

Introduction
Tubulointerstitial nephritis (TIN) is frequently encountered as a cause of acute kidney injury, and can be attributed to a variety of causes, including medications, autoimmune disease, infection, glomerular disease, metabolic disease, and others.1 Although diseases of immune complex deposition affecting the glomeruli are broadly recognized, immune complex deposition in the tubular basement membranes (TBMs) is usually assumed to be secondary to a glomerulocentric process, such as lupus nephritis.2 However, a variety of autoimmune and even infectious processes, including anti-LRP2 nephropathy/anti-brush border antibody TIN (ABBA-TIN),3,4 IgG4-related TIN,5 idiopathic hypocomplementemic TIN,6 drug-induced immune complex TIN,7,8 and polyomavirus nephropathy,9 can present primarily with TBM immune complex deposition and manifest histologically as TIN. Further complicating the issue is that some of these disorders can have modest glomerular immune complex deposition, which can act as a “red herring” and suggest a glomerulocentric process with secondary TBM deposits. This report details a case of anti-LRP2 nephropathy resulting in end-stage kidney disease, and highlights the diagnostic difficulty faced by nephrologists and pathologists in the identification of this rare but likely underrecognized disease.
Case Presentation
Clinical History and Initial Laboratory Data
A 76-year-old Caucasian male with long-standing diabetes (HbA1c 7.0%–8.5%), hypertension, and stage IV chronic kidney disease presented to his nephrologist with a serum creatinine of 8 mg/dl, which increased from a baseline of 2.7 mg/dl 6 months earlier. He had no history of autoimmune disease. His chronic kidney disease was presumptively attributed to diabetic nephropathy and arterionephrosclerosis and he had never undergone a kidney biopsy. There was no known history of nephrotoxic medication usage, including nonsteroidal anti-inflammatory drugs. He was sent to the hospital and was found to have proteinuria of 1.1 g per 24 hours. Additional studies demonstrated a positive antineutrophil antibody titer (1:160). Anti-double-stranded DNA was not performed, but complement levels were normal and there was no clinical evidence of systemic lupus erythematosus. The patient was started on dialysis. A kidney biopsy was performed.
Kidney Biopsy
Light microscopy showed a total of 11 glomeruli, of which 4 glomeruli (36%) were globally sclerosed. The remaining glomeruli showed ischemic changes, including wrinkling of the capillary tuft and periglomerular fibrosis. The glomeruli showed no mesangial or endocapillary hypercellularity, and no subepithelial “spikes” were identified on Jones silver stain. There was no significant mesangial sclerosis. The proximal tubules showed extensive attenuation with loss of the brush borders. Patchy mild interstitial inflammation was present, but plasma cells were not prominent. There was diffuse and frequently marked interstitial fibrosis and tubular atrophy (Figure 1a). The proximal TBMs exhibited significant thickening, imparting an atrophic appearance on initial light microscopic evaluation (Figure 1b). The arteries showed moderate intimal fibrosis, and the arterioles exhibited diffuse arteriolar hyalinosis.
Figure 1.

(a) On low power, the kidney biopsy showed extensive attenuation of the proximal tubules with loss of the brush borders, patchy mild interstitial inflammation, thickening of the tubular basement membranes, and diffuse and frequently marked interstitial fibrosis and tubular atrophy (periodic acid–Schiff [PAS] stain, original magnification ×100). (b) On higher power, the thickened proximal tubular basement membranes resemble atrophic changes, but exhibit an atypical “smudgy” appearance. The proximal tubular epithelial cells demonstrate severe tubular injury with loss of brush borders (PAS stain, original magnification ×400).
By immunofluorescence microscopy, there was diffuse granular to confluent TBM deposits staining for IgG (Figure 2a), C3, and kappa and lambda light chains. The predominant IgG subtype was IgG4, but small amounts of IgG1 and IgG2 were also present. In addition, a few glomeruli showed segmental granular capillary wall staining for IgG (1+) and kappa (1+) and lambda (1+) light chains (Figure 2b), but many glomeruli were negative. Interstitial plasma cells were not prominent and no cytoplasmic IgG4 staining of plasma cells was noted.
Figure 2.

(a) The proximal tubular basement membranes exhibit diffuse “smudgy” immunofluorescence staining for IgG. (b) A few glomeruli showed segmental granular capillary wall immunofluorescence staining for IgG. (c) Conspicuous electron-dense deposits were present in the proximal tubular basement membranes by electron microscopy. (d) Segmental subepithelial electron-dense deposits with intervening glomerular basement membrane “spikes” were present by electron microscopy.
Electron microscopy showed numerous large collections of electron-dense deposits in the proximal TBMs without an identifiable substructure (Figure 2c). The distal tubules were spared. Interstitial or vascular deposits were also not identified. Two glomeruli demonstrated segmental subepithelial electron-dense deposits with intervening glomerular basement membrane “spikes” (Figure 2d). A few vague mesangial deposits were also identified. The podocyte foot processes showed focal effacement, but most remained preserved.
Additional Studies
Given the preceding findings suggestive of an immune complex tubulointerstitial nephritis, additional studies were performed to test for the presence of ABBAs. A full description of these methods is available in the Supplementary Methods. Of note, there was not sufficient biopsy tissue remaining to perform LRP2 immunofluorescence staining.
ABBA Detection by Indirect Immunofluorescence
Indirect immunofluorescence microscopy demonstrated the presence of serum IgG antibodies reactive against the brush border of normal human kidney (titer: 1:640), confirming the presence of serum anti-brush border antibodies (Figure 3a).
Figure 3.

(a) Indirect immunofluorescence shows the patient’s serum staining the renal tubular brush border in a frozen section of normal human kidney (×100). (b–d) Colocalization of the patient’s serum IgG (b, green) and anti-LRP2 (c, red) double stained by multistep indirect immunofluorescence on frozen sections of normal human kidney tissue. Confocal microscopy of merged image demonstrates colocalization of IgG and LRP2 (d, orange).
Colocalization of LRP2 and ABBA IgG
Normal human kidney was incubated with both patient serum and an antibody against the C-terminal portion of the human LRP2 protein in a multistep indirect immunofluorescence assay and observed by confocal microscopy, which confirmed LRP2 as the target of the anti-brush border antibodies (Figure 3b–d).
ABBA Immunoprecipitation
Immunoprecipitation of human tubular extract with patient serum, ABBA-positive serum, and normal human control serum was performed. Blotting with anti-LRP2 antibody demonstrated the presence of LRP2 in the patient and ABBA-positive samples, which was absent in the normal human sample (Figure 4).
Figure 4.
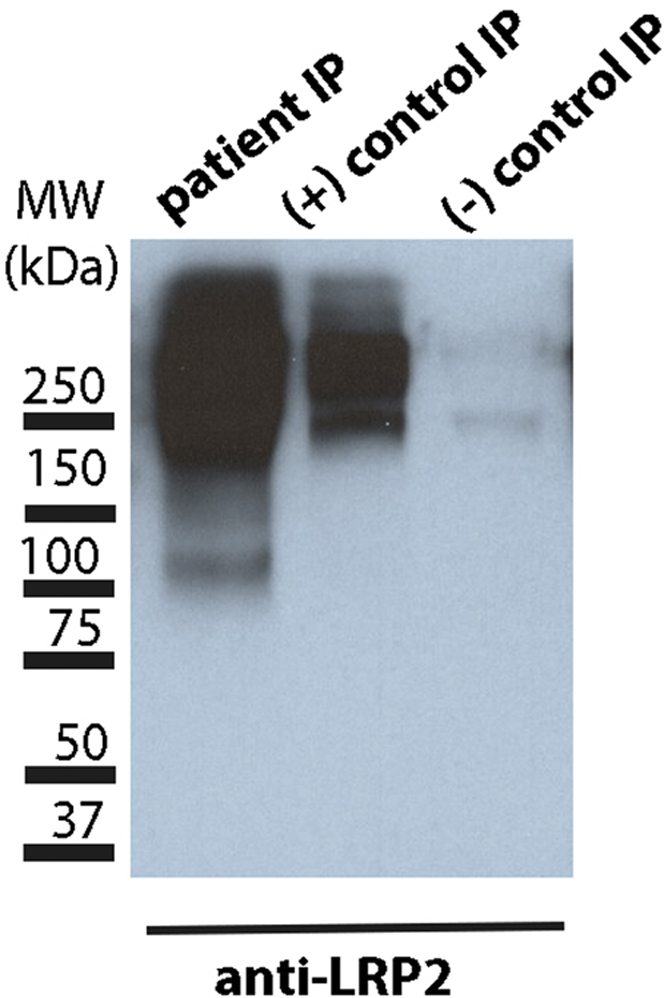
Immunoprecipitate (IP) with human tubular extract (HTE) and patient serum (lane 1), positive ABBA serum (lane 2), and normal human serum (lane 3), run on polyacrylamide gel electrophoresis and blotted for anti-LRP2. Both patient serum and positive control pulled down LRP2 from HTE compared with normal human serum.
Clinical Follow-Up
The patient remains on dialysis without recovery of renal function.
Discussion
In comparison with glomerular immune complex deposition, TBM immune complex deposition is much less frequently encountered in the renal biopsy. Most TBM immune complex deposition is observed in the setting of lupus nephritis, which typically manifests with predominant glomerular immune complexes with or without TBM, vascular, and interstitial immune complexes. Given the widespread immune complex deposition across the different compartments of the kidney, “full-house” immunofluorescence staining, and characteristic clinical and laboratory findings, differentiating lupus nephritis from other causes of TBM immune complex deposition is generally straightforward, but lupus with predominant TBM immune complex deposition (largely sparing the glomeruli) has occasionally been described.2,S1–S3 When immune complexes are present either solely or disproportionately in the TBMs, as in our patient, it raises the possibility of a number of uncommon immune complex TINs.
IgG4-related TIN occurs in the setting of systemic IgG4-related disease, a steroid-sensitive autoimmune condition characterized by storiform fibrosis and IgG4-dominant plasma cell–rich inflammation in multiple organ systems.S4,S5 Immunofluorescence demonstrates IgG4+ plasma cells and granular TBM immune complex deposition for IgG and C3 in the proximal and distal convoluted tubules. A subset of patients also has concomitant phospholipase A2 receptor–negative membranous nephropathy demonstrating IgG4-dominant subepithelial deposits.5,S6
Idiopathic hypocomplementemic TIN occurs predominantly in older men with low serum complement levels and renal dysfunction but no evidence of lupus.6,S7 Immunofluorescence shows granular deposits of IgG and often C3 in the TBMs and interstitium. There may be overlap with IgG4-related TIN, but glomeruli are not involved, and a mass-forming lesion is not present, unlike many cases of IgG4-related TIN. Drug-induced immune complex TIN is extremely rare but has been described in the setting of nonsteroidal anti-inflammatory drug use and shows prominent interstitial inflammation with granular IgG and C3 TBM immune complexes.8 Relatedly, so-called “giant cell tubulitis with TBM deposits” has been described in association with aprotinin, a drug used to reduce bleeding during cardiac and liver surgeries, and shows similar findings with the addition of multinucleated giant cells cuffing TBMs.7 Polyomavirus nephropathy also occasionally shows focal granular deposits of IgG, C3, and C4d in TBMs.9
Our patient did not have storiform fibrosis, plasma cell–rich interstitial inflammation, and the immune complex deposition was limited to the proximal tubules, making the diagnosis of IgG4-related TIN unlikely. The presence of glomerular immune complex deposition; normal complement levels; and the absence of relevant medication, transplantation, or immunosuppression history led us to exclude idiopathic hypocomplementemic TIN, drug-induced immune complex TIN, and polyomavirus nephropathy. Ruling out these disorders led us to favor a diagnosis of anti-LRP2 nephropathy, which was subsequently confirmed by indirect immunofluorescence demonstrating the presence of serum ABBAs colocalizing with LRP2 and by the ability of the patient’s serum, but not control serum, to immunoprecipitated LRP2 from an extract of human tubular proteins.
ABBA-TIN has rarely been described in the literature, with the first report published by Rosales et al.3 in 2016, and a series of 10 patients was added by Larsen et al.4 in 2018 in which LRP2 was first identified as the target antigen. Only a few additional case reports of this entity have since been published, including a case of anti-LRP2 nephropathy with abundant IgG4-positive plasma cells and 2 cases associated with lymphomatous infiltrates within the kidney.S8–S11 Also, 2 case reports from 1981 describe patients with immune complex TIN, mild glomerular subepithelial immune complex deposition, and antibodies against brush border proteins, likely representing ABBA-TIN.S12,S13 The disease occurs predominantly in elderly Caucasian men, manifesting with progressive renal insufficiency, bland urine sediment, and normal complement levels.
The renal pathology in ABBA-TIN is characterized by diffuse proximal tubular injury with loss of brush borders, thickening of the proximal TBMs, and a variable degree of mixed interstitial inflammation without prominent numbers of plasma cells. The degree of interstitial fibrosis and tubular atrophy is frequently severe. The glomeruli may be normal or may show segmental thickening and/or subepithelial “spikes” on Jones stain. Immunofluorescence shows prominent and often confluent granular proximal TBMs deposits for C3 and IgG (frequently the IgG4 subtype). C1q staining is negative. The glomeruli may show segmental granular capillary wall deposits for IgG and C3. Electron microscopy demonstrates electron-dense deposits in the proximal TBMs with sparing of the distal tubules. Glomerular subepithelial immune complex deposits may be present in a segmental distribution, which could also result in a misleading diagnosis of membranous nephropathy.
Of the 16 reported cases of ABBA-TIN in the contemporary literature, 9 developed end-stage kidney disease or died within a few months of presentation. One patient achieved stabilization of serum creatinine, resolution of proteinuria, and decrease in ABBA titer after 12 months of treatment with prednisone and cyclophosphamide.4 Interestingly, the patient described by Rosales et al.3 underwent renal transplantation 4 years after the initial diagnosis of ABBA-TIN, and developed biopsy- and serology-proven recurrence of ABBA-TIN only 7 weeks later. Subsequent plasma exchange and immunosuppressive treatment resulted in a decreased ABBA titer, but the creatinine continued to increase and a repeat allograft biopsy showed worsening of interstitial inflammation and interstitial fibrosis.
Larsen et al.4 convincingly identified LRP2, also known as megalin or gp330, as the specific brush border antigen that is targeted in ABBA-TIN. In the kidney, megalin is localized to the apical microvillous border of proximal tubular epithelial cells and plays an important role in protein reabsorption.S14 Interestingly, megalin was previously implicated as the autoantigenic target in a well-known rat model of human membranous nephropathy (Heymann nephritis)S15,S16; however, megalin is not expressed by podocytes in humans and is not generally believed to play a role in human membranous nephropathy.S14,S17 The anti-LRP2/megalin antibodies targeting the proximal tubular brush borders in ABBA-TIN are thought to induce tubular damage. The mechanism of immune complex deposition in the basally located TBMs is not well-understood, but could be related to tubular reabsorption of the proteins and return through the basolateral aspect of the cells. The presence of sparse glomerular subepithelial deposits, which do not stain for LRP2, is also poorly understood. However, the identification LRP2/megalin is significant, because serum levels can conceivably be measured and followed to assess response to treatment and detect recurrence. To our knowledge, no cases of LRP2-negative ABBA-TIN have been reported, but it is possible that antibodies against other brush border antigens, such as cubilin, which is also expressed on human podocytes, could be present in some cases.
Further investigation is needed to better elucidate the mechanistic underpinnings and clinicopathologic behavior of anti-LRP2. Larger studies assessing the presence or absence of ABBAs in other diseases would be helpful in understanding if these autoantibodies alone are sufficient to cause disease, or whether an additional trigger is required. Given the similarities in immune complex localization and IgG subtype, could a subset of IgG4-related TIN could actually represent ABBA-TIN in which serum ABBAs or anti-LRP2 staining have not been assessed? Seventy-nine percent of IgG4-related TIN cases are associated with extrarenal manifestations,S18 which contrasts with ABBA-TIN, but it is possible that some of the remaining 21% truly represent ABBA-TIN. Although LRP2 staining was negative in 7 cases of IgG4-related TIN used as controls by Larsen et al.,4 further investigation is needed in a larger sample of IgG4-related TIN cases. ABBAs have rarely been described in other diseases, such as membranous nephropathyS19 and inflammatory bowel disease.S20,S21 Autoantibodies to LRP2 have been described in autoimmune thyroiditisS22 and multiple rheumatic diseases, including rheumatoid arthritis, systemic lupus erythematosus, systemic sclerosis, osteoarthritis, and Behcet’s disease,S23 but not all of these patients had renal dysfunction and the renal tissue was not biopsied. More thorough investigation of ABBAs in other nonrenal tissues also would be of interest.
In conclusion, we report a case of anti-LRP2 nephropathy in a patient with end-stage kidney disease. Immune complex TIN is rare outside of lupus nephritis but is important to recognize and classify correctly, as the underlying etiologies have various natural histories, treatments, and propensity to recur in potential future allografts (Table 1). Anti-LRP2 nephropathy is likely underrecognized both clinically and during biopsy interpretation.
Table 1.
Key teaching points
|
|
|
|
ABBA, anti-brush border antibody; TBM, Tubular basement membrane; TIN, tubular interstitial nephritis.
Disclosure
All the authors declared no competing interests.
Footnotes
Supplementary Material
References
- 1.Raghavan R., Eknoyan G. Acute interstitial nephritis - a reappraisal and update. Clin Nephrol. 2014;82:149–162. doi: 10.5414/CN108386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Park M.H., D'Agati V., Appel G.B. Tubulointerstitial disease in lupus nephritis:relationship to immune deposits, interstitial inflammation, glomerular changes, renal function, and prognosis. Nephron. 1986;44:309–319. doi: 10.1159/000184012. [DOI] [PubMed] [Google Scholar]
- 3.Rosales I.A., Collins A.B., do Carmo P.A. Immune Complex tubulointerstitial nephritis due to autoantibodies to the proximal tubule brush border. J Am Soc Nephrol. 2016;27:380–384. doi: 10.1681/ASN.2015030334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Larsen C.P., Trivin-Avillach C., Coles P. LDL receptor-related protein 2 (megalin) as a target antigen in human kidney anti-brush border antibody disease. J Am Soc Nephrol. 2018;29:644–653. doi: 10.1681/ASN.2017060664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cornell L.D. IgG4-related kidney disease. Curr Opin Nephrol Hypertens. 2012;21:279–288. doi: 10.1097/MNH.0b013e32835265ac. [DOI] [PubMed] [Google Scholar]
- 6.Kambham N., Markowitz G.S., Tanji N. Idiopathic hypocomplementemic interstitial nephritis with extensive tubulointerstitial deposits. Am J Kidney Dis. 2001;37:388–399. doi: 10.1053/ajkd.2001.21320. [DOI] [PubMed] [Google Scholar]
- 7.Chang A., Peutz-Kootstra C.J., Kowalewska J. Giant cell tubulitis with tubular basement membrane immune deposits: a report of two cases after cardiac valve replacement surgery. Clin J Am Soc Nephrol. 2006;1:920–924. doi: 10.2215/CJN.02201205. [DOI] [PubMed] [Google Scholar]
- 8.Dixit M.P., Nguyen C., Carson T. Non-steroidal anti-inflammatory drugs-associated acute interstitial nephritis with granular tubular basement membrane deposits. Pediatr Nephrol. 2008;23:145–148. doi: 10.1007/s00467-007-0585-0. [DOI] [PubMed] [Google Scholar]
- 9.Bracamonte E., Leca N., Smith K.D. Tubular basement membrane immune deposits in association with BK polyomavirus nephropathy. Am J Transplant. 2007;7:1552–1560. doi: 10.1111/j.1600-6143.2007.01794.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.