

Abstract
Esophageal adenocarcinoma (EAC) is a major cause of cancer related morbidity and mortality in Western countries. The incidences of EAC and its precursor Barretts esophagus (BE) have increased substantially in the last four decades. Current care guidelines recommend that endoscopy be used for the early detection and monitoring of patients with BE, however, the efficacy of this approach is unclear. In order to prevent the increasing morbidity and mortality from EAC, there is a tremendous need for early detection and surveillance biomarker assays that are accurate, low-cost, and clinically feasible to implement. The last decade has seen remarkable advances in the development of minimally invasive molecular biomarkers, an effort led in large part by the Early Detection Research Network (EDRN). Advances in multi-omics analysis, the development of swallowable cytology collection devices, and emerging technology have led to promising assays that are likely to be implemented into clinical care in the next decade. In this review, an updated overview of the molecular pathology of BE and EAC and emerging molecular biomarker assays, as well as the role of EDRN in biomarker discovery and validation, will be discussed.
Keywords: Barrett’s esophagus, esophageal squamous cell carcinoma, esophageal adenocarcinoma, mutation, epigenetic, DNA methylation, microRNA, proteomics, biomarkers
Introduction
Symptoms of long-standing gastroesophageal reflux disease (GERD) have historically been the main clinical features used to identify people at risk of having Barretts esophagus (BE), who are then advised to undergo endoscopic assessment. It is clear that many people with BE have no history of GERD, which is one of several major reasons behind the lack of success of current BE screening and surveillance programs for preventing esophageal adenocarcinoma (EAC)1. With regards to strategies to identify BE at high risk of progressing to EAC, the presence or absence of BE with or without dysplasia on histologic review is currently the only biomarker used clinically for risk stratification and directing treatment2, 3. This dearth of well-studied biomarkers and the reliance on reflux symptoms and endoscopic findings of dysplasia had led to what Reid called the “paradox” of BE management.4 In this paradox, Reid notes several frustrating epidemiologic facts: 1) a large number of individuals with BE are asymptomatic, 2) nearly 50% develop EAC without associated GERD symptoms, 3) 95% of EACs arise without a prior diagnosis of BE, and 4) nearly 80% of EAC arise without a prior diagnosis of GERD1,4. Furthermore, the vast number of people with BE detected by endoscopy will not progress to EAC and instead will die of unrelated causes, which reflects the late age of occurrence of most EACs. In fact, the majority of people with BE are more likely to die from complications of cardiac disease than from EAC5. With these insights, several areas of active research in molecular biology are underway to resolve the “paradox” of BE and are likely to lead to more effective approaches to identifying and managing those patients with BE. Through efforts of EDRN investigators as well as others, a number of promising markers have been identified, however, currently there are only a limited number of biomarkers to precisely identify patients with BE and those at high-risk of progression to EAC.
With recent advances in genomics (i.e. next-generation sequencing), epigenomics, proteomics, and microarray technology, many potential diagnostic and prognostic molecular biomarkers have been identified at the level of DNA, RNA, and individual proteins. These technologies have been used to characterize the molecular profiles of BE and EAC and to advance our understanding of the molecular alterations that define BE, dysplasia, and EAC. They have also led to the recent identification of promising biomarkers that will likely impact clinical care in the next decade, if not sooner.
BE and EAC Overview
Barretts esophagus (BE), which is specialized small intestinal metaplastic epithelium of the esophagus, is a precursor to esophageal adenocarcinoma (EAC), a cancer that has increased dramatically in the last 40 years. Most, if not all, EAC originates in Barrett’s esophagus (BE). EAC appears to arise via a metaplasia-dysplasia-carcinoma sequence whereby Barrett’s metaplasia can progress to low-grade dysplasia, then high-grade dysplasia before becoming intramucosal carcinoma and finally invasive carcinoma6, 7. Advances in endoscopic therapy over the past two decades have made it feasible to intervene at the dysplastic stage to prevent the progression to EAC without resorting to esophagectomy, which has substantial post-operative long-term morbidity.
Classes of Molecular Alterations in BE and EAC: Characterization of Frequency of Alterations and Their Biology
Genomic Alterations
A comprehensive analysis of somatic mutations in EAC using whole-exome sequencing and whole-genome sequencing has been recently performed8. (See Figure 1). The investigators analyzed 149 EAC tumor-normal matched fresh-frozen samples and identified a series of significantly mutated genes, including “classical” tumor-driver genes, such as TP53, CDKN2A, SMAD4, ARID1A and PIK3CA, as well as new candidate driver genes, such as SPG20, TLR4, ELMO1 and DOCK2 among others. Chromosomal instability and copy number alterations have been found in BE and EAC1, 9. Paulson et al identified 9p loss encompassing the p16/CDKN2A locus in BE, high-grade dysplasia (HGD) and EAC cases, losses of chromosome 5q, 13q and 18q in HGD and EAC, and high-level amplification at ERBB2 on chromosome 17q in EAC10.
Figure 1:
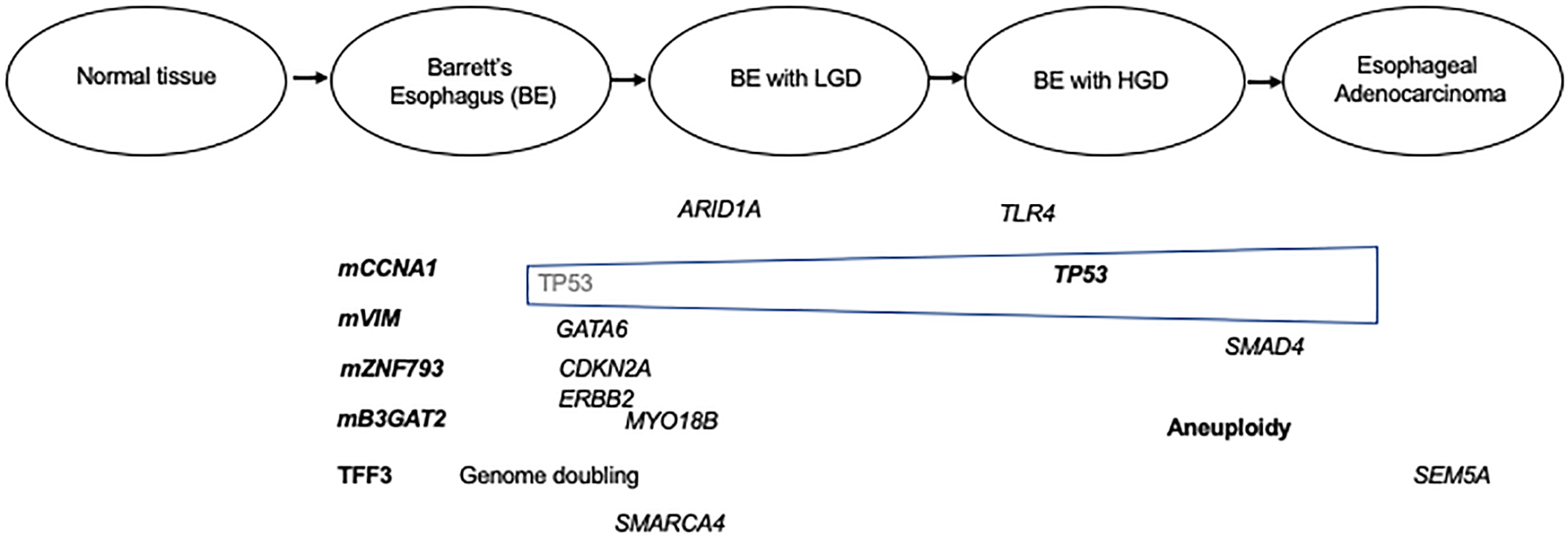
Representation of the Barrett’s esophagus to esophageal adenocarcinoma progression sequence and accompanying genomic and epigenomic alterations. The alterations show are a representative, but not complete list of genes affected by mutation, amplification, or aberrant methylation. Those alterations shown to be candidate biomarkers are in bold text.
More recent studies of BE have revealed an unexpected number of pathogenic variants of a number of oncogenes and tumor suppressor genes in approximately 10–20% of BE cases. An analysis of 25 matched cases of BE and EAC using directed exome next generation sequencing revealed common tumor suppressor gene mutations, with few oncogene mutations and genomic alterations present11. This study found that mutations in TP53 and SMAD4 were the most prevalent mutations in BE and that two pathways of BE progression appeared to be present. One pathway involves TP53 mutations and genomic doubling and may lead to the majority of EAC cases (>60%), while the other pathway involves the serial accumulation of mutations and is enriched for lesions with SMAD4 and CDKN2A alterations. Mutation analyses have shown that with the exception of TP53 and SMAD4, genes altered in BE and EAC do not display differential mutation rates between BE and EAC, even for bona fide tumor suppressors such as CDKN2A (30%) and ARID1A (15%), and others including KMT2D, MYO18B, UNC13C, FBXW7, ATM, FAT2, LRP1B, SMARCA4, etc (all <5%). TP53 mutations have been found in less than 5% of nondysplastic BE; whereas, 70% of cases of HGD and EAC were TP53 mutant11, 12 These results suggest that genetic alterations beyond TP53 and SMAD4 are not likely to yield clinically useful biomarkers for BE risk stratification.
Epigenomic Alterations
Epigenetic alterations, such as DNA hypermethylation in the promoter regions of genes, have also been identified in BE and EAC and are found in the majority of BE and EAC cases13–15. (See Figure 1.) EDRN funded studies by Grady and colleagues have shown that factors including aging smoking and obesity may play a role in the formation of these epigenetic alterations16, 17. Hypermethylated genes include known tumor-suppressor genes, such as APC, CDKN2A (p16INK4a), RUNX3, MGMT, CDH1, and SFRP family members among others13. A subset of the hypermethylated genes are believed to play a driver role in driving the formation of EAC, but many appear to be BE and EAC specific passenger alterations13, 15. Aberrant methylation of classic tumor suppressor genes such as CDKN2A and MGMT has been correlated with loss of mRNA and protein expression in the metaplasia-dysplasia-carcinoma sequence of Barrett’s esophagus18, 19. Recently, through EDRN support, Yu et al identified four methylation subtypes of EAC and BE through genome-wide DNA methylation profiling15. The high methylator subtype (HM) had more activating events in ERBB2 and a higher global mutation loa, compared to the other subtypes. In addition, this study uncovered a novel molecular mechanism by which EAC cells activate the oncogenic ERBB2/EGFR signaling pathway via epigenetically silencing the tyrosine phosphatase non-receptor 13 (PTPN13), specifically in the HM subtype.
Of relevance to biomarker discovery, a large number of genes and loci have been identified as high frequency targets of aberrant methylation in BE and EAC15. Although the functional significance of these methylated genes is still not clear, these DNA methylation events have proved to be highly promising as biomarkers of BE, as discussed below. In summary, he published studies to date suggest aberrant DNA methylation is a common molecular mechanism that mediates the development of esophageal cancer and that aberrantly methylated genes and loci are very promising biomarkers for BE and EAC.
MicroRNA alterations
MicroRNAs are small noncoding RNA molecules of approximately 20 nucleotides that appear to play important roles in diverse cellular processes during carcinogenesis. There is a continually growing number of studies focusing on the potential biological roles of microRNAs (miRNA/miR) in esophageal cancer development20, 21. For example, several studies have shown overexpression of miR-192 during BE→EAC progression14. miR-192 is a downstream target of TP53 and plays a tumor-suppressor role through cell-cycle arrest22. From a clinical perspective, an interesting finding is that altered miRNAs can be detected in the blood of patients with esophageal cancer23, which suggests they may be readily accessible molecular markers for early detection and monitoring chemotherapeutic responsiveness.24. However, the studies published to date have often produced conflicting results, likely secondary in large part to the wide spread use of non-validated analysis methods that are not robust and reproducible. This lack of consistency among studies has substantially limited progress in this area of research and in the use of microRNAs as biomarkers,
Protein alterations
In addition to alterations in genomic DNA, the epigenome, and microRNA expression, aberrant protein expression has also been noted in BE and EAC. These aberrantly expressed proteins for the most part play an unclear role in the pathogenesis of BE and EAC, but they have been shown to be useful as biomarkers for BE. Immunostain assays for two proteins, TFF3 and TP53, have been shown to robust markers for non-dysplastic BE and advanced dysplasia, respectively25 and are discussed in more detail in a following section.
Novel Methods for BE screening and surveillance
BE screening markers
Genetic and epigenetic alterations occurring in Barrett esophagus (BE) and early stage esophageal cancer have the potential to be used as early detection biomarkers. As noted above, candidate early detection markers include somatic mutations, aberrantly methylated genes, overexpressed miRNAs as well as deregulated proteins.
Somatic variants, deletions and rearrangements
As noted earlier, gene mutations arise in the BE→EAC progression sequence and affect a substantially greater proportion of BE with dysplasia and EAC cases compared to non-dysplastic BE cases. This class of molecular alteration was the first type studied in BE and EAC and has shown potential to be biomarkers for BE and EAC26. Chromosomal instability and copy number alterations have been found in BE and EAC1. Paulson et al identified 9p loss encompassing the p16/CDKN2A locus in BE, high-grade dysplasia (HGD) and EAC cases and losses on chromosome 5q, 13q and 18q in HGD and EAC, and high-level amplification at ERBB2 on chromosome 17q in EAC10. In addition, genome-wide association studies have identified common variants that are associated with genetic susceptibility to BE27. Dong et al developed a polygenic risk score using genomic variants and found Individuals in the highest quartile of risk, based on genetic factors (PRS), had a 2-fold higher risk of BE (odds ratio, 2.22; 95% confidence interval, 1.89–2.60) or EAC (odds ratio, 2.46; 95% confidence interval, 2.07–2.92) than individual in the lowest quartile of risk. When they combined data on demographic or lifestyle factors with data on GERD symptoms identified patients with BE with an AUC of 0.793 and patients with EAC with an AUC of 0.74528.
A subset of these candidate genomic DNA based biomarkers have been assessed in case-control clinical studies, including abnormal DNA ploidy, alterations in DNA copy number (based on fluorescence in-situ hybridization (FISH))29–32, gene mutations, loss of heterozygosity of specific DNA loci33, and measurements of clonal diversity in the BE tissue34. These molecular alterations have been shown in early phase studies to serve as adjunctive markers to delineate the degree of dysplasia (e.g., use of FISH probes for C-MYC to confirm HGD or carcinoma31) or to further risk stratify patients at greatest risk for progression to EAC (e.g., loss of ploidy associates with a 38.7% increased relative risk of developing EAC29). Unfortunately, genetic alterations do not appear to be of value as BE screening biomarkers because of their lowprevalence in BE cases. In contrast, TP53 mutations appear to have potential to be EAC screening biomarkers12.
Aberrantly methylated genes
Aberrantly methylated genes and DNA loci have been shown to be robust biomarkers for use in cancer care and prevention for a variety of cancers. Studies largely conducted by EDRN investigators over the last 3 years have shown methylated DNA biomarkers to be the most promising class of BE and EAC biomarkers to date. Through the EDRN, Markowitz and colleagues recently demonstrated that methylated VIM has a high sensitivity for detecting esophageal adenocarcinomas (EAC) and Barrett’s esophagus (BE), and that it even exceeded the robust sensitivity for detecting colon cancer that they had already shown35. The identification of methylated VIM DNA as a biomarker of BE suggested the potential for biomarker based early detection of BE and EAC. This finding prompted Markowitz’s team to develop a “molecular cytology” assay for methylated VIM in DNA samples from esophageal cytology brushings obtained during endoscopies of 322 individuals, divided into training and validation cohorts36. The assay showed 91% sensitivity for detecting BE, BE with dysplasia, and EAC at 93% specificity, with essentially identical results obtained in both the training and validation cohorts36 To further improve performance of a BE detection assay, they conducted a genome-wide analysis of DNA methylation in BE tissue samples using reduced representation bisulfite sequencing and found methylated CCNA1 DNA as a second BE biomarker with performance in both training and validation cohorts similar to methylated VIM36. When combined, the two-marker panel of methylated VIM and methylated CCNA1 DNAs detected 95% of BE, BE and dysplasia and EAC cases at 91% specificity, including detecting 96% of BE with dysplasia and 96% of EAC36.
To advance this biomarker panel toward a practical method for early BE detection, Markowitz, Chak, and colleagues developed and engineered a swallowable balloon based device for obtaining targeted non-endoscopic brushings of the distal esophagus36. To use the device, patients swallow a vitamin pill sized capsule that contains the balloon and is attached to a thin silicone catheter connected to an external syringe. After passage into the stomach, the balloon is inflated with air injected through the catheter and then pulled back into the esophagus to brush the gastro-intestinal junction plus a 6 cm length of distal esophagus. Removal of air via the catheter inverts the balloon back into the capsule, thereby protecting the distal esophagus sample from further dilution and from potential contamination by methylated DNA present in the proximal esophagus and oropharynx. In a clinical trial of 86 subjects, examination with the balloon could be completed in less than 5 minutes with 95% of subjects stating they would recommend the procedure to others36. Analysis for methylated VIM and methylated CCNA1 of DNA samples extracted from the balloon demonstrated 90% sensitivity for detecting non-dysplastic BE with 92% specificity36 providing practical demonstration of a biomarker based approach for detecting this asymptomatic precursor for EAC. In 2019 Lucid Diagnostics received FDA approval for commercial manufacture of the balloon device under the tradename EsoCheck. The combination of the balloon device and the methylated DNA panel is currently being further validated by testing in a nationwide multi-center clinical trial as well as undergoing commercial development under the tradename EsoGuard.
Additional promising BE markers have been identified and validated by others, including the lab of William Grady, an EDRN investigator. Yu et al discovered two genes, B3GAT2 and ZNF793, that are aberrantly methylated in BE. Clinical validation studies confirmed B3GAT2 and ZNF793 methylation levels were significantly higher in BE samples (median 32.5% and 33.1%, respectively) than in control tissues (median 2.29% and 2.52%, respectively; P < 0.0001 for both genes) and that gene-specific MethyLight assays could accurately detect BE (P < 0.0001 for both) in endoscopic brushing samples with mZNF793 having a sensitivity of 70% and specificity of 100% for BE37. These markers show promise to further improve the performance of a methylated gene panel for BE screening.
In addition to the Esocheck device, other swallowable cytology collection devices are being assessed and currently being evaluated for use in BE screening assays, such as the ‘Cytosponge’ and “Esophacap” devices, which are both swallowed capsules that degrade in the stomach to release a sponge tethered to a string38–40. Unlike the Esocheck device, these devices sample the entire esophagus and oropharynx, which increases the potential impairing biomarker performance. Similar to the Esocheck device, they capture esophageal cells which can later be analyzed for particular molecular changes associated with BE and/or dysplasia40. Using a Cytosponge based assay, Chettouh et al discovered and assessed hypermethylated TFPI2, TWIST1, ZNF345 and ZNF569 as potential BE screening markers. Methylated TFPI2 was shown to achieve the best sensitivity in both the pilot and validation Cytosponge cohorts (85% and 79%, respectively, AUC 0.88)41.
In summary, these studies have established that methylated DNA has emerged as a promising new biomarker class that will enable practical non-endoscopic screening and early detection of BE, an approach with potential to reduce the steadily increasing mortality from EAC. These developments have been vigorously supported by the NCI EDRN program and embody the EDRN’s vision for the potential of biomarkers to enable early cancer detection and to reduce cancer mortality.
Protein alterations
A number of proteins are differentially expressed in BE and EAC compared to the normal esophagus. Lao-Sirieix and colleagues surveyed three publicly available microarray datasets to identify putative biomarkers present in BE but absent from normal esophagus and gastric mucosa39. They identified TFF3 and DDC as the most promising candidate biomarkers for BE. Validation studies demonstrated TFF3 as the highest performing biomarker. The authors consequently developed an immunostain assay based on TFF3 in esophageal cytology samples for BE. In a case-control clinical study, they found that TFF3 positive cytology samples collected using the Cytosponge had a reasonable sensitivity (87%) and specificity (92%) for detection of BE segments greater than 3cm in length39. This TFF3 BE detection assay is being furthered assessed in the actively enrolling BEST-3 clinical trial. (See below)
BE Biomarker Clinical Trials
There are currently a number of clinical trials assessing different combinations of these swallowable cytology collection devices and selected biomarkers assays for the early detection of BE, BE with dysplasia and EAC. (See Table 1) Trials that are actively recruiting at the time of this publication include the following:
Table 1:
Validated BE early detection markers
| Biomarker | Method | Study Design | AUC | Sensitivity | Specificity | Ref. |
|---|---|---|---|---|---|---|
| TFF3 | IHC (cytosponge) | BEST-2: Case-control (N=1110) | 80% | 92% | 42 | |
| mVIM and mCCNA1 | bsNSG (Esocheck device) | Case-control Validation cohort (N=86) | 90% | 92% | 36 | |
| mB3GAT2 | methyLight PCR (endoscopic brushings) | Case-control Validation cohort (N=66) | 0.95 | 80% | 86% | 37 |
| mZNF793 | methyLight PCR (endoscopic brushings) | Case-control Validation cohort (N=66) | 0.96 | 80% | 93% | 37 |
| mTFPI2 | methyLight PCR (cytosponge) | Case-control Validation cohort (N=278) | 0.88 (0.84–0.91 | 82% | 96% | 41 |
| mTWIST1 | methyLight PCR (cytosponge) | Case-control validation cohort (N=278) | 81% (0.77–0.86) | 70% | 93% | 41 |
IHC=immunohistochemistry, bsNSG=bisulfite next generation sequencing
Table 1 below summarizes BE biomarkers that have been evaluated in clinical cohorts.
1) NCT02560623, Highly Discriminant Methylated DNA Markers for the Non-endoscopic Detection of Barrett’s Esophagus. Primary site: Mayo Clinic, Principal Investigator Prasad G. Iyer.
2) NCT00288119; Genetic Determinants of Barrett’s Esophagus and Esophageal Adenocarcinoma (FBE); Primary site: Case Western Reserve University, Principal Investigator: A.Chak (supported by the NCI, EDRN and BETRNet)
3) NCT02890979; Swallowable Sponge Cell Sampling Device and Next Generation Sequencing in Detecting Esophageal Cancer in Patients With Low or High Grade Dysplasia, Barrett Esophagus, or Gastroesophageal Reflux Disease; Primary center: Oregon Health Sciences University, PI: James Dolan
4) BEST 3: BMC Cancer. 2018 Aug 3;18(1):784. doi: 10.1186/s12885-018-4664-3.
BE surveillance and risk prediction markers
BE is associated with approximately 4X increased risk of EAC, which has led to the recommendation that patients with BE undergo regular endoscopic surveillance3. However, only 0.1–0.3% of people with Barrett’s esophagus will progress to high-grade dysplasia or EAC each year, thus, a biomarker (or biomarker panel) would be of great clinical utility if it can accurately risk stratify high risk patients with BE who are likely to progress from those low risk BE patients who are unlikely to develop EAC43. Such a marker could potentially spare the great majority of individuals with a diagnosis of BE from the cost, inconvenience, and risks of regular endoscopic surveillance. Being placed in a ‘low-risk’ group might also reduce the feelings of anxiety about developing EAC that have been shown to be associated with a diagnosis of BE44.
The search for accurate risk stratification markers for BE is an area of intense investigation that has led to identification of a number of promising risk biomarkers. To date, none of these markers have proven adequate to be used in the clinical setting, although immunostaining assays for p53 and aneuploidy appear highly promising3.
Methylated DNA markers
In a retrospective study, EDRN investigator Steve Meltzer compared BE patients who progressed to HGD or EAC to those who did not, using hypermethylated CDKN2A (OR 1.74, 95% CI 1.33 – 2.20), RUNX3 (OR 1.80, 95% CI 1.08 – 2.81), and HPP1 (OR 1.77, 95% CI 1.06 – 2.81), which were associated with an increased risk of progression. Age, BE segment length, and hypermethylation of other genes (TIMP3, APC, or CRBP1) were not found to be independent risk factors45. A follow-up study using these same epigenetic markers in combination with three clinical parameters (gender, BE segment length (SL), and pathologic assessment) demonstrated this multi-parameter method could stratify BE patients into high, intermediate, and low risk for progression to HGD or EAC. This tissue based assay has not been adopted into routine clinical use to date46. In a later iteration of this approach, this risk assessment tool was expanded to include additional genes previously shown to be hypermethylated in BE and/or EAC, most of which have been described in the previous section, to generate an eight-marker risk-of-progression panel. In a retrospective analysis of 145 nonprogressors and 50 progressors, this panel predicted progression with a sensitivity of ~50% when the specificity was set at 90%47. None of these candidates have advanced to phase III or IV biomarker trials.
MicroRNA alterations
MicroRNAs (miRNA/miR) are a class of small non-coding RNAs that are often abnormally expressed in cancer. Expression profiles of miRNAs have been used to characterize molecular subtypes of cancers, and as prognostic and predictive markers for certain cancers. By employing high-throughput techniques, such as microarrays and next generation sequencing, a number of recent studies have identified candidate miRNAs as markers of malignant progression of BE.
In studies of Barretts esophagus, dysplasia and esophageal adenocarcinoma, miR-196a, miR-192, miR-194, miR-106b, miR-25, mi-93, let-7c, miR-200,miR-203, −205, −192, −215, and miR-196b have shown incremental increases in expression with each step of progression from normal esophagus to metaplasia to dysplasia and carcinoma48,49.50,51,53.54. In a pilot phase 2 cross-sectional study, Bansal and colleagues compared miRNA expression signatures in metaplasia tissues from BE patients with or without dysplasia/cancer, and identified miR-15b, −203, and −21 as being discriminatory between BE patients with and without dysplasia/cancer, which suggested their potential utility for risk stratification52. More recently, Leidner and colleagues comprehensively characterized miRNA alterations during progressive stages of EAC55. They found 26 miRNAs that are highly and frequently deregulated in BE and EAC when compared to paired normal esophageal squamous tissue55. They identified miR-31 and −375 as potential markers of progression during early and late stages of tumorigenesis, respectively. In an independent study, Wu et.al confirmed miR-375 as a miRNA being downregulated exclusively in cancers, additionally supporting its role as a marker of cancer progression in BE21.
Although significant progress has been made in characterizing miRNA alterations in BE and EAC, there are still substantial limitations of the existing data, most notably being the lack of a consensus miRNA signature of cancer risk across the different studies. This is likely a consequence of studies with small sample sizes, inherent variations among study populations, differing methods for detecting miRNAs, and cellular heterogeneity in BE and EAC. In addition, progress in this field has been impeded by the poor reproducibility of study results, which reflects the lack of robust and reliable detection methods and the lack of sufficient attention to the confounding effects of preanalytical variables. In addition, the differences between disease and normal states are often suboptimal for development of robust biomarkers. These limitations will need to be overcome for microRNA based biomarkers to be clinically useful.
Clonal alterations and LOH (loss of heterozygosity)
Maley, Reid and colleagues have conducted numerous studies describing the relationship between clonal diversity and clonal expansions and the risk of BE progression. One prospective study of 268 BE patients evaluated whether clonal expansions during the progression of BE leads to homogenous cell populations or results in clonal diversity34. The authors found that patients with greater clonal diversity had greater risk of progression to EAC (p<0.001). In a follow-up study, this group compared clonal diversity in 79 BE progressors and 169 non-progressors over 20,425 person-months of follow-up, finding that non-progressors had types of chromosomal instability (small localized deletions involving fragile sites and 9p loss/copy neutral LOH) that generated relatively little genetic diversity56. Meanwhile, individuals that progressed to EAC developed chromosome instability with initial gains and losses, genomic diversity, and selection of somatic chromosomal alterations followed by catastrophic genome doublings. These data suggest that molecular testing to assess risk of progression in BE may need to incorporate assessment of structural genomic alterations and multiple foci of BE from individual patients and that such an assay could then be used as a risk prediction biomarker.
In another study that was a retrospective cohort study of high-risk patients who had a history of biopsy confirmed HGD without EAC, endoscopic brushing specimen were analyzed by FISH probes targeting 8q24 (MYC), 9p21 (CDKN2A), 17q12 (ERBB2), and 20q13 (ZNF217). The presence of polysomy was associated with a significantly higher risk of developing EAC within 2 years (14.2%), compared with patients with a non-polysomic FISH result (1.4%, P < 0.001)32.
Altered TP53 Expression and TP53 mutation
Altered TP53 tissue expression is the most promising risk stratification biomarker to date and has near-term potential to be used in clinical care. A large number of case-control studies have suggested that overexpression of TP53 in BE tissue indicates an increased risk for EAC, especially for BE with low grade dysplasia.
In the last 10 years, a series of studies by investigators at Erasmus MC University Medical Center found that increased TP53 expression in BE, determined by immunohistochemistry (IHC), preceded development of HGD/EAC by several years and that TP53 expression was an important risk factor for HGD/EAC with a hazard ratio (HR) of 6.5 (95% CI: 2.5–17.1)57, 58 In this largest study to date, TP53 immunostaining (N=635 patients, 12,000 biopsies), overexpression and complete loss significantly associated with the risk of neoplastic progression after adjusting for age, gender, BE length, and esophagitis (relative risk [RR] 5.6 [95% CI 3.1–10.3] and RR 14.0 [95% CI 5.3–37.2], respectively). However, only 49% of patients who progressed had aberrant TP53 immunostaining, which significantly limits its potential clinical utility. Furthermore, in a nested case control study by an independent group of investigators that used a registry of BE patients in Ireland, TP53 protein overexpression did not predict progression in a multivariate analysis30
Currently, TP53 is not routinely recommended for risk stratification but the British Society of Gastroenterology does have a grade B recommendation to test TP53 by IHC to clarify an equivocal histologic diagnosis of dysplasia3. The low sensitivity of this assay and concerns about reproducibility of the assay are still major concerns about its use in the clinic.
TissueCypher
The TissueCypher™ (Cernostics) is a quantitative, multiplexed biomarker imaging assay. It uses 14 epithelial and stromal biomarkers (K20, Ki-67, BETA-CATENIN, p16INK4a, AMACR, p53, HER2/neu, CDX-2, CD68, NF-kBp65, COX-2, HIF1a, CD45RO, and CD1a). In a multi-institutional case–control study, a 3-tier 15-feature classifier was identified in a training set (N=183) and tested in a validation set (N=183). The classifier stratified patients into low-, intermediate-, and high-risk classes [HR, 9.42; 95% confidence interval, 4.6–19.24 (high-risk vs. low-risk); P <0.0001]. It also provided independent prognostic information that outperformed predictions based on pathology analysis, segment length, age, sex, or TP53 overexpression59. This assay is a promising tissue based prediction assay for progression to HGD or EAC but requires further evaluation in prospective studies in appropriate populations to determine its clinical utility.
Blood based assays
Blood, stool or saliva biomarker based assays, in principal, are an ideal screening or surveillance method given the easy access of samples and safety of collection. A number of candidate blood-based biomarkers, including methylated DNA, circulating microRNAs, metabolite panels and peptides have been identified in small, retrospective, in vitro and non-human trials, although to date none have been evaluated in prospective clinical trials65–68. Most recently, in a proof of principle study Qin et demonstrated that a 5 methylated DNA biomarker panel (FER1L4, ZNF671, ST8SIA1,TBX15, ARHGEF4) used in a plasma based assay achieved an AUC of 0.93 (95% CI,0.89–0.96) on best-fit and 0.81 (95% CI, 0.75–0.88) on cross validation. At 91% specificity, the panel detected 74% of esophageal cancer (EAC and esophageal squamous cell cancer) overall, and 43%, 64%, 77%, and 92% of stages I, II, III, and IV, respectively69. (See Table 2.)
Table 2:
Candidate BE risk stratification markers
| Biomarker | Study Design | Sample Size | Outcome |
|---|---|---|---|
| Abnormal DNA ploidy, 9pLOH, 17pLOH34 | Prospective Cohort | N=243 | RR=38.7 |
| (95% CI 10.8–138.5) | |||
| Aneuploidy, tetraploidy60 | Retrospective analysis | N=322 | RR=11 (95% CI 5.5–21) |
| LOH by FISH: 17p13.161 | Retrospective analysis of surveillance cohort | N=151 | 5% of NDBE |
| 9% of LGD | |||
| 46% of HGD | |||
| CNA and LOH by FISH: | Prospective | N=138 | LGD: sens 70%, spec 89% |
| 8q24, 9p21, 17q11.2, and 20q13.262 | HGD: sens 84%, spec 93% | ||
| EAC: sens 94%, spec 93% | |||
| Hypermethylation of CDKN2A, RUNX3, HPP163 | Retrospective and longitudinal | N=53 | CDKN2A OR 1.74 |
| RUNX3 OR 1.8 | |||
| HPP1 OR 1.77 | |||
| Jin methylated gene panel64 | Retrospective, multi-center, double-blinded | N=50 progressors | AUC=0.843 at 2 years |
| N=145 non-progressors | AUC=0.829 at 4 years | ||
| Tissue Cypher59 | Case-Control multi-center | N=145 non-processors, N=45 progressors | OR 9.4 High vs. Low risk (95% CI 2.65, 33.28) OR 2.35 Intermediate vs. Low risk (95% CI 0.66, 8.41) |
Conclusions
Remarkable advances in early detection assays and technologies have occurred over the last decade. The most promising class of biomarkers for BE early detection is based on aberrantly methylated DNA. The EDRN has played a central role in the discovery and development of BE early detection assays that use non-endoscopic minimally invasive devices. Progress in the development of minimally invasive biomarkers for EAC and for predicting the risk for EAC in BE patients has also been made but no markers to date have been validated for use in clinical care. There is great promise that the next decade will see the advent of this next generation of BE screening assays in the clinic and that well validated assays for the detection of EAC will be determined. The EDRN and its investigators has played and will undoubtedly continue to play a central role in BE and EAC biomarker research.
Acknowledgements:
These studies were supported by funding from the NIH:UO1CA152756 (WMG), RO1CA194663, P30CA015704, U54CA163060 (WMG), UO1CA086402, UO1CA182940 to WMG, P50CA150964 (SDM) and UO1CA152756 to SDM, R50CA233042 to MY, and U54CA163060 and P30DK097948 to AC. Funding is also provided by the Cottrell Family Fund and Listwin Foundation to WMG.
Footnotes
Conflicts of Interest:
W. M. Grady is an advisory board member for Freenome, Guardant Health, and SEngine. He is an investigator in a clinical trial sponsored by Janssen Pharmaceuticals. S. D. Markowitz receives income related to patents licensed to Exact Sciences, has founders shares and stock options in LucidDx, serves on the board of directors of LucidDx, serves as a consultant to LucidDx, has sponsored research with LucidDx, and has a royalty interest in patents licensed to LucidDx. A. Chak is an equity holder and advisor for Lucid Diagnostics, consultant for Interpace Diagnostics, nd receives research support from C2Therapeutics/Pentax Inc.
References
- 1.Reid BJ. Early events during neoplastic progression in Barrett’s esophagus. Cancer Biomark 2010;9:307–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.American Gastroenterological A, Spechler SJ, Sharma P, et al. American Gastroenterological Association medical position statement on the management of Barrett’s esophagus. Gastroenterology 2011;140:1084–91. [DOI] [PubMed] [Google Scholar]
- 3.Fitzgerald RC, di Pietro M, Ragunath K, et al. British Society of Gastroenterology guidelines on the diagnosis and management of Barrett’s oesophagus. Gut 2014;63:7–42. [DOI] [PubMed] [Google Scholar]
- 4.Reid BJ, Li X, Galipeau PC, et al. Barrett’s oesophagus and oesophageal adenocarcinoma: time for a new synthesis. Nat Rev Cancer 2010;10:87–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Solaymani-Dodaran M, Card TR, West J. Cause-specific mortality of people with Barrett’s esophagus compared with the general population: a population-based cohort study. Gastroenterology 2013;144:1375–83, 1383 e1. [DOI] [PubMed] [Google Scholar]
- 6.Sharma P Clinical practice. Barrett’s esophagus. N Engl J Med 2009;361:2548–56. [DOI] [PubMed] [Google Scholar]
- 7.Pennathur A, Gibson MK, Jobe BA, et al. Oesophageal carcinoma. Lancet 2013;381:400–12. [DOI] [PubMed] [Google Scholar]
- 8.Dulak AM, Stojanov P, Peng S, et al. Exome and whole-genome sequencing of esophageal adenocarcinoma identifies recurrent driver events and mutational complexity. Nat Genet 2013;45:478–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu N, Wang C, Ng D, et al. Genomic characterization of esophageal squamous cell carcinoma from a high-risk population in China. Cancer Res 2009;69:5908–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paulson TG, Maley CC, Li X, et al. Chromosomal instability and copy number alterations in Barrett’s esophagus and esophageal adenocarcinoma. Clin Cancer Res 2009;15:3305–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stachler MD, Taylor-Weiner A, Peng S, et al. Paired exome analysis of Barrett’s esophagus and adenocarcinoma. Nat Genet 2015;47:1047–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weaver JM, Ross-Innes CS, Shannon N, et al. Ordering of mutations in preinvasive disease stages of esophageal carcinogenesis. Nat Genet 2014;46:837–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaz AM, Grady WM. Epigenetic biomarkers in esophageal cancer. Cancer Lett 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shah AK, Saunders NA, Barbour AP, et al. Early diagnostic biomarkers for esophageal adenocarcinoma--the current state of play. Cancer Epidemiol Biomarkers Prev 2013;22:1185–209. [DOI] [PubMed] [Google Scholar]
- 15.Yu M, Maden SK, Stachler M, et al. Subtypes of Barrett’s oesophagus and oesophageal adenocarcinoma based on genome-wide methylation analysis. Gut 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaz AM, Wong CJ, Varadan V, et al. Global DNA methylation patterns in Barrett’s esophagus, dysplastic Barrett’s, and esophageal adenocarcinoma are associated with BMI, gender, and tobacco use. Clin Epigenetics 2016;8:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Curtius K, Wong CJ, Hazelton WD, et al. A Molecular Clock Infers Heterogeneous Tissue Age Among Patients with Barrett’s Esophagus. PLoS Comput Biol 2016;12:e1004919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Salam I, Hussain S, Mir MM, et al. Aberrant promoter methylation and reduced expression of p16 gene in esophageal squamous cell carcinoma from Kashmir valley: a high-risk area. Mol Cell Biochem 2009;332:51–8. [DOI] [PubMed] [Google Scholar]
- 19.Kuester D, El-Rifai W, Peng D, et al. Silencing of MGMT expression by promoter hypermethylation in the metaplasia-dysplasia-carcinoma sequence of Barrett’s esophagus. Cancer Lett 2009;275:117–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Song JH, Meltzer SJ. MicroRNAs in pathogenesis, diagnosis, and treatment of gastroesophageal cancers. Gastroenterology 2012;143:35–47 e2. [DOI] [PubMed] [Google Scholar]
- 21.Wu X, Ajani JA, Gu J, et al. MicroRNA expression signatures during malignant progression from Barrett’s esophagus to esophageal adenocarcinoma. Cancer Prev Res (Phila) 2013;6:196–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Braun CJ, Zhang X, Savelyeva I, et al. p53-Responsive micrornas 192 and 215 are capable of inducing cell cycle arrest. Cancer Res 2008;68:10094–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Komatsu S, Ichikawa D, Takeshita H, et al. Circulating microRNAs in plasma of patients with oesophageal squamous cell carcinoma. Br J Cancer 2011;105:104–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kurashige J, Kamohara H, Watanabe M, et al. Serum microRNA-21 is a novel biomarker in patients with esophageal squamous cell carcinoma. Journal of Surgical Oncology 2012;106:188–192. [DOI] [PubMed] [Google Scholar]
- 25.Bansal A, Fitzgerald RC. Biomarkers in Barrett’s Esophagus: Role in Diagnosis, Risk Stratification, and Prediction of Response to Therapy. Gastroenterol Clin North Am 2015;44:373–390. [DOI] [PubMed] [Google Scholar]
- 26.Paulson TG, Reid BJ. Focus on Barrett’s esophagus and esophageal adenocarcinoma. Cancer Cell 2004;6:11–6. [DOI] [PubMed] [Google Scholar]
- 27.Su Z, Gay LJ, Strange A, et al. Common variants at the MHC locus and at chromosome 16q24.1 predispose to Barrett’s esophagus. Nat Genet 2012;44:1131–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luo C, Tao Y, Zhang Y, et al. Regulatory network analysis of high expressed long non-coding RNA LINC00941 in gastric cancer. Gene 2018;662:103–109. [DOI] [PubMed] [Google Scholar]
- 29.Galipeau PC, Li X, Blount PL, et al. NSAIDs modulate CDKN2A, TP53, and DNA content risk for progression to esophageal adenocarcinoma. PLoS Med 2007;4:e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bird-Lieberman EL, Dunn JM, Coleman HG, et al. Population-based study reveals new risk-stratification biomarker panel for Barrett’s esophagus. Gastroenterology 2012;143:927–35 e3. [DOI] [PubMed] [Google Scholar]
- 31.Rygiel AM, Milano F, Ten Kate FJ, et al. Assessment of chromosomal gains as compared to DNA content changes is more useful to detect dysplasia in Barrett’s esophagus brush cytology specimens. Genes Chromosomes Cancer 2008;47:396–404. [DOI] [PubMed] [Google Scholar]
- 32.Brankley SM, Halling KC, Jenkins SM, et al. Fluorescence in situ hybridization identifies high risk Barrett’s patients likely to develop esophageal adenocarcinoma. Dis Esophagus 2016;29:513–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reid BJ, Blount PL, Rubin CE, et al. Flow-cytometric and histological progression to malignancy in Barrett’s esophagus: prospective endoscopic surveillance of a cohort. Gastroenterology 1992;102:1212–9. [PubMed] [Google Scholar]
- 34.Maley CC, Galipeau PC, Finley JC, et al. Genetic clonal diversity predicts progression to esophageal adenocarcinoma. Nat Genet 2006;38:468–73. [DOI] [PubMed] [Google Scholar]
- 35.Moinova H, Leidner RS, Ravi L, et al. Aberrant vimentin methylation is characteristic of upper gastrointestinal pathologies. Cancer Epidemiol Biomarkers Prev 2012;21:594–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moinova HR, LaFramboise T, Lutterbaugh JD, et al. Identifying DNA methylation biomarkers for non-endoscopic detection of Barrett’s esophagus. Sci Transl Med 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu M, O’Leary RM, Kaz AM, et al. Methylated B3GAT2 and ZNF793 Are Potential Detection Biomarkers for Barrett’s Esophagus. Cancer Epidemiol Biomarkers Prev 2015;24:1890–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Z, Kambhampati S, Cheng Y, et al. Methylation Biomarker Panel Performance in EsophaCap Cytology Samples for Diagnosing Barrett’s Esophagus: A Prospective Validation Study. Clin Cancer Res 2019;25:2127–2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lao-Sirieix P, Boussioutas A, Kadri SR, et al. Non-endoscopic screening biomarkers for Barrett’s oesophagus: from microarray analysis to the clinic. Gut 2009;58:1451–9. [DOI] [PubMed] [Google Scholar]
- 40.Kadri SR, Lao-Sirieix P, O’Donovan M, et al. Acceptability and accuracy of a non-endoscopic screening test for Barrett’s oesophagus in primary care: cohort study. BMJ;341:c4372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chettouh H, Mowforth O, Galeano-Dalmau N, et al. Methylation panel is a diagnostic biomarker for Barrett’s oesophagus in endoscopic biopsies and non-endoscopic cytology specimens. Gut 2018;67:1942–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cancer Genome Atlas Research N, Analysis Working Group: Asan U, Agency BCC, et al. Integrated genomic characterization of oesophageal carcinoma. Nature 2017;541:169–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Spechler SJ. Barrett esophagus and risk of esophageal cancer: a clinical review. JAMA 2013;310:627–36. [DOI] [PubMed] [Google Scholar]
- 44.Chiba T, Marusawa H, Ushijima T. Inflammation-associated cancer development in digestive organs: mechanisms and roles for genetic and epigenetic modulation. Gastroenterology 2012;143:550–63. [DOI] [PubMed] [Google Scholar]
- 45.Schulmann K, Sterian A, Berki A, et al. Inactivation of p16, RUNX3, and HPP1 occurs early in Barrett’s-associated neoplastic progression and predicts progression risk. Oncogene 2005;24:4138–48. [DOI] [PubMed] [Google Scholar]
- 46.Sato F, Jin Z, Schulmann K, et al. Three-Tiered Risk Stratification Model to Predict Progression in Barrett’s Esophagus Using Epigenetic and Clinical Features. PLoS One 2008;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jin Z, Cheng Y, Gu W, et al. A multicenter, double-blinded validation study of methylation biomarkers for progression prediction in Barrett’s esophagus. Cancer Res 2009;69:4112–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maru DM, Singh RR, Hannah C, et al. MicroRNA-196a Is a Potential Marker of Progression during Barrett’s Metaplasia-Dysplasia-Invasive Adenocarcinoma Sequence in Esophagus. American Journal of Pathology 2009;174:1940–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Luzna P, Gregar J, Uberall I, et al. Changes of microRNAs-192, 196a and 203 correlate with Barrett’s esophagus diagnosis and its progression compared to normal healthy individuals. Diagn Pathol 2011;6:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Revilla-Nuin B, Parrilla P, Lozano JJ, et al. Predictive value of MicroRNAs in the progression of barrett esophagus to adenocarcinoma in a long-term follow-up study. Ann Surg 2013;257:886–93. [DOI] [PubMed] [Google Scholar]
- 51.Leidner RS, Fu P, Clifford B, et al. Genetic abnormalities of the EGFR pathway in African American Patients with non-small-cell lung cancer. J Clin Oncol 2009;27:5620–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bansal A, Lee IH, Hong X, et al. Feasibility of mcroRNAs as biomarkers for Barrett’s Esophagus progression: a pilot cross-sectional, phase 2 biomarker study. Am J Gastroenterol 2011;106:1055–63. [DOI] [PubMed] [Google Scholar]
- 53.Smith CM, Watson DI, Leong MP, et al. miR-200 family expression is downregulated upon neoplastic progression of Barrett’s esophagus. World J Gastroenterol 2011;17:1036–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fassan M, Volinia S, Palatini J, et al. MicroRNA expression profiling in human Barrett’s carcinogenesis. Int J Cancer 2011;129:1661–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Leidner RS, Ravi L, Leahy P, et al. The microRNAs, MiR-31 and MiR-375, as candidate markers in Barrett’s esophageal carcinogenesis. Genes Chromosomes Cancer 2012;51:473–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li X, Galipeau PC, Paulson TG, et al. Temporal and spatial evolution of somatic chromosomal alterations: a case-cohort study of Barrett’s esophagus. Cancer Prev Res (Phila) 2014;7:114–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kerkhof M, Steyerberg EW, Kusters JG, et al. Aneuploidy and high expression of p53 and Ki67 is associated with neoplastic progression in Barrett esophagus. Cancer Biomark 2008;4:1–10. [DOI] [PubMed] [Google Scholar]
- 58.Sikkema M, Kerkhof M, Steyerberg EW, et al. Aneuploidy and overexpression of Ki67 and p53 as markers for neoplastic progression in Barrett’s esophagus: a case-control study. Am J Gastroenterol 2009;104:2673–80. [DOI] [PubMed] [Google Scholar]
- 59.Critchley-Thorne RJ, Duits LC, Prichard JW, et al. A Tissue Systems Pathology Assay for High-Risk Barrett’s Esophagus. Cancer Epidemiol Biomarkers Prev 2016;25:958–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Reid BJ, Prevo LJ, Galipeau PC, et al. Predictors of progression in Barrett’s esophagus II: baseline 17p (p53) loss of heterozygosity identifies a patient subset at increased risk for neoplastic progression. Am J Gastroenterol 2001;96:2839–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rygiel AM, van Baal JW, Milano F, et al. Efficient automated assessment of genetic abnormalities detected by fluorescence in situ hybridization on brush cytology in a Barrett esophagus surveillance population. Cancer 2007;109:1980–8. [DOI] [PubMed] [Google Scholar]
- 62.Brankley SM, Wang KK, Harwood AR, et al. The development of a fluorescence in situ hybridization assay for the detection of dysplasia and adenocarcinoma in Barrett’s esophagus. J Mol Diagn 2006;8:260–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Streppel MM, Lata S, DelaBastide M, et al. Next-generation sequencing of endoscopic biopsies identifies ARID1A as a tumor-suppressor gene in Barrett’s esophagus. Oncogene 2014;33:347–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kandoth C, McLellan MD, Vandin F, et al. Mutational landscape and significance across 12 major cancer types. Nature 2013;502:333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Matsuzaki J, Suzuki H. Circulating microRNAs as potential biomarkers to detect transformation of Barrett’s oesophagus to oesophageal adenocarcinoma. BMJ Open Gastroenterol 2017;4:e000160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Buas MF, Gu H, Djukovic D, et al. Candidate serum metabolite biomarkers for differentiating gastroesophageal reflux disease, Barrett’s esophagus, and high-grade dysplasia/esophageal adenocarcinoma. Metabolomics 2017;13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kelly BD, Miller N, Healy NA, et al. A review of expression profiling of circulating microRNAs in men with prostate cancer. BJU Int 2013;111:17–21. [DOI] [PubMed] [Google Scholar]
- 68.Chiam K, Wang T, Watson DI, et al. Circulating Serum Exosomal miRNAs As Potential Biomarkers for Esophageal Adenocarcinoma. J Gastrointest Surg 2015;19:1208–15. [DOI] [PubMed] [Google Scholar]
- 69.Qin Y, Wu CW, Taylor WR, et al. Discovery, Validation, and Application of Novel Methylated DNA Markers for Detection of Esophageal Cancer in Plasma. Clin Cancer Res 2019;25:7396–7404. [DOI] [PMC free article] [PubMed] [Google Scholar]