

Abstract
The recent accumulation of cancer genomic data provides an opportunity to understand how a tumor’s genomic characteristics can affect its responses to drugs. This field, called pharmacogenomics, is a key area in the development of precision oncology. Deep learning (DL) methodology has emerged as a powerful technique to characterize and learn from rapidly accumulating pharmacogenomics data. We introduce the fundamentals and typical model architectures of DL. We review the use of DL in classification of cancers and cancer subtypes (diagnosis and treatment stratification of patients), prediction of drug response and drug synergy for individual tumors (treatment prioritization for a patient), drug repositioning and discovery and the study of mechanism/mode of action of treatments. For each topic, we summarize current genomics and pharmacogenomics data resources such as pan-cancer genomics data for cancer cell lines (CCLs) and tumors, and systematic pharmacologic screens of CCLs. By revisiting the published literature, including our in-house analyses, we demonstrate the unprecedented capability of DL enabled by rapid accumulation of data resources to decipher complex drug response patterns, thus potentially improving cancer medicine. Overall, this review provides an in-depth summary of state-of-the-art DL methods and up-to-date pharmacogenomics resources and future opportunities and challenges to realize the goal of precision oncology.
Keywords: deep learning, precision oncology, pharmacogenomics, cancer, drug discovery
Introduction
Advances in high-throughput technologies have led to a rapid accumulation of different types of cancer-related omics data, such as tumor mutations, transcriptomes, methylomes, proteomes and microbiomes. These enormous data resources have rapidly transformed translational research and clinical practice in cancer diagnosis and management. As a result, tumors are now routinely profiled for DNA and RNA alterations and are matched with therapeutic options, making precision oncology a reality for some patients [1–4]. These early successes demonstrate the potential of genomics to help address remaining formidable clinical challenges, such as predicting and preemptively preventing drug resistance. The first systematic pharmacogenomic study of cancer was carried out by the National Cancer Institute (NCI) that molecularly profiled and tested drug response of 60 human cancer cell lines (CCLs), known as the NCI-60 panel [5, 6]. The data unveiled links between high-dimensional omics to mechanisms of drug sensitivity and resistance [5–9]. Using genomic data to make discoveries requires extracting a statistically meaningful pattern from tens of thousands of features. For instance, a single RNA sequencing experiment can generate expression values for over 50 000 entities, including coding and non-coding genes. This high-dimensional structure usually requires a significant sample size for providing adequate statistical power, an effort typically beyond the capacity of a single research lab. Fortunately, consortium efforts such as The Cancer Genome Atlas (TCGA) [10–14] and the Cancer Cell Line Encyclopedia (CCLE) [15] have spearheaded models where large amounts of data are generated and shared using standard protocols. These resources range from characterization of CCLs, primary and metastatic tumors to probing cellular vulnerabilities of CCLs, creating a diverse portfolio for integrative analyses [16–18].
Machine learning (ML) approaches have been used to learn hidden patterns behind genomic data related to cancer. Unlike conventional statistical methods that analyze data based on assumptions of data distributions, ML methods rely on predefined ‘learning models’ or learning the data models (or data representation) de novo from large, complex and heterogeneous genomic data [19]. Among ML methods, deep learning (DL) architecture commonly refers to an artificial neural network (ANN) with multiple hidden layers. DL can model complex non-linear relationships and is widely used in image and voice processing, speech recognition, natural language processing and complex data modeling. The use of DL to analyze genomic data is still in its early phase, mostly for investigating nucleic acid sequences and the binding proteins [20–23], dimension reduction in single-cell RNA-seq analysis [24, 25], tumor classification [26, 27] or predicting survival outcomes [28–30]. DL algorithms have also been used to predict drug responses [31, 32] and drug synergy [33].
Reviews of general DL applications in bioinformatics and computational biology, DNA/RNA sequence analyses (genetic variations and regulatory effects) and general pharmacology have been published (see selected reviews in Table 1). However, a comprehensive review focused on genomic aspects of DL applications in cancer pharmacogenomics (i.e., gene mutations, gene expression, etc.) is still lacking (Figure 1A). Here, we systematically surveyed data resources of genomics and pharmacogenomics and their corresponding DL applications (either published or our in-house analyses) for critical pharmacogenomics topics. Since current cancer therapeutics and trials focus on a specific cancer type/subtype, or genomic alterations independent of cancer type (also known as ‘genome-driven oncology’) [34], such as NCI Molecular Analysis for Therapy Choice (NCI-MATCH) trial [35], here we discuss the use of DL for
Table 1.
Selected reviews of DL in pharmacologic research
| Year | Topics | References |
|---|---|---|
| General DL and bioinformatics | ||
| 2015 | DL fundamentals | [19] |
| 2016 | DL in bioinformatics | [146] |
| 2016 | DL in computational biology | [38] |
| DNA/RNA sequence analyses | ||
| 2018 | Genetic variations and regulatory networks | [40] |
| 2019 | Genetic variations on gene regulatory mechanisms | [147] |
| Pharmacology and healthcare | ||
| 2018 | DL for healthcare | [39] |
| 2018 | Genetic variations, patient stratification by medical records and drug target discovery | [148] |
| 2019 | ML and DL for drug discovery | [149] |
| Chemoinformatics for drug design | ||
| 2016 | ML and DL studies on various pharmaceutical topics (solubility, toxicity, etc.) | [73] |
| 2018 | DL models for drug design | [74] |
| 2018 | Chemical descriptors and tools/databases | [76] |
| 2018 | Chemical fingerprints and descriptors for drug design | [75] |
Figure 1.
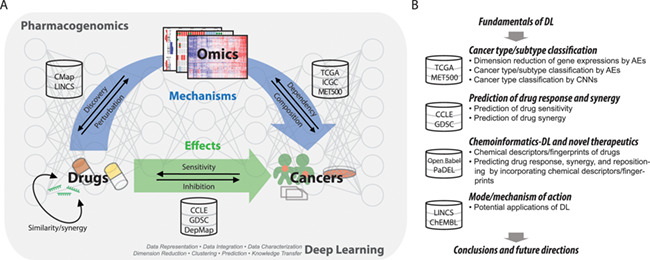
Pharmacogenomics in cancer and overview of this review. (A) Cancer pharmacogenomics and DL. Cancer pharmacogenomics has three interconnected key components: genomics, drugs and patient outcomes and responses. Emerging large data resources are enabling DL-based study of cancer pharmacogenomics and the achievement of precision oncology. (B) Overview of the study. The study sought to comprehensively review data resources of cancer genomics and pharmacogenomics and demonstrate how DL learning can be applied to achieve several major goals of pharmacogenomics and precision oncology.
cancer type/subtype classification and
predictions of drug response and synergy based on cancer genomic data.
Accurate classifications of cancer types or subtypes facilitate understanding of disease processes and possible identification of treatment targets and thus enable discovery of drugs that target a specific class of cancers. However, identifying the best drug(s) requires understanding of tumor genomics, regardless of the cancer type. In light of novel therapeutics and drug mechanisms, here we also comprehensively survey
drug repositioning and discovery that incorporate chemoinformatics descriptors and/or fingerprints of drugs and
resources to analyze drugs’ mechanisms and modes of action.
Figure 1B illustrates the goals of this review. Specifically, we aim to provide a reference of resources for investigators entering the field of pharmacogenomics and highlight the potential of DL to enrich the discipline of pharmacogenomics and to accelerate the pursuit of precision oncology.
Fundamentals of DL
Conventional ANNs and DL models
ANN is one of the most powerful and classic ML methods that imitate humans’ brain functioning and decision-making through multiple layers of interconnected nodes. These nodes are linked via algebraic equations to create stacked layers of neurons termed hidden layers [36]. Information flows from the input layer through a hidden layer and activates some nodes in the hidden layer. ANN model parameters are determined by a learning process, usually through back propagation. ANNs achieve ideal performance if the data provided for training have relatively recognizable internal patterns and large sample sizes. Compared to other classic ML methods, ANNs increase the search space for adjustable parameters and omit assumptions of data models such as linearity and variable independence. Typical ANN models have limited number of hidden layers due to computational complexity and limited training samples.
With the methodological and technological advances in the past two decades, efficient training of deep ANNs with multiple hidden layers, or DL, has become possible. The depth of DL and new model architectures further increases the search space to capture intricate patterns and data representations much better than conventional ANNs (reviewed in [19]). Furthermore, the multi-layered design enables many flexible and diverse DL architectures to tackle different optimization tasks. Here we introduce several core layers of DL and combinations of core layers into typical DL models.
Core DL layers
DL-based research reviewed here typically use architectures that can be broken down to three core layers (Figure 2A):
Figure 2.
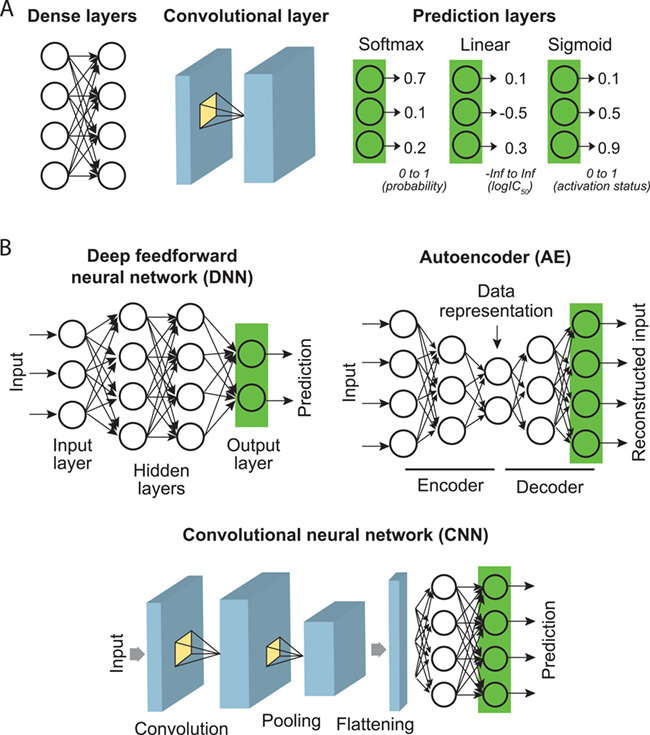
Core DL layers and typical model architectures for pharmacogenomics studies. (A) Core DL layers. (B) Combinations of core layers into the simplest form of a DL model (DNN), dimension-reducing network (AE) and DL learning patterns from inputs (CNN).
‘Dense layers’, also known as fully connected layers, are the simplest form of DL layer that connects every neuron of a layer to every neuron of another layer.
‘Convolutional layers’ slide multiple learnable filters (kernels; representing patterns) through an image-like input to calculate cross-correlations between each filter and sections of the input and determine whether patterns exist locally in the image. After optimization, each filter typically recognizes a local data pattern.
‘Output layers’ generate the output of a DL with desired numerical properties. Based on the prediction goal, output layers (or prediction layers) can take different activation functions to yield outputs with desired numerical properties. These include ‘softmax’, to yield positive values for output neurons that sum up to 1 such that it can be interpreted as prediction probabilities (e.g., cancer types); ‘linear’ activation, to generate an unbounded continuous value [e.g., log-transformed half-maximal inhibitory concentration (IC50)]; ‘ReLU’ (rectified linear unit) activation, which is similar to ‘linear’ activation but truncates negative values (to zeros; e.g., IC50); and ‘sigmoid’ which yields a logistic output between 0 and 1.
Combining core layers into typical DL architectures
Core layers are building blocks of DL models with supervised, semi-supervised or unsupervised designs. Supervised models find the mapping process of labeled datasets (i.e., data with ground truth), while unsupervised methods work on unlabeled datasets to unveil properties of the data. Semi-supervised methods address situations where only a small portion of dataset is labeled. Various DL models in these three classes are being developed for different purposes with biological datasets. Here we introduce several typical architectures frequently applied to genomic and/or pharmacogenomic data.
‘Deep feedforward neural networks’ (DNNs) are the simplest yet powerful DL architecture that is composed of multiple fully connected layers and an output layer (Figure 2B). DNNs can be easily configured to perform simple prediction and regression tasks.
‘Autoencoders’ (AEs) are unsupervised DL models that learn the most informative representation (compression) of the input data. Their goal is to find an identity function that compresses inputs into shorter coding vectors and reconstructs the original data from the coding vectors [37]. As shown in Figure 2B, a typical AE is composed of a dimension-reducing encoder network and a dimension-reconstructing decoder network. As a result, outputs of the bottleneck layer that captures crucial information regarding input data can be used for different purposes, such as sample classification and noise reduction.
‘Convolutional neural networks’ (CNNs) are one of the most powerful breakthroughs in DL and have been adapted to many data intensive fields, such as computer vision and natural language processing. CNNs employ convolution layers that learn local patterns underlying images or sequences for recognition or prediction (Figure 2B). In medical informatics and bioinformatics researches, CNNs have been applied to computer-aided diagnosis and sequence-based analysis, such as motif discovery and variant analysis, as reviewed in [38–40]. When analyzing high-dimensional genomics data related to pharmacogenomics, this powerful model has only very recently been applied to the prediction of cancer types—but as far as we know, not yet to other tasks.
In the following sections, we review state-of-the-art applications of DNNs, AEs and CNNs to address important pharmacogenomic topics and further challenges and opportunities.
Classifying cancer types and subtypes using genomics profiles
Data resources
With advances in high-throughput sequencing and efforts of international consortia, several large-scale datasets of pan-cancer cancer genomics have made tremendous contributions to our understanding of cancer heterogeneity. Table 2 summarizes the most representative data resources. In the past decade, the pan-cancer atlas generated by TCGA has covered almost all types of DNA- and RNA-derived genomics data for 33 kinds of adult cancers, based on over 11 000 pairs of tumor and normal tissues. Harmonized data can be easily accessed through the Genomic Data Commons (GDC) web portal or the R/Bioconductor package TCGAbiolinks [41] (Table 2). Using TCGA data, a recent collection of studies published by the Cell Press comprehensively cataloged cell-of-origin patterns, oncogenic processes and oncogenic signaling pathways in a pan-cancer setting, as reviewed in [42]; papers are accessible at https://www.cell.com/pb-assets/consortium/pancanceratlas/pancani3/index.html. In addition, a group at the University of Michigan Comprehensive Cancer Center sequenced tumor samples from 500 adult patients with 30 types of cancers who had metastases in 22 different organs (the MET500 cohort) [43].
Table 2.
Resources for pan-cancer genomics profiles and tools
| Resource | Data type | Profiling platform | Sample size | Description | Link | References |
|---|---|---|---|---|---|---|
| Adult cancers | ||||||
| TCGA (The Cancer Genome Atlas) | Clin, CNA, GEX, Methyl, miEX, SNV | Microarray, NGS | ~11 300 | Mostly primary tumors of 33 cancers | Individual cancers: https://portal.gdc.cancer.gov/ Merged pan-cancer data: https://gdc.cancer.gov/node/905/ Also downloadable by an R/Bioconductor package TCGAbiolinks [41] | [150] |
| MET500 | CNA, SNV | NGS | 500 | Metastatic tumors of 30 cancers | https://met500.path.med.umich.edu/ | [43] |
| Pediatric cancers | ||||||
| TARGET (Therapeutically Applicable Research to Generate Effective Treatments) | Clin, GEX, miEX, SNV | NGS | ~3200 (according to the GDC Data Portal accessed in May 2018) | 6 pediatric cancers (according to the GDC Data Portal accessed in May 2018) | https://portal.gdc.cancer.gov/ Also downloaded by an R/Bioconductor package TCGAbiolinks [41] | [44] |
| PedPanCan (Pediatric Pan-Cancer study) | SNV | NGS | 961 | 24 pediatric cancers | http://www.pedpancan.com | [45] |
| Cancer cell lines | ||||||
| CCLE (Cancer Cell Line Encyclopedia) | CNA, GEX, RPPA, SNV | Microarray, NGS | ~1500 | https://portals.broadinstitute.org/ccle Also accessible through the Cancer Dependency Map (DepMap): https://depmap.org/portal/ | [15, 151] | |
| Curations | ||||||
| ICGC (International Cancer Genome Consortium) | Clin, CNA, GEX, Methyl, miEX, SNV | Curation | ~24 000 | Curation of 80+ international cancer projects, including TCGA and TARGET | http://icgc.org/ | [46] |
| COSMIC (Catalogue of Somatic Mutations in Cancer) | CNA, SNV | Curation | Summarization of cancer-related mutations across 32 000+ tumors and cancer cells curated from 25 000 papers | https://cancer.sanger.ac.uk/cosmic | [48] | |
| Pan-cancer data visualization | ||||||
| TumorMap | 2D maps | Curation | Visualization of TCGA, TARGET, etc. | https://tumormap.ucsc.edu/ | [47] | |
| Gene signatures and biological pathways | ||||||
| MSigDB (Molecular Signatures Database | Genes sets | Curation | ~17 800 gene sets | Genes sets of cytobands, curations, motifs, computation, Gene Ontologies, oncogenic signatures and immunology. | http://software.broadinstitute.org/gsea/msigdb/index.jsp | [52–54] |
| Pathway Commons | Biological pathways | Curation | 4000+ pathways | Collection of biological pathways from 20+ databases, including KEGG and Reactome | https://www.pathwaycommons.org/ | [152] |
| NDEx (Network Data Exchange) | Biological networks | Curation | Interactive database that allows users to query, visualize, upload, share and distribute biological networks | www.ndexbio.org/ | [153] | |
| Normal tissues | ||||||
| GTEx (Genotype-Tissue Expression) | GEX | NGS | ~11 700 | Expression profiles of 53 non-diseased tissues across ~1000 individuals that can be used as normal controls for cancer studies | https://gtexportal.org/home/ | [154, 155] |
Clin, clinical data; CNA, copy number alteration; GEX, gene expression; Methyl, methylation; miEX, miRNA expression; NGS, next-generation sequencing; RPPA, reverse phase protein array; SNV, single nucleotide variant.
For childhood cancers, the Therapeutically Applicable Research to Generate Effective Treatments (TARGET) project has so far comprehensively profiled ~1700 pediatric tumors (including leukemia, Wilms tumor, neuroblastoma and osteosarcoma) [44]. Another European project, the Pediatric Pan-Cancer (PedPanCan) study, has analyzed genetic alterations of ~1000 samples of 24 molecular types of cancers [45]. The St. Jude PeCan Data Portal (https://pecan.stjude.cloud/home) provides interactive visualizations of pediatric cancer mutations identified by these resources.
Efforts have been devoted to curate and process these datasets into large data resources. For instance, the International Cancer Genome Consortium (ICGC) [46] has collected and provided a data portal for profiles of more than 24 000 tumors from over 80 large cancer projects, including TCGA and TARGET. Using dimension reduction methods of t-distributed stochastic neighbor embedding (t-SNE) and principal component analysis (PCA), the UCSC Genomics Institute developed an interactive browser, called the TumorMap, that visualizes two-dimensional clusters of TCGA and TARGET samples and allows users to map and explore their samples on these maps [47]. In addition, the Catalogue of Somatic Mutations in Cancer (COSMIC) is the world’s largest database of somatic mutations and variants [copy number alterations (CNAs) and differentially expressed genes, etc.] of human cancers [48].
Taken together, these integrative pan-cancer datasets have unveiled a comprehensive landscape of adult and pediatric cancers that is huge enough for DL-based studies. In the next section, we survey a collection of published AE-based models for dimension reduction/visualization and different CNN configurations for classification of cancer types using pan-cancer gene expression data. For a comprehensive evaluation, we also implemented several regular and specific AE-based models for classifying cancer types.
Dimension reduction of gene expression profiles by AEs and biological knowledge-regularized AEs
AEs are well suited to learn data representation and perform dimension reduction of complex data. In genomics, AEs have been successfully used to cluster single-cell RNA-seq data that typically include up to 10 000 samples [24, 25]. Apart from AEs made of dense layers, the hierarchy of Gene Ontology (GO) has been used to regularize AEs, so that neurons represented GO terms and edges between neurons were configured according to hierarchical associations between corresponding GOs [49]. The model, named DCell, was applied to model the functional hierarchy of a cell. Such a model is also called a ‘visible’ neural network (VNN), as opposed to so-called ‘black-box’ DL, for the output of each neuron represents the activation state of a GO term and can be easily visualized and interpreted.
To demonstrate the application of this approach to pan-cancer gene expression profiles with relatively limited sample sizes, we tested several differently configured AEs for dimension reduction of TCGA data [fully connected AE (FC-AE1); architecture in Figure 3A]. Transcripts per million values of 15 931 genes in 8070 tumors (17 different cancer types with a sample size ≥200) were downloaded from the TumorMap. We ran a hyper-parameter optimization method called hyperas [50] to determine the optimal AE architecture (number of neurons at each layer, 15 931, 1000, 500, 1000 and 15 931; Figure 3A). Samples were randomly split by 90% and 10% for training and validation, respectively, to avoid control overfitting. We also adopted the early stopping strategy to stop the training when the validation loss did not further improve. To illustrate the information captured by this classic AE, we used t-SNE to visualize the output of each layer and compared it to classical methods, including PCA and non-negative matrix factorization (NNMF). The three methods performed comparably well in preserving inter-cancer differences underlying gene expression profiles, even though the dimensions were reduced by 96.9% (Figure 3A and B). We also compared the results to our recently proposed Gene Superset AE (GSAE) model, which incorporated prior knowledge of gene sets and biological pathways [51]. Briefly, GSAE regularizes an AE by known gene–gene interactions curated in the Molecular Signatures Database (MSigDB) [52–54], i.e., input genes involved in a pathway or a similar function are linked to a node at the second (gene-set) layer (Figure 3C). Such regularization extracts biologically meaningful data from high-throughput genomic profiles and greatly improves the convergence and efficiency of AEs [51]. Applying a default GSAE (15 931 genes – 2334 gene sets – 200 gene supersets – 2334 reconstructed gene sets – 15 931 reconstructed genes) to the pan-cancer data, we showed that the built-in regularization constraint preserved the representation of high-dimensional expression profiles (Figure 3C). We utilized the Davies-Bouldin index (DB index) [55] and the average ratio between mean intra-cluster distances to mean inter-cluster distances across cancer classes [distance ratio (DR) index] to quantify the richness of information captured by these dimension reduction methods by comparing intra- and inter-cancer distances. A small index value represents high intra-cancer similarity. The 500 bottleneck nodes of FC-AE1 (Figure 3A) and the 200 gene superset nodes of GSAE (Figure 3C) captured richer cancer type specific information than the top 500 components identified by PCA and NNMF (Figure 3B; DB index, 1.80 and 1.89 versus 2.23 and 3.05; DR index, 0.46 and 0.52 versus 0.71 and 0.85). However, computation time of PCA and NNMF (2.3 and 1.2 CPU hours using MATLAB functions) was shorter than AE (5.4 CPU hours using the CPU version of TensorFlow, or <5 minutes with a 96× CPU-core server). Similar to DCell, GSAE is essentially a VNN model, as scores of its gene-set nodes directly indicate the activation or repression of pathways/functions. At the layer of gene-superset nodes, we can observe the interactions between functions and their contribution to each cancer type [51].
Figure 3.
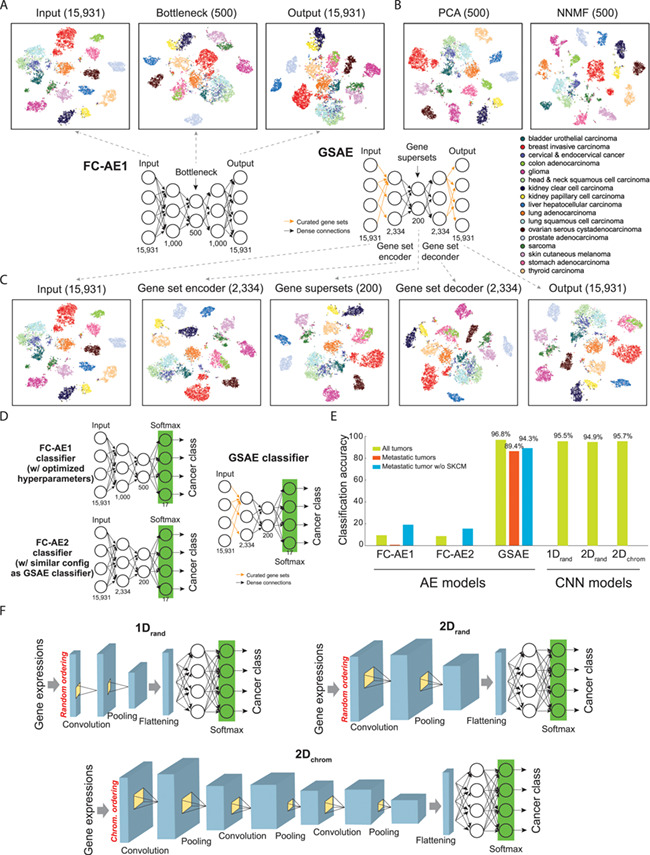
Classifying cancer types using pan-cancer gene expression data. (A) Dimension reduction by fully connected AEs (FC-AE) of 17 cancer types. (B) Dimension reduction by PCA and NNMF. (C) Architecture of GSAE that incorporates curated gene sets into an AE and t-SNE visualization of outputs at each layer. (D) Architectures of models classifying cancer types of primary and metastatic tumors by linking the bottleneck layer of AEs to a classification layer. Performance of a GSAE classifier was compared to two AEs, an FC-AE1 classifier with hyperparameter optimization (as shown in A) and an FC-AE2 classifier that introduced full connections to the GSAE classifier. (E) Performance of AE and CNN-based classifiers of cancer types. Performance of FC-AE1, FC-AE2 and GSAE was assessed by our in-house analysis of 17 largest cancer types using 10-fold cross validation. Performance of 1Drand and 2Drand was reported by [58] on all 33 cancer types by 5-fold cross validation; 2Dchrom was evaluated by [59] on all 33 cancer types by a 10-fold cross validation, where subscript ‘chrom’ denotes genes ordered by chromosomal position and ‘rand’ for genes randomly ordered. (F) Architectures of different embedding methods of gene expression data and CNN models for classifying cancers proposed by [58] and [59].
Classification of cancer types and subtypes by regularized AEs
We then tested whether the representations learned by fully connected AE and GSAE could be used to classify cancer primary or metastatic status. For this purpose, the bottleneck layer of each AE was connected to a 17-neuron prediction layer with a softmax activation, so that each input sample was classified to one of the 17 cancer types (GSAE classifier; Figure 3D). Here we analyzed two fully connected AEs, one optimized by hyperas (FC-AE1 classifier) and the other obtained by introducing full connections between neurons configured identically to GSAE (FC-AE2 classifier; Figure 3D). We performed a 10-fold cross-validation to test each model. In each iteration, 80% of samples were used to train a model with 10% validation to enable early stopping; the remaining 10% were used to test classification accuracy. Here the accuracy was measured by the number of samples that were classified to the correct cancer type divided by the total number of samples. The two fully connected AEs could not be successfully trained for most of the 10 iterations (accuracy, 9.4% and 8.7%; Figure 3E), largely due to the large numbers of weights to be estimated (16.4 and 37.7 million in the FC-AE1 and FC-AE2 classifiers, respectively). Regularization introduced by GSAE markedly reduced the number of weights to 0.65 million and improved the classification accuracy to 96.8% (Figure 3E), suggesting the necessity of incorporating biological knowledge to improve the efficiency of learning.
We also tested the ability of AE to classify the primary site of metastatic tumors. Since most metastatic tumors profiled by TCGA were derived from skin cutaneous melanoma (SKCM; 366 out of 388, or 94.3%), we constructed two models to eliminate potential biases, one classifying all metastatic samples and the other for non-SKCM samples only. The metastatic samples to be classified were excluded from model training. As a result, the GSAE-based network accurately classified the primary sites of these tumors (accuracy, 89.4% for all metastatic samples and 86.4% for the samples excluding SKCM; Figure 3E). Besides the classification of cancer types, Danaee et al. [56] classified tumors versus normal RNA-seq samples by applying a support vector machine (SVM) or simple neural network to the bottleneck-layer outputs of stacked denoising AEs (accuracy, 97.0–98.3%).
Applying an analog network of the GSAE classifier in breast cancer, our recent study also performed four breast cancer subtype classification (basal, Her2, luminal A and luminal B) with an overall accuracy of 88.8% and highly comparable sensitivity and specificity (sensitivity for each subtype, 0.84–0.96; specificity, 0.91–1.00) [51]. Here the subtypes were assessed by the PAM50 gene signature [57] using TCGA RNA-seq data [11]. The results demonstrated a potential application of such a model to classification tasks much more challenging than cancer types.
Classification of cancer types by CNNs
CNNs are recently adopted to classify cancers using gene expression profiles. To generate ‘pseudo-images’ for CNN models to learn from, two studies embedded pre-filtered genes (typically around 10 000) onto one- or two-dimensional maps by random [58] or chromosomal orders [59] and colored each element (or gene) according to abundance of expression (Figure 3F). Thus, expression data of each tumor were converted to an ‘expression image’ and fed into a CNN model. These image embedding methods achieved very similar performance using TCGA data even when used by different CNN models composed of 1–3 convolutional layers (accuracy from cross-validations, 94.9–95.6%; Figure 3E), comparable to regularized AE-based classification. Comparably, using DNA methylation profiles, a recent study evaluated several CNN model configurations and achieved 84.3–92.9% accuracy of classifying 33 cancer types [60]. Altogether, published and our in-house analyses demonstrate the capability of dimension-reducing DL, including AEs regularized by biological knowledge and CNNs, in learning representations of high-dimensional gene expression and other omics data that capture rich information for classifying cancers. We note that TCGA is by far the largest harmonized dataset of cancer genomics. Thus, the aforementioned published and our in-house models were mostly tested using hold-out and/or cross-validations of the TCGA dataset, not in independent datasets where patient selection, clinical parameters annotation and sample preparation may vary drastically. Future works that comprehensively incorporate data resources of Table 2 to implement a more robust DL model and a comprehensive evaluation are warranted.
Predicting drug response and drug synergy of cancer
Data resources and ML methods
CCLE is one of the earliest attempts to conduct systematic high-throughput screening of anti-cancer compounds [15] (Table 3). A total of 504 CCLs were treated with 24 drugs. A dose-response curve was generated for each CCL–drug pair by measuring cellular response (from 0 to 1) across treatment concentrations. Drug sensitivity was measured by the IC50 and area under the curve (AUC). Recently, the Genomics of Drug Sensitivity in Cancer (GDSC) project assayed ~1000 CCLs for their response to 265 anti-cancer drugs [61, 62].
Table 3.
Resources for drug response and drug synergy
| Resource | Measurement of response | Number of treatments | Number of cell lines | Link | References |
|---|---|---|---|---|---|
| Drug response | |||||
| CCLE (Cancer Cell Line Encyclopedia) | IC50 and AUC | 24 drugs | 504 | https://portals.broadinstitute.org/ccle/data | [15] |
| GDSC (Genomics of Drug Sensitivity in Cancer) | IC50 and AUC | 265 drugs | 991 | Supplemental information of [61]; also accessible through the DepMap: https://depmap.org/portal/ | [61, 62] |
| Drug synergy | |||||
| NCI-DREAM (Dialogue on Reverse Engineering Assessment and Methods) | IC20 and ranks | 91 pairs (13 drugs) | 1 (OCI-LY3 cell line) | https://www.synapse.org/NCI_DREAM | [65] |
| OncoPolyPharmacology Screen | VHSA and VBliss scores | 583 pairs (38 drugs) | 39 | Supplemental information of [66] | [66] |
| NCI-ALMANAC (National Cancer Institute-A Large Matrix of Anti-Neoplastic Agent Combinations) | NCI ComboScore | 5232 pairs (104 drugs) | 60 (NCI-60 panel) | https://dtp.cancer.gov/ncialmanac/ | [67] |
AUC, area under the dose response curve; IC20, concentration of drugs needed to kill 20% of cancer cells.
In search of an algorithm for predicting drug sensitivity based on cancer genomics, a collaboration between the NCI and the Dialogue on Reverse Engineering Assessment and Methods (DREAM) project launched a community challenge in 2012 (DREAM7 Challenge). Eventually 44 ML algorithms were evaluated [63]. Each algorithm was trained on response data of 28 drugs in 35 breast CCLs and tested in 18 independent CCLs. Prediction performance was evaluated by a weighted, probabilistic c-index (wpc-index) that measures the similarity between predicted and real ranks of CCLs responding to a drug (drug-centric design). The wpc-index was shown to be highly concordant to the Spearman correlation coefficient ρ [63]. The best-performing algorithm integrated multi-omics and incorporated biological pathways by a non-linear regression model that outperformed all other kernel and regression-based methods. The group reached a consensus that gene expression data have the highest prediction power compared to other single-omics methods, but prediction performance increases when multiple omics data are integrated [63]. Similarly, later ML methods used the GDSC data [61, 62] to identify logic optimization of pairs of alterations (AND/OR operations) predictive of IC50; incorporating different types of genomics data into elastic net regression or random forests models achieved better performance [61]. A network-based method further incorporated the GDSC data, human protein–protein interactome and gene modules derived from mutation and gene expression profiles of TCGA, to prioritize anti-cancer drugs [64].
The heterogeneity of cancer enables the development of resistance to a single drug. Researchers, thus, have studied the synergistic effects of two drugs on cancer cells. The same NCI-DREAM project, DREAM7 Challenge, also issued a challenge to predict the activity of pairs of drugs in 2012 [65]. The challenge evaluated a total of 32 learning algorithms, mostly focused on the similarity/dissimilarity among drugs, using experimentally verified synergistic effects of 91 pairs of drugs against the OCI-LY3 human diffuse large B-cell lymphoma cell line (Table 3). Although the accuracy of these methods was not optimal (largely due to the limited sample size), the challenge demonstrated early promise for predicting drug synergy by ML methods.
Recently, the OncoPolyPharmacology Screen tested cell viabilities for 583 drug combinations of 38 drugs in 39 CCLs [66]. In addition, a benchmark dataset was recently derived by the NCI-ALMANAC (A Large Matrix of Anti-Neoplastic Agent Combinations) project on over 5000 pairs of approved anti-cancer drugs against 60 well-characterized (NCI-60) CCLs [67]. These resources have stimulated recent developments in DL methods to predict drug synergy incorporating genomic data from cancer cells and chemical properties of drugs.
DL models for predicting drug responses of cancer cells and tumors
Different DL models have been designed to analyze genomics profiles of CCLs and to predict response to anti-cancer drugs: one type of DL model predicted CCL–single drug relationships using genomics of CCLs and molecular descriptor/fingerprint describing the drug (Figure 4A, left), and the other simultaneously predicted responses to multiple drugs in CCLs without fingerprinting drugs (Figure 4A, right). Chemical fingerprints are reviewed later in the section ‘Chemoinformatics-facilitated DL models for drug repositioning and discovery.’ Given the same drug screening data, the former type of DL uses 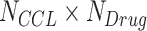 samples, while the latter can be trained on only
samples, while the latter can be trained on only 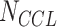 samples. Thus, they require very different training strategies to achieve optimal performance.
samples. Thus, they require very different training strategies to achieve optimal performance.
Figure 4.
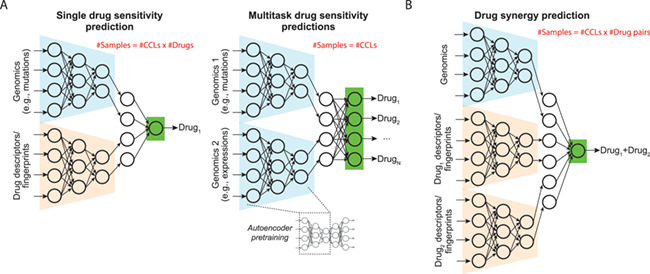
DL models for predicting drug response and synergy. (A) Models for predicting drug sensitivity of a single drug and simultaneous comparisons of multiple drugs. The former model is composed of subnetworks learning from a sample’s genomics profile and chemical descriptor/fingerprint of a drug and yields a drug response score. The latter takes only genomics profiles of samples as input. Each node at the output layer predicts response to a drug. (B) Model for predicting drug synergy. The model learns from genomics of a sample and descriptors/fingerprints of two drugs and outputs a synergy score.
Cancer Drug Response profile scan (CDRscan) is a representative model of the first type of DL models [31]. It is one of the earliest approaches to integrate compound fingerprints with genomic profiles using a DL model. In CDRscan, a model was proposed that combined mutation status of a CCL (28 328 positions in 567 cancer genes extracted from COSMIC) and PaDEL fingerprints of a drug (with 3072 binary features) to yield an IC50 value. CDRscan was trained using 152 594 instances composed of 787 CCLs and 244 anticancer drugs using GDSC data. It achieved much higher prediction performance (mean coefficient of determination R2, 0.84) than conventional ML methods, such as random forest and SVM (R2, 0.70 and 0.56) [31].
While the second type of DL design can simultaneously predict the IC50 of many drugs, it is trained on a much smaller sample size and thus requires a specialized training strategy. We recently proposed a method called DeepDR (Deep learning for Drug Response) [32] that learned from mutation status (18 281 genes) and gene expression profiles (15 363 genes) of 622 CCLs to simultaneously predict IC50 values of 265 anti-cancer drugs screened by GDSC. DeepDR has three sub-networks: an encoder (dimension-reducing) network for data representation of mutations, a similar encoder network for expression data and a prediction network that concatenates outputs of the two encoders and yields 265 IC50 values (Figure 4A, right panel). To facilitate model capability and convergence, we designed a transfer learning scheme between CCLs and tumors with a large number of samples. An AE was trained for each type of genomics data using ~8000 tumors from TCGA to effectively embed tumor-specific characterization into the AE. Parameters (numbers of neurons, edges and weights) of the encoder sub-network were applied to initialize the corresponding encoder network of DeepDR. The entire DeepDR model (including the encoders) was then re-trained using drug screening data from CCLs to optimize predicted drug response. Such a transfer learning design achieved a better prediction performance (CCL-centric Pearson and Spearman ρ of 0.74–0.95 and 0.70–0.92) than a model of similar architecture without transferring data from tumors, as well as linear regression and SVM [32]. Furthermore, this design enabled a biologically meaningful application of the DeepDR model to predict tumors. Application of DeepDR to TCGA data confirmed well-known drug targets (such as epidermal growth factor receptor inhibitors in non-small cell lung cancer and tamoxifen in estrogen receptor-positive breast tumors) and identified novel drugs for further investigation. Alternatively, Matlock et al. [68] have proposed a unique stacking model, in which outputs of DL of individual omics are stacked at the last layer to predict drug sensitivity.
Performance comparison between DL and ML models
For the prediction of drug sensitivity, DL-based models (CDRscan and DeepDR) were shown to outperform simple conventional ML methods, such as linear regression, random forest and SVM, by training errors and/or correlation coefficients metrics [31, 32]. We note that the datasets, training/testing designs and performance measures of CDRscan, DeepDR and sophisticated ML models proposed in the DREAM challenge are quite different, impeding a direct and comprehensive comparison of their performance. CDRscan was trained using 95% of CCL-drug pairs; virtually all CCLs and drugs were seen by the model during the training process. With a focus on predicting new samples (i.e., unscreened CCLs and tumors) that are foreign to the model, DeepDR was trained (90% of CCLs) and tested (10%) using two independent sets of CCLs over all drugs. We re-evaluated the prediction by a drug-centric measure of performance in order to compare to the results of the DREAM challenge. The performance of DeepDR (mean Pearson and Spearman ρ across 265 drugs over 64 CCLs, 0.35 and 0.30) was better than all ML models proposed in the DREAM challenge (resampled Spearman ρ across 28 drugs over 18 CCLs, −0.02 to 0.22 for 44 models). Our data demonstrate the power of DL models to learn from large datasets and warrant further systematic implementation and evaluation of sophisticated ML methods, as well as model interpretation and visualization, to better assessing these ML and DL models.
DL models for predicting drug synergy of cancer cells
DL models have been proposed for drug synergy. They typically take inputs of genomic data of a CCL and descriptors/fingerprints of two drugs of interest (Figure 4B). A DNN model, DeepSynergy, was proposed and used Open Babel [69] fingerprints of drugs and gene expression profiles [33]. The model was trained using the OncoPolyPharmacology data and achieved a Pearson ρ of 0.73 between predicted and original synergy scores. DeepSynergy outperformed many ML methods, including gradient boosting machines, RF, SVM and elastic nets by metrices of mean squared error, Pearson ρ, areas under the receiver operating characteristic curve and precision-recall curve. The AuDNNsynergy (Deep Neural Network Synergy model with Autoencoders) model extended the DeepDR model by introducing an additional genomics profile (CNAs) and molecular fingerprints of two drugs to the prediction sub-network [70]. The model was trained using the same dataset as DeepSynergy and achieved similar correlations of drug combinations (Spearman ρ, 0.56 to 0.81). Alternatively, Xia et al. developed a DL prediction machine using gene expression, miRNA expression and proteomic features, as well as molecular descriptors and fingerprints of two drugs [71]. The model was trained using a more recent and larger synergy screen conducted by NCI-ALMANAC and achieved very high prediction performance (Pearson and Spearman ρ, 0.97 and 0.97).
Chemoinformatics-facilitated DL models for drug repositioning and discovery
Another area of pharmacogenomics research with potential for DL application is drug discovery and development [72]. The application harnesses several strengths of DL to (i) reposition existing drugs by learning similarities between cancer cells and between drugs, (ii) improve clinical performance by identifying synergistic drug combinations and (iii) develop novel compounds by using quantitative structure–activity relationship (QSAR) and refining tool compounds to improve therapeutic index. We note here that QSAR does not consider underlying molecular characteristics of the cells being treated and the interaction between genomics and drugs. We include this topic for it is a very active field in ML and DL that can greatly accelerate the discovery of novel drugs in the near future. More in-depth summaries of DL applications in drug design and activity prediction of ligand–protein interaction can be found in previous reviews [73–76] (Table 1).
A critical step in integrating compound structures into DL models is the representation (chemical descriptors) of compounds using numerical features extracted from chemical structures, similar to those used for compound similarity and activity comparison studies [77]. Common definitions of chemical descriptors are (i) molecular weights, bond counts, fragment counts, etc. (0D and 1D descriptors); (ii) topological descriptors or other graph invariants (2D descriptors); and (iii) geometrical descriptors such as 3D-MoRES [78], and autocorrelation and surface-volume descriptors (3D descriptors). Slightly more abstract, but easily handleable by computers, are molecular fingerprints: binary strings that represent the presence or absence of particular substructure keys of a molecule. The fingerprint (the fixed-length bit-string) contains bits in which each bit represents the absence (0) or presence (1) of a chemical characteristic of molecules. Binary bits are also used to encode discrete variables (similarly for the continuous variables using their ranges). Since different descriptors or descriptor groups can be assigned to different locations within the bit-string, different fingerprint systems can be established, based on different dictionary-assisted bit-string assignment, to group particular functions or fragments within the fingerprint. Table 4 provides a shortlist of such systems or software packages to generate chemical fingerprints. For efficiently processing chemical information, Simplified Molecular Input Line Entry System (SMILES) [79] encodes molecular graphs of compounds into a human-readable line notation of short ASCII (American Standard Code for Information Interchange) strings. The SMILES notations of ~96 million compounds can be downloaded from PubChem [80–83] and processed by most of the aforementioned fingerprint/descriptor software.
Table 4.
Chemoinformatics software to analyze molecular descriptors and fingerprints
| Software | Descriptions | Link | References |
|---|---|---|---|
| CDK | Open-source modular Java libraries for chemoinformatics | https://sourceforge.net/projects/cdk | [156] |
| rcdk | R interface to CDK | https://cran.r-project.org/web/packages/rcdk | [157] |
| ChemmineR | Chemoinformatics package for analyzing drug-like small molecule data in R | https://www.bioconductor.org/packages/release/bioc/vignettes/ChemmineR/inst/doc/ChemmineR.html | [158] |
| Open Babel | Support for more than 100 chemical file formats, fingerprint generation, property determination, similarity and substructure search, structure generation and molecular force fields. Available in C++ with Python, Perl, Java, Ruby, R, etc. | http://openbabel.org | [69] |
| Mordred | 1825 descriptors based on RDKit | https://github.com/mordred-descriptor | [159] |
| RDKit | Major chemoinformatics tool with capability of handling of molecular data, fingerprints, substructure and similarity search and many other functions | http://www.rdkit.org | |
| PaDEL | 1875 descriptors (1444 1D and 2D descriptors and 431 3D descriptors) and 12 types of fingerprints (total 16 092 bits) | http://www.yapcwsoft.com/dd/padeldescriptor/ | [160] |
| KNIME | Graphic development environment that has plugins for chemoinformatics (CDK, RDKit, etc.) and ML modules | https://www.knime.org | [161, 162] |
| PubChem | Fingerprints of 881 bits in length for 2D structures | ftp://ftp.ncbi.nlm.nih.gov/pubchem/specifications/pubchem_fingerprints.txt | [80–83] |
ML models incorporating chemoinformatics to predict drug sensitivity and synergy
It remains challenging to integrate chemical fingerprints into state-of-the-art high-throughput techniques such as genomics, transcriptomics, proteomics, metabolomics and perhaps high-dimensional data sources such as medical imaging to predict drug response. Apart from the aforementioned DL models (CDRscan [31], DeepSynergy [33] and DL designed for the NCI-ALMANAC dataset [71]), many network-based ML methods have been proposed to predict drug sensitivity and synergy (see a review article [84]). Liu et al. [85] formulated drug-response prediction as a recommender system—since similar cell lines and drugs should theoretically exhibit similar responses—and then used the K most similar neighbors (cell line or drug) to predict the unknown ones. Zhang et al. [86] proposed a dual-layer cell line-drug network model, which integrates both cell-line similarity networks (using gene expression data) and drug similarity networks (using PaDEL fingerprints), to predict drug response significantly better than an elastic net model (Pearson ρ > 0.6 with observed responses for most drugs). Similarly, Wei et al. [87] used gene expression for cell-line similarity and Open Babel [69] for drug similarity to build a cell line-drug complex network, and then inferred drug response with a weighted prediction model. In addition, Menden et al. [88], in one of the earliest studies that integrated chemical information from drugs and molecular information from cellular responses, used neural networks and random forest regression models to predict IC50 values of drugs for a given CCL. Recently, Cheng et al. [89] developed a network-based method for predicting drug synergy by incorporating both drug target–disease protein and protein–protein networks. Altogether, these methods demonstrated the feasibility of incorporating chemical fingerprints of drugs and network-based integration with large genomics data to facilitate drug development.
DL models for drug repositioning
Several DL models have been developed to reposition drugs for their anti-cancer capability. Aliper et al. [90] proposed a pioneering DL model that took the input of transcriptomic perturbation signatures of 678 drugs against three CCLs at two different time points derived by The Library of Integrated Network-based Cellular Signatures (LINCS) [91] (see ‘Resources for investigating mechanisms and modes of action for treatments’) without using compound fingerprints. The model achieved superior accuracy of predicting 12 therapeutic categories of Medical Subject Headings over SVM, and incorporation of pathway information into the DL model improved the performance even further. Another study applied a simple DNN to learn low-dimensional representations of the LINCS transcriptomic dataset and utilized the representations to identify functionally similar drugs that complemented structural similarities [92]. Recently, Zeng et al. [93] proposed a network-based DL model for drug repositioning, named deepDR. deepDR integrated multiple networks of drug–disease, drug–side effect, drug–target and drug–drug associations by a multi-modal AE and then used a variational AE (VAE) to incorporate known drug–disease pairs and make recommendations of efficacious drug repositioning. deepDR assessed chemical similarities between drugs by Open Babel fingerprints [69]. The model was tested using curated databases, including DrugBank [94], repoDB [95] and the ClinicalTrials.gov database (https://clinicaltrials.gov/), and deepDR outperformed a broad panel of ML methods. Though deepDR was not developed specifically for cancer therapeutics, we expect its application to cancer datasets can improve discovery and development of anti-cancer drugs. Besides DL models, readers may refer to other extensive review articles on conventional ML methods for drug repositioning [96] and network-based drug repositioning [97].
DL models for novel drug discovery
Many studies have described drug discovery methods that formalize chemical information using QSAR models to virtually screen for novel biologically active compounds [98]. The QSAR models are trained to learn the relationship between chemical properties (such as structures) and experimentally determined biological activities (i.e., cellular response) of a compound, without considering specific molecular characteristics of the cells being treated. Early QSAR models were developed using various conventional ML methods, such as SVM [99–101], support vector regression [102], and a ranking method that directly optimizes the prioritization of compounds [103]. The earliest series of DL methods tackling this task utilized recurrent neural networks (RNNs) for data of a sequential nature, such as natural language processing [104]. For the task of de novo drug design, RNNs are typically trained to generate new molecule structures with desired biochemical properties by iteratively: (i) generating molecules (e.g., a SMILES code), (ii) scoring molecules based on desired bioactivity (predicted property of the SMILES code) and (iii) searching for better molecules (a refined SMILES code) [105]. RNNs were integrated with dimension-reducing AEs or VAE for novel molecules that carry desired properties [106, 107]. Lately, reinforcement learning, a technique that is widely used to fine-tune a DL model by assigning a ‘reward’ signal to the model during model optimization, is incorporated into many RNN models to improve the generation of chemical structures [108–110]. Case studies suggested that 93–95% of the molecules generated by these methods are chemically valid [108–110]. Besides RNNs, a class of AEs that can generate new samples by learning the distribution of data, namely adversarial AEs (AAEs), has been adopted for de novo drug design [82, 111]. A proof-of-concept study trained an AAE model using NCI-60 data of the MCF7 cell line and identified potential anti-cancer drugs by screening ~72 million compounds of the PubChem [80–83]. Very recently, Zhavoronkov et al. [112] built a generative tensorial reinforcement learning (GENTRL) model by incorporating VAE with reinforcement learning to optimize synthetic feasibility, compound novelty and biological activity for de novo drug design. GENTRL successfully designed novel inhibitors of DDR1, a kinase target implied in fibrosis, that were successfully synthesized, validated in cell-based assays and showed favorable response in a mouse pharmacokinetics study. The design, synthesis and experimental validation of these drugs were completed in 46 days, tremendously shortening the drug discovery cycle. Altogether, these exciting results point to the tremendous potential of DL models, especially generative models, to generate and screen for novel drugs that can be hardly matched by conventional ML methods, and we expect they can greatly accelerate cancer drug discovery and development. For benchmarking ML and future DL models on drug discovery, the Pande group at Stanford University established an open-source DL framework (DeepChem; https://deepchem.io/) and multiple curated datasets and evaluation matrices in MoleculeNet [113].
It is worth noting that these promising successes of novel drug discovery using DL models do not take the genomics of cells or tumors being treated into consideration. We expect future research will integrate these drug discovery models with drug response prediction machines based on genomics profiles, such as CDRscan and DeepDR, to achieve the goal of personalized drug discovery.
Resources for investigating mechanisms and modes of action for treatments
To realize the promise of personalized oncology, pharmacological research has sought to understand the mechanisms and modes of action of drugs. The former focuses on the biochemical binding of a drug to its targeting proteins; the latter emphasizes the functional changes in cells upon exposure to a drug. Mechanism-of-action studies focus on how bioactive compounds interact with a targeting enzyme or receptor in the cell and produce their pharmacologic effects. Elucidating mechanisms of action helps to (i) predict which patients may have better responses to treatment and (ii) discover novel therapies.
ChEMBL by EMBL-EBI is the largest database of biochemical activities (~15 million) with an interactive web interface that enables users to search by drugs, target proteins and cells or tissues (Table 5) [114–116]. Other databases focus on the targeted gene variants and proteins, such as PharmGKB (Pharmacogenetics Knowledge Base) [117], Cancer Therapeutics Response Portal (CTRP) [118–120], DrugBank [94], Therapeutic Target Database (TTD) [94], Search Tool for Interacting Chemicals (STITCH) [121] and OncoKB [122]. OncoKB classifies actionable genes by the level of evidence, i.e. levels 1 and 4 contain FDA-approved and biologically proven gene variant–drug pairs, respectively, with the potential to support optimal treatment decisions. In addition to gene variants, the CTRP group tested correlations between basal gene expression levels and the response of 481 compounds on 860 CCLs measured by both IC50 values and area under the dose-response curve [118–120].
Table 5.
Resources for the mechanism and mode of action of chemical and genetic perturbations
| Resource | Genomics target | Number of perturbations or compound–gene associations | Number of cell lines/tumors | Description | Link | References |
|---|---|---|---|---|---|---|
| Mechanisms of action | ||||||
| ChEMBL | Proteins | ~15 Million activities between ~1.7 million compounds and ~11 500 target genes (version 23) | The largest data resource with comprehensive search interface of associations between ligands and protein targets curated from ~30 data sources | https://www.ebi.ac.uk/chembl/ | [114–116] | |
| PharmGKB (Pharmacogenetics Knowledge Base) | Gene alterations; PD and PK pathways | ~20 000 variant–drug pairs of 641 drugs | Curation of variant—PD/PK pathways—drug with clinical annotations | https://www.pharmgkb.org/ | [117] | |
| CTRP (Cancer Therapeutics Response Portal) | GEX | 481 compounds | 860 cell lines | Correlation between basal expression levels of ~19 000 genes and AUC of 481 compounds across cell lines | https://portals.broadinstitute.org/ctrp/ | [118–120] |
| OncoKB | Gene alterations | 86 drugs to ~3900 variants on 477 genes | 60 cancer types | Curated classification of level-1 (FDA-approved) to level-4 (biologically proven) drug targeting variants | http://oncokb.org | [122] |
| Modes of action | ||||||
| Library of Integrated Network-Based Cellular Signatures (LINCS) | GEX (L1000 array: ~1000 assayed and ~11 000 inferred genes); proteins (P100 array: ~100 phosphorylated peptides) | Phase I data (completed): ~20 000 chemical and ~8000 genetic perturbations with several doses and time points | Phase I: 76 cell lines Phase II: 41 Union: 98 | ~1 300 000 (Phase I) and ~350 000 (Phase II) basal and post-treatment GEX profiles normalized to ~476 000 and ~118 000 gene signatures | Cloud-based analysis platform: https://clue.io/ Data Portal: http://lincsportal.ccs.miami.edu/dcic-portal/ Phase I data also available at: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE92742 Phase II data also available at: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE70138 Proteomic data: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE101406 MATLAB, Python, R tools for data processing [123]: https://github.com/cmap | [91] |
| Phase II (version Mar 2017): ~1800 chemical perturbations with several doses and time points | ||||||
| Molecular Signatures Database (MSigDB) – C2.CGP collection (Chemical and Genetic Perturbations) | GEX (significantly changed genes) | ~3400 chemical and genetic perturbations | Curated gene sets of genes changed with chemical and genetic perturbations | http://software.broadinstitute.org/gsea/msigdb/index.jsp | [52–54] | |
AUC, area under the dose response curve; GEX, gene expression; PD, pharmacodynamics; PK, pharmacokinetics.
At the molecular level, the mode of action can be investigated by changes in gene expression profiles associated with drug treatment, such as the signatures curated in the MSigDB (Table 5) [52–54]. The LINCS Project uses a customized microarray, called L1000, to profile baseline and post-treatment gene expression profiles at different time points and doses of treatments with replicates, as an extension of the Connectivity Map (CMap) project. The LINCS Pilot Phase I completed in 2013 generated 1.3 million profiles of ~20 000 chemical (small molecules) and genetic (shRNAs) perturbations in 76 human CCLs (Table 5) [91]. To boost the scale and accessibility of LINCS data, the Production Phase II is currently generating perturbation signatures on more CCLs and improving data coordination and integration. So far, LINCS has generated and provided normalized data on 1.7 million gene expression profiles in a GCTx file format and created software packages available across multiple platforms to efficiently store and access the huge datasets [123] (Table 5). Subramanian et al. [91] applied ML to project and visualize these perturbation signatures and illuminated the mode of action of previously unannotated drugs, revealing promising candidates for clinical trials. Facilitated by the LINCS dataset, a study benchmarked a variety of ML methods, simple DNNs and graph CNN (a CNN architecture that learns from network data) for the prediction of primary sites/subtypes and mechanism of actions [124]. Recently, Deep Compound Profiler (DeepCOP) was developed to predict the perturbation of gene expression by treatments of small-molecule drugs [125].
Despite the development of such comprehensive resources on mechanisms and modes of drug action, only a limited number of DL studies have been carried out using these emerging resources, partly due to the complexity of response time points and treatment dosages. For future studies, our aforementioned dimension reduction models can be used to learn data representation and functional activation underlying expressional profiles associated with different perturbations at different doses and time points. Also, a DL-based incorporation of perturbation signatures (e.g., LINCS)/mechanism of actions (ChEMBL) and drug sensitivity (GDSC)/drug synergy (NCI-ALMANAC) approach may yield insights into the cause of drug resistance, further improve prediction performance and ultimately move forward towards precision oncology.
Conclusions and future directions
We have extensively reviewed data resources that facilitate pharmacogenomic studies and demonstrated how DL methods can be designed to analyze such data, with the ultimate goal of informing precision oncology approaches. Most of the work we have described was based on relatively simpler forms of DL, such as DNNs and AEs. Two rapidly evolving models, CNN and Generative Adversarial Network (GAN), have been extensively used for DNA/RNA sequence analyses [20, 21, 126, 127], but remain very limited in pharmacogenomics applications except for classifying cancer types. We expect these two classes of DL will leap forward when new advances in genomic data embedding method that systematically converts genomic data into informative images are achieved.
For the multi-layered nature of DL, it has much higher learnability but lower interpretability than conventional ML methods; this is the reason why DL machines are often criticized for being black boxes. Such an issue remains to be addressed before DL models move to clinical applications and before government regulation scrutinization [128]. However, as we reviewed in this article, visible models, such as DCell and GSAE, address the problem by embedding prior biological knowledge into model architecture. Biological interpretation (corresponding to the term ‘visualization’ in DL for image analysis) becomes possible by reading the activation states of neurons. Besides, many DL models have been developed to interpret the knowledge learned by neural networks by investigating the relationship between input and output data of a model [129–131] (reviewed in [132]). For instance, interpretation of a cancer type prediction DL model by the saliency map [130] allowed a comprehensive search for potential cancer marker genes and functions [58]. We anticipate that advances in model interpretation will shed light into the black box of DL.
Current large-scale screens for drug sensitivity and drug synergy are mostly performed using cultured CCLs. Although studies have demonstrated that a broad collection of CCLs resembles genomic or pathway alterations found in primary tumors [15, 61, 133–135], it has long been questioned whether in vitro models fully recapitulate primary tumors’ heterogeneity and microenvironment for clinically relevant drug response prediction and drug discovery [136–138]. We expect that the gap between CCLs and primary tumors can be bridged by transfer learning [139]. As demonstrated in the DeepDR study, a transfer learning model sequentially trained on tumor genomics data without any information of drug response (unlabeled data) and drug response data of cell lines (labeled) learned from both datasets and capable of predicting drug response in tumors as reported in actual clinical testing [32]. Such a transfer learning scheme may be further optimized by incorporating data of emerging development of patient-derived xenograft-based screens (reviewed in [140–142]).
The high dimension of genomic features (typically 10 000–30 000 features) requires huge sample sets to successfully train a DL model, such as the well-managed TCGA project, yet it is extremely challenging to collect such datasets in clinical settings and costly to generate genomic profiles. For instance, the use of ‘liquid biopsies’ is an emerging tool for minimally invasive early detection of cancers [143, 144]. While DL was used to capture cancer-related mutations [145], the lack of a large-scale, yet carefully designed collection of liquid biopsies limits more innovative applications. We expect that future breakthroughs in DL methods will comprehensively incorporate biological knowledge into DL models and address the rapid accumulation of pharmacogenomics, genomics and biomedical data to pave the way forward toward precision oncology.
Key points
We present a comprehensive review of genomics and pharmacogenomics data resources that enable DL-based studies towards the goal of precision oncology.
DL models, such as AE and CNN-based models, achieved accurate classifications of cancer types/subtypes by using gene expression and other omics profiles.
DL models can accurately predict drug response and synergy based on cancer genomics and molecular fingerprints of drugs.
DL models incorporating chemoinformatics descriptors and/or fingerprints of drugs show early promise for drug repositioning and discovery.
DL has the potential to be applied to additional pharmacogenomic data resources to study the mechanisms and/or modes of drug action.
Authors’ contributions
YCC, HHC, AG, MM, SZ, YH and YC conceived the study. YCC, AG, MM and YC summarized data resources. YCC and HHC performed data analysis. YCC, SZ, YH and YC interpreted the data. YCC, HHC, AG, MM, SZ, YH and YC wrote and approved the final version of the paper.
Competing interests
The authors declare that they have no competing interests.
Yu-Chiao Chiu PhD is a postdoctoral fellow at the Greehey Children’s Cancer Research Institute at the University of Texas Health San Antonio. His postdoctoral research is focused on developing deep learning models for pharmacogenomic studies.
Hung-I Harry Chen PhD received his PhD from the Department of Electrical and Computer Engineering of the University of Texas at San Antonio, where he participated in this study. His doctoral research was developing computational methods for characterizing gene expression profiles. He is currently a bioinformatics scientist at NGM Biopharmaceuticals, Inc.
Aparna Gorthi PhD is an AACR-AstraZeneca START/NCATS TL1 postdoctoral fellow at the Greehey Children’s Cancer Research Institute. Her research is focused on combining computational modeling for pharmacogenomics with benchtop experiments to elucidate the molecular basis and treatment strategies for pediatric cancers.
Milad Mostavi is a graduate student towards the doctoral degree in the Department of Electrical and Computer Engineering at the University of Texas at San Antonio. His research area is in developing deep learning models with the aim to address existing genomics problems.
Siyuan Zheng PhD is an Assistant Professor in the Department of Population Health Sciences, Greehey Children’s Cancer Research Institute at the University of Texas Health San Antonio. He specializes in cancer genomics, and his research is focused on understanding genomic instability in adult and pediatric cancer.
Yufei Huang PhD is a Professor in the Department of Electrical and Computer Engineering at the University of Texas at San Antonio. His current research interests include uncovering the functions of mRNA methylation using high-throughput sequencing technologies, microRNA functions and target identification, brain–machine interaction using EEG data, and deep learning algorithms and application.
Yidong Chen PhD is a Professor in the Department of Population Health Sciences and the director of Computational Biology and Bioinformatics at Greehey Children’s Cancer Research Institute at the University of Texas Health San Antonio. His research interests include bioinformatics methods in next-generation sequencing technologies, integrative genomic data analysis, genetic data visualization and management and genetic network modeling in translational cancer research.
Funding
This research and this article’s publication costs were supported partially by the National Cancer Institute (NCI) Cancer Center Shared Resources (NIH-NCI P30CA54174 to YC, RP190346 to YC and YH, and RR170055 to SZ), National Institutes of Health (NIH) (CTSA 1UL1RR025767–01 to YC, and R01GM113245 to YH), Cancer Prevention and Research Institute of Texas (CPRIT) (RP160732 to YC, RP190346 to YC and YH, and RR170055 to SZ) and San Antonio Life Sciences Institute (SALSI Innovation Challenge Award 2016 to YH and YC; SALSI Postdoctoral Research Fellowship to YCC). AG is supported by the National Center for Advancing Translational Sciences of the National Institutes of Health TL1 Translational Science Training award (TL1TR002647) and the American Association for Cancer Research-AstraZeneca Stimulating Therapeutic Advances through Research Training grant (18-40-12-GORT). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
References
- 1. Roychowdhury S, Chinnaiyan AM. Translating cancer genomes and transcriptomes for precision oncology. CA Cancer J Clin 2016;66:75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vargas AJ, Harris CC. Biomarker development in the precision medicine era: lung cancer as a case study. Nat Rev Cancer 2016;16:525–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kumar-Sinha C, Chinnaiyan AM. Precision oncology in the age of integrative genomics. Nat Biotechnol 2018;36:46–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ashley EA, Butte AJ, Wheeler MT, et al. Clinical assessment incorporating a personal genome. Lancet 2010;375:1525–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Weinstein JN, Myers TG, O'Connor PM, et al. An information-intensive approach to the molecular pharmacology of cancer. Science 1997;275:343–9. [DOI] [PubMed] [Google Scholar]
- 6. Scherf U, Ross DT, Waltham M, et al. A gene expression database for the molecular pharmacology of cancer. Nat Genet 2000;24:236–44. [DOI] [PubMed] [Google Scholar]
- 7. Butte AJ, Tamayo P, Slonim D, et al. Discovering functional relationships between RNA expression and chemotherapeutic susceptibility using relevance networks. Proc Natl Acad Sci U S A 2000;97:12182–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Potti A, Dressman HK, Bild A, et al. Genomic signatures to guide the use of chemotherapeutics. Nat Med 2006;12:1294–300. [DOI] [PubMed] [Google Scholar]
- 9. Reinhold WC, Varma S, Sunshine M, et al. RNA sequencing of the NCI-60: integration into CellMiner and CellMiner CDB. Cancer Res 2019;79:3514–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. International Cancer Genome C, Hudson TJ, Anderson W, et al. International network of cancer genome projects. Nature 2010;464:993–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cancer Genome Atlas N Comprehensive molecular portraits of human breast tumours. Nature 2012;490:61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Genomes Project C, Auton A, Brooks LD, et al. A global reference for human genetic variation. Nature 2015;526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sudmant PH, Rausch T, Gardner EJ, et al. An integrated map of structural variation in 2,504 human genomes. Nature 2015;526:75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. MacArthur J, Bowler E, Cerezo M, et al. The new NHGRI-EBI catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res 2017;45:D896–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Barretina J, Caponigro G, Stransky N, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012;483:603–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Noor AM, Holmberg L, Gillett C, et al. Big data: the challenge for small research groups in the era of cancer genomics. Br J Cancer 2015;113:1405–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen B, Butte AJ. Leveraging big data to transform target selection and drug discovery. Clin Pharmacol Ther 2016;99:285–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lachmann A, Torre D, Keenan AB, et al. Massive mining of publicly available RNA-seq data from human and mouse. Nat Commun 2018;9:1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. LeCun Y, Bengio Y, Hinton G. Deep learning. Nature 2015;521:436–44. [DOI] [PubMed] [Google Scholar]
- 20. Alipanahi B, Delong A, Weirauch MT, et al. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat Biotechnol 2015;33:831–8. [DOI] [PubMed] [Google Scholar]
- 21. Zhou J, Troyanskaya OG. Predicting effects of noncoding variants with deep learning-based sequence model. Nat Methods 2015;12:931–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schwessinger R, Suciu MC, McGowan SJ, et al. Sasquatch: predicting the impact of regulatory SNPs on transcription factor binding from cell- and tissue-specific DNase footprints. Genome Res 2017;27:1730–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang Y, An L, Xu J, et al. Enhancing Hi-C data resolution with deep convolutional neural network HiCPlus. Nat Commun 2018;9:750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lin C, Jain S, Kim H, et al. Using neural networks for reducing the dimensions of single-cell RNA-Seq data. Nucleic Acids Res 2017;e156:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tian T, Wan J, Song Q, et al. Clustering single-cell RNA-seq data with a model-based deep learning approach. Nat Mach Intell 2019;1:191–8. [Google Scholar]
- 26. Xu Y, Jia Z, Wang LB, et al. Large scale tissue histopathology image classification, segmentation, and visualization via deep convolutional activation features. BMC Bioinformatics 2017;18:281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mobadersany P, Yousefi S, Amgad M, et al. Predicting cancer outcomes from histology and genomics using convolutional networks. Proc Natl Acad Sci U S A 2018;115:E2970–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ching T, Zhu X, Garmire LX. Cox-nnet: an artificial neural network method for prognosis prediction of high-throughput omics data. PLoS Comput Biol 2018;14:e1006076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Katzman JL, Shaham U, Cloninger A, et al. DeepSurv: personalized treatment recommender system using a Cox proportional hazards deep neural network. BMC Med Res Methodol 2018;18:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chaudhary K, Poirion OB, Lu L, et al. Deep learning-based multi-omics integration robustly predicts survival in liver cancer. Clin Cancer Res 2018;24:1248–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chang Y, Park H, Yang HJ, et al. Cancer drug response profile scan (CDRscan): a deep learning model that predicts drug effectiveness from cancer genomic signature. Sci Rep 2018;8:8857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chiu YC, Chen HH, Zhang T, et al. Predicting drug response of tumors from integrated genomic profiles by deep neural networks. BMC Med Genomics 2019;12:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Preuer K, Lewis RPI, Hochreiter S, et al. DeepSynergy: predicting anti-cancer drug synergy with deep learning. Bioinformatics 2018;34:1538–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hyman DM, Taylor BS, Baselga J. Implementing genome-driven oncology. Cell 2017;168:584–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Brower V. NCI-MATCH pairs tumor mutations with matching drugs. Nat Biotechnol 2015;33:790–1. [DOI] [PubMed] [Google Scholar]
- 36. Glorot X, Bengio Y. Understanding the difficulty of training deep feedforward neural networks In: Proceedings of the Thirteenth International Conference on Artificial Intelligence and Statistics, 2010, 249–56.
- 37. Baldi P. Autoencoders, unsupervised learning, and deep architectures In: Proceedings of ICML Workshop on Unsupervised and Transfer Learning, 2012, 37–49.
- 38. Angermueller C, Parnamaa T, Parts L, et al. Deep learning for computational biology. Mol Syst Biol 2016;12:878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Miotto R, Wang F, Wang S, et al. Deep learning for healthcare: review, opportunities and challenges. Brief Bioinform 2018;19:1236–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Telenti A, Lippert C, Chang PC, et al. Deep learning of genomic variation and regulatory network data. Hum Mol Genet 2018;27:R63–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Colaprico A, Silva TC, Olsen C, et al. TCGAbiolinks: an R/bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res 2016;e71:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hutter C, Zenklusen JC. The cancer genome atlas: creating lasting value beyond its data. Cell 2018;173:283–5. [DOI] [PubMed] [Google Scholar]
- 43. Robinson DR, Wu YM, Lonigro RJ, et al. Integrative clinical genomics of metastatic cancer. Nature 2017;548:297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ma X, Liu Y, Liu Y, et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature 2018;555:371–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Grobner SN, Worst BC, Weischenfeldt J, et al. The landscape of genomic alterations across childhood cancers. Nature 2018;555:321–7. [DOI] [PubMed] [Google Scholar]
- 46. Consortium ICG International Cancer Genome Consortium Publications, http://icgc.org/icgc/publications.
- 47. Newton Y, Novak AM, Swatloski T, et al. TumorMap: exploring the molecular similarities of cancer samples in an interactive portal. Cancer Res 2017;77:e111–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Forbes SA, Beare D, Bindal N, et al. COSMIC: high-resolution cancer genetics using the catalogue of somatic mutations in cancer. Curr Protoc Hum Genet 2016;91:10 11 11–37. [DOI] [PubMed] [Google Scholar]
- 49. Ma J, Yu MK, Fong S, et al. Using deep learning to model the hierarchical structure and function of a cell. Nat Methods 2018;15:290–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pumperla M. Keras + Hyperopt: A Very Simple Wrapper for Convenient Hyperparameter Optimization, 2016.
- 51. Chen HH, Chiu YC, Zhang T, et al. GSAE: an autoencoder with embedded gene-set nodes for genomics functional characterization. BMC Syst Biol 2018;12:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Liberzon A, Subramanian A, Pinchback R, et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011;27:1739–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Liberzon A, Birger C, Thorvaldsdottir H, et al. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst 2015;1:417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Davies DL, Bouldin DW. A cluster separation measure. IEEE Trans Pattern Anal Mach Intell 1979;PAMI-1:224–7. [PubMed] [Google Scholar]
- 56. Danaee P, Ghaeini R, Hendrix DA. A deep learning approach for cancer detection and relevant gene identification. Pac Symp Biocomput 2017;22:219–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Parker JS, Mullins M, Cheang MC, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol 2009;27:1160–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mostavi M, Chiu Y-C, Huang Y, et al. Convolutional neural network models for cancer type prediction based on gene expression. arXiv preprint arXiv 2019;1906:07794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lyu B, Haque A. Deep learning based tumor type classification using gene expression data In: Proceedings of the 2018 ACM International Conference on Bioinformatics, Computational Biology, and Health Informatics. Washington, DC: ACM, 2018, 89–96. [Google Scholar]
- 60. Chatterjee S, Iyer A, Avva S, et al. Convolutional neural networks in classifying cancer through DNA methylation. arXiv preprint arXiv 2018;1807:09617. [Google Scholar]
- 61. Iorio F, Knijnenburg TA, Vis DJ, et al. A landscape of pharmacogenomic interactions in cancer. Cell 2016;166:740–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yang W, Soares J, Greninger P, et al. Genomics of drug sensitivity in cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res 2013;41:D955–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Costello JC, Heiser LM, Georgii E, et al. A community effort to assess and improve drug sensitivity prediction algorithms. Nat Biotechnol 2014;32:1202–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Cheng F, Lu W, Liu C, et al. A genome-wide positioning systems network algorithm for in silico drug repurposing. Nat Commun 2019;10:3476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bansal M, Yang J, Karan C, et al. A community computational challenge to predict the activity of pairs of compounds. Nat Biotechnol 2014;32:1213–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. O'Neil J, Benita Y, Feldman I, et al. An unbiased oncology compound screen to identify novel combination strategies. Mol Cancer Ther 2016;15:1155–62. [DOI] [PubMed] [Google Scholar]
- 67. Holbeck SL, Camalier R, Crowell JA, et al. The National Cancer Institute ALMANAC: a comprehensive screening resource for the detection of anticancer drug pairs with enhanced therapeutic activity. Cancer Res 2017;77:3564–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Matlock K, De Niz C, Rahman R, et al. Investigation of model stacking for drug sensitivity prediction. BMC Bioinformatics 2018;19:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. O'Boyle NM, Banck M, James CA, et al. Open Babel: an open chemical toolbox. J Chem 2011;3:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zhang T, Zhang L, Payne PR, et al. Synergistic drug combination prediction by integrating multi-omics data in deep learning models. arXiv preprint arXiv 2018;1811:07054. [DOI] [PubMed] [Google Scholar]
- 71. Xia F, Shukla M, Brettin T, et al. Predicting tumor cell line response to drug pairs with deep learning. BMC Bioinformatics 2018;19:486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ramsundar B, Kearnes S, Riley P, et al. Massively multitask networks for drug discovery. arXiv preprint arXiv 2015;1502:02072. [Google Scholar]
- 73. Ekins S. The next era: deep learning in pharmaceutical research. Pharm Res 2016;33:2594–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Chen H, Engkvist O, Wang Y, et al. The rise of deep learning in drug discovery. Drug Discov Today 2018;23:1241–50. [DOI] [PubMed] [Google Scholar]
- 75. Hessler G, Baringhaus KH. Artificial intelligence in drug design. Molecules 2018;23: 2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Rifaioglu AS, Atas H, Martin MJ, et al. Recent applications of deep learning and machine intelligence on in silico drug discovery: methods, tools and databases. Brief Bioinform 2018: 1–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Todeschini R, Consonni V. Molecular Descriptors for Chemoinformatics: Volume I: Alphabetical Listing/Volume II: Appendices, References. Weinheim: John Wiley & Sons, 2009. [Google Scholar]
- 78. Devinyak O, Havrylyuk D, Lesyk R. 3D-MoRSE descriptors explained. J Mol Graph Model 2014;54:194–203. [DOI] [PubMed] [Google Scholar]
- 79. Weininger D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J Chem Inf Comput Sci 1988;28:31–6. [Google Scholar]
- 80. Kim S, Thiessen PA, Bolton EE, et al. PubChem substance and compound databases. Nucleic Acids Res 2016;44:D1202–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kim S. Getting the most out of PubChem for virtual screening. Expert Opin Drug Discovery 2016;11:843–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Kadurin A, Aliper A, Kazennov A, et al. The cornucopia of meaningful leads: applying deep adversarial autoencoders for new molecule development in oncology. Oncotarget 2017;8:10883–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kim S, Chen J, Cheng T, et al. PubChem 2019 update: improved access to chemical data. Nucleic Acids Res 2019;47:D1102–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Tsigelny IF. Artificial intelligence in drug combination therapy. Brief Bioinform 2019;20:1434–48. [DOI] [PubMed] [Google Scholar]
- 85. Liu H, Zhao Y, Zhang L, et al. Anti-cancer drug response prediction using neighbor-based collaborative filtering with global effect removal. Mol Ther Nucleic Acids 2018;13:303–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Zhang N, Wang H, Fang Y, et al. Predicting anticancer drug responses using a dual-layer integrated cell line-drug network model. PLoS Comput Biol 2015;e1004498:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Wei D, Liu C, Zheng X, et al. Comprehensive anticancer drug response prediction based on a simple cell line-drug complex network model. BMC Bioinformatics 2019;20:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Menden MP, Iorio F, Garnett M, et al. Machine learning prediction of cancer cell sensitivity to drugs based on genomic and chemical properties. PLoS One 2013;e61318:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Cheng F, Kovacs IA, Barabasi AL. Network-based prediction of drug combinations. Nat Commun 2019;10:1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Aliper A, Plis S, Artemov A, et al. Deep learning applications for predicting pharmacological properties of drugs and drug repurposing using transcriptomic data. Mol Pharm 2016;13:2524–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Subramanian A, Narayan R, Corsello SM, et al. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell 2017;171:1437–52 e1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Donner Y, Kazmierczak S, Fortney K. Drug repurposing using deep embeddings of gene expression profiles. Mol Pharm 2018;15:4314–25. [DOI] [PubMed] [Google Scholar]
- 93. Zeng X, Zhu S, Liu X, et al. deepDR: a network-based deep learning approach to in silico drug repositioning. Bioinformatics 2019:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Wishart DS, Feunang YD, Guo AC, et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res 2018;46:D1074–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Brown AS, Patel CJ. A standard database for drug repositioning. Sci Data 2017;4:170029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Yella JK, Yaddanapudi S, Wang Y, et al. Changing trends in computational drug repositioning. Pharmaceuticals (Basel) 2018;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Cheng F, Hong H, Yang S, et al. Individualized network-based drug repositioning infrastructure for precision oncology in the panomics era. Brief Bioinform 2017;18:682–97. [DOI] [PubMed] [Google Scholar]
- 98. Kubinyi H. Quantitative structure-activity relationships (QSAR) and molecular modelling in cancer research. J Cancer Res Clin Oncol 1990;116:529–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Warmuth MK, Liao J, Rätsch G, et al. Active learning with support vector machines in the drug discovery process. J Chem Inf Comput Sci 2003;43:667–73. [DOI] [PubMed] [Google Scholar]
- 100. Jorissen RN, Gilson MK. Virtual screening of molecular databases using a support vector machine. J Chem Inf Model 2005;45:549–61. [DOI] [PubMed] [Google Scholar]
- 101. Geppert H, Horváth T, Gärtner T, et al. Support-vector-machine-based ranking significantly improves the effectiveness of similarity searching using 2D fingerprints and multiple reference compounds. J Chem Inf Model 2008;48:742–6. [DOI] [PubMed] [Google Scholar]
- 102. Prakash O, Khan F. Cluster based SVR-QSAR modelling for HTS records: an implementation for anticancer leads against human breast cancer. Comb Chem High Throughput Screen 2013;16:511–21. [DOI] [PubMed] [Google Scholar]
- 103. Agarwal S, Dugar D, Sengupta S. Ranking chemical structures for drug discovery: a new machine learning approach. J Chem Inf Model 2010;50:716–31. [DOI] [PubMed] [Google Scholar]
- 104. Mikolov T, Karafiát M, Burget L, et al. Recurrent neural network based language model In: Eleventh Annual Conference of the International Speech Communication Association, 2010.
- 105. Segler MHS, Kogej T, Tyrchan C, et al. Generating focused molecule libraries for drug discovery with recurrent neural networks. ACS Cent Sci 2018;4:120–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Gomez-Bombarelli R, Wei JN, Duvenaud D, et al. Automatic chemical design using a data-driven continuous representation of molecules. ACS Cent Sci 2018;4:268–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Blaschke T, Olivecrona M, Engkvist O, et al. Application of generative autoencoder in de novo molecular design. Mol Inform 2018;37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Olivecrona M, Blaschke T, Engkvist O, et al. Molecular de-novo design through deep reinforcement learning. J Chem 2017;9:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Popova M, Isayev O, Tropsha A. Deep reinforcement learning for de novo drug design. Sci Adv 2018;4: eaap7885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Stahl N, Falkman G, Karlsson A, et al. Deep reinforcement learning for multiparameter optimization in de novo drug design. J Chem Inf Model 2019;59:3166–76. [DOI] [PubMed] [Google Scholar]
- 111. Kadurin A, Nikolenko S, Khrabrov K, et al. druGAN: an advanced generative adversarial autoencoder model for de novo generation of new molecules with desired molecular properties in silico. Mol Pharm 2017;14:3098–104. [DOI] [PubMed] [Google Scholar]
- 112. Zhavoronkov A, Ivanenkov YA, Aliper A, et al. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat Biotechnol 2019. [DOI] [PubMed] [Google Scholar]
- 113. Wu Z, Ramsundar B, Feinberg Evan N, et al. MoleculeNet: a benchmark for molecular machine learning. Chem Sci 2018;9:513–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Bento AP, Gaulton A, Hersey A, et al. The ChEMBL bioactivity database: an update. Nucleic Acids Res 2014;42:D1083–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Davies M, Nowotka M, Papadatos G, et al. ChEMBL web services: streamlining access to drug discovery data and utilities. Nucleic Acids Res 2015;43:W612–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Gaulton A, Hersey A, Nowotka M, et al. The ChEMBL database in 2017. Nucleic Acids Res 2017;45:D945–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Hewett M, Oliver DE, Rubin DL, et al. PharmGKB: the pharmacogenetics knowledge base. Nucleic Acids Res 2002;30:163–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Basu A, Bodycombe NE, Cheah JH, et al. An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell 2013;154:1151–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Seashore-Ludlow B, Rees MG, Cheah JH, et al. Harnessing connectivity in a large-scale small-molecule sensitivity dataset. Cancer Discov 2015;5:1210–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Rees MG, Seashore-Ludlow B, Cheah JH, et al. Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat Chem Biol 2016;12:109–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Szklarczyk D, Santos A, von Mering C, et al. STITCH 5: augmenting protein–chemical interaction networks with tissue and affinity data. Nucleic Acids Res 2016;44:D380–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Chakravarty D, Gao J, Phillips SM, et al. OncoKB: a precision oncology knowledge base. JCO Precis Oncol 2017;2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Enache OM, Lahr DL, Natoli TE, et al. The GCTx format and cmap {Py, R, M, J} packages: resources for optimized storage and integrated traversal of annotated dense matrices. Bioinformatics 2019;35:1427–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. McDermott M, Wang J, Zhao WN, et al. Deep learning benchmarks on L1000. Gene expression data. IEEE/ACM Trans Comput Biol Bioinform 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Woo G, Fernandez M, Hsing M, et al. DeepCOP—deep learning-based approach to predict gene regulating effects of small molecules. Bioinformatics 2019:1–6. [DOI] [PubMed] [Google Scholar]
- 126. Zeng H, Edwards MD, Liu G, et al. Convolutional neural network architectures for predicting DNA-protein binding. Bioinformatics 2016;32:i121–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Killoran N, Lee LJ, Delong A, et al. Generating and designing DNA with deep generative models. arXiv preprint arXiv 2017;1712:06148. [Google Scholar]
- 128. US Food and Drug Administration Proposed Regulatory Framework for Modifications to Artificial Intelligence/Machine Learning (AI/ML)-Based Software as a Medical Device (SaMD)-Discussion Paper and Request for Feedback, White Oak, 2019. [Google Scholar]
- 129. Yosinski J, Clune J, Nguyen A, et al. Understanding neural networks through deep visualization. arXiv preprint arXiv 2015;1506:06579. [Google Scholar]
- 130. Simonyan K, Vedaldi A, Zisserman A. Deep inside convolutional networks. Visualising image classification models and saliency maps. arXiv preprint arXiv 2013;1312:6034. [Google Scholar]
- 131. Samek W, Binder A, Montavon G, et al. Evaluating the visualization of what a deep neural network has learned. IEEE Trans Neural Netw Learn Syst 2017;28:2660–73. [DOI] [PubMed] [Google Scholar]
- 132. Q-s Z, Zhu S-c. Visual interpretability for deep learning: a survey. Front Info Tech Electron Eng 2018;19:27–39. [Google Scholar]
- 133. Tsherniak A, Vazquez F, Montgomery PG, et al. Defining a cancer dependency map. Cell 2017;170:564–76 e516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Behan FM, Iorio F, Picco G, et al. Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature 2019;568:511–6. [DOI] [PubMed] [Google Scholar]
- 135. Goodspeed A, Heiser LM, Gray JW, et al. Tumor-derived cell lines as molecular models of cancer pharmacogenomics. Mol Cancer Res 2016;14:3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Gillet JP, Calcagno AM, Varma S, et al. Redefining the relevance of established cancer cell lines to the study of mechanisms of clinical anti-cancer drug resistance. Proc Natl Acad Sci U S A 2011;108:18708–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Gillet JP, Varma S, Gottesman MM. The clinical relevance of cancer cell lines. J Natl Cancer Inst 2013;105:452–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Wilding JL, Bodmer WF. Cancer cell lines for drug discovery and development. Cancer Res 2014;74:2377–84. [DOI] [PubMed] [Google Scholar]
- 139. Pan SJ, Yang Q. A survey on transfer learning. IEEE Trans Knowl Data Eng 2010;22:1345–59. [Google Scholar]
- 140. Herter-Sprie GS, Kung AL, Wong KK. New cast for a new era: preclinical cancer drug development revisited. J Clin Invest 2013;123:3639–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Day CP, Merlino G, Van Dyke T. Preclinical mouse cancer models: a maze of opportunities and challenges. Cell 2015;163:39–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Pompili L, Porru M, Caruso C, et al. Patient-derived xenografts: a relevant preclinical model for drug development. J Exp Clin Cancer Res 2016;35:189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Aravanis AM, Lee M, Klausner RD. Next-generation sequencing of circulating tumor DNA for early cancer detection. Cell 2017;168:571–4. [DOI] [PubMed] [Google Scholar]
- 144. Cohen JD, Li L, Wang Y, et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018;359:926–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Kothen-Hill ST, Zviran A, Schulman RC, et al. Deep Learning Mutation Prediction Enables Early Stage Lung Cancer Detection in Liquid Biopsy, In: International Conference on Learning Representations, 2018.
- 146. Min S, Lee B, Yoon S. Deep learning in bioinformatics. Brief Bioinform 2016;18:851–69. [DOI] [PubMed] [Google Scholar]
- 147. Eraslan G, Avsec Z, Gagneur J, et al. Deep learning: new computational modelling techniques for genomics. Nat Rev Genet 2019;20:389–403. [DOI] [PubMed] [Google Scholar]
- 148. Kalinin AA, Higgins GA, Reamaroon N, et al. Deep learning in pharmacogenomics: from gene regulation to patient stratification. Pharmacogenomics 2018;19:629–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Vamathevan J, Clark D, Czodrowski P, et al. Applications of machine learning in drug discovery and development. Nat Rev Drug Discov 2019;18:463–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Network TCGA TCGA Research Network Publications, https://cancergenome.nih.gov/publications.
- 151. Cancer Cell Line Encyclopedia C, Genomics of Drug Sensitivity in Cancer C . Pharmacogenomic agreement between two cancer cell line data sets. Nature 2015;528:84–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Cerami EG, Gross BE, Demir E, et al. Pathway Commons, a web resource for biological pathway data. Nucleic Acids Res 2011;39:D685–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Pratt D, Chen J, Welker D, et al. NDEx, the network data exchange. Cell Syst 2015;1:302–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Consortium GT. The genotype-tissue expression (GTEx) project. Nat Genet 2013;45:580–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Consortium GT, Laboratory DA, Coordinating Center -Analysis Working G, et al. Genetic effects on gene expression across human tissues. Nature 2017;550:204–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Steinbeck C, Hoppe C, Kuhn S, et al. Recent developments of the chemistry development kit (CDK)—an open-source java library for chemo- and bioinformatics. Curr Pharm Des 2006;12:2111–20. [DOI] [PubMed] [Google Scholar]
- 157. Guha R. Chemical Informatics Functionality in R. J Stat Softw 2007;18:1–16. [Google Scholar]
- 158. Cao Y, Charisi A, Cheng LC, et al. ChemmineR: a compound mining framework for R. Bioinformatics 2008;24:1733–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Moriwaki H, Tian YS, Kawashita N, et al. Mordred: a molecular descriptor calculator. J Chem 2018;10:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Yap CW. PaDEL-descriptor: an open source software to calculate molecular descriptors and fingerprints. J Comput Chem 2011;32:1466–74. [DOI] [PubMed] [Google Scholar]
- 161. Berthold MR, Cebron N, Dill F, et al. KNIME-the Konstanz information miner: version 2.0 and beyond. AcM SIGKDD Explorations Newsletter 2009;11:26–31. [Google Scholar]
- 162. Beisken S, Meinl T, Wiswedel B, et al. KNIME-CDK: workflow-driven cheminformatics. BMC Bioinformatics 2013;14:257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Guha R, Bender A. Computational Approaches in Cheminformatics and Bioinformatics. John Wiley & Sons, 2011. [Google Scholar]
- 164. Pirhadi S, Sunseri J, Koes DR. Open source molecular modeling. J Mol Graph Model 2016;69:127–43. [DOI] [PMC free article] [PubMed] [Google Scholar]