

Abstract
The intrinsic fluorescence properties of two related pyrido[1,2-a]benzimidazole antimalarial compounds suitable for cellular imaging of the human malaria parasite Plasmodium falciparum without the need to attach extrinsic fluorophores are described. Although these compounds are structurally related, they have been shown by confocal microscopy to not only accumulate selectively within P. falciparum but to also accumulate differently in the organelles investigated. Localization to the digestive vacuole and nearby neutral lipids were observed for compound 2 which was shown to inhibit hemozoin formation using a cellular fractionation assay indicating that, this is a contributing mechanism of action. By contrast, compound 1, which differs from compound 2 by the replacement of the imidazole[l,2-a:4,5-b’]dipyridine core with the benzimidazole core as well as the presence of Cl substituents, shows very different localisation patterns and shows no evidence of hemozoin inhibition, suggesting a different mechanism of antimalarial action. Docking profiles of both compounds on the hemozoin surface further provided insight into their mechanisms of action.
Graphical Abstract
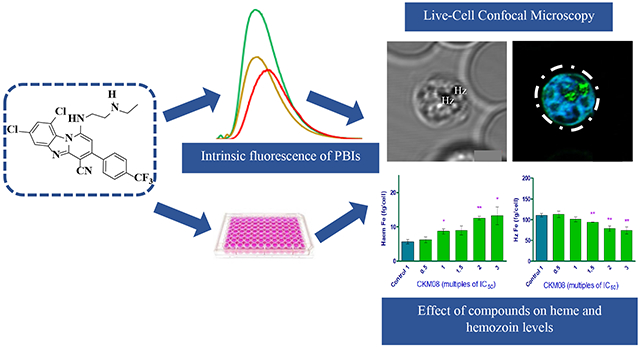
Intrinsic fluorescence properties, docking and a hemozoin inhibition assay were employed to study the mechanism of action of two structurally related pyrido-[1,2-a]benzimidazole derivatives.
Introduction
Malaria remains a major cause of morbidity and mortality globally. According to the 2019 World Health Organisation report, an estimated 228 million cases of malaria occurred worldwide, with the most affected being pregnant women and children under five years in Africa.1 Efforts to reduce transmission of the disease (such as the use of insecticide-treated bed nets) and the success of artemisinin-based combination therapies have helped reduce the incidence rate of malaria from 71 to 57 cases per 100 population at risk between 2010 to 2017.1 However, progress in combating the disease has recently stalled as a result of the emergence of the human malaria parasite Plasmodium falciparum resistance to available treatments. Consequently, there is an urgent need to fuel the antimalarial drug discovery pipeline with safe and efficacious drugs with novel modes of action and activity across multiple stages of the parasite’s life cycle.
Earlier, we identified pyrido[1,2-a]benzimidazole (PBI) compounds as a novel class of antimalarial agents.2, 3 Two representatives of this class of potent antimalarials, compounds 1 and 2 are depicted in Figure 1. Compound 1 showed good in vitro activity against P. falciparum sensitive (NF54) and multidrug resistant strains (K1) with IC50 values in the low nanomolar range.3 It also showed significant activity against late-stage gametocytes indicating the potential of this compound as a dual-acting antimalarial. Although compound 2 did not show activity in the nanomolar range, it showed moderate activity against both the sensitive and resistant strains of the parasite.3 Despite the fact that these compounds induced the desired phenotypic response (antiplasmodium activity) against the parasite, little is known about their mechanism of action (MoA) although inhibition of hemozoin formation was previously suggested as a contributing MoA for some related derivatives.3 While the phenotypic whole-cell screening approach to drug discovery has been relatively successful in delivering first-in-class drugs over the last decade4, it is burdened with challenges in the mechanistic deconvolution of hits and/or leads. However, the emergence of a variety of approaches and new technologies in chemical proteomics, genomics and fluorescent drug localization studies have aided in the identification and validation of relevant targets of phenotypic leads and understanding their MoA.5-8 The primary goal of chemical proteomics is to identify protein binding partners of bioactive compounds that serve as viable and druggable targets in drug discovery and development. This approach utilises integrated knowledge in chemistry, biology, structural biology, biochemistry and bioinformatics to obtain information about the binding site of bioactive molecules and to provide suitable starting points for the development of drug screening systems based on the structure of target proteins.9, 10 On the other hand, the genomics approach to target identification utilises the vast information obtained from the completion of the human genome sequencing project11 and the genome sequencing of different organisms, to understand complex biological systems of human diseases and to help identify and validate new targets.12, 13 one of the useful techniques in genomics, tailored to understanding the mechanism of resistance (MoR) and the MoA of small molecules is resistance selection studies. In case of Plasmodium falciparum, resistant mutants of the parasite are raised by their continuous exposure to large concentrations of the drug. Whole-genome sequencing then reveals genes that are mutated and by so doing provide insight into their MoR and also their MoA.14, 15 While these approaches have contributed successfully to studying the MoA of small molecules through target identification, fluorescent live-cell imaging tools over the years have provided unique insights into the effects of small molecules on many dynamic processes that occur in Plasmodium falciparum. This it does in intact cells under conditions that cause minimum disruptions to the parasite’s life processes. The major advantage of live-cell fluorescence imaging is its ability to differentiate between spatial signals from the parasite and those from the surrounding infected red blood cells such that, even subcellular organelles, including the nucleus and the parasite’s acidic digestive vacuole can be illuminated.16
Figure 1:

Chemical structures of 1 and 2. Representatives of the pyrido[1,2-a]benzimidazole (PBI) antimalarial compounds
In this study, we sought to utilise drug localization studies towards gaining insight into the MoA (s) of antimalarial PBIs exemplified by compounds 1 and 2. These compounds, which differ in substitutions on the left-hand side aromatic ring (Figure 1, Ring A), where the benzimidazole core in compound 1 in the presence of two Cl substituents is replaced by an imidazo[1,2-a:4,5-b’]dipyridine core in compound 2. These compounds were rationally selected from the series of compounds synthesized to date as they represent unique structural features and substitution patterns to facilitate probing the possibility of a mechanistic switch. As a starting point towards understanding the MoA of this class of compounds, we initially sought to employ drug localization studies to investigate the subcellular accumulation of the target compounds in P. falciparum. We hypothesized that a systematic investigation of their subcellular localization within the parasite would provide insight into the compounds’ MoA. Fluorescence is generally observed in highly conjugated polycyclic aromatic compounds that are capable of undergoing electronic transitions. This results in the intrinsic fluorescent properties of compounds. In the absence of this property, extrinsic fluorescent dyes are attached to the compounds to allow for their fluorescent imaging. While most fluorescent imaging protocols have required the derivatization of the target compounds by attaching extrinsic fluorophores,17, 18 there have been growing concerns that the presence of these extrinsic fluorophores may change the MoA of the compounds under study and hence interfere with their subcellular accumulation in the parasite even when they retain antiplasmodium activity. Also, attaching external fluorophores requires the need for structure activity relationship studies to ascertain the most appropriate site for attachment and results in the need for additional synthetic steps. Most fluorophores are either too expensive, too big or generally not suitable for the molecule under study, making it a daunting task to find the appropriate fluorophore suitable for each molecule. Thus the intrinsic fluorescent properties of the study compounds are advantageous in circumventing these drawbacks.
We describe, for the first time, the intrinsic fluorescent properties of compounds 1 and 2 suitable for the live-cellular imaging of P. falciparum. We observed using confocal microscopy intense localization of these compounds without causing any significant photodamage to the cells. We further performed docking experiments and a cellular heme fractionation assay to support our findings from the fluorescent localization studies.
Experimental Procedures
Reagents
Unless otherwise stated, all compounds were purchased from Sigma-Aldrich or Combi-Blocks and used without further purification. Solvents were generally purchased from Science World. Bovine hemin (Fe(III)PPIX-Cl) was obtained from Fluka. NP-40 was obtained from Pierce Biotechnology (Rockford, IL, USA). Deionized and purified water (dH2O) was provided by a Millipore Direct-Q3 water purification system. DRAQ 5, LysoTracker Red, ER-Tracker Red, and MitoTracker Deep Red were obtained from Molecular Probes (Oregon, USA) and later Thermo Fisher Scientific. Nile Red was purchased from Sigma-Aldrich. Absorption spectra were recorded on a Shimadzu UV-1800 spectrophotometer and were baseline-corrected while fluorescence spectra were measured using a Varian Cary Eclipse spectrofluorometer. Assay plates were read using a SpectraMax 340 PC 384 Absorbance Microplate Reader (Molecular Devices).
Fluorescence profiling of target compounds.
The fluorescence profile for both compounds was obtained using the Varian Cary Eclipse spectrofluorimeter in four different solvents, dichloromethane (DCM), dimethyl sulfoxide (DMSO), methanol and water. 5 μM solutions of both compounds were prepared, and their ultraviolet-visible profile obtained at 25 °C from which the absorption maxima and the molar extinction coefficients were obtained. The absorption maxima of each compound was used as a suitable wavelength for the excitation of the compounds, (Excitation at 420 nm) from which an emission profile was obtained. The effect of pH on the fluorescence intensity and wavelength of both compounds was assessed in 20 mM HEPES buffer at Ph 2.5 - 8.5.
Fluorescent Live-cell Imaging.
Nunc Lab-Tek II 8-well chamber slide (Thermo Fisher Scientific ) with no.1.5 cover glass was coated with a 150 μL poly-L-lysine solution for 10 mins after which, the excess solution was removed, and the chamber slide was air-dried. Human erythrocytes infected with P. falciparum chloroquine-sensitive strain (NF54) were cultured, according to Trager and Jensen.19 5 μL of the harvested parasitized erythrocyte pellets were resuspended in 5mL Ringer’s solution (pH 7.5). 100 μL aliquot of the suspended cells were placed in each well of the chamber slide and incubated for 30 mins to allow cells to adhere to the glass chamber. Excess Ringer’s solution was removed alongside all non-adhering parasitized red blood cells. The cells were washed twice with Ringer’s solution after which a new solution containing the appropriate concentration of drugs and organelle markers was added. Confocal microscopy was performed using a Zeiss laserscanning microscope (LSM880) with an Airyscan detector. Plan- Apochromat 63x/1.40 Oil DIC M27 objective lens was used to keep laser transmission as low as possible to prevent phototoxicity to the cells. Images were captured and processed using Zeiss ZEN software (Carl Zeiss Microscopy GmbH).
Parasite cellular heme speciation assay.
Ring stage PfNF54 parasites (5% parasitemia and 2% hematocrit) were synchronized and incubated with varying concentrations of compounds 1 and 2 as well as no compound, as a control, for 36h, after which the mature trophozoites were isolated by saponin lysis. The isolated trophozoites were resuspended in 100 μL phosphate-buffered saline (PBS) and accurately transferred to a round-bottomed, 96-well 0.5mL plate (Axygen Scientific) referred to as the “stock plate”. A “counting plate” was prepared by adding 10 μL of the resuspended trophozoites to a fluorescence-activated cell sorting (FACS) diluent (PBS pH 7.5 containing 0.125 % (v/v) glutaraldehyde and 0.5 % (v/v) DNase) to a final volume of 200 μL and refrigerated at 4 °C. Cell counts were analysed using flow cytometry on a Becton Dickinson (BD) AccuriTM C6 Plus using SSC/FL1530nm with BD AccuriTM C6 Plus software. In a flat-bottomed, 96-well plate samples were prepared by diluting 20 μL of the solution from the counting plate with 160 μL of 1 x SYBR Green I in PBS. To this, 20 μL of TrucountTM beads (BD) were added such that a fixed amount of beads were contained in a final volume of 200 μL. The plate was then incubated in the dark at 37 °C for 30 minutes and mixed well prior to reading on the flow cytometer. Typically, between 10 000 total events were counted for each sample. The concentration of cells in the wells were calculated according to the equation:
Where:
CF = concentration of cells per mL
T = number of trophozoites gated
B = number of TrucountTM beads gated
CB = concentration of fluorescent beads in each well per mL (calibrated bead count according to supplier stipulation)
The data acquired was analysed using FlowJo Software (V10).
The stock plate was stored at −20 °C. To determine the amount of hemoglobin, heme and hemozoin a series of cellular fractionation steps were involved. The samples in the stock plate were thawed to promote cell membrane lysis; to this, 100 μL of water was added, and the samples were sonicated for 5 mins. A solution of HEPES buffer (50 μL, 0.02 M, pH 7.5) was added and the sample centrifuged at 3600 rpm for 20 mins. The supernatant of the solution was collected and transferred to an adjacent set of wells on the same plate. To the supernatant, 50 μL of 4% SDS was added, sonicated for 5 min and incubated at room temperature for 30 minutes. Finally, to the supernatant, 50 μL of 0.3 M NaCl and 25% pyridine (v/v) in 0.2 M HEPES pH 7.5 were added and 200 μL of this transferred to a flat-bottomed, 96-well plate termed the “reading plate”. To the pellet remaining, 50 μL water, 50 μL 4 % SDS were added and resuspended well. The plate was then sonicated for 5 min and incubated at room temperature for 30 min to allow the free haem to be solubilised. Thereafter were added 50 μL 0.2 M HEPES pH 7.5, 50 μL 0.3M NaCl and 25 % pyridine and the plate was centrifuged at 3600 rpm for 20 min. The supernatant was transferred to an adjacent set of well on the same plate and diluted to 400 μL with water. Of this solution, 200 μL was transferred to the reading plate. The remaining pellet was treated with 50 μL of water and 0.3 M NaOH and sonicated for 15 minutes to solubilise the hemozoin. The plate was then incubated for 20 minutes at room temperature. Finally, 50 μL of 0.2 M HEPES buffer, 50 μL of 0.3 M HCl and 50 μL 25% pyridine solution were added, and the supernatant was diluted with water to a final volume of 400 μL. Of this, 200 μL was transferred to the reading plate. The UV-visible spectra of these fractions were recorded between 400 nm and 415 nm on a multi-well plate reader (Spectramax 340PC, Molecular Devices). Percentages of the three fractions; hemoglobin, heme and hemozoin fractions, were determined from the absorbance values. The total heme in each of these samples was quantified using a standard curve. GraphPad Prism (v5) was used to analyse the final data set and perform the significance tests.
Docking studies.
Compounds 1 and 2 were docked against the β-haematin surface using the previously published 3D crystal structure.20 The Fe-O bonds were treated as zero-order bonds to constrain the input geometry. The receptor grid encompassed the (100) and (001) faces of the crystal. The protonation states of both the crystal and the ligands were generated at pH 4.8 ± 0.5 using Epik. The ligands were minimized using Schrodinger’s OPLS3e force field. The output conformers were docked using Grid-based ligand docking with energetics (Glide extra precision). Epik state penalties were added to the docking score and intramolecular hydrogen bonds were rewarded. Planarity of conjugated π groups was also enhanced.
Results and Discussion
Fluorescence profiling of compounds 1 and 2 showed that both compounds possessed similar photophysical properties. Their absorption maxima were observed at 420 nm and fluorescence emission following excitation at this wavelength was observed at 480 nm. We attribute these fluorescent properties of the compounds to the extended conjugation ensuring a continuous delocalization of electrons from the benzimidazole core to the trifluoromethyl phenyl substituent. These compounds exhibited small solvatochromic shifts from 480-500 nm in their fluorescent emission over four solvents. The most intense fluorescence emission was observed in non-polar environments while intensity in aqueous solutions was relatively weaker. This is advantageous in detecting both neutral and phospholipid bilayers. At the same time, the moderate fluorescence intensity of these compounds in aqueous media implies that, should they accumulate in the digestive vacuole, their fluorescence signals would not overwhelm the detection of signals from other secondary sites of accumulation. Also, the fluorescence intensity of both compounds was significantly affected by the changes in pH. Fluorescence intensity was highest at pH 4.5 (Figure 2) where the compounds are fully protonated, and this also represents the biologically relevant pH in the digestive vacuole of Plasmodium falciparum.
Figure 2:

Effect of pH changes on the fluorescence emission of Compound 2, measured in 20 mM HEPES buffer at pH 2 - 8.5. Compound concentration was kept at 10 μ m
During the intraerythrocytic stage of the parasite’s life cycle, it degrades nearly all its host cell’s hemoglobin to obtain nutrients. This catabolic process occurs in the acidic, oxygen-rich, lysosome-like digestive vacuole (DV).21, 22 As a consequence of this process, large quantities of heme are released into the DV as a by-product. Due to the toxicity of heme to the survival of the parasite, it quickly sequesters the released heme by converting it into an insoluble and relatively unreactive microcrystalline dimer known as hemozoin.20, 23, 24 The heme detoxification pathway is a unique target for antimalarial chemotherapy, as demonstrated by the 4-aminoquinolines.25 This pathway is specific to the Plasmodium parasite and resistance to scaffolds known to target this pathway did not arise from the changes in the heme detoxification pathway but rather mutations in the DV membrane transporter, Plasmodium falciparum chloroquine resistance transporter (PfCRT).26 Also, studies have shown that several chloroquine analogues remain potent against chloroquine-resistant strains of the parasite, while still targeting this pathway.27-29 For these reasons, the heme detoxification pathway is still regarded as a viable target for antimalarial drug discovery. Chloroquine has been the most successful antimalarial to target this pathway.30 It is a neutral, weak base that distributes evenly in the cytoplasm of the parasite. However, in acidic medium, the compound’s basic side chain is protonated, leading to its accumulation in the DV through a process known as ion trapping.31 Also, intermolecular interactions permit chloroquine to bind to heme and interfere with its biocrystallization to hemozoin. This causes heme to build up to levels that lead to the parasite’s death. Solution state proton NMR has earlier revealed the prominent π–π interaction between Fe(III)PPIX and chloroquine.32 However, de Villiers et al. were the first to provide an example of a crystal structure of Fe(III)PPIX with an antimalarial, halofantrine, that inhibits the formation of hemozoin.33 They were able to show that just like chloroquine, most aryl methanol compounds inhibiting the formation of hemozoin possess hydrophobic aromatic rings which interact with the porphyrin ring of heme through π–π-stacking. They also showed evidence of hydrogen bonding between the propionate group of Fe(III)PPIX and the protonated nitrogen of the compounds studied as well as coordination of the deprotonated hydroxyl groups to the iron (III) centre of the heme monomer33-35 Chloroquine, on the other hand appears to interact by π stacking and hydrogen bonding.36 Previous studies have also shown that, chloroquine and other quinoline-type antimalarials inhibit the nucleation and growth of β-hematin crystals by adsorbing onto their 001 surface through a porphyrin acid to quinoline amine salt bridge and through the intercalation of the quinoline rings between the aromatic groups of β-hematin.37-39 Consequently, we hypothesized that both compounds 1 and 2 could be acting through this pathway since both compounds possess hydrophobic aromatic groups, a basic side chain with protonatable nitrogen in acidic medium (Figure 3).
Figure 3:

Structural similarities between compound 1 and chloroquine, revealing the presence of aromatic hydrophobic rings (Blue), basic amino side chains (Red) and chloro-substituted aromatic rings in both compounds (Purple).
To further investigate this hypothesis, we employed docking to predict whether either or both compounds are likely β-hematin inhibitors. Compounds were docked against the β-hematin surface using the 3D crystal structure previously published by Pagola et al20 ( Figure 4 and 5). We observed intermolecular interactions between compound β and the crystal surface of β-hematin. The benzo[4,5]imidazo[1,2-a]pyridine group of this compound interacts through π–π stacking with the porphyrin ring of β-hematin. Its basic amino side chain also forms a hydrogen bond with the propionate group of β-hematin when protonated at pH 4.5, suggesting it as a possible inhibitor of β-hematin (Figure 4). A similar docking pose was observed for compound 2; however, the presence of the pyridine ring in this compound accords it a second site for protonation in acidic media. The pyridine ring present in compound 2 differentiates it from compound 1. At pH 4.5, both the secondary amine on the side chain and the basic nitrogen in the pyridine ring are protonated, allowing for diprotonation in compound 2 (Figure 5B). This phenomenon also occurs in chloroquine, and allows for diprotonation in this compound as well.40 However in compound 2, the π–π-stacking interaction between the porphyrin ring of β-hematin and the heteroaromatic ring of the compound is lost due to changes in the orientation of the compound at the active site in the diprotonated state (Figure 5B). The two compounds were tested in the in vitro cell-free β-hematin inhibitions assay (BHIA) according to Lu et al41 with both compounds exhibiting good β-hematin inhibiting activity represented by values comparable to that of chloroquine.3 This further suggests β-hematin inhibition as a plausible MoA for the compounds.
Figure 4:

Predicted binding mode with the 001 face of β-hematin showing hydrogen bonding interactions between protonated nitrogen (monoprotonated) on the basic side chain of compound 1 and the propionate group of heme at pH 4.5 (1.8 Å) and π–π-stacking interactions shown between the porphyrin ring of heme and the tricyclic aromatic hydrophobic ring of compound 1 (3.5, 4.0 Å).
Figure 5:

Predicted binding mode of compound 2 to the 001 face of β-hematin showing hydrogen bonding interactions between protonated nitrogen (A) monoprotonated and (B) di-protonated on the basic side chain of the compound and the propionate group of heme at pH 4.5 (1.8, 1.7 Å). π–π-stacking interactions shown between the porphyrin ring of heme and the tricyclic aromatic hydrophobic ring of compound 2 (3.5, 3.8 Ȧ)
Although docking studies and the BHIA assay provided some insight into the possible MoA of these compounds, these experiments are cell-free and designed to only predict and mimic conditions in the Plasmodium parasite. Hence, we subjected both compounds to live-cell confocal microscopy in P. falciparum-infected erythrocytes to shed more light on the accumulation of the compounds in vitro using their intrinsic fluorescence properties. Initial fluorescence confocal microscopy of P. falciparum-infected erythrocytes revealed that the compounds selectively accumulate within erythrocytes infected with P. falciparum. Meanwhile, no fluorescent signal was observed for unparasitized red blood cells (Figure 6). This reiterates the specificity of both compounds to the Plasmodium parasite. It is to be noted that only trophozoites and schizonts were studied. This is because, the heme detoxification pathway is dominant in the trophozoite stage, as it is at this stage of the parasite’s life cycle that nearly 80% of the host haemoglobin is degraded for nutrient derivation, hence developing a distinct DV. During this stage, all organelles are fully developed, making their visualization easier by confocal microscopy. However, besides the digestive vacuole, several other organelles such as the endoplasmic reticulum (ER), mitochondrion, and plastids (apicoplast) play important roles during the intraerythrocytic stage and unlike the DV, these organelles persist in all stages of the intraerythrocytic cycle of the parasite making them equally viable targets. Hence, we investigated the accumulation of the compounds in the ER, mitochondrion, nucleus and the parasite’s lipid bodies.
Figure 6:

Panels A and B represent P. falciparum-infected erythrocytes incubated with compounds 1 and 2, respectively, with arrows showing regions of intense accumulation around hemozoin (Hz) and the differential interference contrast is represented as the DIC. Panel C represents an uninfected erythrocyte incubated with both compounds. Scale bar: 5 μm.
To investigate in detail the subcellular accumulation of these compounds, the cells were treated with LysoTracker Red, a dye which is believed to accumulate in the membranes of acidic organelles like the DV18 and DRAQ5, an anthraquinone dye with high affinity for double-stranded DNA and hence suitable to serve as a nuclear marker.42 We also used MitoTracker Deep Red fluorescent dye to illuminate the mitochondrion of the parasite since its chloromethyl moiety reacts with the thiol-containing cysteine residues in the mitochondria allowing it to diffuse across membranes and localize in the mitochondria.43 To establish the localization of both compounds in neutral and phospholipids, we treated the cells with Nile Red. This lipophilic dye is effective in staining lipids in cells. In aqueous media, Nile Red is entirely quenched; however, in non-polar environments and lipid-rich environments, it shows intense fluorescence.44 All fluorescent dyes were selected carefully to avoid crosstalk between the dyes and the compounds under study.
Once the target compounds were incubated with LysoTracker Red, we expected to see intense fluorescence colocalization in the DV between the compounds and the tracker dye, assuming both compounds inhibit the formation of hemozoin. However, no colocalization was observed between compound 1 and LysoTracker Red. The tracker dye accumulated in the digestive vacuole shown by the white arrow in figure 6 but no significant accumulation was observed for compound 1 suggesting that contrary to the earlier hypothesis, the compound’s site of accumulation is not in the digestive vacuole and its primary action is not to inhibit the formation of hemozoin. It should be noted that the fluorescence emission of this compound is at least 0.2-fold greater in acidic environments than basic and neutral environments. Hence, the concern that the compound could undergo pH quenching in the digestive vacuole is highly unlikely.
Compound 2, on the other hand, showed significant colocalization with LysoTracker Red with regions of intense accumulations around the hemozoin crystals. This suggests that unlike compound 1, significant amounts of compound 2 permeate the digestive vacuole membrane and accumulates in the acidic digestive vacuole. This, we believe, is due to the compound being diprotonated as predicted by docking studies, allowing it to accumulate in the digestive vacuole to concentrations high enough to observe through pH trapping. Also, once diprotonated, compound 2 still binds efficiently to the hemozoin surface. Although both compounds possess structural features that interact with β-hematin and prevent its growth as shown by the docking, only compound 2 accumulates significantly in the parasite’s DV. This is evident in the live-cell imaging. Aside from accumulation in the digestive vacuole, both compounds showed association with the parasite’s membranous structures (Figure 7). However, no co-localization was observed between either compound and the nuclear marker DRAQ5, suggesting that these compounds do not act in the nucleus of the parasite (Figure 8).
Figure 7:

Live-cell confocal microscopy of P. falciparum-infected erythrocyte, treated with Compounds 1, 2 ( blue) and LysoTracker Red (green). White arrows indicate areas of intense localization of the dye and regions of overlap shown in the merged image. Scale bars: 2 μm.
Figure 8:

Live-cell confocal microscopy showing co-localization of compounds 1 and 2 (blue) with the nuclear marker DRAQ5 (red) Scale bars: 2 μm.
In the Plasmodium parasite, neutral lipids have been shown to carry out important functions during the asexual blood stage of the parasite’s life cycle. Fitch and co-workers have previously shown that neutral lipid mixtures enhance the formation of β-hematin in vitro and that these lipid structures are closely associated with the digestive vacuole where the formation of hemozoin occurs.45 Using electron microscopy and Nile Red fluorescence, Sullivan et al. showed ultrastructural evidence of the presence of neutral lipids in the in vivo site of hemozoin formation.46 Therefore we decided to investigate the colocalization of our compounds with Nile Red. Although this tracker dye illuminates both phospholipid membranes and neutral lipid bodies in mature Plasmodium parasites, we could differentiate fluorescence emissions arising from the neutral lipid structures from those from the phospholipids using confocal microscopy. Interestingly, we observed punctate spherical structures illuminated by the Nile Red at an excitation that only detects neutral lipids. These neutral lipid structures were observed to only colocalize with compound 2 and not with compound 1. They were observed to lie in the cytoplasm, near the hemozoin crystals, possibly in intimate contact with the DV as shown by arrows in the second panel of Figure 9. We suspect that colocalization with the neutral lipids may be one of the mechanisms through which compound 2 is transported into the DV where it accumulates and disrupts the biocrystallisation of heme to hemozoin. However, this phenomenon was not observed for compound 1. The compound showed no colocalisation with the neutral lipid bodies and this goes to further suggest that this compound’s action is not in the digestive vacuole as seen for compound 2 and that the subtle change in substituents has led to substantial changes in intracellular localization as observed by confocal fluorescence microscopy and a possible change in the MoA of the compounds To investigate the effect of these compounds on the levels of heme and hemozoin in the parasite cultured in vitro, we employed the heme fractionation assay previously described by Combrinck et al.47 Exposure of P. falciparum to compound 1 had no effect on the levels of heme and hemozoin (Figure 10, Panel A). This is consistent with the fact that this compound does not localize in the digestive vacuole of the parasite, as shown by the fluorescent live-cell microscopy earlier discussed. At increasing concentrations of compound 1, both heme and hemozoin levels remained constant. This confirms that although compound 1 has all the structural properties required to interfere with the conversion of heme to hemozoin, it does not accumulate in the DV to levels that can cause any significant inhibition of this pathway. Hence this compound has a different mechanism of action from hemozoin inhibition. However, contrary to compound 1, we observed a significant change in the levels of both heme and hemozoin when the parasite was exposed to increasing concentrations of compound 2. The levels of heme increased steadily with a concomitant decrease in hemozoin levels as a function of dose. This demonstrates that the exposure of P. falciparum to this compound causes the build-up of toxic heme, which leads to the death of the parasite. Therefore, unlike compound 1, compound 2 acts through the inhibition of hemozoin formation. We also observed that, compared to chloroquine, compound 2 has a less pronounced effect on heme and hemozoin levels in the parasite at increasing concentrations.47, 48 (Figure 10, Panel B). This suggests that there might be other contributory mechanisms to the action of the compound, and this merits further investigation. As earlier stated, compound 1 is about 5 times more active against both the sensitive and resistance strains of Plasmodium falciparum. Based on this and the results from the hemozoin inhibition assay, we are further convinced that compound 1’s target is outside the digestive vacuole and that its action and target (s) is clearly different from that of compound 2. We believe that compound 1 acts at lower doses at which sufficient accumulation in the digestive vacuole does not occur to inhibit hemozoin formation. Also, the loss of π–π-stacking interaction observed between the porphyrin ring of β-hematin and the heteroaromatic ring of compound 2 once it is diprotonated could account for the lesser activity of the compound in comparison to known drugs that target the pathway. However, it is intriguing to note that two structurally similar compounds, with subtle changes in substitution, could have different MoA (s). This further debunks the common assumption that compounds of the same class have the same mechanism of action and the same biological targets and supports the view that, every change in substitution can potentially cause a change in the mechanism of action.
Figure 9:

Live-cell confocal microscopy showing co-localization between Nile Red (red) and compounds 1 and 2 (blue) Scale bars: 1 μm.
Figure 10:

Heme fractionation profiles of compounds 1/CKM06 (A) and 2/CKM08 (B). The amount of “free” heme Fe and hemozoin (Hz) Fe at increasing concentrations of both compounds. Significance levels are depicted with the asterisks; Where * = p<0.5, ** = p < 0.1 and *** = p < 0.05
Conclusions
In summary, we report for the first time the intrinsic fluorescence properties of previously described antimalarial PBIs (compounds 1 and 2), which are suitable for probing their subcellular accumulation in P. falciparum. Live-cell microscopy showed no fluorescence localization of either compound in the nucleus of the parasite, suggesting that the compounds’ action is not in the nucleus. Accumulation in the digestive vacuole of the parasite was selective for compound 2 only and not compound 1. This shows the difference in localization between the two structurally similar compounds. We also showed the association of compound 2 with neutral lipid aggregates, which have been shown to mediate the formation of hemozoin. This observation suggests that only this compound has an effect in the digestive vacuole of the parasite, although docking studies showed that both compounds are able to bind to the surface of hemozoin. We also showed via the heme fractionation assay that, at increasing dose, compound 2 showed direct evidence of inhibition of hemozoin formation. In contrast, compound 1 showed no impact on the levels of either heme or hemozoin. Altogether, these results support hemozoin inhibition as a mechanism through which compound 2 acts while suggesting the possibility of additional contributing mechanisms. Compound 1, on the other hand, has been shown to act through a different, as yet unknown, mechanism, suggesting that a slight change in substitution on the chemotype can lead to different MoA (s) and hence different targets in the Plasmodium parasite.
The concept of mechanisms and molecular targets of both compounds have still not been fully understood; however, results shown here suggest that structurally related compounds from the same series will not necessarily have the same molecular target or act through the same pathway. Further work will investigate the effect of these compounds on the genome of the parasite by employing resistance selection and whole-genome sequencing approaches. This is expected to provide some insight into the mechanism of resistance of the parasite and potentially also their MoA.
Supplementary Material
Acknowledgements
We thank Professor Dirk Lang and Susan Cooper of the Confocal and Light Microscope Imaging Facility Unit, University of Cape Town, for their contribution and help with the confocal microscope. We also acknowledge our funders, Medicines for Malaria Venture (MMV) and Merck KGaA for supporting this work. The South African Medical Research Council and South African Research Chairs Initiative of the Department of Science and Innovation, administered through the South African National Research Foundation, are gratefully acknowledged for support (K.C). TJE, LG and LA acknowledge funding by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under grant number 5R01AI143521. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Conflicts of interest
The authors declare no conflict of interest
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
Notes and references
- 1.W. H. Organisation, World Malaria report, 2018.
- 2.Ndakala AJ, Gessner RK, Gitari PW, October N, White KL, Hudson A, Fakorede F, Shackleford DM, Kaiser M, Yeates C, Charman SA and Chibale K, Journal of Medicinal Chemistry, 2011, 54, 4581–4589. [DOI] [PubMed] [Google Scholar]
- 3.Singh K, Okombo J, Brunschwig C, Ndubi F, Barnard L, Wilkinson C, Njogu PM, Njoroge M, Laing L, Machado M, Prudêncio M, Reader J, Botha M, Nondaba S, Birkholtz L-M, Lauterbach S, Churchyard A, Coetzer TL, Burrows JN, Yeates C, Denti P, Wiesner L, Egan TJ, Wittlin S and Chibale K, Journal of Medicinal Chemistry, 2017, 60, 1432–1448. [DOI] [PubMed] [Google Scholar]
- 4.Swinney DC and Anthony J, Nature Reviews Drug Discovery, 2011, 10, 507–519. [DOI] [PubMed] [Google Scholar]
- 5.Bantscheff M and Drewes G, Bioorganic & Medicinal Chemistry, 2012, 20, 1973–1978. [DOI] [PubMed] [Google Scholar]
- 6.Guiguemde WA, Shelat AA, Bouck D, Duffy S, Crowther GJ, Davis PH, Smithson DC, Connelly M, Clark J, Zhu F, Jiménez-Díaz MB, Martinez MS, Wilson EB, Tripathi AK, Gut J, Sharlow ER, Bathurst I, Mazouni FE, Fowble JW, Forquer I, McGinley PL, Castro S, Angulo-Barturen I, Ferrer S, Rosenthal PJ, DeRisi JL, Sullivan DJ, Lazo JS, Roos DS, Riscoe MK, Phillips MA, Rathod PK, Van Voorhis WC, Avery VM and Guy RK, Nature, 2010, 465, 311–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hart CP, Drug Discovery Today:, Volume 10, Number 7–Number 7.15676292 [Google Scholar]
- 8.Hartwig CL, Lauterwasser EMW, Mahajan SS, Hoke JM, Cooper RA and Renslo AR, Journal of Medicinal Chemistry, 2011, 54, 8207–8213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Han S-Y and Hwan Kim S, Archiv der Pharmazie, 2007, 340, 169–177. [DOI] [PubMed] [Google Scholar]
- 10.Jeffery DA and Bogyo M, Current Opinion in Biotechnology, 2003, 14, 87–95. [DOI] [PubMed] [Google Scholar]
- 11.Witkowski J, Nature, 2010, 466, 921–922. [Google Scholar]
- 12.Steven Zheng XF and Chan T-F, Drug Discovery Today, 2002, 7, 197–205. [DOI] [PubMed] [Google Scholar]
- 13.Das RK, Samanta A, Ghosh K, Zhai D, Xu W, Su D, Leong C and Chang Y-T, Interdisciplinary Bio Central, 2011, 3, 3.1–3.16. [Google Scholar]
- 14.Sidhu ABS, Sun Q, Nkrumah LJ, Dunne MW, Sacchettini JC and Fidock DA, Journal of Biological Chemistry, 2007, 282, 2494–2504. [DOI] [PubMed] [Google Scholar]
- 15.Nzila A and Mwai L, Journal of Antimicrobial Chemotherapy, 2010, 65, 390–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dzekunov SM, Ursos LMB and Roepe PD, Molecular and Biochemical Parasitology, 2000, 110, 107–124. [DOI] [PubMed] [Google Scholar]
- 17.Chan L, Teo J, Tan K, Sou K, Kwan W and Lee C-L, Molecules, 2018, 23, 2635–2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Woodland JG, Hunter R, Smith PJ and Egan TJ, Organic & Biomolecular Chemistry, 2017, 15, 589–597. [DOI] [PubMed] [Google Scholar]
- 19.Trager W and Jensen J, Science, 1976, 193, 673–675. [DOI] [PubMed] [Google Scholar]
- 20.Pagola S, Stephens PW, Bohle DS, Kosar AD and Madsen SK, Nature, 2000, 404, 307–310. [DOI] [PubMed] [Google Scholar]
- 21.Sullivan DJ, International Journal for Parasitology, 2002, 32, 1645–1653. [DOI] [PubMed] [Google Scholar]
- 22.Francis SE, Sullivan DJ, Goldberg and Daniel E, Annual Review of Microbiology, 1997, 51, 97–123. [DOI] [PubMed] [Google Scholar]
- 23.Egan TJ and Ncokazi KK, Journal of Inorganic Biochemistry, 2005, 99, 1532–1539. [DOI] [PubMed] [Google Scholar]
- 24.Slater AF, Swiggard WJ, Orton BR, Flitter WD, Goldberg DE, Cerami A and Flenderson GB, Proceedings of the National Academy of Sciences, 1991, 88, 325–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Slater AFG and Cerami A, Nature, 1992, 355, 167–169. [DOI] [PubMed] [Google Scholar]
- 26.Fidock DA, Nomura T, Talley AK, Cooper RA, Dzekunov SM, Ferdig MT, Ursos LMB, bir Singh Sidhu A, Naudé B, Deitsch KW, Su X.-z., Wootton JC, Roepe PD and Wellems TE, Molecular Cell, 2000, 6, 861–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cogswell FB, Krogstad DJ, De D and Krogstad FM, The American Journal of Tropical Medicine and Hygiene, 1996, 55, 579–583. [DOI] [PubMed] [Google Scholar]
- 28.Ridley RG, Hofheinz W, Matile H, Jaquet C, Dorn A, Masciadri R, Jolidon S, Richter WF, Guenzi A, Girometta MA, Urwyler H, Fluber W, Thaithong S and Peters W, Antimicrobial agents and chemotherapy, 1996, 40, 1846–1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Biot C, Glorian G, Maciejewski LA, Brocard JS, Domarle O, Blampain G, Millet P, Georges AJ, Abessolo H, Dive D and Lebibi J, Journal of Medicinal Chemistry, 1997, 40, 3715–3718. [DOI] [PubMed] [Google Scholar]
- 30.Egan TJ, Ross DC and Adams PA, FEBS Letters, 1994, 352, 54–57. [DOI] [PubMed] [Google Scholar]
- 31.Wongsrichanalai C, Pickard AL, Wernsdorfer WH and Meshnick SR, The Lancet Infectious Diseases, 2002, 2, 209–218. [DOI] [PubMed] [Google Scholar]
- 32.de Dios AC, Casabianca LB, Kosar A and Roepe PD, Inorganic Chemistry, 2004, 43, 8078–8084. [DOI] [PubMed] [Google Scholar]
- 33.de Villiers KA, Marques HM and Egan TJ, Journal of Inorganic Biochemistry, 2008, 102, 1660–1667. [DOI] [PubMed] [Google Scholar]
- 34.de Villiers KA, Gildenhuys J and le Roex T, ACS Chemical Biology, 2012, 7, 666–671. [DOI] [PubMed] [Google Scholar]
- 35.Gildenhuys J, Sammy CJ, Müller R, Streltsov VA, le Roex T, Kuter D and de Villiers KA, Dalton Transactions, 2015, 44, 16767–16777. [DOI] [PubMed] [Google Scholar]
- 36.Kuter D, Streltsov V, Davydova N, Venter GA, Naidoo KJ and Egan TJ, Journal of Inorganic Biochemistry, 2016, 154, 114–125. [DOI] [PubMed] [Google Scholar]
- 37.Buller R, Peterson ML, Almarsson Ö and Leiserowitz L, Crystal Growth & Design, 2002, 2, 553–562. [Google Scholar]
- 38.Solomonov I, Osipova M, Feldman Y, Baehtz C, Kjaer K, Robinson I,K, Webster GT, McNaughton D, Wood BR, Weissbuch I and Leiserowitz L, Journal of the American Chemical Society, 2007,129, 2615–2627. [DOI] [PubMed] [Google Scholar]
- 39.L'Abbate FP, Müller R, Openshaw R, Combrinck JM, de Villiers KA, Hunter R and Egan TJ, European Journal of Medicinal Chemistry, 2018, 159, 243–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chinappi M, Via A, Marcatili P and Tramontano A, PLoS ONE, 2010, 5, e14064–e14064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu W-J, Wicht KJ, Wang L, Imai K, Mei Z-W, Kaiser M, El Sayed IET, Egan TJ and Inokuchi T, European Journal of Medicinal Chemistry, 2013, 64, 498–511. [DOI] [PubMed] [Google Scholar]
- 42.Edward R, Molecules and Cells, 2009, 27, 391–396. [DOI] [PubMed] [Google Scholar]
- 43.Melhuish WH, The Journal of Physical Chemistry, 1961, 65, 229–235. [Google Scholar]
- 44.Maier O, Oberle V and Hoekstra D, Chemistry and physics of lipids, 2002, 116, 3–18. [DOI] [PubMed] [Google Scholar]
- 45.Fitch CD, Cai G.-z., Chen Y-F and Shoemaker JD, Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease, 1999, 1454, 31–37. [DOI] [PubMed] [Google Scholar]
- 46.Pisciotta JM, Coppens I, Tripathi AK, Scholl PF, Shuman J, Bajad S, Shulaev V and Sullivan DJ, Biochemical Journal, 2007, 402, 197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Combrinck JM, Fong KY, Gibhard L, Smith PJ, Wright DW and Egan TJ, Malaria Journal, 2015, 14, 253–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fong KY and Wright DW, Future Medicinal Chemistry, 2013, 5, 1437–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.