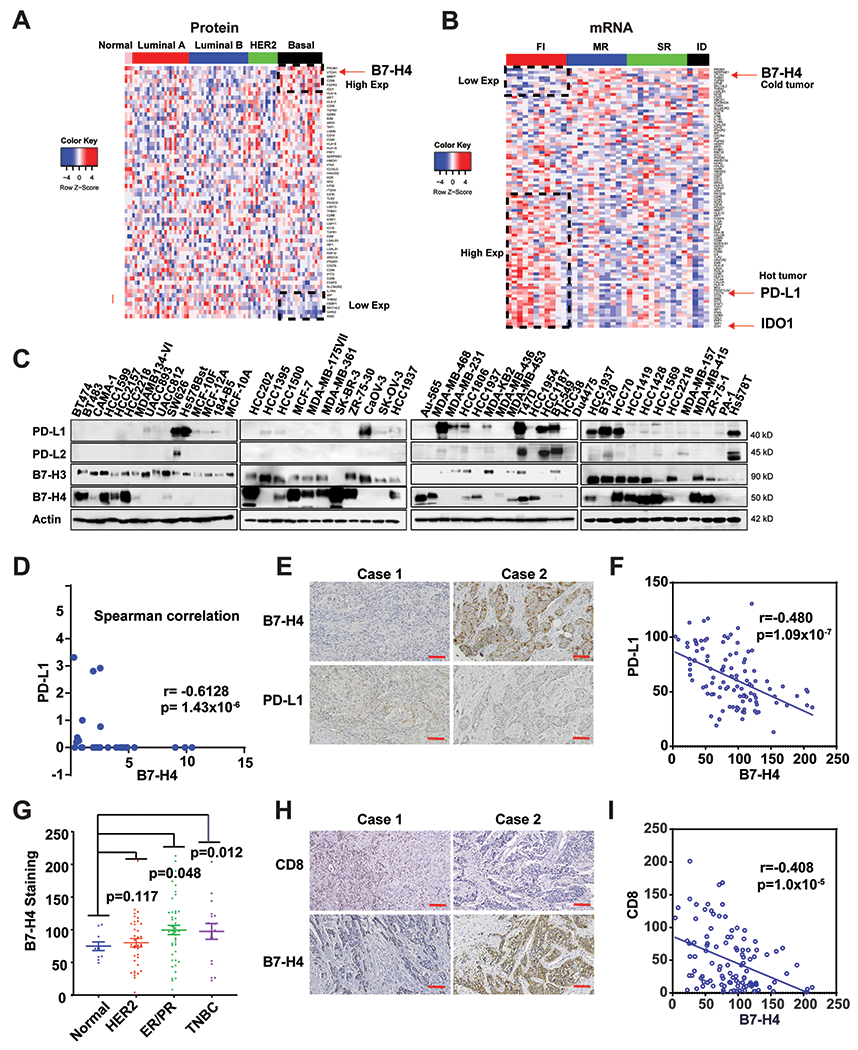
(A) Proteomic analysis of 59 immune-relevant proteins in TCGA samples of PAM50-defined intrinsic subtypes including 25 basal-like, 29 luminal A, 33 luminal B, and 18 HER2-enriched tumors, along with three normal breast tissue samples. The genes (rows) are sorted according to the difference between the average proteomic level in basal subtype and the average proteomic in the other cancer types. (B) Heatmaps depicting expression of immune-relevant genes (mRNA) in the bulk tumor in FI (fully inflamed), SR (stroma restricted), MR (margin restricted) and ID (immune desert) TNBC (n=37). (C) The protein expression of PD-L1, PD-L2, B7-H3 and B7-H4 in 45 breast and 4 ovarian cancer cell lines were measured by immunoblot. (D) Expression of PD-L1 and B7-H4 in the indicated cell lines was quantified using ImageLab. Spearman correlation indicates B7-H4 expression is negatively correlated with PD-L1 expression in the test cancer lines (r = −0.6128, p =1.43x10−6). (E-I) Tissue array of 110 breast invasive ductal carcinoma (including 46 cases of ER/PR positive, 37 cases of Her2 positive and 17 cases of TNBC and 10 adjacent normal tissue specimens) were subjected to immunohistochemistry. (E) Representative paired immunohistochemical staining of B7-H4 and PD-L1. (F) Statistical analysis of immunohistochemical staining indicates B7-H4 expression is negatively correlated with PD-L1 expression in breast cancer tissues (r = −0.480, p =1.09x10−7). (G) The B7-H4 staining in the tissue array was quantified based on the subtypes including ER+/PR+, HER2+, TNBC and normal breast tissue samples. (H) Representative paired immunohistochemistry of B7-H4 and CD8. (I) Statistical analysis of immunohistochemically staining indicates that CD8 T cell number is negatively correlated with B7-H4 expression in breast cancer tissues (r = −0.408, p =1.00x10−5).
Recent studies have subdivided TNBC into four subsets based on the tumor immune microenvironment (TIME): “fully inflamed” (FI), “margin-restricted”, “stroma-restricted” (SR) and “immune desert” (ID). ID and MR are both poorly infiltrating tumors with signatures of fibrosis. SR subtype is defined by the infiltration of CD8+ T cells in the stroma and low IFN signature. Only the FI TNBC subtype has a proinflammatory microenvironment defined by a type I IFN gene signature, CD8+ T cell infiltration in the tumor epithelium, and good outcomes (19). We compared gene expression in FI vs. non-fully inflamed tumors (MR+SR+ID). In 38 TNBC patient specimens from the GEO database (Accession number: GSE88847), the mRNAs coding for the aforementioned immune-relevant proteins upregulation of B7-H4 expression was associated with the non-fully inflamed phenotype (Fig. 1B), contrasting with high expression of CD274 (best known as PD-L1) and IDO-1 in hot tumors, consistent with another report (19).
We then systematically measured the protein expression levels of PD-L1, PD-L2, B7-H3 and B7-H4 in a panel of 45 breast and 4 ovarian cell lines (Fig. 1C). B7-H3 and PD-L1 but neither B7-H4 nor PD-L2 were expressed by normal breast cells (MCF-10A, MCF010F, 184-B5 and MCF-12A). In contrast, B7-H4 was detected in 60.9% (25/41) of the tested breast cancer cell lines (among which 10 are TNBC cell lines), compared with 10% breast cancer cells for PD-L2 expression and 25% for PD-L1 expression. Intriguingly, the expression of B7-H4 was inversely correlated with PD-L1 expression in these cell lines (Spearman correlation analysis) (Fig. 1D and Supplementary Table. 1). An immunohistochemistry array performed on a tissue array of 110 breast invasive ductal carcinomas confirmed the inverse correlation between B7-H4 and PD-L1 expression (Fig. 1E & F). As compared to normal breast tissue, B7-H4 was increased in hormone receptor-positive (ER+/PR+) and TNBC (Fig. 1G). Many groups have reported a negative correlation between B7-H4 expression and T-cell infiltration, including in breast invasive ductal carcinomas, uterine endometrioid adenocarcinomas, human esophageal squamous cell carcinoma, and clear cell ovarian cancer (20–23), though some reports are controversial (24–26). In our study, we observed a negative correlation between B7-H4 expression and CD8 T cell infiltration based on the tissue array (Fig. 1H & I). Taken together, our results suggest that B7-H4 is overexpressed in TNBC, especially in immune-cold tumors.
B7-H4 glycosylation antagonizes its ubiquitination and stabilizes the protein.
Previous studies reported that B7-H4 mRNA is transcribed in multiple peripheral tissues in both mouse and human, while its protein expression is low in normal tissues (3,27,28). This discrepancy between the mRNA and protein levels of B7-H4 in normal tissues suggests a role for post-transcriptional regulation of B7-H4. Intrigued by the accumulation of B7-H4 in TNBC (Fig. 1A & C & G), we decided to investigate the posttranslational modifications of this protein. The electrophoretic mobility of B7-H4 corresponds to a protein of ~ 50 kDa, with additional bands around 40 kDa and 28 kDa (the predicted size of unglycosylated B7-H4), as determined by immunoblot. Protein extracts from HCC1954, MDA-MB-231 and SKBR3 cells were treated with PNGase F, a peptide:N-glycosidase F, revealing that removal of asparagine-linked carbohydrates leads to the disappearance of the 40 and 50 kDa bands and an accentuation of the 28 kDa band (Fig. 2A). Thus, differential N-glycosylation leads to the generation of at least three forms of B7-H4 with high glycosylation (approximately 50 kDa), less glycosylation (approximately 40 kDa) and no glycosylation (28 kDa). Next, we examined the protein stability of B7-H4 by means of a pulse-chase assay with cycloheximide (CHX), an inhibitor of new protein biosynthesis. The less-glycosylated and unglycosylated forms of B7-H4 had a faster turnover rate (half-life <2h) than the highly glycosylated form (half-life >6h) (Fig. 2B), suggesting that glycosylation enhances the stability of B7-H4 protein.
Figure 2. B7-H4 is tightly regulated by both glycosylation and ubiquitination.
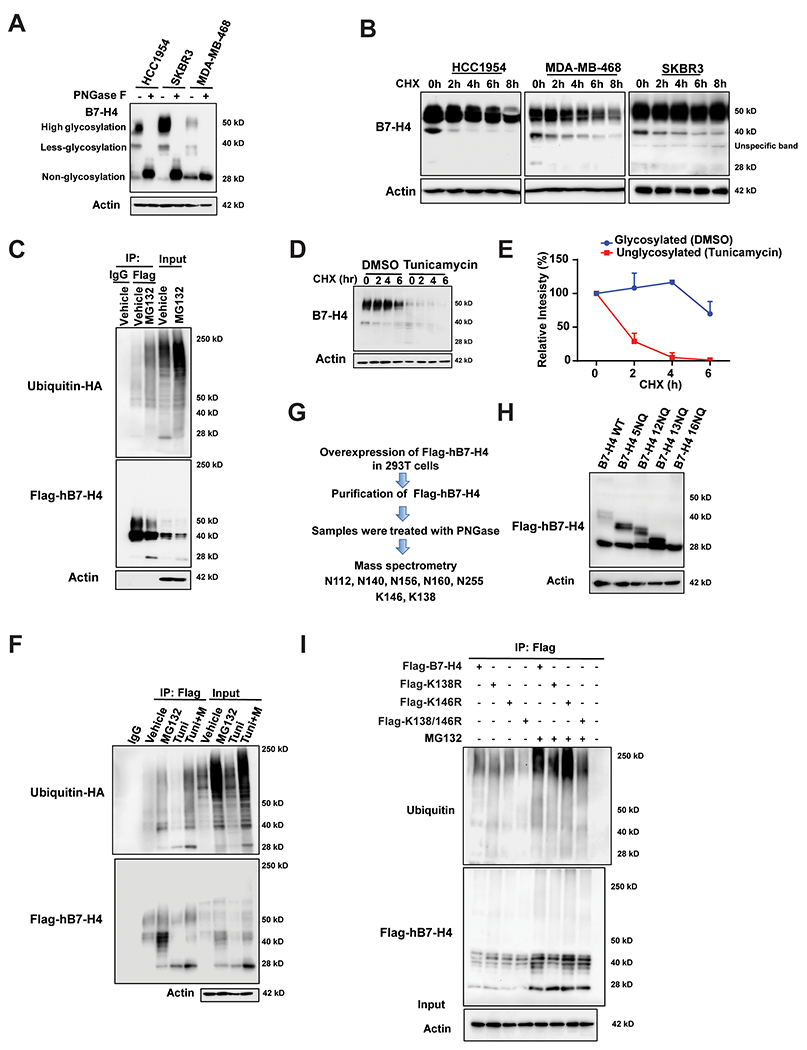
(A) HCC1954, SKBR3 and MDA-MB-468 were treated with PNGase F followed by immunoblot analysis. (B) Pulse-chase analysis for HCC1954, SKBR3 and MDA-MB-468 cells. Cells were treated with 100 μg/ml cycloheximide at the indicated time point. B7-H4 levels were measured by immunoblotting. Actin was used as a loading control. (C) Ubiquitination assay. Flag-hB7-H4 were transfected into 293T cells in the presence or absence of proteasome inhibitor MG132. Then Flag-B7-H4 was immunoprecipitated by anti-Flag M2-beads followed by immunoblot using antibody against ubiquitin. (D) Deglycosylation of B7-H4 enhances its turnover. MDA-MB-468 were treated with 10 μg/ml N-glycosylation inhibitor tunicamycin for 24 h followed by pulse-chase with 100 μg/ml cycloheximide. B7-H4 protein levels at the indicated time points were monitored by immunoblot analysis. (E) The intensity of the 50 kDa form of B7-H4 in DMSO-group vs the 25 kDa form of B7-H4 in the tunicamycin group after the treatment with cycloheximide in MDA-MB-468 cells was quantified using ImageLab. (F) Glycosylation of hB7-H4 antagonizes its ubiquitination. 293T cells were transfected with Flag-hB7-H4 in the presence or absence of MG132 and/or tunicamycin. Flag-hB7-H4 was then immunoprecipitated followed by immunoblotting using anti-ubiquitin antibody. (G) The identification of glycosylation sites on hB7-H4. Flag-hB7-H4 was transfected into 293T cells followed by affinity capture purification in the absence or presence of PNGase F. The indicated glycosylation and ubiquitination sites have been identified by mass spectrometry analysis. (H) A series of glycosylation sites mutants were constructed and validated by sequencing followed by the transfection into 293T cells and immunoblotting. (I) Flag-tagged hB7-H4 wildtype and the mutants (K138R, K146R and K138/K146R (2KR)) were transfected into 293T cells. B7-H4 ubiquitination was conducted in the presence or absence of MG132.
The proteasome inhibitor MG132 significantly increased the abundance of the non-glycosylated form of B7-H4. This, along with the fact that MG132 increased ubiquitin-conjugates of B7-H4 (Fig. 2C), suggested that B7-H4 turnover is regulated by the ubiquitin-proteasome pathway. Next, we examined the impact of glycosylation on B7-H4 stability by treating cells with N-glycosylation inhibitor tunicamycin, followed by a pulse-chase assay with CHX. As tunicamycin decreased the glycosylation of B7-H4, it increased the relative abundance of non-glycosylated B7-H4 (Fig. 2D & E), while simultaneously enhanced the turnover of B7-H4. The effect of glycosylation on B7-H4 ubiquitination was confirmed in two additional breast cancer cell lines by immunoblot (Supplementary Fig. 1A) and the ubiquitination assay (Fig. 2F and Supplementary Fig. 1B), showing that the inhibition of N-glycosylation enhanced B7-H4 ubiquitination.
Identification of glycosylation and ubiquitination sites on B7-H4
To identify the glycosylation and ubiquitination sites on hB7-H4, Flag-tagged hB7-H4 protein was ectopically expressed in 293T cells, and then purified by affinity capture approach in the absence or presence of PNGase F, followed by mass spectrometry. This led to the identification of a panel of N-linked glycosylation sites at asparagine (N) residues N112, N140, N156, N160, N255 of B7-H4 (NP_078902.2), as well as that of two ubiquitination sites at lysine (K) residues K146 and K138 (Fig. 2G and Supplementary Fig. 1C & D). These two ubiquitination sites were only detected in the presence of PNGase F, indicating that N-linked glycosylation at the asparagine sites may interfere with ubiquitination at the lysine residues. In the next step, we mutated the aforementioned glycosylation sites. Mutation of the five identified asparagines to glutamine residues in B7-H4 (5NQ, amino acids 112, 140, 156, 160, and 255) partially reduced B7-H4 protein glycosylation, as reflected by its shift to higher electrophoretic mobility (Fig. 2H). We also mutated all 16 asparagine residues found in B7-H4 to glutamine residues (16NQ: amino acids 47, 112, 119, 140, 142, 156, 160, 190, 196, 202, 205, 216, 220, 221, 229, and 255), confirming that this manipulation completely blocked B7-H4 glycosylation (Fig. 2H). Next, CHX chase experiments were performed with these mutants, revealing that hB7-H4-16NQ had an increased turnover rate compared with that of B7-H4 wildtype or other mutants (Supplementary Fig. 1E). The 16NQ mutation also led to a marked increase in ubiquitinated B7-H4 compared with B7-H4 wildtype, both in the absence or presence of MG132 treatment (Supplementary Fig. 1F). To investigate whether misfolding of B7-H4 16NQ induces its degradation, cells were treated with the HSP90 inhibitor 17-AAG. Immunoblot analysis showed that 17-AAG, which interrupts folding of HSP90-client proteins, does not affect B7-H4 16NQ degradation in the absence or presence of cycloheximide (Supplementary Fig. 1G), indicating that mutant 16NQ protein stability or degradation is independent from HSP90-facilitated protein folding and degradation. Finally, we examined the effect of mutating K146 and K138 to arginine (R) residues on B7-H4 ubiquitination and degradation. CHX pulse chase experiments indicated that hB7-H4 K146R modestly delayed B7-H4 degradation, whereas dual K138/K146R mutation (hB7-H4-2KR) significantly inhibited the degradation of B7-H4 (Supplementary Fig. 1H). hB7-H4-2KR also reduced B7-H4 ubiquitination, confirming the impact of these two sites on B7-H4 degradation and ubiquitination (Fig. 2I).
Identification of E3 ligase and glycosyltransferases that govern B7-H4 protein stability
In the next step, we sought to determine the ubiquitin E3 ligases and glycosyltransferases affecting B7-H4 stability. A Flag-B7-H4 construct was stably expressed in MDA-MB-468 cells and the B7-H4 protein was purified by affinity capture purification (Fig. 3A), followed by mass spectrometrometic identification of the B7-H4 interactome. Within this interactome, we found the ubiquitin E3 ligase autocrine motility factor receptor (AMFR) and several glycosyltransferases such as UDP-glucose:glycoprotein glucosyltransferase (UGGG1), several OST complex subunits such as STT3A, RPN1 and RPN2 (Fig. 3A), as well as, p97, which interacts with AMFR to participate in endoplasmic reticulum (ER) associated degradation (ERAD). Using a similar experimental approach, the glycosyltransferases RPN1, RPN2, and UGGG1 were confirmed to bind B7-H4 in 293T cells (Supplementary Fig. 2A). Co-immunoprecipitation assays performed on breast cancer cells confirmed the interactions of B7-H4 with AMFR, STT3A, UGGG1 and HSP90 (Fig. 3B). Furthermore, immunostaining followed by confocal microscopy revealed the colocalization of B7-H4 and AMFR or that of B7-H4 and STT3A in the cytoplasm (Fig. 3C), a colocalization that was further corroborated by means of the Duolink proximity ligation assay (Fig. 3D). Of note, the B7-H4 and AMFR expression levels as determined by immunohistochemistry showed an inverse correlation for an array of 110 specimens of breast invasive ductal carcinomas (r =-0.3464, p =2.1x10−4) (Supplementary Fig. 2B & C), suggesting a potential role for AMFR in controlling B7-H4 expression. Conversely, B7-H4 and STT3A protein levels were positively correlated with each other in this tissue array (r = 0.3030, p =1.29x10−3) (Supplementary Fig. 2B–D), in line with a possible role for STT3A in stabilizing B7-H4.
Figure 3. Identification of the E3 ligase and glycosyltransferases of B7-H4 that govern B7-H4 protein stability and function.
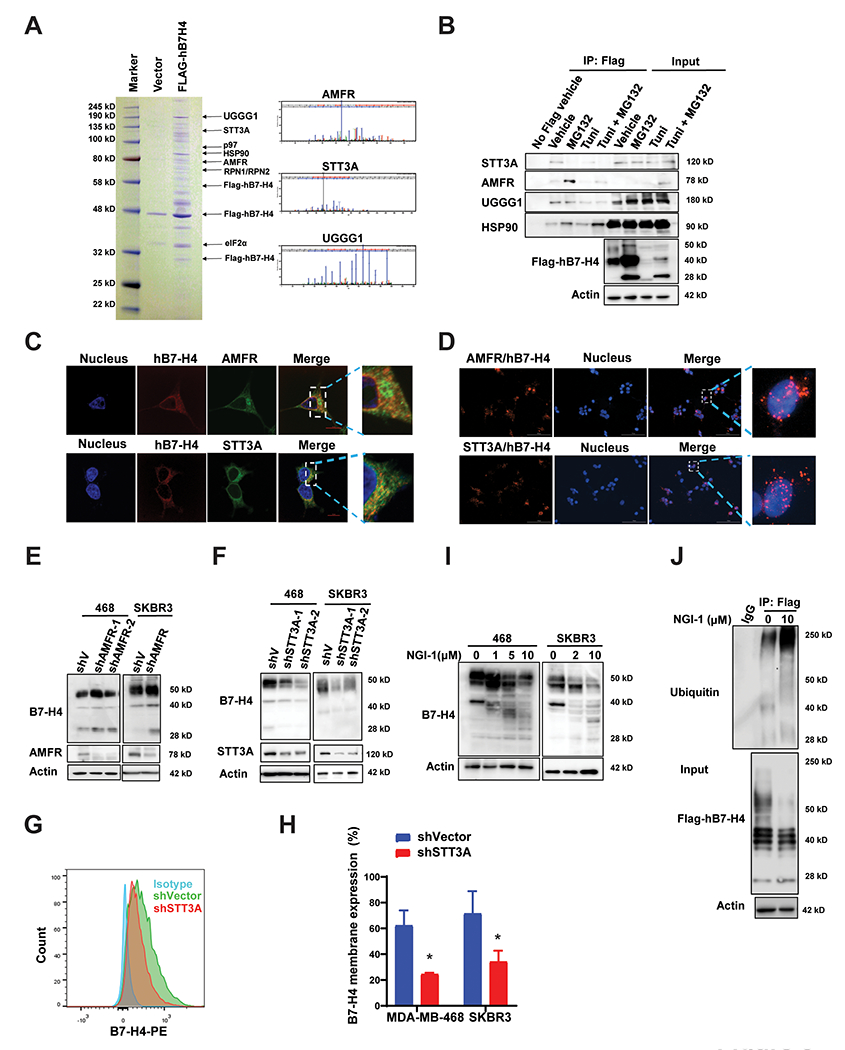
(A) Stable expression of Flag-hB7-H4 was engineered to MDA-MB-468 cells (MDA-MB-468-Flag-hB7-H4). B7-H4 complex was then purified followed by mass spectrometry analysis. Coomassie blue staining of the purified B7-H4 immunocomplex is shown. The ubiquitin E3 ligase AMFR and several glycosyltransferases including STT3A, RPN1, RPN2, and UGGG1 were identified, and the representative spectra of AMFR, STT3A and UGGG1 are shown. (B) Validation of biochemical interactions of B7-H4 with AMFR, STT3A, UGGG1 as well as HSP90. MDA-MB-468-Flag-hB7-H4 cells were utilized for immunoprecipitation using anti-Flag M2-beads in the presence or absence of MG132 and/or tunicamycin. The interactions of AMFR, STT3A, UGGG1 as well as HSP90 with B7-H4 were measured by immunoblot. (C) Double immunofluorescence staining hB7-H4-Flag with AMFR or STT3A in MDA-MB-468-Flag-hB7-H4 cells followed by the confocal microscope (Scale bar =10 μm). (D) MDA-MB-468-Flag-hB7-H4 cells were subjected to duolink in situ PLA assay with specific Flag mouse antibody and AMFR or STT3A rabbit antibody (Scale bar = 100 μm). Red dots indicate the binding of the indicated two proteins. (E) AMFR knockdown results in upregulation of B7-H4. Stable knockdown of AMFR in MDA-MB-468 and SKBR3 were established. The expression of AMFR and B7-H4 were examined by immunoblotting. (F) STT3A knockdown leads to downregulation of B7-H4. STT3A stable knockdowns in MDA-MB-468 cells were established. The expression of STT3A and B7-H4 were examined by immunoblotting. (G) Decreased membrane B7-H4 in STT3A knockdown cells. MDA-MB-468-shVector, MDA-MB-468-shSTT3A, SKBR3-shVector, and SKBR3-shSTT3A cells were stained with PE anti-human B7-H4 antibody followed by flow cytometry. Representative images are shown. (H) The quantification of membrane staining of B7-H4 in MDA-MB-468-shVector, MDA-MB-468-shSTT3A, SKBR3-shVector, and SKBR3-shSTT3A cells are shown. (I) MDA-MB-468 and SKBR3 cells were treated with 10 μM OST inhibitor NGI-1 for 24 h. The expression of B7-H4 was examined by immunoblotting. (J) Blockade of B7-H4 glycosylation by NGI-1 enhances B7-H4 ubiquitination. 293T cells were transfected with Flag-hB7-H4 in the presence or absence of 10 μM NGI-1 for 24 h. Then Flag-hB7-H4 was immunoprecipitated followed by immunoblotting using antibody against ubiquitin.
Next, we generated two AMFR knockdown cell lines using two non-overlapping shRNAs targeting AMFR. AMFR knockdown led to an increase of B7-H4 protein as compared to the vector control (pLKO) (Fig. 3E). Transfection-enforced overexpression of AMFR wildtype (AMFR-WT-Flag) increased B7-H4 ubiquitination, while the AMFR mutant with ring-domain mutation C356G/H361A (AMFR-RM-Flag, which loses its ubiquitin ligase activity) (29) failed to do so, confirming that AMFR functions as a critical E3 ubiquitin ligase for B7-H4 (Supplementary Fig. 2E).
Glycosylation involves several enzymatic steps including steps involving the OST complex, glucosidases, ER mannosidases, and UGGG1 in the ER (17). The knockdown of the OST catalytic subunit STT3A by two distinct non-overlapping shRNAs resulted in the reduction of B7-H4 protein levels (Fig. 3F). Flow cytometry of membrane B7-H4 staining indicated that the level of membrane bound B7-H4 is significantly decreased with STT3A knockdown in MDA-MB-468 and SKBR3 cells (Fig. 3G & H). These experiments imply that the protein stability of B7-H4 is governed by an interplay between AMFR-mediated ubiquitination and STT3A-dependent glycosylation. Indeed, the knockdown of two other subunits of OST complex RPN1 and RPN2 (Supplementary Fig. 2F & G) also resulted in reduced B7-H4 glycosylation. UGGG1 is an ER-sessile enzyme that selectively reglucosylates unfolded glycoproteins, thus providing quality control for protein transport out of the ER (30). The knockdown of UGGG1 by two independent shRNAs led to decreased expression of B7-H4 glycosylation in two breast cancer cell lines (Supplementary Fig. 2H), indicating that UGGG1 also contributes to B7-H4 glycosylation.
NGI-1, an aminobenzamide-sulfonamide inhibitor, was initially identified from a cell-based high-throughput screen and lead compound optimization campaign targeting the catalytic subunit STT3A/B of oligosaccharyltransferase (OST) (31). NGI-1 inhibited the glycosylation of B7-H4 and reduced its expression in two breast cancer cells as determined by immunoblot (Fig. 3I). NGI-1 also increased the ubiquitination of B7-H4 (Fig. 3J). NGI-1 was reported to block epidermal growth factor receptor (EGFR) signaling in NSCLC and enhance glioma radiosensitivity (31–33). We examined the level of EGFR expression in MDA-MB-468, SKBR3, and mouse breast cancer cells 4T1 cells and E0771 cells by flow cytometry (Supplementary Fig. 3A & B) and immunoblot (Supplementary Fig. 3C) comparing the effect of NGI-1 on B7-H4 and EGFR. We observed that NGI-1 has a modest effect on inhibiting EGFR in HCC1954, MDA-MB-231, and SKBR3 cells, with no effect on EGFR in MDA-MB-468 cells. On the other hand, NGI-1 significantly inhibited B7-H4 in all tested breast cancer cell lines (Supplementary Fig. 3D). EGFR expression is undetectable in 4T1 cells, indicating that 4T1 is an ideal breast cancer cell line to study the effect of NGI-1 on B7-H4 to exclude a possible bias from EGFR. We also compared the effect of glycosylation on the stability of B7-H4 or EGFR by cycloheximide pulse-chase. STT3A knockdown (Supplementary Fig. 3E–G) and pretreatment with 1 µM NGI-1 (Supplementary Fig. 3H–J) increased protein turnover of B7-H4 but not EGFR. These experiments indicated that glycosylation affects the turnover of the B7-H4 but not EGFR. In conclusion, the genetic or pharmacological inhibition of glucosyltransferase STT3A indistinguishably increases the E3 ligase AMFR-mediated ubiquitination and degradation of B7-H4.
Molecular analysis of the interaction between B7-H4 and the E3 ligase AMFR
To identify the domains of AMFR and B7-H4 responsible for their interaction, we constructed a series of Flag-tagged B7-H4 and Myc-tagged AMFR truncation mutants (Fig. 4A–D), co-transfected them into cells and performed co-immunoprecipitation experiments. This approach led to the prediction that the domain comprising amino acid residues 153-241 (corresponding to the Ig-like C2-type domain) in B7-H4 (Fig. 4B) and the RING domain comprising residues 302-379 on AMFR are implicated in the AMFR-B7-H4 interaction (Fig. 4D). Next, we conducted a series of docking and molecular dynamics (MD) simulation. The X-ray structure of the B7-H4 Ig-like V-type domain (PDB: 4GOS) (34) and PD-L1 (PDB: 5JDR) (35), which shares 23.9% sequence identity with B7-H4, were used as templates in homology modeling. The AMFR RING domain (the structure of which has been determined) was docked onto the predicted B7-H4 structure, yielding a molecular model of the interactions between the AMFR RING domain mainly and the B7-H4 Ig-like C2-type domain (Fig. 4E–J). In order to assess the stability of these complex conformations, we performed MD simulations for the top five clusters among the top 100 ZDOCK docking poses, where the root-mean-square deviations (RMSDs) in the atomic positions of AMFR RING domain were evaluated by structurally aligning the generated conformers after superposing their Ig-like C2-type domain. A low RMSDs profile over 120 ns simulation time confirmed that the RING domain was highly stable in the bound conformation of the top 1 cluster (Fig. 4F, see the trajectory in Supplementary Movie). Fig. 4G & J displays the time evolution of the most persistent inter-residue interactions at the interface between AMFR RING domain and B7-H4 Ig-like C2-type domain in the 120 MD simulations of the top 2 clusters. The two zinc ions, Zn2+ I and Zn2+II, stably coordinated four residues each: H358, C356, C375, and C378; and H361, C341, C344, and C364, respectively (35); these sites have been shown (but not labeled) in Fig. 4 E & H as sticks. Six out of these eight residues are from three flexible AMFR RING-type Zinc finger loops which stabilized upon binding of the two Zinc ions, and most of the residues of these loops are involved in interactions with B7-H4 Ig-like C2-type domain, especially those are vicinity to the eight coordinated Cys/His residues (Fig. 4G & J). We also observed that the Zinc ions form strong salt bridge interactions with negatively charged residues (e.g., D237 and E200) in B7-H4 Ig-like C2-type domain in the 120 ns simulations. The B7-H4 Ig-like C2-type domain has more negatively charged residues than that of the V-type domain. This helps us rationalize why AMFR binds onto the B7-H4 Ig-like C2-type domain rather than the Ig-like V-type domain. B7-H4 N49, N156, N160 and N229 are close to the binding interfaces observed in MD simulations. These results support our hypothesis that glycosylation is likely to inhibit the ubiquitination by interfering with the complex formation between AMFR RING domain and B7-H4, through these glycosylation sites.
Figure 4. Mapping of molecular domains/motifs and structure-based modeling and simulations reveal critical regions and interfacial interactions involved in the complex formation between B7-H4 and E3 ligase AMFR.
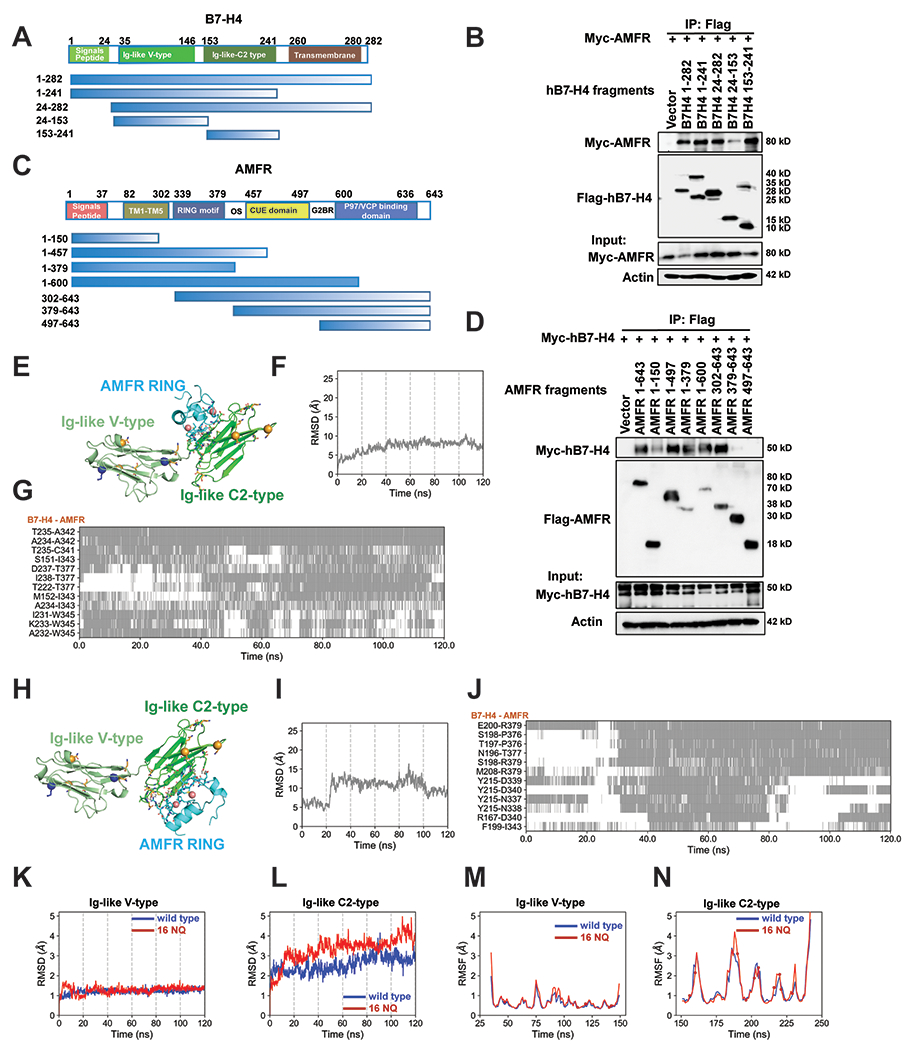
(A) Schematic diagram of human B7-H4 domains and strategy to engineer a series of B7-H4 deletion mutants. (B) Mapping of B7-H4 regions (sequence ranges) involved in interactions with AMFR. The interactions between Myc-AMFR and the displayed Flag-hB7-H4 fragments were examined by co-IP experiments in 293T cells. (C) Schematic diagram of human AMFR domains/motifs and strategy to engineer a series of AMFR deletion mutants. Abbreviations: TM (transmembrane domain), RING motif (E3 ligase activity), OS (Oligomerization domain), CUE domain (Couples Ubiquitin molecules to ER degradation), G2BR (Ube2G2 binding region), VIM (p97/VCP interacting motif). (D) Mapping of AMFR domains/motifs that interact with B7-H4. The interactions between Myc-hB7-H4 and the displayed Flag-AMFR fragments were examined by co-IP experiments in 293T cells. The representative structures of the top 1 cluster of the ZDOCK docking poses between B7-H4 and AMFR RING domain are shown in (E). AMFR RING, B7-H4 Ig-like V type and Ig-like C2-type domains are in cyan, pale green and green. Ubiquitination sites (K138 and K146) are shown as blue sticks with alpha-carbon atoms highlighted in blue spheres. All asparagines are shown as orange sticks, and the alpha-carbon atoms of five identified asparagines N112, N140, N156, N160, N255 (not shown in the structure) are highlighted in orange spheres. The two ZN2+ ions form coordination bonds are shown as salmon spheres. Residues in the binding interfaces from AMFR and B7-H4 are shown as cyan and green sticks, respectively. Time evolution of the RMSDs of AMFR RING domain in the 120 ns MD simulations of the complex formed with B7-H4 using the start points in (E) is shown in (F). The RMSD was evaluated after structurally aligning the conformers observed during MD trajectories with respect to the B7-H4 Ig-like C2-type domain. The corresponding time evolution of residue-residue interactions between B7-H4 and AMFR RING domain residues are shown in (G). Regions shaded in gray refer to time intervals during which the indicated atom pairs (ordinate) made interfacial contacts. The representative structures of the top 2 cluster of the ZDOCK docking poses between B7-H4 and AMFR RING domain are shown in (H). Time evolution of the RMSDs of AMFR RING domain in the 120 ns MD simulations of the complex formed with B7-H4 using the start points in (H) is shown in (I). The corresponding time evolution of residue-residue interactions between B7-H4 and AMFR RING domain residues are shown in (J).The RMSDs profiles of the B7-H4 Ig-like (K) V-type and (L) C2-type domains are shown in blue and red curves for B7-H4 wildtype and 16NQ mutant, respectively. The corresponding RMSFs values of residues in the two domains are shown in (M) and (N), respectively. The spheres on the red curves indicate the positions of the 16NQ mutations. The RMSDs and RMSFs values for the 16NQ mutant are slightly higher than those of the wildtype in the C2-type domain, though the difference is not statistically significant. Flexibilities (RMSDs and RMSFs) of the Ig-like V-type domain of B7-H4 wildtype and16NQ mutant are quite similar.
We also evaluate the stability of B7-H4 wildtype and mutant 16NQ (Fig. 4K–N) by MD simulations. The differences in RMSDs and root-mean-square fluctuations (RMSFs) profiles of Ig-like V-type domain between B7-H4 wildtype and 16NQ were marginal (Fig. 4K & M). Higher values of RMSDs and RMSFs for the 16NQ Ig-like C2-type domain were observed but the differences were not significant (Fig. 4L & N). This suggests that the mutant 16NQ is flexible compared to B7-H4 wildtype in the Ig-like C2-type domain, while the entire structure is still stable.
B7-H4 inhibits doxorubicin-induced ICD through regulating PERK/eIF2α/CALR axis.
Immunogenic cell death is defined by the release and exposure of damage-associated molecular patterns (DAMPs) from and on dying tumor cells, thus initiating an anticancer immune response (13). One major DAMP is CALR, a protein that usually locates to the lumen of the endoplasmic reticulum but then translocates to the cell surface when ICD is induced and acts as an ‘eat-me’ signal to facilitate the phagocytosis of portions of the tumor cell by immature DCs (14,36). The phosphorylation of eukaryotic translation initiation factor 2 subunit alpha (eIF2a) is a critical event in the molecular cascade leading to CALR exposure on the plasma membrane (15). The B7-H4 interactome includes ER chaperones as well as eIF2α (Fig. 3A and Supplementary Fig. 2A), suggesting a possible connection between B7-H4 and ICD. Results from clinical studies unveiled a critical role for ICD with regards to the success of chemotherapy with anthracyclines such as doxorubicin (DOX) (13,37). Camsirubicin (Cam, GPX-150) is a non-cardiotoxic doxorubicin analog (38). We observed that DOX-or Cam-induced cell death, reduced cell viability, and clone formation of MDA-MB-468 breast cancer cells was not affected by NGI-1 (Supplementary Fig. 4A–E) in conditions in which NGI-1 reduced B7-H4 expression. Cam also increased the exposure of CALR and HSP90 on the cell surface (Supplementary Fig. 4F–H). However, NGI-1 significantly increased the DOX-induced expression of cell surface CALR and that of other DAMPs including HSP90 and HSP70 on the cell surface, as detectable by immunofluorescence and flow cytometry (Fig. 5A & B and Supplementary Fig. 5A–I). In addition, DOX, NGI-1 alone, or in combination induced the release of another DAMP, the high-mobility group protein B1 (HMGB1) visualize when fused to green-fluorescent protein (GFP) from the nuclei of biosensor cells lines in vitro (Supplementary Fig. 5J).
Figure 5. B7-H4 inhibits doxorubicin-induced ICD through regulating the PERK/eIF2a/CALR axis.
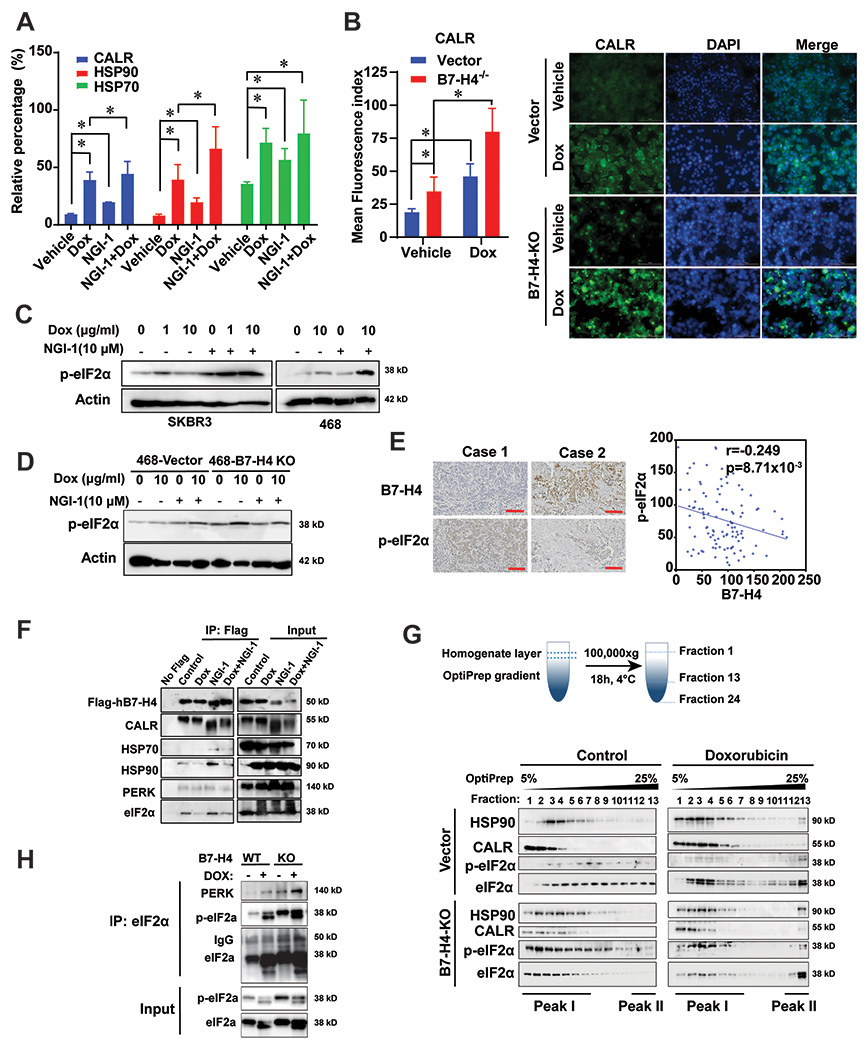
(A) SKBR3 cells were treated with 10 μM doxorubicin and/or 10 μM NGI-1 for 24 h. Membrane CALR, HSP70 and HSP90 were measured by flow cytometry. (B) MDA-MB-468-vector and MDA-MB-468-B7-H4 knockout cells were established and treated with 5 μM doxorubicin for 24 h. Immunofluorescence staining of the immunogenic cell death markers CALR on the cell surface was performed. Mean fluorescence index of CALR was quantified by ImageJ. Representative images are shown. (C-D) SKBR3, MDA-MB-468, MDA-MB-468-vector, MDA-MB-468-B7-H4 knockout cells were treated with 1 or 10 μM doxorubicin and/or 10 μM NGI-1 for 24 h. p-eIF2a and actin were examined by immunoblotting. Scale bar, 100 μm. (E) Representative paired immunohistochemistry staining of B7-H4 and phospho eIF2α (Ser51) in tissue array BC081120. Statistical analysis of immunohistochemical staining indicates B7-H4 expression is negatively correlated with p-eIF2α expression in breast cancer (r = −0.249, p =8.71x10−3). (F) MDA-MB-468-Flag-hB7-H4 were treated in the presence or absence of doxorubicin (10 μM) and/or NGI-1 (10 μM). Then Flag-hB7-H4 was immunoprecipitated followed by immunoblot. The indicated proteins were examined. (G) Schematic diagram of the procedure of OptiPrep density gradient assay with 24 collected fractions from low to high density is shown. MDA-MB-468-vector and MDA-MB-468-hB7-H4 knockout cells were treated with 10 μM doxorubicin for 24 hr followed by OptiPrep density gradient assay. HSP90, CALR, eIF2α and p-eIF2α in fraction 1 to 13 were examined by immunoblotting. (H) eIF2a was immunoprecipitated in fraction 13 in both MDA-MB-468-vector and MDA-MB-468-B7-H4 knockout cells followed by immunoblotting. PERK, eIF2α and p-eIF2α were examined.
MDA-MB-468-B7-H4 knockout (KO) cells engineered by CRISP/Cas9 technology (Supplementary Fig. 5B) exposed more CALR and HSP90 on their surface when treated with DOX than control cells transfected with an irrelevant gRNA (Fig. 5B and Supplementary Fig. 5F–G). Conversely, CALR was significantly reduced in HCC1954 cells that overexpressed transgenic B7-H4 compared with the control cells (Supplementary Fig. 5C & H & I). Of note, NGI-1 significantly enhanced the DOX-induced phosphorylation of eIF2α in both SKBR3 and MDA-MB-468 cells (Fig. 5C). Moreover, eIF2α hyperphosphorylation was observed in DOX-treated MDA-MB-468-B7-H4 knockout cells (Fig. 5D), suggesting that B7-H4 acts as an endogenous inhibitor of eIF2α phosphorylation. In support of this interpretation, the expression of B7-H4 inversely correlated with the phosphorylation of eIF2α in the tissue microarray of 110-breast invasive ductal carcinomas (Fig. 5E).
To investigate how B7-H4 affects eIF2α phosphorylation, we treated Flag-hB7-H4-expressing cells with doxorubicin and/or NGI-1 and then immunoprecipitated the flagged protein, followed by immunoblot detection of eIF2α, eIF2α kinase 3 (EIF2AK3, best known as the ER stress kinase PERK), CALR, HSP70 and HSP90. This experiment revealed that B7-H4 interacts with eIF2α, PERK, CALR and HSP90. Intriguingly, eIF2α was released from the B7-H4 protein complex following DOX treatment (Fig. 5F).
To further investigate the alteration of B7-H4 protein complex in the presence or absence of DOX, we conducted an Iodixanol (OptiPrep) gradient analysis. DOX induced the formation of novel molecular complex peak (peak II) including eIF2α, CALR and HSP90 compared with the control (Fig. 5G). Co-IP experiments performed on this new complex revealed increased binding between eIF2α and PERK upon DOX treatment, and this effect was more pronounced in cells lacking B7-H4 expression (Fig. 5H).
Taken together, these results suggest that B7-H4 can bind to eIF2α, thus inhibiting its phosphorylation by PERK. Upon the treatment with DOX, eIF2α is released from this inhibitory interaction, allowing for its phosphorylation and the subsequent translocation of DAMPs to the plasma membrane.
B7-H4 subverts spontaneous and doxorubicin-induced immunosurveillance
In the context of ICD, surface-exposed CALR, HSP70 and HSP90 are potent ‘eat-me’ signals for DCs, thus facilitating the presentation of tumor-associated antigens to cytotoxic T lymphocytes. Moreover, the co-culture of malignant cells succumbing to ICD and immature DCs induces DC maturation. We wondered whether inhibition of DAMP exposure by B7-H4 would affect this pathway. Mouse B7-H4 expression in 4T1 cells was undetectable by immunoblot (Supplementary Fig. 6A). Real-time quantitative PCR showed that the Ct (cycle threshold) of mouse B7-H4 in 4T1 cells was more than 37 cycle, indicating no expression of mouse B7-H4 in 4T1 cells. Thus, mouse 4T1 breast cancer cells were stably transfected with vector or mouse B7-H4 (Supplementary Fig. 6A). The cells were then labeled with 5-(and-6)-(((4-chloromethyl)benzoyl)amino) tetramethylrhodamine (CMTMR), and treated with NGI-1 and/or DOX, and then confronted with DCs isolated from the spleens of BALB/c mice. The DCs were identified by immunostaining for CD11c. B7-H4 overexpression led to reduced phagocytosis (measured by the co-occurrence of CMTMR and CD11c) of the cancer cells by DCs, unless the cancer cells were treated with both NGI-1 and DOX (Figure 6A & B, Supplementary Fig. 6B & C). Of note, mB7-H4-overexpression was also linked to relatively reduced induction of the DC maturation markers CD86 and MHC class II (I-A/I-E) (Supplementary Fig. 6D & E).
Figure 6. B7-H4 and its ubiquitination or glycosylation-deficient mutants profoundly alter tumor growth and doxorubincin-induced immunogenic cell death.
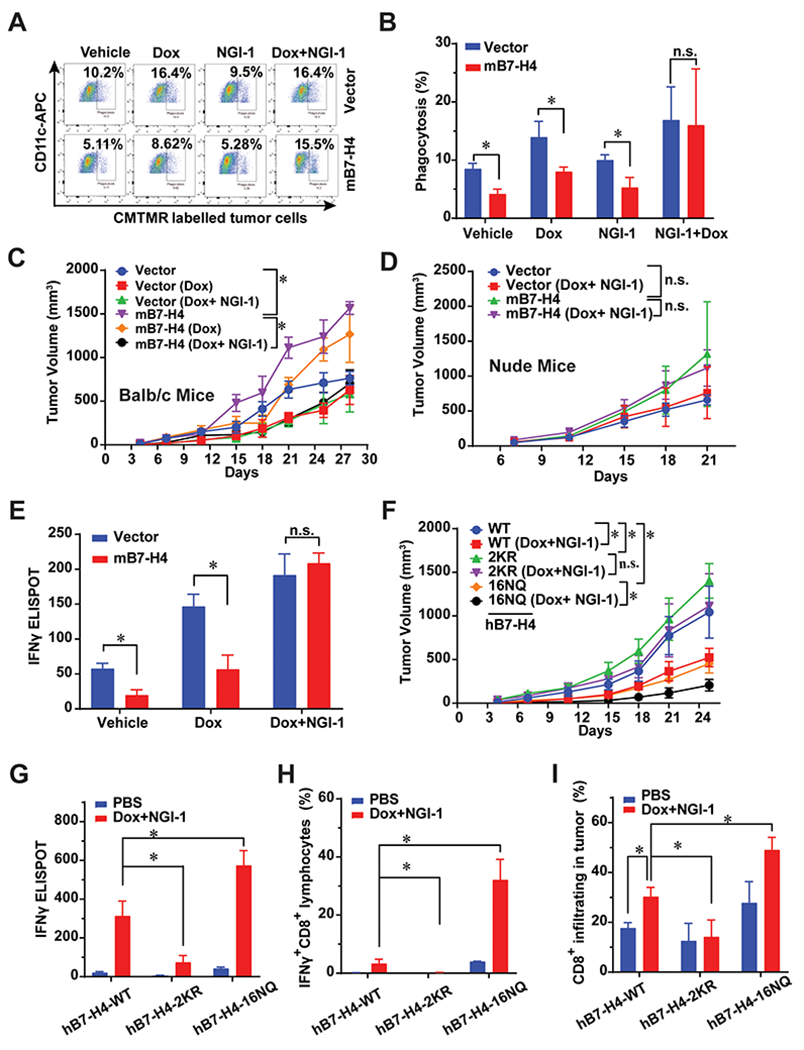
(A-B) In vitro phagocytosis of co-culture of mouse DCs and tumor cells. 4T1-vector and 4T1-B7-H4 cells were treated with doxorubicin (25 μM) or NGI-1 (10 μM) for 24 hour and co-cultured with the purified CD11c positive cells for 2 hours at a ratio of 1: 1, and then subjected to flow cytometry. n=3 mice per group. (C) In vivo vaccination assay. 4T1-vector or 4T1-B7-H4 cells were treated with doxorubincin (Dox, 25 μM) alone or in combination with NGI-1 (10 μM) for 24 h. Then these cells (106 per mice) were orthotopically injected into the right fourth mammary gland of the BALB/c mice (the vaccination step). PBS was used in the non-vaccinated group. One week later, all mice were rechallenged with live 4T1-vector or 4T1-B7-H4 cells (3 × 105 per mouse) of the same kind as the vaccination step in the left fourth mammary gland (the challenge step). The tumor growth was monitored twice per week. n=8 mice per group. (D) Failure of nu/nu BALB/c to mount an immune response against Dox/NGI-1 treated 4T1-vector or 4T1-B7-H4 cells. Nude mice were inoculated with Dox/NGI-1 treated 4T1-V or B7-H4 cells 1-2 weeks before the injection of live 4T1-vector or B7-H4 cells into the opposite flank, the tumor growth was monitored. n=6 mice per group. (E) 4T1-vector or 4T1-B7-H4 cells were treated with doxorubincin (Dox, 25 μM) alone or in combination with NGI-1 (10 μM) for 24 h. These cells were then orthotopically injected into the right fourth mammary gland of the BALB/c mice (the vaccination step). PBS was used in non-vaccinated control group. One or two week later, all mice were challenged with injection of live 4T1-vector or 4T1-B7-H4 cells (the same kind cells as used in the vaccination step) in the left fourth mammary gland (the challenge step). On day 28, mouse spleens of BALB/C mice were harvested and followed by ELISPOT. Quantification of IFNγ ELISPOT is shown. n=3 mice per group. (F-I) In vivo vaccination assays were performed with 4T1-hB7-H4, 4T1-hB7-H4-2KR, and 4T1-hB7-H4-16NQ cells. BALB/c mice were inoculated with Dox/NGI-1-treated 4T1-hB7-H4, 4T1-hB7-H4-2KR, and 4T1-hB7-H4-16NQ cells 1-2 weeks (the vaccination step, PBS was used in the non-vaccinated group) before the injection of the live cells of the same kind (4T1-hB7-H4, 4T1-hB7-H4-2KR and 4T1-hB7-H4-16NQ cells) into the opposite mammary gland (the challenge step), (F) the tumor growth was monitored. n=10 mice per group. (G) On day 28, mouse spleens were harvested and followed by IFNγ ELISPOT. Quantification of IFNγ ELISPOT is shown. n=3 mice per group. PBS is used in non-vaccinated group. (H) On day 28, mouse spleens were harvested and followed by flow cytometry of staining IFNγ and CD8. Quantification of IFNγ+CD8+cells is shown. n=3 mice per group. PBS is used in non-vaccinated control group. (I) On day 28, mouse tumors in the mammary gland were harvested and digested followed by flow cytometry by detecting CD45 and CD8. n=3 mice per group. PBS is used in non-vaccinated control group. Quantification of CD8+ infiltrating cells is shown.
To determine the effects of B7-H4 in DOX-induced ICD in vivo, we performed vaccination experiments in which immunocompetent BALB/c mice or T cell-deficient nu/nu mice were first injected with cells cultured with DOX and/or NGI-1 and then were challenged one week later with the same kind of live cells injected into the opposite mammary gland (Fig. 6C–D and Supplementary Fig. 6F). In non-vaccinated mice, 4T1-B7-H4 tumors grew faster than 4T1-vector alone in immunocompetent (but not in immunodeficient) mice. 4T1-B7-H4 tumor growth was transiently delayed in immunocompetent mice vaccinated with DOX-treated cells, in line with the fact that 4T1 cells are poorly immunogenic. Intriguingly, combined DOX/NGI-1 treatment yielded a more potent vaccine than DOX treatment alone (Fig. 6C). In contrast, no differences were observed between the non-vaccinated and the vaccinated groups in nude mice (Fig. 6D), in line with the interpretation that T cells are required for the DOX/NGI-1-elicited immunogenic effect.
Interferon gamma (IFNγ) production by CD8+ cytotoxic T cells is essential for tumor growth control after activation of the immune response by cancer cells that succumb to ICD. The frequency of splenic CD8+ IFNγ+ T cells significantly decreased in mice bearing 4T1-B7-H4 tumors compared to mice with 4T1-vector tumors, as determined by immunofluorescence detection of intracellular IFNγ among CD8+ T lymphocytes. CD8+/IFNγ+ T cells were remarkably increased in mice vaccinated with DOX+NGI-1-treated 4T1-B7-H4 cells than in mice vaccinated with DOX only-treated cells (Supplementary Fig. 6G). The enzyme-linked immunospot (ELISPOT) assays confirmed these results at the level of splenic IFNγ production (Fig. 6E), and immunohistochemistry at the level of CD8+ infiltration of the tumors (Supplementary Fig. 6H). In each case, DOX-treated mB7-H4-overexpressing cancer cells induced a relatively poor CD8+ IFNγ+ T cell immune response (compared to parental cancer cells expressing little or no B7-H4) unless they were treated with both DOX and the B7-H4 degradation-inducing drug NGI-1.
Modulation of B7-H4 glycosylation and ubiquitination affects tumor growth and doxorubicin-induced immunogenic cell death.
4T1 breast cancer cells were engineered to stably express human(h) B7-H4 wildtype, the ubiquitination-deficient hB7-H4-2KR mutant or glycosylation-deficient hB7-H4-16NQ mutant (Supplementary Fig. 7A). No differences in cell growth and proliferation were observed in 4T-1-hB7-H4, 4T-1-hB7-H4-2KR and 4T-1-hB7-H4-16NQ cells (Supplementary Fig. 7B). The mutant B7-H4-16NQ protein (which cannot be glycosylated) was more restricted to the cytosol, while B7-H4 and B7-H4-2KR (which cannot be ubiquitinated) were localized in both the cytosol and the cell membrane (Supplementary Fig. 7C). B7-H4-16NQ was able to localize to the cell membrane, but at significantly lower levels than B7-H4-WT (Supplementary Fig. 7D & E), suggesting that the mutant retains partial functional activities of B7-H4 in terms of the membrane localization. Moreover, as compared to the unmutated hB7-H4, the glycosylation-deficient hB7-H4-16NQ mutant exhibited increased binding to AMFR, but lost binding to HSP90 (Supplementary Fig. 7F), indicating AMFR is crucial for the regulation of B7-H4 16NQ protein abundance.
In contrast, when inoculated into immunocompetent hosts, hB7-H4-2KR tumors progressed more quickly than 4T1-B7-H4 tumors, contrasting with the delayed growth of hB7-H4-16NQ tumors compared with 4T1-B7-H4 tumors. hB7-H4-WT and 16NQ tumor growth were significantly reduced in response to vaccination with cell treated with DOX + NGI-1. In contrast, hB7-H4-2KR tumors did not respond to vaccination with cells treated with DOX + NGI-1 (Fig. 6F and Supplementary Fig. 7G).
Splenic CD8+ IFNγ+ T cells were increased in mice bearing 4T1-hB7-H4-16NQ tumors, whereas CD8+ IFNγ+ T cells were decreased in 4T1-hB7-H4-2KR tumor bearers compared with that of 4T1-B7-H4 group responding to the vaccination with Dox + NGI-1. Similar results were obtained with ELISPOT assay (Fig. 6G & H and Supplementary Fig. 7H). Notably, we observed a two-fold increase of tumor infiltrating CD8 T cells in 4T1-hB7-H4-16NQ tumors compared with that of the 4T1-B7-H4 group. No significant difference in CD8 density between the vaccination group and non-vaccination group was observed for 4T1-hB7-H4-2KR tumors (Fig. 6I). Altogether, these results confirm that the abolition of B7-H4 glycosylation (hB7-H4-16NQ) and ubiquitination (hB7-H4-2KR) affected tumor cell immunogenicity in a positive and negative fashion, respectively.
The anti-tumor effects of combined treatment with DOX and NGI-1 are exacerbated by PD-L1 blockade
Previous in vivo vaccination experiments showed that combined DOX/NGI-1 treatment yields a more potent vaccine than DOX treatment alone (Fig. 6C), indicating that NGI-1 combined with DOX could prevent tumor growth in settings of tumor cell vaccinations. Therefore, we investigated whether NGI-1 combined with immunogenic chemotherapy has antitumor efficacy against established tumors. The PD-1 pathway regulates activated T cells at the later stages of an immune response, primarily in peripheral tissues (39). B7-H4 is thought to inhibit T cell responses at an early stage (40). We showed that NGI-1 inhibits B7-H4 significantly, and also inhibits glycosylation of PD-L1 (Supplementary Fig. 3D). However, NGI-1 also increased unglycosylation of PD-L1 (Supplementary Fig. 3D), indicating that the blocking of the PD-L1 signaling pathway might still be required. We therefore examined the effect of triple combination of NGI-1, immunogenic chemotherapy, and anti-PD-L1 blocking antibody in mouse orthotopic tumor models of 4T1, where the basal mouse PD-L1 was knocked out followed by adding back human PD-L1 gene (Fig. 7A–D and supplementary Fig. 8A–E) and E0771 (Fig. 7E–G and supplementary 8F–I). Cam shares similar immunogenicity with Dox as we aforementioned and has been utilized in the triple combination.
Figure 7. The anti-tumor efficacy of the combination of NGI-1 and a doxorubicin analog are enhanced by PD-L1 blockade.
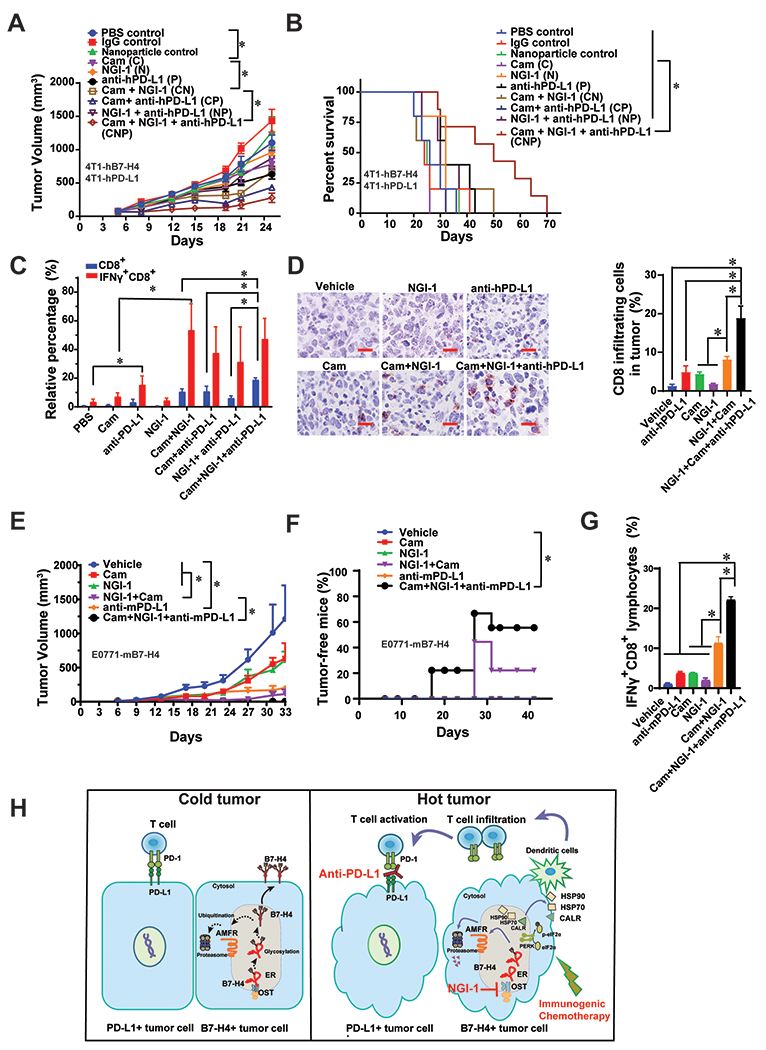
(A) 4T1-hPD-L, where the basal mouse PD-L1 was knocked out followed by adding back human PD-L1 gene, and 4T1-hB7-H4 cells were orthotopically injected into the left fourth mammary fat pad and allow to grow around 100 mm3, followed by injection of Cam (25 mg/kg, i.p.) for 4 times, NGI-1 nanoparticle (10 mg/kg, i.v.) as well as PD-L1 antibody durvalumab (5 mg/kg, i.p.) for 3 times. The tumor growth was monitored twice per week. n=8 mice per group. (B) Survival curve of the mice of the combination of Cam, NGI-1 nanoparticle and durvalumab. n=8 mice per group. (C) On day 18, mouse tumors were harvested, and digested followed by flow cytometry of staining IFNγ and CD8. Quantification of IFNγ+CD8+ or CD8+ cell in the tumor mass is shown. n=3 mice per group. (D) Tumor tissues were subjected to the immunohistochemically staining with anti-CD8 antibody. n=3 mice per group. The representative images are shown. Scale bar, 20 μm. Quantification of CD8+ infiltrating cells is shown. (E) E0771-mB7-H4 cells (1×105) were orthotopically injected into the left fourth mammary fat pad. When the tumors were visible on day 6, Cam (5 mg/kg, i.p.) was injected on day 6, 8, 10, and 12; NGI-1 nanoparticles (10 mg/kg, i.v.) were injected on day 7, 9, 11, and 12; and the anti-mPD-L1 antibody (5 mg/kg, i.p.) was injected on day 8, 10, and 12. The tumor growth was monitored twice per week. n=9 mice per group. (F) The percentage of tumor free mice was evaluated every week for 42 days (n=9). Log-rank (Mantel-Cox) test was performed for statistical analysis. (G) On day 27, spleens were harvested followed by flow cytometry of staining IFNγ and CD8. Quantification of IFNγ+CD8+ or CD8+ cells in the spleens is shown. n=3-5 mice per group. (H) The proposed working model of the combination of immunogenic chemotherapy, NGI-1 and PD-L1 blockade.
Human PD-L1-expressing mouse tumor cells have been previously utilized to study therapeutic activity and mechanism of the action of anti-human PD-L1 in mice. The hPD-L1 mouse model system has facilitated the pre-clinical investigation of efficacy and immune modulatory function of various forms of anti-hPD-L1 antibodies (41,42). Given that intertumoral and intratumor heterogeneity is a key challenge in cancer medicine (43), and based on our observation of an inverse correlation between B7-H4 and PD-L1 expression in breast cancer cells and tissues (Fig. 1D–F). For 4T1 orthotopic mouse model, we injected a 1:1 ratio mixture of 4T1-hPD-L1 (42) and 4T1-B7-H4 cells (Supplementary Fig. 8A) into the left fourth mammary fat pad and allow the tumor to grow to ~ 100 mm3, followed by injection of Cam, NGI-1-releasing nanoparticles and or the PD-L1 antibody durvalumab (Supplementary Fig. 8B–E). The administration of Cam at 25 mg/kg (but not that of NGI-1 nanoparticles or durvalumab) caused a 5% weight loss at week 4, but this weight loss was not further accentuated by the co-treatments (Supplementary Fig. 8C). No obvious heart toxicity was observed in these groups (Supplementary Fig. 8E). The combination of Cam+NGI-1+durvalumab significantly decreased tumor growth (Fig. 7A), reduced tumor weight (Supplementary Fig. 8D), and enhanced mouse survival (Fig. 7B) compared to the single treatments. We also observed that NGI-1 treatment alone does not directly alter the percentage of T cells or immature myeloid cells or mature dendritic cells (Supplementary Fig. 8I) or affect CD8 T cells infiltration in tumors (Fig. 7C & D), but it does inhibit both α-2,3 linkage (MAL II lectin staining) and α-2,6 linkage (SNA lectin staining) in the tumor and its microenvironment (Supplementary Fig. 8J & K). The frequency of CD8+ T cells, in particular CD8+ IFNγ+ cells, was significantly increased by the combination of Cam+NGI-1+durvalumab compared with the monotherapies (Fig. 7C & D).
In addition, we generated E0771 cells expressing mouse-B7-H4 expression for experimental use (Supplementary Fig. 8F). An E0771-mB7-H4 syngeneic C57BL6 mouse model was established by mammary gland orthotopic injection of E0771-mB7-H4 cells, followed by injection of Cam, NGI-1-releasing nanoparticles, and/or anti-mPD-L1 (clone:10F.9G2) (Supplementary Fig. 8G). The administration of Cam at 5 mg/kg, NGI-1 nanoparticles and anti-mPD-L1 did not cause weight loss (Supplementary Fig. 8H). The combination of Cam+NGI-1+anti-mPD-L1 significantly decreased tumor growth (Fig. 7E) and caused the disappearance of more than 50% of E0771-mB7-H4 tumors (Fig. 7F). The frequency of CD8+ IFNγ+ T cells in spleens was significantly increased by the combination of Cam+NGI-1+anti-mPD-L1 compared with each monotherapy (Fig. 7G). Taken together, the above results suggest that the suppression of B7-H4 by NGI-1 may enhance the efficacy of PD-L1-targeted immunotherapy combined with immunogenic chemotherapy.
DISCUSSION
Based on unbiased bioinformatic analyses of immune-related proteins, we identified B7-H4 to be accumulated in immune-cold subtypes of TNBC tumor, and that B7-H4 expression negatively correlated with that of PD-L1. We also demonstrated that posttranslational modifications of B7-H4 dictate tumor immunogenicity in the context of DOX-induced immunogenic cell death. Pharmacological inhibition of B7-H4 glycosylation by NGI-1 facilitated ubiquitination of B7-H4, thereby causing B7-H4 depletion and de-inhibition of ICD pathways, allowing the tumors to respond to a combination of ICD-eliciting chemotherapy and PD-L1 blockade (Fig. 7H). This study provides a new paradigm to develop strategies to treat immune-cold TNBC patients.
The mechanisms rendering tumors immune-cold are manifold, including the lack of tumor antigen, defects in antigen presentation, absence of T cell activation, and deficient homing of immune cells to the tumor bed (2). Chemotherapy-induced ICD converts malignant cells into vaccines and may improve T cell priming, ultimately facilitating the T cell-mediated attack of residual cancer cells (13,37). Other therapeutic approaches to transform cold tumors into hot ones include using: oncolytic viruses; tumor vaccines; CD40 agonistic antibodies; cytokines; anti-angiogenic therapy; adoptive T cell therapy including chimeric antigen receptor (CAR) T cells, or T cell recruiting bi-specific antibodies (2).
B7-H4 is well demonstrated as a transmembrane protein that is expressed by tumor-infiltrating myeloid cells (44). However, the function of B7-H4 expressed by cancer cells is less understood. B7-H4 has been reported to be present in the plasma membrane (45), cytosol (22) and nucleus (46). We observed a strong intracellular localization of B7-H4 in tumor cells. Although the receptor of membrane B7-H4 remains unknown or controversial (47), it is generally recognized that cell surface-associated B7-H4 inhibits T cell function (40). The function of the cytosolic pool of B7-H4 has been unclear. Our study suggests that cytosolic B7-H4 contained in malignant cells functions as a negative regulator of ICD. This effect apparently resides in the capacity of B7-H4 to inhibit the PERK-mediated phosphorylation of eIF2α that is required for the exposure CALR at the cell surface as a ‘danger’ or ‘eat-me’ signal. Thus, B7-H4 subverts the entire cascade that usually allows ICD-associated DAMPs to elicit tumor cell phagocytosis by DCs, DC maturation, and T cell recruitment to the tumor site. Fractionation analyses as well as co-IP experiments suggest that B7-H4 directly or indirectly interacts with eIF2α. This either may result in conformational changes of eIF2α that subsequently block its phosphorylation or sterically prevent eIF2α to bind to its kinases (such as PERK), a possibility that awaits further investigation.
Our studies uncovered a hidden B7-H4 protein turnover mechanism that can be ignited by the removal of N-linked glycosylation. B7-H4 is degraded through the ubiquitin-proteasome pathway especially after the enzymatic removal or the pharmacological or genetic inhibition of glycosylation. We found that the previously described glycosylation of B7-H4 (27) involves the hetero-oligomeric enzyme OST complex including its catalytic subunit STT3A. In addition, B7-H4 can be reglucosylated by UGGG1 to facilitate its appropriate folding. We demonstrated that the glycosylation of B7-H4 within the ER ultimately determines the cellular abundance of the protein. Mass spectrometry identified five asparagine residues as N-linked glycosylation sites. However, the mutagenesis of these sites was not sufficient to generate an entirely glycosylation-deficient mutant, perhaps because additional asparagine residues could compensate for the function of the mutated sites. Thus, we generated unglycosylated B7-H4 mutant (B7-H4-16NQ). The mutant 16NQ is more flexible than B7-H4 wildtype in the Ig-like C2-type domain, though the whole structure is still stable by MD simulations. The 16NQ retains partial ability for trafficking to the membrane. Protein stability of 16NQ is independent from the HSP90-facilitated protein folding and degradation pathway; rather, it involves the AMFR-mediated proteasome-ubiquitin pathway, possibly because glycosylation deficiency of the 16NQ B7-H4 facilitates the binding with AMFR.
Several strategies have been developed to target B7-H4 in cancer treatment, including antibodies, CART cells and drug conjugates. B7-H4 is reported to be an unstable cell surface antigen, and hence may not be suitable as a target of antibody-dependent cellular cytotoxicity or monoclonal antibodies (48). Here, we delineate an alternative therapeutic approach to target B7-H4 through glycosylation inhibition by NGI-1. NGI-1 suppresses the activity of OST through a direct and reversible interaction with both catalytic subunits, though does not cause a complete disruption of glycosylation (as this would occur with tunicamycin), a fact that may explain its reduced toxicity as compared to other, more complete glycosylation inhibitors (31). NGI-1 is reported to block EGFR signaling in NSCLC and glioma (31–33). Our study suggests NGI-1 inhibits B7-H4 more potently than EGFR in all tested breast cancer cell lines. Whether NGI-1 efficiently targets other proteins awaits further investigation.
One critical challenge in cancer immunotherapy research is to develop more accurate pre-clinical models that may translate to human. In our present work, to examine the anti-tumor effects of combined treatment with DOX and NGI-1 in association with PD-L1 blockade, we have utilized a mouse orthotopic tumor models of 4T1-hPD-L1, where the basal mouse PD-L1 was knocked out followed by adding back human PD-L1 gene. Ideally, a mouse model that tests human immunologic genes in mice by knocking in the human gene could provide more precise result. Coincidently, 4T1 mouse breast cancer cells do not express B7-H4, which provides convenience to test the impact of B7-H4 in DOX-induced immunogenic cell death in this work. Although the “humanization” of mouse cancer cells with human molecules is a possible strategy for studying the effects of therapeutic agents targeting such molecules, this system has some drawbacks resulting from incompatibilities between the two species, including immune responses to xenoantigens. Our results need to be further validated by more physiological relevant models in the future translation study.
In summary, this study revealed a previously unidentified role for B7-H4 in determining local immunosuppression in the context of TNBC. B7-H4 protein stability is regulated by molecular crosstalk between ubiquitination and glycosylation, which subsequently dictates the efficacy of immunogenic (ICD-inducing) chemotherapy. Targeting B7-H4 for destruction by inhibiting its glycosylation, combined with immunogenic chemotherapy and PD-L1 blockade, could be an efficient strategy to treat TNBC, in particular when B7-H4 is abundantly expressed.
METHODS
Bioinformatics analysis
The proteomic data are provided by TCGA BRCA samples (18). The samples are grouped by their PAM50 subtypes. The genes are sorted according to the difference between the average proteomic level in Basal subtype and the average proteomic level across the other samples. The heatmap was generated with R (version, 3.3.3).
The Accession numbers GSE88847 (for bulk tumor gene expression) in GEO database is retrieved and processed using GEOquery package which including 38 therapy-naïve TNBC patients. The TNBC subgroups (referred to as TIME subtypes) based on differential localization of CD8+ T cells were requested from the author (19). Gene expression from the listed immune-related proteins was obtained with R (version, 3.3.3). All software dependencies are available in the Comprehensive Repository R Archive Network (CRAN) (https://cran.r-project.org/) or on Bioconductor (https://www.bioconductor.org/).
Cell Lines and Cell Culture
HEK293T, the authenticated breast cancer cell panel (#30-4500K), and the authenticated ovarian cancer cell panel (#TCP-1021) were obtained from ATCC (American Type Culture Collection, Manassas, VA) in 2017. U2OS-HMGB1-GFP/H2B-RFP cells were provided by Dr. Guido Kroemer (49) in 2018. 4T1-hPD-L1 cells, where the basal mouse PD-L1 was knocked out followed by adding back human PD-L1 gene, were provided by Dr. Mien-Chie Hung (41) in 2019. All cells were maintained in the media suggested by ATCC such as 10% FBS, 100 units/ml streptomycin and 100 units/ml penicillin. All cells were cultured at 37 °C in a humidified atmosphere containing 5% CO2. All the cell lines were routinely tested for mycoplasma infections. All cell lines were Mycoplasma tested every 3 months using MycoProbe Mycoplasma Detection Kit (R&D Systems). The length of time between cell line thawing and use in experiment does not exceed 1 month (two or more passages). All cell lines were authenticated by STR DNA fingerprinting every 1 to 2 years and by visual inspection prior to experiments and freezing of cell line stocks in central cell bank.
Chemicals and antibodies
Camsirubicin was kindly provided by Monopar therapeutics Corporation. PNGase F was purchased from New England Biolabs Inc. Cycloheximide (CHX), doxorubicin, MG132, tunicamycin, Brefeldin A, puromycin dihydrochloride and polyethyleneimine (PEI; branched – average Mw ∼800, average Mn ∼600) were purchased from Sigma Aldrich, NGI-1 and Durvalumab were purchased from MedChem Express. PEG (5000)-b-PLA (10,000) (Diblock Polymer) was purchased from Polysciences, Inc.
The following antibodies were used: B7-H4 (Cell signaling,14572) Flag (Cell signaling, 8146 or 14793), Myc (Cell signaling, 2272), eIF2a (Cell signaling, 5324), p-eIF2a (Cell signaling, 3398), HSP70 (Cell signaling, 4873), Hsp90 (Cell signaling, 4877), CALR (Cell signaling, 12238), Normal IgG (Cell signaling, 2729), ubiquitin (Cell signaling, 43124), AMFR (Cell signaling, 9590), PERK (Cell signaling, 5683), eIF2a (Sigma, HPA084885), AMFR (Sigma,HPA077835), B7-H4 (Sigma, HPA054200), STT3A (proteintech, 12034), mouse B7-H4 (R&D, AF2154), RPN1 (Bethyl, A302-026A), RPN2 (Bethyl, A304-265A), STT3A (Bethyl, A305-531A) and UGGG1 (Bethyl, A305-530A), CD45 Rat anti Mouse, Brilliant Violet 711 (BD, ThermoFisher Scientific, 563709), IFNγ Rat anti Mouse, Brilliant Violet 711 (BD, ThermoFisher Scientific, 564336), CD8a Rat anti Mouse, APC, (BD, ThermoFisher Scientific, 553035), I-A/I-E Rat anti-Mouse, Brilliant Violet 711 (BD, Thermo Fisher Scientific, 563414), CD86 Rat anti-Mouse, PE-Cy7 (BD, Thermo Fisher Scientific, 560582), CD11c Hamster anti-Mouse, APC, (Thermo Fisher Scientific, 550261), PE Mouse anti-Human B7-H4 (BD, 562507), FITC Rat anti-Mouse CD4 (BD, 553046), APC-Cy7 labeled anti-Mouse CD11b (BD, 557657), PE-Cy7 labeled anti-Mouse Ly-6G and Ly-6C (Gr-1) (BD, 552985), Anti-rat IgG (H+L), (Alexa Fluor(R) 647 Conjugate) (Cell Signaling Technology, 4418), Anti-rabbit IgG (H+L), F(ab’)2 Fragment (Alexa Fluor(R) 647 Conjugate) (Cell Signaling Technology, 4414), Rabbit IgG Isotype Control (Cell signaling Technology, 3452), (Alexa Fluor(R) 488 Conjugate) (Cell Signaling Technology, 4340S), Anti-rabbit IgG (H+L), F(ab’)2 Fragment (Alexa Fluor(R) 488 Conjugate (Cell Signaling Technology, 4412S).
Tissue microarray, immunohistochemistry and image analysis
The Breast cancer tissue array with adjacent normal breast tissue BC081116d which contains 110 cases (15 mm tissue cores for each tissue) were purchased from US biomax (Derwood, MD). Immunohistochemically staining was carried out following standard streptavidin-biotin-peroxidase complex method. Briefly, section was deparaffinized, and nonspecific bindings were blocked with 10% normal goat serum for 30 min. The section was then incubated with antibody overnight at 4 °C. Antibodies including B7-H4 (Sigma-Aldrich HPA054200), PD-L1 (E1L3N) (Cell Signaling Technology 14772S), STT3A (Fisher Scientific PIPA556550), AMFR (Fisher Scientific PIPA512051), Sambucus Nigra Lectin (SNA, Vector Lab, B-1305-2), Maackia Amurensis Lectin II (MAL II, Vector Lab, B-1265-1), hCD8α (Cell signaling, 70306), mCD8α (Cell signaling, 98941) and Phospho eIF2α (Ser51) (D9G8) (1:50; Cell Signaling Technology 3398S). After PBS washes, sections were incubated with biotinylated secondary and ABC kit Vectastain PK-6100 from Vector Labs. DAB (DAKO) was used to visualize the reaction, followed by counterstaining with Hematoxylin. After immunostaining, the sections were scanned at 20x magnification at standardized settings using a Nanozoomer (Hamamatsu) by a single investigator who was not informed of the clinical characteristics.
The whole slide image was initially acquired in the NDPI File Format and further semiquantitative analyzed by open-source software QuPath v0.2.0-m4 (50). The software was run on a standard off-the-shelf laptop computer. The H-score (or “histo” score) of B7-H4, CD8α, PD-L1, STT3A, AMFR and Phospho-eIF2α (Ser51) staining in tumor cells were evaluated by TMA algorithms. H-score is assigned using the following formula: H-score=3x % of strongly staining cells+2x% of moderately staining cells+1x % of weakly staining cells, giving a score range of 0-300.
Plasmids and Transfection
The N-Flag tag human B7-H4 (HG10738-NF), N-Myc tag human B7-H4 (HG10738-NM), mouse-B7-H4-C-Flag tag (MG50017-CF), and AMFR-C-Myc tag (MG59687-CM) plasmids were ordered from Sino Biological Inc. pcDNA-AMFR-C-Flag (Plasmid #62370) and pCDNA-AMFR-Flag RING mutant C356G H361A C-Flag (#61751) (29) were purchased from Addgene. For plasmids transfection, cells were plated to form a 50-70% confluent culture. The HEK293T cells were transfected using Lipofectamine 2000 (Invitrogen).
LentiCas9-Blast (Plasmid #52962) was purchased from Addgene. All shRNA and CRISPR clones were purchased from Sigma Aldrich. For CRISPR B7-H4 knockout using sanger clone HS5000001377 and HS5000001378, lentiCRISPRv2 puro (Addgene, Plasmid #98290) was used for the vector control. UGGG1 (SHCLNG-NM_198899, TRCN0000110450 and SHCLNG-NM_198899, TRCN0000110453), STT3A (SHCLNG-NM_008408, TRCN0000093714 and SHCLNG-NM_008408, TRCN0000093716), AMFR (SHCLNG-NM_001144, TRCN0000003374 and SHCLNG-NM_001144, TRCN0000368947). RPN1 (SHCLNG-NM_002950, TRCN0000421570), and RPN2 (SHCLNG-NM_002951, TRCN0000159696).
Fragment construction and site-directed mutagenesis
Indicated B7-H4 and AMFR fragments were amplified by PCR. All amplified products were further cloned into the pIRES vector with Flag-HA or HA tag. Fragment constructs were confirmed by DNA sequencing. B7-H4 2KR, 5NQ, 12NQ, 13NQ, and16NQ mutations were introduced using mutagenic primers (QUICKCHNGE MULTISITE KIT, Agilent Technologies, Santa Clara, CA, USA). Mutants were confirmed by sequence analysis.
Lentiviral construction, generation and Infection
hB7-H4, hB7-H4-2KR and hB7-H4-16NQ were generated by PCR amplification and were subcloned into pENTER-D-TOPO (Invitrogen) as an entry clone. In vitro recombination between an entry clone (containing a gene of interest flanked by attL sites) and a destination vector was performed to construct lentivirus expression vector. Clones with the right sequence were chosen.
Lentiviral particles are generated by transfection of the expression plasmid (i.e., pLenti-hB7-H4, hB7-H4-2KR and hB7-H4-16NQ), plus the packaged lentiviral particles (pVSV-G, pRRE, and pRSV-REV) were collected, mixed with polybrene, and added into 293-T cells using Lipofectamine 2000 (Life technologies). Culture media were harvested 48 hours after transfection, filtered through 0.45 μm filters. 4T1 cells were transduced with the lentivirus. Upon infection, the stable cell lines were established by selecting with 2-5 µg/ml puromycin.
Enzymatic deglycosylation and glycosylation sites identification
Deglycosylation experiments were performed on protein extracts from the indicated cells using Peptide N-Glycosidase F (New England Biolabs, Inc, Beverly, MA) as per the manufacturer’s directions. Briefly, denaturing reaction conditions were used for PNGase F treatment, as follows. Cell lysates are denatured in PNGaseF denaturing buffer (5% SDS and 10% 2-mercaptoethanol) for 10 minutes at 100 oC. Denatured lysates are then cooled and mixed with 1/10 volume each of concentrated PNGaseF reaction buffer, 0.5 M sodium phosphate pH 7.5, and 10% NP-40. Samples are then digested with PNGaseF for 1 hour at 37oC. The reaction is stopped by the addition of sample buffer containing 2-mercaptoethanol. The reaction is then incubated at 100 oC for 5 minutes before immunoblotting.
Purification of B7-H4 complex (affinity capture purification)
MDA-MB-468 cells stable expression Flag-B7-H4 were lysed with NP40 buffer (1% NP40, 10% glycerol, 25 mM Tris-HCl (pH 7.9) and protease inhibitor cocktails. B7-H4 interacting proteins were purified by immunoprecipitation on anti-Flag M2 beads (Cat: Sigma, A2220) and washing four times with TBST buffer (137 mM NaC1, 20 mM Tris-HCl [pH 7.6],0.1% Tween-20). The complex was eluted with 3×Flag peptide (Sigma, F4799) in TBS buffer. Flag-B7-H4 protein was purified by immunopurification on anti-Flag M2 beads (Sigma). The elute was then separated on SDS-PAGE followed by Coomassie blue staining. The bands were cut out for mass spectrum analysis. For the glycosylation sites, the elute was treated with Peptide N-Glycosidase F followed by SDS-PAGE. The bands were cut out at the bottom of stacking gel before running into the separating gel for mass spectrometry analysis.
Cell proliferation assay
Cells were seeded in 96-well plates at the concentration of 5000 cells per well and incubated at 37 °C for 3 days. After 3 days, the media with treatments were dumped and 100 μl of phenol red free DMEM with 10% CCK8 were added in each well and incubated for 1 h. The absorbance of light with wavelength of 450 nm was measured with 800TS Absorbance Reader from the Biotek.
Immunoblot analysis
Cells were lysed with 1× cell lysis buffer (Cell Signaling Technology, 9803) containing protease inhibitor (Sigma-Aldrich, P8340) on ice for 10 min, homogenized by passing through a 21-gauge needle, and centrifuged at 14,000 g for 15 min at 4°C to pellet the cell debris. Protein was quantified using the BCA assay (Thermo Fisher Scientific, 23225) and 20 μg of each sample was resolved on SDS-PAGE and transferred to nitrocellulose membrane. Membranes were blocked with TBST containing 5% skim milk for 1 h and incubated overnight at 4°C with various primary antibodies followed by incubation with goat anti-rabbit or anti-mouse IgG HRP secondary antibody (Cell Signaling Technology, 7074 or 7076) at room temperature for 1 hr Chemiluminescence substrate was applied using SuperSignal™ West Pico Chemiluminescent Substrate (Thermo Fisher Scientific, 34080) and blots were analyzed using the ChemiDoc™ Touch Imaging System (Bio-Rad). Semi-quantification of data was performed using Image Lab (Bio-Rad).
Immunoprecipitation analysis
Cells were lysed at 4°C in ice-cold immunoprecipitation assay buffer (Cell Signaling Technology, 9806) and cell lysates were cleared by brief centrifugation (13,000 g, 15 min). Concentrations of proteins in the supernatant were determined using the BCA assay. For immunoprecipitation assay, cell lysate was incubated with anti-Flag M2 beads or IP-proved antibodies overnight at 4 °C on a rotator, followed by the addition of protein A/G plus agarose to the reaction for 2 h at 4 °C. To detect ubiquitin conjugates, the lysates were denatured in lysis buffer containing 1% SDS to disrupt any protein-protein interactions, and then diluted ~10 fold prior to IP followed by immunoblot. Prior to immunoprecipitation, samples containing equal amounts of proteins were pre-cleared with protein A agarose beads (Santa Cruz Biotechnology, sc-2027; 4°C, 3 h), and subsequently incubated with various irrelevant IgG or specific antibodies in the presence of protein A agarose beads for 2 h or overnight at 4°C with gentle shaking. After five washes with lysis buffer supplemented with protease inhibitor mixture, complexes were released from the anti-Flag M2 beads by boiling for 5 min in 2x SDS-PAGE loading buffer.
Flow cytometry
Single-cell suspensions were stained with relevant mAbs and then washed twice with cold PBS. Intracellular IFNγ staining under stimulation with 10 ng/ml PMA, 100 ng/ml ionomycin plus 10 μg/ml BFA for 4 hr, was performed according to the manufacturer’s instruction (BD Bioscience) and were stained with surface markers and DEAD™ Fixable Violet Dead Cell Stain Kit (Thermo Fisher Scientific, # L34963) was used before fixation in 4% formaldehyde for 10 min at room temperature. Cells were washed with ice-cold PBS containing 2% BSA, followed by the second wash step with ice-cold PBS. Cells were resuspended in 80% methanol and incubated for 30 min at −20 °C. The pellet was washed twice with ice-cold PBS. Annexin V Alexa Fluor™ 647 Ready Flow Conjugate (Thermo Fisher Scientific, #R37175) and DAPI staining were used to measure cell death. Cells were washed with ice-cold PBS containing 2% BSA and analyzed by flow cytometry. Samples were conducted on a BD FACSymphony A5-Laser Analyzer, and data were analyzed with FlowJo software.
Phagocytosis and DC maturation assay
The cells isolated from spleen of three naïve mice were harvested, followed by the purification of CD11c positive cells using Cd11c Microbeads UltraPure (Miltenyi Biotec, # 130-108-338). Tumor cells were stained with CellTracker Orange CMTMR (Thermo Fisher Scientific, # C2927) at 37°C for 30 minutes, and then treated w/o doxorubicin (25 μM) or NGI-1 (10 μM) for 24 hours. These tumor cells were co-cultured with the CD11c positive cells for 2 hours at a ratio of 1: 1, and then subjected to flow cytometry (phagocytosis assay). For dendritic maturation assay, tumor cells were co-cultured with the CD11c positive cells for 2 hours at a ratio of 1: 1, and then subjected to flow cytometry (13).
OptiPrep density gradient protein fractionation assay
To prepare the iodixanol gradient, a 50% (w/v) and 5% (w/v) solution of iodixanol were made by diluting the stock solution (60% (w/v) aqueous iodixanol from StemCell Technologies with 0.25 M sucrose, 6 mM EDTA, 60 mM Tris-HCl pH7.4. The gradient was formed by gradient makers and loaded into a 14x89 mm polyallomer tube (Beckman Coulter). Cells were lysed with 0.5% NP40, and 5mg protein was overlaid onto the top of the gradient, and centrifugation performed at 100,000x g for 18h at 4 degree. 24 individuals 0.5 ml gradient fractions were collected with increasing density.
Immunofluorescence
Cells grown on coverslip were fixed with 4% paraformaldehyde in PBS for 10 min. The membrane staining of the indicated proteins was performed without membrane permeabilization by 0.1% Triton X-100 otherwise stated. The indicated antibodies were incubated with cells 4°C overnight. After three washes with PBSB, cells were incubated with Alexa 488 anti-rabbit /594-anti-mouse IgG diluted in PBSB for 1 h. Cells were washed, mounted with UltraCruz DAPI containing mounting medium (Santa Cruz), viewed, and photographed under Nikon A1 (Nikon) with the software of NIS-Elements. The fluorescence of Dox alone was also checked.
Duolink Proximity ligation assay
The colocalization of B7-H4, AMFR, STT3A and CALR was analyzed by the proximity ligation assay by using Duolink In Situ PLA kit (Sigma-Aldrich, Saint Louis, MO) and following manufacturer’s protocol. In brief, cells were plated on glass coverslips. Then the cells were fixed with 4% formaldehyde followed by permeabilization (0.1% Triton X-100). Cells were then incubated with blocking buffer followed by primary antibody overnight at 4 °C. Cells were then incubated with PLA probes. Add ligation mix to cells for 1 h at 37 °C. Add the amplification mix for 2 h at 37 °C and wash the cells twice. Mount the coverslips using the Duolink® In Situ Mounting Medium followed by imaging the cells with BioTek LionHeart automated fluorescence microscope.
Structure modeling and docking analysis
Structural models for B7-H4 Ig-like V-type 1 and V-type 2 domains were generated by comparative/homology modeling, searching the Swiss-Model server. Protein-protein docking simulations for AMFR RING domain (PDB: 2LXH) and the modeled B7-H4 structure were performed using the ZDOCK server Docking poses were ranked based on the ZDOCK 3.0.2’s scoring function and the top 100 binding poses were clustered using K-means clustering with a cutoff RMSD of 5 Å into five clusters. Docking poses with the highest scores in each cluster were selected to conduct two independent 120 ns unbiased MD simulations to further evaluate the stability of the predicted binding poses. NAMD package and CHARMM36m force field were used in MD simulations.
In vivo vaccination assay
BALB/C and nu/nu mice (6-week-old female) were purchased from Charles River. Stable expression of empty-vector 4T1 (4T1-vector) or B7-H4 wildtype of 4T1 (4T1-B7-H4) cells were treated with doxorubicin (Dox; 25 μM) alone or in combination with NGI-1 (10 μM) for 24 h. Then the cells were collected and orthotopically injected (106 cells per mouse) into the right fourth mammary gland of immunocompetent BALB/c mice. PBS was used in non-vaccinated control group. One week later, all mice were rechallenged with the same kind of live 4T1-vector or 4T1-B7-H4 cells (3 × 105 per mouse) in the left fourth mammary gland. The tumor growth was monitored twice per week. All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of Northwestern University.
In vivo therapeutic assay
NGI-1 nanoparticle was prepared as reported (33). Briefly, polyethylene glycol (PEG)-b-Polylactic acid nanoparticles were synthesized using diblock polymer (Polysciences, # 25018) and Polyethyeleneimine (Sigma-Aldrich, #408719) using a nanoprecipitation technique. Polymer was dissolved in DMSO and was added drop-wise to deionized water to create an NP suspension followed by Amicon Ultracell 100k centrifugal filter unit, and centrifuged at 4,000 g at 4°C for 30 minutes. For drug-loaded NPs, NGI-1 was dissolved in DMSO at a concentration of 50 mg/ml, and PEI was dissolved in DMSO at 50 mg/ml. The NGI-1 and PEI solutions were then mixed at a 6:1 ratio (by weight) of PEI:drug and then added to the PLA-PEG solution at a 10% ratio of NGI-1:PLA-25 PEG by weight.
Mixture of 4T1-hPD-L1 (41) (2 × 105) and 4T1-B7-H4 (2 × 105) cells at a ratio of 1:1 or E0771-mB7-H4 (1 × 105) were orthotopically injected into the left fourth mammary fat pad followed by injection of Camsirubicin intraperitoneally at dose of 25 mg/kg or 5 mg/kg for total 4 times. NGI-1 nanoparticle was then injected intravenously at 10 mg/kg for four times, followed by the administration of PD-L1 antibody durvalumab or inVivoMAb anti-mPD-L1(10F.9G2) (BioX Cell, Cat: BE0101) intraperitoneally at 5 mg/kg for total 3 times. The tumor growth was monitored twice per week. All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of Northwestern University.
ELISPOT assay
For detection of IFNγ-secreting cells, spleens were harvested at day 20 after tumor cell injection and stimulated with 10 ng/ml PMA, 100 ng/ml ionomycin ex vivo with 2 × 105 cells/well for 24 hr. ELISPOT assay was performed with Mouse IFNγ ELISpot Kit (Thermo Fisher Scientific, #EL485) according to the manufacturer’s instruction. The numbers and diameters of spots were counted in duplicate, and calculated by an automatic ELISPOT counter (CTL ImmunoSpot® S5 Macro Analyzer, Cellular Technology Limited, OH, USA).
Statistical analysis
Unless specified, results were expressed as mean ± SEM. Experiments were performed at least twice unless otherwise specified. Statistical analyses were performed using SPSS (SPSS, Chicago, IL, USA), GraphPad Prism (ver.8, GraphPad Software, San Diego, CA) or R software (version, 3.3.3). The significance of the differences in the assays was analyzed by Student’s t test or one or two-way ANOVA, followed by Tukey’s multiple comparisons test. Comparison of survival curves or tumor-free curve was performed using Log-rank (Mantel-Cox) test. Pearson correlation or Spearman correlation was used to study the correlation between two molecules. A value of p< 0.05 was considered significant.
Data availability
All data are available from the authors upon reasonable request.
Supplementary Material
STATEMENT OF SIGNIFICANCE.
This work unravels the regulation of B7-H4 stability by ubiquitination and glycosylation, which affects tumor immunogenicity, particularly regarding immune-cold breast cancers. The inhibition of B7-H4 glycosylation can be favorably combined with immunogenic chemotherapy and PD-L1 blockade to achieve superior immuno-infiltration of cold tumors, as well as improved tumor growth control.
ACKNOWLEDGEMENTS
We are grateful to Drs. Wade Harper and Jianping Jin for kindly providing the TAP purification vector. Ziyi Fu participated part of plasmid construction and experimental help. We appreciate the proteomic core at the Feinberg School of Medicine Northwestern University for Mass Spectrometry Analysis and post data analysis (NCI CCSG P30 CA060553 and P41 GM108569). This work was also supported by the Northwestern University Pathology Core Facility, RHLCCC Flow Cytometry Facility, Image CORE, Animal Resources Facility and a Cancer Center Support Grant (NCI CA060553). We thank all members of Wan, Bahar, Kepp, Hung and Kroeme laboratories for their helpful discussion. We also acknowledge Dr. Qiang Hu in Roswell Park Comprehensive Cancer Center for kindly helping with bioinformatics analysis. We are thankful to Monopar Therapeutics for kindly providing us Camsirubicin. This work was supported by the Northwestern University Zell scholar fund and part of effort for YW is covered by NIH R01CA202963. Support from NIH grants P01 DK096990 and P41 GM103712 is gratefully acknowledged by IB. GK is supported by the Ligue contre le Cancer (équipe labellisée); Agence National de la Recherche (ANR) – Projets blancs; ANR under the frame of E-Rare-2, the ERA-Net for Research on Rare Diseases; AMMICa US23/CNRS UMS3655; Association pour la recherche sur le cancer (ARC); Association “Le Cancer du Sein, Parlons-en!”; Cancéropôle Ile-de-France; Chancelerie des universités de Paris (Legs Poix), Fondation pour la Recherche Médicale (FRM); a donation by Elior; European Research Area Network on Cardiovascular Diseases (ERA-CVD, MINOTAUR); Gustave Roussy Odyssea, the European Union Horizon 2020 Project Oncobiome; Fondation Carrefour; Institut National du Cancer; Inserm; Institut Universitaire de France; LeDucq Foundation; the LabEx Immuno-Oncology (ANR-18-IDEX-0001); the RHU Torino Lumière; the Seerave Foundation; the SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); and the SIRIC Cancer Research and Personalized Medicine (CARPEM).
Funding resource: (1) Northwestern University Zell scholar fund, and (2) CA202963
Footnotes
Disclosure of Potential Conflicts of Interest
The authors declare no potential conflicts of interest.
REFERENCES
- 1.Marra A, Viale G, Curigliano G. Recent advances in triple negative breast cancer: the immunotherapy era. BMC medicine 2019;17(1):90 doi 10.1186/s12916-019-1326-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bonaventura P, Shekarian T, Alcazer V, Valladeau-Guilemond J, Valsesia-Wittmann S, Amigorena S, et al. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front Immunol 2019;10:168 doi 10.3389/fimmu.2019.00168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sica GL, Choi IH, Zhu G, Tamada K, Wang SD, Tamura H, et al. B7-H4, a molecule of the B7 family, negatively regulates T cell immunity. Immunity 2003;18(6):849–61. [DOI] [PubMed] [Google Scholar]
- 4.Prasad DV, Richards S, Mai XM, Dong C. B7S1, a novel B7 family member that negatively regulates T cell activation. Immunity 2003;18(6):863–73 doi 10.1016/s1074-7613(03)00147-x. [DOI] [PubMed] [Google Scholar]
- 5.Zang X, Loke P, Kim J, Murphy K, Waitz R, Allison JP. B7x: a widely expressed B7 family member that inhibits T cell activation. Proc Natl Acad Sci U S A 2003;100(18):10388–92 doi 10.1073/pnas.1434299100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Podojil JR, Miller SD. Potential targeting of B7-H4 for the treatment of cancer. Immunol Rev 2017;276(1):40–51 doi 10.1111/imr.12530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, et al. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell 2005;123(2):321–34 doi 10.1016/j.cell.2005.08.032. [DOI] [PubMed] [Google Scholar]
- 8.Willingham SB, Volkmer JP, Gentles AJ, Sahoo D, Dalerba P, Mitra SS, et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc Natl Acad Sci U S A 2012;109(17):6662–7 doi 10.1073/pnas.1121623109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu B, Guo H, Xu J, Qin T, Guo Q, Gu N, et al. Elimination of tumor by CD47/PD-L1 dual-targeting fusion protein that engages innate and adaptive immune responses. mAbs 2018;10(2):315–24 doi 10.1080/19420862.2017.1409319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barkal AA, Brewer RE, Markovic M, Kowarsky M, Barkal SA, Zaro BW, et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature 2019;572(7769):392–6 doi 10.1038/s41586-019-1456-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao J, Zhong S, Niu X, Jiang J, Zhang R, Li Q. The MHC class I-LILRB1 signalling axis as a promising target in cancer therapy. Scandinavian journal of immunology 2019;90(5):e12804 doi 10.1111/sji.12804. [DOI] [PubMed] [Google Scholar]
- 12.Barkal AA, Weiskopf K, Kao KS, Gordon SR, Rosental B, Yiu YY, et al. Engagement of MHC class I by the inhibitory receptor LILRB1 suppresses macrophages and is a target of cancer immunotherapy. Nature immunology 2018;19(1):76–84 doi 10.1038/s41590-017-0004-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Casares N, Pequignot MO, Tesniere A, Ghiringhelli F, Roux S, Chaput N, et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med 2005;202(12):1691–701 doi 10.1084/jem.20050915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer 2012;12(12):860–75 doi 10.1038/nrc3380. [DOI] [PubMed] [Google Scholar]
- 15.Bezu L, Sauvat A, Humeau J, Gomes-da-Silva LC, Iribarren K, Forveille S, et al. eIF2alpha phosphorylation is pathognomonic for immunogenic cell death. Cell Death Differ 2018;25(8):1375–93 doi 10.1038/s41418-017-0044-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tannous A, Pisoni GB, Hebert DN, Molinari M. N-linked sugar-regulated protein folding and quality control in the ER. Seminars in cell & developmental biology 2015;41:79–89 doi 10.1016/j.semcdb.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vembar SS, Brodsky JL. One step at a time: endoplasmic reticulum-associated degradation. Nature reviews Molecular cell biology 2008;9(12):944–57 doi 10.1038/nrm2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mertins P, Mani DR, Ruggles KV, Gillette MA, Clauser KR, Wang P, et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 2016;534(7605):55–62 doi 10.1038/nature18003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gruosso T, Gigoux M, Manem VSK, Bertos N, Zuo D, Perlitch I, et al. Spatially distinct tumor immune microenvironments stratify triple-negative breast cancers. J Clin Invest 2019;129(4):1785–800 doi 10.1172/JCI96313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou L, Ruan M, Liu Y, Zhu Y, Fu D, Wu K, et al. B7H4 expression in tumor cells impairs CD8 T cell responses and tumor immunity. Cancer Immunology, Immunotherapy 2020;69(2):163–74 doi 10.1007/s00262-019-02451-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen LJ, Sun J, Wu HY, Zhou SM, Tan Y, Tan M, et al. B7-H4 expression associates with cancer progression and predicts patienťs survival in human esophageal squamous cell carcinoma. Cancer Immunol Immunother 2011;60(7):1047–55 doi 10.1007/s00262-011-1017-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miyatake T, Tringler B, Liu W, Liu SH, Papkoff J, Enomoto T, et al. B7-H4 (DD-O110) is overexpressed in high risk uterine endometrioid adenocarcinomas and inversely correlated with tumor T-cell infiltration. Gynecol Oncol 2007;106(1):119–27 doi 10.1016/j.ygyno.2007.03.039. [DOI] [PubMed] [Google Scholar]
- 23.Pagnotti GM, Atkinson RM, Romeiser J, Akalin A, Korman MB, Shroyer KR. B7-H4 is Inversely Correlated With T-Cell Infiltration in Clear Cell but Not Serous or Endometrioid Ovarian Cancer. Appl Immunohistochem Mol Morphol 2019;27(7):515–22 doi 10.1097/pai.0000000000000608. [DOI] [PubMed] [Google Scholar]
- 24.Quandt D, Fiedler E, Boettcher D, Marsch W, Seliger B. B7-h4 expression in human melanoma: its association with patients’ survival and antitumor immune response. Clin Cancer Res 2011;17(10):3100–11 doi 10.1158/1078-0432.CCR-10-2268. [DOI] [PubMed] [Google Scholar]
- 25.Bregar A, Deshpande A, Grange C, Zi T, Stall J, Hirsch H, et al. Characterization of immune regulatory molecules B7-H4 and PD-L1 in low and high grade endometrial tumors. Gynecologic Oncology 2017;145(3):446–52 doi 10.1016/j.ygyno.2017.03.006. [DOI] [PubMed] [Google Scholar]
- 26.MacGregor HL, Garcia-Batres C, Sayad A, Elia A, Berman HK, Toker A, et al. Tumor cell expression of B7-H4 correlates with higher frequencies of tumor-infiltrating APCs and higher CXCL17 expression in human epithelial ovarian cancer. Oncoimmunology 2019;8(12):e1665460–e doi 10.1080/2162402X.2019.1665460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Salceda S, Tang T, Kmet M, Munteanu A, Ghosh M, Macina R, et al. The immunomodulatory protein B7-H4 is overexpressed in breast and ovarian cancers and promotes epithelial cell transformation. Exp Cell Res 2005;306(1):128–41 doi 10.1016/j.yexcr.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 28.Ou D, Wang X, Metzger DL, Ao Z, Pozzilli P, James RF, et al. Suppression of human T-cell responses to beta-cells by activation of B7-H4 pathway. Cell Transplant 2006;15(5):399–410. [PubMed] [Google Scholar]
- 29.Liu Y, Soetandyo N, Lee JG, Liu L, Xu Y, Clemons WM Jr., et al. USP13 antagonizes gp78 to maintain functionality of a chaperone in ER-associated degradation. eLife 2014;3:e01369 doi 10.7554/eLife.01369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arnold SM, Fessler LI, Fessler JH, Kaufman RJ. Two homologues encoding human UDP-glucose:glycoprotein glucosyltransferase differ in mRNA expression and enzymatic activity. Biochemistry 2000;39(9):2149–63 doi 10.1021/bi9916473. [DOI] [PubMed] [Google Scholar]
- 31.Lopez-Sambrooks C, Shrimal S, Khodier C, Flaherty DP, Rinis N, Charest JC, et al. Oligosaccharyltransferase inhibition induces senescence in RTK-driven tumor cells. Nat Chem Biol 2016;12(12):1023–30 doi 10.1038/nchembio.2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lopez Sambrooks C, Baro M, Quijano A, Narayan A, Cui W, Greninger P, et al. Oligosaccharyltransferase Inhibition Overcomes Therapeutic Resistance to EGFR Tyrosine Kinase Inhibitors. Cancer Res 2018;78(17):5094–106 doi 10.1158/0008-5472.CAN-18-0505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baro M, Lopez Sambrooks C, Quijano A, Saltzman WM, Contessa J. Oligosaccharyltransferase Inhibition Reduces Receptor Tyrosine Kinase Activation and Enhances Glioma Radiosensitivity. Clin Cancer Res 2019;25(2):784–95 doi 10.1158/1078-0432.CCR-18-0792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jeon H, Vigdorovich V, Garrett-Thomson SC, Janakiram M, Ramagopal UA, Abadi YM, et al. Structure and cancer immunotherapy of the B7 family member B7x. Cell Rep 2014;9(3):1089–98 doi 10.1016/j.celrep.2014.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang F, Wei H, Wang X, Bai Y, Wang P, Wu J, et al. Structural basis of a novel PD-L1 nanobody for immune checkpoint blockade. Cell Discovery 2017;3(1):17004 doi 10.1038/celldisc.2017.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kepp O, Senovilla L, Vitale I, Vacchelli E, Adjemian S, Agostinis P, et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology 2014;3(9):e955691 doi 10.4161/21624011.2014.955691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garg AD, More S, Rufo N, Mece O, Sassano ML, Agostinis P, et al. Trial watch: Immunogenic cell death induction by anticancer chemotherapeutics. Oncoimmunology 2017;6(12):e1386829 doi 10.1080/2162402X.2017.1386829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Van Tine BA, Agulnik M, Olson RD, Walsh GM, Klausner A, Frank NE, et al. A phase II clinical study of 13-deoxy, 5-iminodoxorubicin (GPX-150) with metastatic and unresectable soft tissue sarcoma. Cancer Med 2019;8(6):2994–3003 doi 10.1002/cam4.2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wei SC, Anang NAS, Sharma R, Andrews MC, Reuben A, Levine JH, et al. Combination anti-CTLA-4 plus anti-PD-1 checkpoint blockade utilizes cellular mechanisms partially distinct from monotherapies. Proc Natl Acad Sci U S A 2019;116(45):22699–709 doi 10.1073/pnas.1821218116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li J, Lee Y, Li Y, Jiang Y, Lu H, Zang W, et al. Co-inhibitory Molecule B7 Superfamily Member 1 Expressed by Tumor-Infiltrating Myeloid Cells Induces Dysfunction of Anti-tumor CD8(+) T Cells. Immunity 2018;48(4):773–86.e5 doi 10.1016/j.immuni.2018.03.018. [DOI] [PubMed] [Google Scholar]
- 41.Huang A, Peng D, Guo H, Ben Y, Zuo X, Wu F, et al. A human programmed death-ligand 1-expressing mouse tumor model for evaluating the therapeutic efficacy of anti-human PD-L1 antibodies. Scientific reports 2017;7:42687- doi 10.1038/srep42687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li CW, Lim SO, Chung EM, Kim YS, Park AH, Yao J, et al. Eradication of Triple-Negative Breast Cancer Cells by Targeting Glycosylated PD-L1. Cancer Cell 2018;33(2):187–201 e10 doi 10.1016/j.ccell.2018.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McGranahan N, Swanton C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017;168(4):613–28 doi 10.1016/j.cell.2017.01.018. [DOI] [PubMed] [Google Scholar]
- 44.John P, Wei Y, Liu W, Du M, Guan F, Zang X. The B7x Immune Checkpoint Pathway: From Discovery to Clinical Trial. Trends in pharmacological sciences 2019;40(11):883–96 doi 10.1016/j.tips.2019.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Simon I, Zhuo S, Corral L, Diamandis EP, Sarno MJ, Wolfert RL, et al. B7-h4 is a novel membrane-bound protein and a candidate serum and tissue biomarker for ovarian cancer. Cancer Res 2006;66(3):1570–5 doi 10.1158/0008-5472.CAN-04-3550. [DOI] [PubMed] [Google Scholar]
- 46.Zhang L, Wu H, Lu D, Li G, Sun C, Song H, et al. The costimulatory molecule B7-H4 promote tumor progression and cell proliferation through translocating into nucleus. Oncogene 2013;32(46):5347–58 doi 10.1038/onc.2012.600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Podojil JR, Chiang MY, Ifergan I, Copeland R, Liu LN, Maloveste S, et al. B7-H4 Modulates Regulatory CD4(+) T Cell Induction and Function via Ligation of a Semaphorin 3a/Plexin A4/Neuropilin-1 Complex. J Immunol 2018;201(3):897–907 doi 10.4049/jimmunol.1700811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kaur G, Janakiram M. B7x-from bench to bedside. ESMO open 2019;4(5):e000554 doi 10.1136/esmoopen-2019-000554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Martins I, Kepp O, Menger L, Michaud M, Adjemian S, Sukkurwala AQ, et al. Fluorescent biosensors for the detection of HMGB1 release. Methods Mol Biol 2013;1004:43–56 doi 10.1007/978-1-62703-383-1_4. [DOI] [PubMed] [Google Scholar]
- 50.Bankhead P, Loughrey MB, Fernandez JA, Dombrowski Y, McArt DG, Dunne PD, et al. QuPath: Open source software for digital pathology image analysis. Sci Rep 2017;7(1):16878 doi 10.1038/s41598-017-17204-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available from the authors upon reasonable request.