

Abstract
Infrared multiphoton dissociation (IRMPD) has been used in mass spectrometry to fragment peptides and proteins, providing fragments mostly similar to collisional activation. Using the 10.6 μm wavelength of a CO2 laser, IRMPD suffers from the relative low absorption cross-section of peptides and small proteins. Focusing on top-down analysis, we investigate different means to tackle this issue. We first reassess efficient sorting of phosphopeptides from nonphosphopeptides based on IR-absorption cross-sectional enhancement by phosphate moieties. We subsequently demonstrate that a myo-inositol hexakisphosphate (IP6) noncovalent adduct can substantially enhance IRMPD for nonphosphopeptides and that this strategy can be extended to proteins. As a natural next step, we show that native phospho-proteoforms of proteins display a distinct and enhanced fragmentation, compared to their unmodified counterparts, facilitating phospho-group site localization. We then evaluate the impact of size on the IRMPD of proteins and their complexes. When applied to protein complexes ranging from a 365 kDa CRISPR–Cas Csy ribonucleoprotein hetero-decamer, a 800 kDa GroEL homo-tetradecamer in its apo-form or loaded with its ATP cofactor, to a 1 MDa capsid-like homo-hexacontamer, we conclude that while phosphate moieties present in crRNA and ATP molecules enhance IRMPD, an increase in the IR cross-section with the size of the protein assembly also favorably accrues dissociation yields. Overall, our work showcases the versatility of IRMPD in the top-down analysis of peptides, phosphopeptides, proteins, phosphoproteins, ribonucleoprotein assemblies, and large protein complexes.
Introduction
Structural characterization of proteins by mass spectrometry is critically dependent on the fragmentation technique used. Collisional activation (CID/HCD)—the most commonly used technique—usually effectively dissociates small peptides and small denatured proteins. However, as the size of native compounds increases, the number of detected CID/HCD backbone cleavages decreases, making CID/HCD less favorable for top-down analysis.1 This has led to the implementation of alternative dissociation techniques for larger proteins and native protein complexes making use of surfaces, electrons, and/or photons.2,3
One alternative activation method to CID/HCD is infrared multiphoton photodissociation (IRMPD). Applied for the first time to proteins—ubiquitin (8.6 kDa) and carbonic anhydrase (29 kDa)—already about 25 years ago,4 IRMPD has significant advantages. These include a high level of control over the energy added by photons, no gas load in the vacuum of the mass analyzer, effective trapping of fragments at low RF voltages in multipoles, and the ability to induce the cleavage of certain disulfide bonds (as illustrated for an intact antibody in Figure S1).3,5,6 Performed using a cost-effective continuous-wave(CW) CO2 laser, IRMPD is commonly described as nonselective—both parent ions and product ions are irradiated and dissociated—which can be beneficial because of the large variety of product ions formed.7 Applied to peptides and phosphopeptides in particular, IRMPD has been found to yield more extensive sequence information than CID/HCD, primarily because of its ability to form secondary and higher order fragments upon the absorption of multiple photons.8 While essentially independent of ion optics, the effectiveness of IRMPD depends on irradiance, the number of excitable chromophores, as well as the channels open for energy redistribution and relaxation. For CO2 laser-driven IRMPD, some common absorbers are the CH3-rocking, O–H bend, P–O, P–O–C, and P–O–P stretch vibrations, while de-excitation mainly involves collisional cooling at the pressures commonly achieved in multipoles.
Compared to peptides and denatured proteins, IRMPD of intact, quasi-native proteins and their assemblies is a more recent development that involves electrospraying compounds from solutions mimicking physiological conditions. To date, IRMPD dissociation into subunits has been demonstrated for avidin (AT, 64 kDa, 4-mer), GroEL (803 kDa, 14-mer), and CS2 hydrolase (189 kDa, 8-mer) on a ToF9 as well as glycogen phosphorylase (193 kDa, 2-mer), glutamate dehydrogenase (GDH, 334 kDa 6-mer), and β-galactosidase (GTD, 464 kDa, 4-mer) on an FT-ICR mass spectrometer.10 While primarily yielding extensive sequence coverage for denatured proteins in conjunction with electron capture dissociation, IRMPD has also been found to yield valuable native top-down information—protein backbone cleavages—as demonstrated by Loo and co-workers for selected compounds.10
Building upon Muddiman’s and Brodbelt’s groups selective 10.6 μm IRMPD of phosphopeptides over nonphosphorylated ones,8,11 here, we chose to revisit the possibility of IRMPD enhancement by phosphate moieties (beyond phosphopeptides) to structurally probe peptides, native proteins, and protein assemblies. This broad focus is motivated by the absorption bands of molecules such as myo-inositol hexakisphosphate,12 adenosine triphosphate (ATP),13 and (deoxy)ribonucleic acids (DNA and RNA)14 about the 10,600 nm ≡ 943 cm–1 excitation line of a CO2 laser. Over the past decade, gas-phase IR action (photodissociation) spectra corresponding to the 800–1900 cm–1 range have been recorded using free-electron lasers for phosphocompounds ranging from protonated phosphothreonine, phosphotyrosine, phosphoserine,15−17 2′-deoxyadenosine-5′-monophosphate, and adenosine-5′-monophosphate cations18 to deprotonated 2′-deoxynucleotide-5′-monophosphate anions19 and nucleotide 5′-triphosphate anions.20 All demonstrate substantial absorption around 943 cm–1. Consequently, following the demonstration that our IRMPD implementation on an EMR Q-Exactive Orbitrap can selectively dissociate phosphopeptides, we highlight the impact of noncovalent phospho-adducts on the IRMPD of peptides and native proteins. Applied to the native phospho-proteoforms of proteins, IRMPD yields distinct and enhanced fragmentation patterns, compared to their unmodified counterparts, facilitating phospho-group site localization. For increasing protein sizes and, therefore, IR absorption cross-sections, we show that 10.6 μm IRMPD preferentially cleaves native proteins at aspartic acid (D, Asp) and glutamic acid (E, Glu) residues concurrently with an extensive concomitant golden ion pair—complementary b and y ions—formation.21 This process, rationalized in terms of direct cleavage of the neighboring peptide bond by the aspartic acid proton, appears enhanced for IRMPD compared to CID/HCD likely due to the favorable absorption of Asp’s and Glu’s carboxylic moiety about 943 cm–1. We conclude by assessing IRMPD for noncovalent complexes and ribonucleoproteins ranging from a few hundred kDa to 1 MDa, making use of the fact that RNA and/or ATP bound to protein assemblies will also enhance IR absorption.
Experimental Section
Materials
Peptide and protein standards were, unless described otherwise, purchased from Sigma-Aldrich (Merck KGaA, Germany). The phosphopeptides were custom-synthesized by Dris El Atmioui in the Huib Ovaa laboratory (LUMC, Leiden). Bora was expressed and phosphorylated in vitro by using the aurora kinase as described previously in ref (22). The CRISPR–Cas complex was provided by the Fineran group and expressed and purified as described in ref (23). The GroEL complex was expressed and purified as described in refs.24−26 The AaLS virus-like capsid was provided by the Hilbert group and expressed and purified as described in ref (27).
While peptides were simply solubilized in aqueous 150 mM NH4OAc (pH 6.8), all analyzed proteins were buffer exchanged to aqueous 150 mM NH4OAc (pH 6.8) using Amicon ultracentrifugal filters (Millipore, Merck KGaA, Germany) with a 10 kDa cutoff (5 kDa for ubiquitin). As typical for native electrospray ionization, protein and peptide concentrations in between 1 and 5 μM were used.
Myo-inositol hexakisphosphate (IP6), also called phytic acid [50% (w/w) solution in H2O, Sigma-Aldrich #593648], was desalted using Supel-Select SCX columns. Immobilization was followed by extensive washing using a 750 mM NH4OAc solution. Elution involved a concentrated formic acid solution (pH 2), followed by neutralization using NH4OH. Following solvent evaporation, the eluate was resolubilized in water. We estimated IP6 losses upon desalting to be on the order of 50%. IP6 was added to concentrated 150 mM ammonium acetate solutions of peptides and proteins, resulting in 100:1 eq/eq excess. The optimal amount of IP6 added depends on the pH, ionic strength, and concentration of the binding peptide or protein. Typical adduct yields are displayed in Figure S7.
Native Top-Down MS
Top-down MS of native complexes was performed on an extended mass range (EMR) Q-Exactive orbitrap28 (Thermo Fisher Scientific, Bremen, Germany), as schematized in Figure 1. All compounds were electrosprayed with homemade gold-coated borosilicate capillaries using voltages in the 1.1–1.7 kV range. All spectra were acquired by setting the noise threshold parameter to 3.64. The standard resolution was 140,000 @ m/z 200. The extended m/z range of the EMR orbitrap used enables us to simultaneously record precursor ions, ejected subunits, high m/z product ions, and peptide fragments. Using IRMPD and low-pressure conditions, we can make full use of the long transient records, resulting in instrumental resolutions enabling isotopic resolution up to a few tens of kDa.
Figure 1.
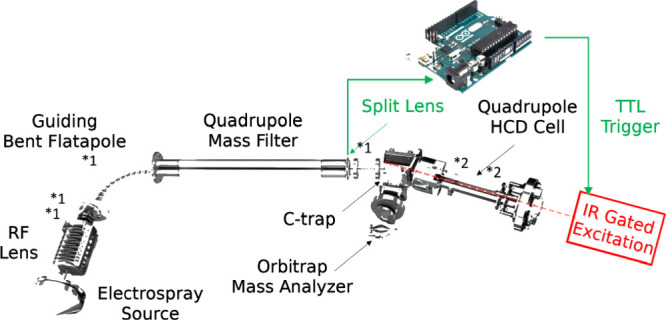
Diagram of the coupling of the IR-laser to the EMR orbitrap. The main parameters altered to optimize detection for large complex subunits and fragments are displayed with an asterisk: *1 for source-side deceleration of the ions of interest and *2 for trapping in and ejection from the HCD cell (details in text).
Implementation of IRMPD
For the IRMPD experiments, precursor ions were transferred to the HCD cell at a low kinetic energy [HCD direct eV setting = 1 (peptide) or 0 (proteins)] to prevent dissociation. The IRMPD mass spectra were acquired using a 10.6 μm gated laser pulse of 80 ms (ti60HS Firestar, Synrad, Mukilteo, WA, U.S.A.) at laser powers reaching 95 W (57 W effective due to a 60% transmission into the HCD cell) as described in the next section. The HCD cell trapping and extraction parameters were optimized for low-nitrogen collision gas pressures to achieve efficient detection of the subunits and fragments. Following mass selection in the linear quadrupole, the ions were transferred and stored in the HCD cell.
For the implementation of IRMPD on the EMR orbitrap, we used a design similar to the one previously used for UVPD and comparable to other CW setups.29,30 A BaF2 viewport was added to the back of the HCD cell adjacent to the C-trap and a BaF2 window in lieu of the HCD cell electrometer. The laser beam divergence was compensated using an adjustable beam reducer consisting of zinc selenide lenses with 50 mm and −25 mm focal lengths. Coaxial adjustment of the 10.6 μm laser beam with the HCD cell multipole, following its reduction by an aperture diameter of 2 mm, was achieved using a two mirrors–periscope configuration, which could be advantageously replaced by an optical fiber coupler.31 As previously described,29 the instrument Split Lens and HCD Exit test points were used to generate TTL trigger pulses using voltage comparators. An Arduino microcontroller was used both to provide the laser with gating TTL pulses and to ensure their synchronization 3 ms after injection into the HCD cell, as determined from processing the signals of the test points. Optimal operation was observed at very low nitrogen pressures (around 5.5 × 10–11 mbar readout on the UHV pressure gauge). Pressure was measured indirectly via monitoring the UHV readouts unscaled for the gas type. Effective pressures in the HCD cell are estimated to be in the 10–5 to 10–4 mbar range. To ensure a long enough observation time window, a detection delay (IonGun Time) of 150 ms was used without significantly affecting the duty cycle at a 140,000 @ m/z 200 resolution characterized by 512 ms transients.
Differently, compared to the setup of Brodbelt and co-workers who implemented IRMPD on a LTQ orbitrap Velos,32 here, no additional gasline was added to the C-trap to compensate for the low HCD cell pressure required to enable IRMPD. We instead optimized the setup to achieve efficient ion transfer at low kinetic energies. Among critical potential differences, the HCD cell DC offset relative to the C-trap—HCD direct eV parameter—was set to 0 (effective offset of 10 V) or 1 (effective offset of 1 V) whenever possible. The HCD cell DC offset of 1 was used to preclude collisional activation of the ions upon entry into the HCD cell and thereby ensure that all fragments detected resulted from the absorption of IR photons. The low pressure in the HCD cell C-trap assembly, although found to affect the transmission and detection of higher m/z precursors and product ions, improved their detection at high mass resolution.
For MS2 experiments on large protein assemblies, precursor ions of a single charge state were isolated using a 50–150 m/z window. MS/MS experiments were performed with the following settings: 1 laser pulse at a 0–95 W CW intensity typically gated to 80 ms. By directing the original beam into the HCD cell, the beam has been measured to retain 60% of its intensity. Consequently, 80 ms laser pulses range from 0 J/pulse (0%) to 4.56 J/pulse (95%) for a beam diameter of 2 mm. Assuming a uniform energy distribution, the irradiances used range from 0 to 1814 W/cm2 for a total pulse duration of 80 ms (fluence between 0 and 145 J/cm2, energy between 0 and 4.56 J/pulse, and % maximum laser intensity from 0 to 95).
Data Analysis
Processing of the fragmentation spectra involved conversion of the raw files to mzML. We used the MSDeisotope python library (Joshua Klein, Boston University CBMS) with minimum_score = 2.0 and mass_error_tolerance = 0.02 for peptides and minimum_score = 10.0 and mass_error_tolerance = 0.02 for all proteins except Bora to generate charge deconvoluted—all ions are 1+—spectra with all the isotopic peaks retained.33,34 For Bora, Xtract, as implemented in Thermo Xcalibur 4.2.28.14 (Thermo Fisher Scientific), was used with S/N 1.5, Fit Factor 60%, and Remainder 5%. The fragment assignment involved LcMsSpectator35 (Pacific Northwest National Laboratory) on the charged deconvoluted spectra generated by the MSDeisotope library. The accuracy threshold was set to 5 ppm for all assignments with the results exported as .tsv files for further analysis. Sequence assignments accommodated the major IRMPD ion types (b, y) without taking into account H2O and NH3 neutral losses, except when explicitly mentioned.
Results
Implementation of IRMPD on a Q-Exactive EMR Orbitrap
The primary obstacle for using IRMPD in a Q-Exactive orbitrap HCD cell, as carried out in the present work, is collisional de-excitation which hinders efficient dissociation at normal operating pressures and temperatures.9 Our implementation of IRMPD on a Q-Exactive EMR orbitrap is schematically depicted in Figure 1. We basically used a design similar to the one we previously reported for UVPD29 (for details, see Experimental Section). For irradiances commonly achieved by a 10–50 W CO2 laser combined with a standard quadrupole ion trap, room-temperature dissociation efficiency drops between 2.7 × 10–4 and 5.3 × 10–4 mbar (uncorrected He pressure) because of effective collisional quenching at these pressures. At pressures in excess of 1 × 10–3 mbar, the dissociation efficiency is essentially zero in the absence of beam focusing.7,36,37 Here, we tackled this issue by operating the HCD cell at quite low static pressures.
Phosphopeptides
As initially shown by Brodbelt et al., IRMPD can selectively fragment phosphorylated over nonphosphorylated peptides because of the larger absorption cross-section of the former at 943 cm–1.8,11,32 To test our IRMPD setup as implemented on the EMR orbitrap, we decided to first reassess these findings. In agreement with these earlier reports, we also observed dramatically enhanced IRMPD fragmentation of a variety of synthetic phosphopeptides over the corresponding nonphosphorylated peptides, as shown in Figures 2, S2, and S3 for the peptides [SVTPKTVTPASSAKT(p)SPAK, SV(p)TPKTV(p)TPAS(p)SAKT(p)SPAK] and Figures S4–S6 [TPA(p)TPTSSAS, GNSP(p)TPVSRW, LSPA(p)TPTSEG]. At irradiances enabling fragmentation, IRMPD-driven metaphosphoric acid loss (−HPO3) led to fragments similar to those observed in HCD/CID. Optimal fragmentation yields reached values ranging from 90 to 100% for all phosphorylated peptides investigated, while this yield remained below 10% for their nonphosphorylated counterparts. Using the maximal irradiance currently available to us, only one nonphosphorylated peptide (GNSPTPVSRW, Figure S5) underwent some IRMPD (fragmentation yield of 35%), while its phosphorylated counterpart fully dissociated under identical conditions. The number of phosphate groups present on a peptide also affected the extent of fragmentation as seen in Figures 2 and S2, where we observed that a quadruply phosphorylated peptide fragments already at much lower irradiances than the corresponding singly phosphorylated peptide, whereas the nonphosphorylated counterpart hardly fragmented even at the highest irradiances. Overall, based on the fragments detected, IR activation at 943 cm–1 very effectively cleaved the backbone of phosphopeptides, suggesting an efficient intramolecular energy redistribution leading to a nonpreferential cleavage site.
Figure 2.

IRMPD of the (a–c) singly phosphorylated SVTPKTVTPASSAKTpSPAK peptide and (d–f) the quadruply phosphorylated SVpTPKTVpTPASpSAKTpSPAK peptide. (a,d) Annotated charge-deconvoluted spectra highlighting fragments resulting from complete HPO3 loss (mass of HPO3 = 79.9663 Da, for a comparison with fragments retaining HPO3 see Figure S2), fragments labeled with * have lost H2O, precursor ions are annotated by a pink dot, (b,e) annotated peptide sequences with y+ ions in red and b+ ions in blue, (c,f) survival yield (blue) as a function of the maximum laser power (0–95% range) for the nonphosphorylated (P.) and the corresponding phosphorylated (phosphoP.) peptides (all fragments are taken into account). The complementary fragment fraction is displayed in green. 100 scans, 1 laser pulse of 80 ms per scan, (a–c) 5.56 × 10–11 mbar and (d–f) <5.00 × 10–11 mbar N2 UHV readout, resolution of 140000 @ m/z 200. Enhancing peptide IRMPD using noncovalently attached IP6 as the chromophore.
Taken together, the results are consistent with the earlier work published by Crowe and Brodbelt using a Finnigan LCQ-Duo ion trap mass spectrometer.8 We considered this data as a clear benchmark to demonstrate that our IRMPD setup on the EMR orbitrap performed well.
Next, we sought to enhance IRMPD for nonphosphopeptides. Photodissociation can be enhanced using nonendogenous chromophores covalently attached to a peptide or protein of interest.38−41 For CO2 laser-driven IRMPD, a chromophore of choice has been 4-methylphosphonophenylisothiocyanate (PPITC) attached to the N-terminus of the peptides.42 As already mentioned by Brodbelt and co-workers, the idea of attaching chromophores via noncovalent interactions is, however, far more appealing and potentially more generally applicable than derivatization.43 We therefore explored here myo-inositol hexakisphosphate as a noncovalent adduct (IP6, C6H18O24P6, 659.861 Da) to enhance the IRMPD of nonphosphorylated peptides. Based on the available literature, IP6 can be expected to interact preferentially with basic residues such as arginine and lysine.44,45 We observed that the IRMPD efficiency was enhanced by about 1 order of magnitude at optimal irradiances. Use of the adduct did not hinder the analysis: we could easily disentangle the precursor ions carrying the adduct both from the bare precursor and fragments ions upon charge deconvolution, as illustrated in Figures 3 and S8. The absence of a detectable phosphate transfer from the adduct to the peptide fragments also considerably facilitated the interpretation of the peptide fragment ion spectra.
Figure 3.

Charge-deconvoluted IRMPD mass spectrum of (a–c) angiotensin I doubly protonated precursor (exp. m/z 978.776) complexed with an IP6 adduct (m/z 1956.545, pink dot) and without (m/z 1296.684, orange dot) (d–f) [Glu1-fibrinopeptide B doubly charged (exp. m/z 1115.772) precursor with an IP6 adduct (m/z 2230.537, pink) and without an IP6 adduct (m/z 1570.676, orange). (a,d) The m/z gap between the ions retaining partially photofragmented IP6, immediately to the left of the precursors with IP6 (pink), and the bare precursors (orange) allows for easy rejection of ions carrying residual IP6 fragments. (b) Annotated angiotensin I and (e) [Glu1]-fibrinopeptide B structures with detected y+ ions in red and b+ ions in blue. (c,f) Relative abundances of precursors with IP6 (red), precursor without IP6 (blue), and sequence informative fragments (green) upon IRMPD of (c) angiotensin I and (f) [Glu1]-fibrinopeptide B, with and without IP6 adduct at different pulse energies. ∼1000 scans, 1 laser pulse of 80 ms per scan, 7.98 × 10–11 mbar N2 UHV readout, 140,000 @ m/z 200 resolution.
The first peptide we investigated (Figure 3a–c) was angiotensin I (Table S1). IRMPD at the maximal laser power (4.56 J/pulse) of angiotensin I doubly protonated ions (m/z 648.858) and yielded barely detectable fragments: all but one had a relative intensity below 0.06% compared to the precursor ion. Photoactivation of the [angiotensin I·IP6] complex (m/z 978.776, 2.4 J/pulse), on the other hand, did lead to extensive fragmentation. Besides the precursor ion (charge-deconv. m/z 1956.545), IRMPD yielded protonated angiotensin I (charge-deconv. m/z 1296.684), b-ions from b2+ to b6+ up to b9+, and the complete y-ion series from y3+ to y9+. In other words, we achieved the full sequence coverage of angiotensin I by IRMPD of the peptide-IP6 noncovalent complex.
As a second example (Figure 3d–f) of IP6-enhanced IRMPD of a peptide, we used human [Glu1]-fibrinopeptide B (glufib). Similar to angiotensin I (Figure 3a–c) and bradykinin (Figure S7), IRMPD of the doubly charged ions (m/z 785.842) at maximal laser power yielded barely detectable fragments, whose relative intensity compared to the precursor ions was, for all but one, below 1.4%. IRMPD of glufib complexed with IP6 (C6H18O24P6, 659.861 Da) led, on the other hand, to extensive fragmentation of glufib. Besides the precursor ion (charge-deconv. m/z 2230.537), IRMPD yielded protonated glufib (charge-deconv. m/z 1570.676), b-ions from b4+ to b11+ with the exception of b6+ and b9+, and the complete y-ion series from y1+ to y11+ with the exception of y5+. Hence, IRMPD of the IP6 complex resulted in full sequence coverage of [Glu1]-fibrinopeptide B. As was the case for all the peptides studied (including bradykinin, Figure S8), mass separation enabled easy rejection of the intact peptides retaining partially dissociated IP6. Data analysis was further considerably facilitated by the absence of peptide fragments, retaining IP6 or fragments thereof.
Enhancing Protein IRMPD Using Noncovalently Attached IP6 as the Chromophore
We next applied this chromophore-assisted approach to native proteins not directly amenable to IRMPD by investigating the enhancement of their 10.6 μm absorption cross-section by the complexation of myo-inositol hexakisphosphate.
Of note, quite a substantial part of all proteins are expected to interact with molecules containing a phosphate group.46 Often, the interacting phosphate takes part in a network of hydrogen bonds with atoms from the backbone forming a geometrically and energetically favorable scaffold.47 A positive electrostatic potential promoting the binding of negatively charged phosphate groups such as those found in DNA and RNA is another common feature.47 In our study of chromophore-enhanced IRMPD of native proteins, we therefore targeted the bacteriocin colicin E9, which is a DNase (Figure 4) harboring a positive patch susceptible to bind DNA and thus possibly also IP6. Indeed, noncovalent complexes of E9 with a single molecule of IP6 could be generally made and ionized by nanoelectrospray. Next, we investigated whether IP6 could enhance absorption and lead to energy transfer to the protein backbone on IRMPD.
Figure 4.

IRMPD of native apo-colicin E9 DNase. (a) Charge-deconvoluted IRMPD MS spectrum of bare E9 [8+], (b) annotated charge-deconvoluted MS spectrum of the E9 protein with IP6 adduct [8+], note the different scales of the y-axis in (a,b). For the IRMPD of the E9 protein with IP6 adduct are depicted in (c) the observed annotated (b, y) fragment ion pairs with y+ ions in red and b+ ions in blue separately normalized. (d) The sequence coverage and (e) the crystal structure with the positive patches in blue [PatchFinderPlus (PFplus), http://pfp.technion.ac.il] and cleavages in yellow (PDB ID: 1fsj). 1000 scans, one pulse of 4.56 J over 80 ms per scan, 6.02 × 10–11 mbar N2 UHV readout, 140,000 @ m/z 200 resolution.
Colicin E9, which is a bacteriocin produced by Escherichia coli, acts against competing E. coli by cleaving their DNA at specific locations. We mass-selected the 8+ charge state of the native apoprotein (without the Zn2+ cofactor), which corresponds to a compact conformation. In Figure 4a, close to no fragments are detected upon IRMPD of bare Colicin E9, while nearly 3 orders of magnitude increase in fragment yield was observed upon IP6 binding (Figure 4b). Interestingly, the most abundant fragments were (b, y) golden pairs (Figure 4c), thereby facilitating the identification. An analysis of the cleavage sites established a weak correlation between the fragments formed and the positive patch that likely serves as the binding site of the IP6 chromophore (displayed as a blue patch in Figure 4e). Similar to peptides, extensive fragmentation of endonuclease Colicin E9 and mAmetrine (Figure S9) occurs under the low-pressure conditions achievable on our instrumentation consistently with intramolecular vibrational energy redistribution (IVR) on timescales significantly shorter than collisional deactivation.
The high dissociation yields and the absence of IP6 adducts on the generated fragments considerably increase the potential of IRMPD for native proteins in the low- to mid-size range.
Phospho-Proteins
Following the use of noncovalent myo-inositol hexakisphosphate (IP6) adducts to enhance the IR absorption cross-section of peptides and native proteins, we focused on proteins harboring different phospho-proteoforms. The potential of IR spectroscopic methods to identify posttranslational modifications has been recently reviewed by Maitre and co-workers.17
Because of the loss of labile phospho-groups on collisional activation in the gas-phase, native phospho-proteoforms are particularly challenging to characterize using thermal activation MS methods, such as CID and IRMPD. Herein, we nevertheless demonstrate that valuable information can be gained from the native top-down analysis of phospho-proteoforms by IRMPD on an orbitrap. Our hypothesis was based on the trend we observed for phosphopeptides. We expect that also for native proteins, the more phosphate groups they carry, the more extensive the IR absorption will be, with consequently richer and more informative fragmentation spectra.
Here, we focused on the 17.5 kDa N-terminal fragment of the mitotic regulator Bora.48−50 Following expression and purification, we phosphorylated Bora in vitro using the aurora A kinase (AurA) as described in detail previously.51 Bora has been determined to display an average of three phospho-groups under stationary (“equilibrium”) reaction conditions. Of the 8 phospho-sites determined for the Bora studied here,22 two, Ser59 and Thr144, have been determined to be specific AurA targets largely complying with the AurA substrate recognition sequence on Bora and therefore highly abundant.52 Two additional moderately phosphorylated sites, Ser4 and Ser123, could also be identified from quantitative liquid chromatography with tandem mass spectrometry experiments.22
As for Colicin E9 DNase (Figure 4), only a minimal fragmentation of Bora (Figures 5, S10, and S11) occurs in the absence of phospho-groups (Figure 5a). A comparison of the charge-deconvoluted IRMPD mass spectra of the three most abundant phospho-Bora signals is displayed in Figure 5b. Under identical IRMPD activation conditions, the fragmentation yields continuously increase from Bora(PO4)2, Bora(PO4)3, to Bora(PO4)4, although not as markedly as between a bare and a phosphorylated compound (Figure 5e). Fragments are primarily b-ions with the most intense high m/z ions formed upon the cleavage of N-terminal or C-terminal of an Asp residue (Figure 5c). Although the fragmentation patterns display marked differences directly related to the number of phospho-groups (Figure 5e), all three are consistent with the phosphorylation of Ser59 unambiguously assigned from Bora(PO4)4 data (Figure 5d). The limited sequence coverage currently precludes unambiguous assignments beyond Ser59.
Figure 5.

Charge-deconvoluted IRMPD mass spectra of Bora. (a) IRMPD on unphosphorylated Bora produces no fragments, (b) IRMPD mass spectra of multiple phosphorylated Bora. (c) Assigned b/y fragment ions for Bora(HPO3)4. (d) Bora(HPO3)4 sequence coverage (only b and y ions) with the phospho-sites marked as red circles. (e) Relative abundances of the precursor without IP6 (blue) and sequence informative fragments (green) upon IRMPD of Bora (P.) and its phospho-proteoforms. 1000 scans, one pulse of 4.56 J over 80 ms per scan, 6.24 × 10–11 mbar N2 UHV readout, 140,000 @ m/z 200 resolution.
In the present section, we have shown that IRMPD can yield structural information beyond amino acid sequence cleavages. Characterizing the structural features responsible for biological function calls for techniques leading to selective dissociation of or near the functional region. One way to achieve this is to design or make use of fragmentation sensitizers directly binding or affecting the region of interest. We have shown that phospho-groups potentially can serve this purpose; other candidates are O-sulfation groups.17
IRMPD of Large Complexes
In this last section, we examined whether IRMPD can be applied to high-mass protein complexes. We investigated the supportive role of phosphate moieties in IRMPD and also hypothesized that possibly very large complexes already absorb sufficiently well because of the very large number of absorbing oscillators present. We thereby pursue the integration of native IRMPD and top-down proteomics initiated for large protein assemblies by Robinson and co-workers9 and Loo and co-workers.10 In short, we demonstrate in this section that the presence of phosphate groups (e.g., also in the form of RNA or ATP) and larger size are indeed beneficial for efficient IRMPD of the 346 kDa CRISPR–Cas Csy (aka type I–F) ribonucleoprotein complex (Figure 6a–c), the 800 kDa GroEL 14-mer chaperone (Figure 6d–g) both with and without ATP co-factors bound, and ultimately, the 1 MDa wt-AaLS virus-like synthetic nanocontainer (Figure S13).
Figure 6.

(a–c) IRMPD and CID/HCD of SCRI104 CRISPR–Cas Csy complex, see Table S2 for additional information. (a) IRMPD of (black envelope, 38+) precursor (pr.) leading to the ejection of Csy3 (green, 21+) with an isotopically resolved peak (inset, 20+) and Cas6f (purple, 17+), as well as a subunit fragment (red, 18+), and the formation of pr.–Cas6f (purple, 40+), pr.–fragment (red, 30+), pr.–Cas6f (purple, 23+), pr.–Csy3 (green, 19+). Collision energy = 0 direct eV, 1.44 J/pulse, p(N2) = 1.29 × 10–10 mbar, resolution 140k @ m/z 200, no micro-scan averaging. (b) CID/HCD of (black envelope, 38+) precursor leading to the ejection of Csy3 dimer (blue, 22+), Csy1 (red, 20+), Csy3 (green, 15+) with zoom-on isotopic distribution (inset, 20+), and Cas6f (purple, 10+). Collision energy = 120 direct eV, no laser, p(N2) = 3.22 × 10–10 mbar, resolution 140k @ m/z 200, 10 micro-scans averaging. (c) Structural model of an analogous CRISPR–Cas complex (PDB ID: 5UZ9). (d–g) IRMPD of GroEL loaded with ATP. (d) Structural model of GroEL (PDB ID: 4AAS). (e) Comparison of the mass spectra of (black) bare GroEL and (blue) GroEL loaded with ATP. (f) IRMPD of bare GroEL for different irradiances and laser shot numbers. Collision energy = 1 direct eV, p(N2) = 1.27 × 10–10 mbar, resolution 140k @ m/z 200, no microscans averaging. (c) Structural model of an analogous CRISPR–Cas complex (PDB ID: 5UZ9). (g) IRMPD of GroEL loaded with ATP at different irradiances. Collision energy = 1 direct eV, p(N2) = 1.61 × 10–10 mbar, resolution 140k @ m/z 200, no micro-scans averaging.
The CRISPR–Cas Csy ribonucleoprotein complex (In Figure 6a–c) grants adaptive immunity to the Pectobacterium atrosepticum SCRI10453 and is capable of homology-directed detection as well as degradation of invading genetic elements.54 A 60-nucleotide crRNA strand serves as the backbone for the subunits of the CRISPR–Cas Csy complex. While the P. atrosepticum SCRI104 CRISPR–Cas Csy complex has been extensively structurally characterized, its crRNA phosphate moiety containing the backbone makes it an ideal candidate to assess the chromophore-enhanced IRMPD of large complexes. In Figure 6a, we illustrate the photodissociation of the precursor ion of mass 346,962 ± 30 Da and charge distribution [42+-35+]. The dissociation was highly asymmetric—the intact subunits leaving the complex took a significant part of the charge with them, suggesting extensive unfolding of the ejected subunits. In Figure 6a, we assign the fragment distribution to two intact subunits—Cas6f [20+-13+] and Csy3 [24+-13+]—as well as their complementary high mass products—pr-Csy3 [23+-16+] and pr-Cas6f [26+-19+] & [40+-36+]. A lower intensity [18+-13+] fragment was detected as well as a high-intensity high mass [33+-25+] product ion which remains currently unassigned. Analysis of fragments generated in a separate experiment led to the detection of a sequence tag corresponding to Csy1 (Figure S12). Interestingly, while the charge distributions of Csy3 and pr-Csy3 complement each other yielding the charge distribution of the precursor, the charge distribution of the Cas6f complementary high mass product (pr-Cas6f) is bimodal. The lower charge pr-Cas6f component matches the detected Cas6f distribution, while the higher charge pr-Cas6f component appears to correspond to Cas6f being ejected close to neutral. The origin of this difference, while currently unknown, is expected to be related either to the coexistence of two competing activation mechanisms (e.g., direct protein activation vs crRNA mediated) or two conformations of the complex. Comparison of IRMPD (Figure 6a) with CID/HCD (Figure 6b) highlights behavioral differences. The charge distributions of ejected subunits are systematically higher for IRMPD than for CID/HCD. Consequently, IRMPD-ejected subunits are concentrated in the low m/z range, where instrumental resolution is optimal and enable isotopic resolution while the complementary high m/z products are shifted to a higher m/z where they overlap less. IRMPD and CID/HCD also differ in the relative abundance of the ejected subunits: the ratio Cas6f to Csy3 is about 10 times higher for IRMPD than for CID/HCD possibly because of phosphate-related local hotspots. Finally, using CID/HCD, we detected the Csy1 subunit intact and a Csy3 dimer, while using IRMPD, we detect Csy1 only as fragments and no dimer. Overall, although both activation methods provide somewhat similar fragment ions, IRMPD does not fully give the same MS/MS spectra as CID/HCD for the SCRI104 CRISPR–Cas Csy complex, and thus, complementary structural information can be extracted.
Next, we subjected the E. coli GroEL chaperonin to IRMPD. GroEL is a 800 kDa homo-14-mer,55 arranged in two heptameric rings stacked back to back (Figure 6d) that has become one of the standard samples used in native MS.56−60 To assist substrate folding, GroEL requires ATP binding, which is known to be highly cooperative.
In Figure 6e–g, we depict and compare the IRMPD mass spectra of bare and ATP-loaded GroEL. As can be seen from the overlapping spectra (Figure 6e) of bare GroEL and GroEL incubated with ATP, up to 7 ATPs can be detected bound to GroEL. While displaying similar IRMPD dissociation patterns (Figure 6f,g)—the GroEL monomers are ejected intact without bound ATP—IRMPD of ATP-loaded GroEL occurs at lower irradiances compared to bare GroEL. About a third of the bare GroEL irradiance is needed to almost completely dissociate ATP-loaded GroEL. It is interesting to note that the charge distributions of the GroEL monomers are bimodal in the high-energy IRMPD mass spectra of the bare and ATP-loaded GroEL ions on par with a sequential ejection of subunits: the first subunit rips more charge from the precursor than the second.
Finally, to further extend the mass range, we proceeded to perform IRMPD of wt-AaLS, a homo-hexacontamer virus-like capsid (60-mer) formed by lumazine synthase (Figure S13). IRMPD of AaLS yields the intact subunit as well as the complementary fragment. While UVPD can lead to both intact ejection and fragmentation of the subunits, IRMPD primarily results into their intact ejection at the irradiances used, very similar to what is observed in CID/HCD.61 The subunits ejected on IRMPD carry off on average more charges than on UVPD under identical conditions.23
Mechanistic Aspects of IRMPD
In summary, we demonstrated that IRMPD of (phospho)peptides, native (phospho)proteins, and protein assemblies is possible in the HCD cell of a Q-Exactive orbitrap mass spectrometer operated at low pressure. In the present section, we attempt to provide a physicochemical rationale for our observations.
Under high-vacuum conditions and in the absence of an IR chromophore, evidence favors that IRMPD occurs by excitation via the coherent quasiresonant-stepwise mechanism.62 In the ∼10–5 to 10–4 mbar HCD cell of a Q-Exactive orbitrap, IRMPD conditions tend to deviate from those required for the coherent quasiresonant-stepwise excitation mechanism to operate. Under standard HCD cell conditions, collisional deactivation completely quenches IRMPD. Adjusting the pressure to an optimal trade-off between ion signal abundance and IRMPD efficiency is therefore essential for optimal operation. Under our pressure and irradiance conditions, both extensive backbone cleavage and partial retention of phospho-groups are observed. We hypothesize that the phospho-groups influence IRMPD in two ways: (1) the total absorption cross-section is increased, leading to a higher vibrational excitation, and (2) phospho-sites can act as localized hotspots which, despite fast IVR, may affect the relative abundance of the detected fragments.
Another factor to account for is carboxylic acid groups. While intrinsic chromophores such as the carboxylic groups of aspartic and glutamic acid enhance 943 cm–1 IR absorption, they are also involved in the formation of so-called golden pairs, an efficient dissociation channel, as inferred from the intensity of the detected fragments.
As compounds’ size increases, the amount of internal energy required to unfold native compounds and/or separate noncovalently bound fragments increases. Because of intramolecular energy redistribution and the requirement for an ever-larger number of collisions or collisions at higher energy, CID becomes ineffective for the top-down characterization of native compounds as compounds’ size increases. Furthermore, with CID/HCD necessarily impacting surface residues more than the buried ones, native top-down characterization of proteoforms by CID/HCD can be hindered by the loss of labile posttranslational modifications located on the compound’s surface. IRMPD, on the other hand, directly benefits from the increase of the 10.6 μm absorption cross-section of proteins with the protein size, the increased number of aspartic and glutamic acid residues, and a rather spatially homogeneous excitation of the residues in the absence of extrinsic chromophores. IRMPD is therefore potentially less likely to induce the loss of labile posttranslational modifications located on the compound’s surface, at irradiances enabling competition between unfolding, backbone fragmentation, and loss of labile groups. Our data suggest that for native compounds, IRMPD (combined or not with CID/HCD) may prove a suitable approach to characterize compounds carrying phospho-groups. A summary of our findings in terms of absorption cross-section is depicted in Figure 7.
Figure 7.
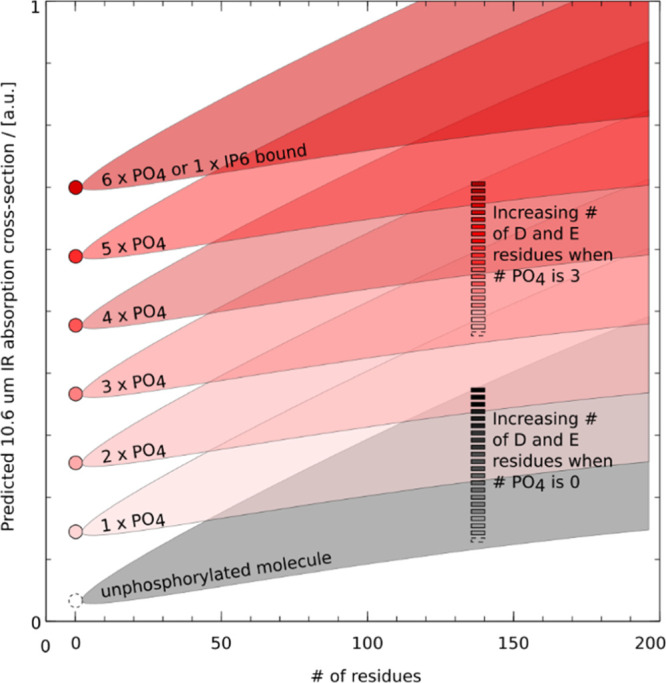
Schematic summary of the impact of molecular size, the number of phospho-groups, and the number of D and E residues on the 10.6 μm absorption cross-section of peptides and proteins. The dependence of the IR absorption cross-section on the # of residues and composition is depicted as a wedge: in the absence of phosphate groups (gray wedge), the 10.6 μm absorption cross-section increases with the # of residues, and all the more so that the fraction of D and E residues is large. On phosphorylation [addition of n × HPO3 resulting in the formation of n phosphate group(s)], an absorption jump is seen: transition from the gray wedge to a red one (or for compounds consisting only of phosphates from the dashed circle to a colored one). As the number of phospho-groups increases, we transition to edges characterized by ever-larger 10.6 μm absorption cross-sections.
Finally, for very large native complexes, achievable irradiances result, despite the increase of the 10.6 μm absorption cross-section with protein size, into a slow energy buildup. In these systems, subunit ejection is the primary dissociation channel with, as demonstrated by Robinson and co-workers,57 an asymmetric charge partitioning, indicative of subunit unfolding. Although largely overlooked, the present results suggest a bright future for the pulsed-CO2 excitation of native compounds.
Conclusions
In the present work, we demonstrate the capabilities of IRMPD implemented in the HCD cell of an orbitrap mass spectrometer for compounds ranging from phosphopeptides, native proteins, to large supramolecular assemblies. Specifically, we address dissociation yield issues by operating our setup at low nitrogen pressure and by compensating for low absorption cross-sections at 10.6 μm by using phosphate-based chromophores.
Additionally, while earlier reported IRMPD work primarily focused on peptides and denatured proteins, we expanded IRMPD substantially by demonstrating its applicability to native phospho- and phospho-adduct proteins. A multiple-fold enhancement of dissociation was recorded for phospho-compounds compared to bare ones. We also showed that preferential cleavages similar to those observed in CID and SID took place. Under optimal IRPMD conditions, phospho-groups are retained, despite the fact that they act as IR-chromophores and, thereby, enhance dissociation. When applied to large protein complexes ranging from a CRISPR–Cas Csy complex, a (ATP-loaded) GroEL complex, to a 1 MDa virus-like capsid, IRMPD leads to the ejection of highly charged intact subunits. The unfolding induced by IRMPD may therefore be beneficial to native top-down proteomics on use in conjunction with activation techniques such as UVPD.
Combined with the recently demonstrated coupling of a CO2 laser to an orbitrap via an IR optical fiber,31 the present results support a bright future for a native top-down chromophore-enhanced IRMPD MS on commercial instruments beyond FT-ICRs.
Acknowledgments
We thank the members of the Heck laboratory for the support, especially Arjan Barendregt. Dris El Atmioui of the Huib Ovaa laboratory (LUMC, Leiden) provided the synthetic phosphopeptides. Donald Hilvert’s group (ETH Zürich, Switzerland) and Stephan Tetter in particular are acknowledged for providing the AaLS sample. Peter C. Fineran’s group (University of Otago, New-Zealand), Robert D. Fagerlund, and Howard W. R. Maxwell in particular are acknowledged for supplying the CRISPR–Cas Csy (type I–F Cascade) complex. We kindly acknowledge Dietmar Reusch and Markus Haberger from Roche, Penzberg, Germany for supplying Trastuzumab. This research received funding through The Netherlands Organization for Scientific Research (NWO), projects ENPPS.LIFT.019.001, SPI.2017.028, and TTW.15575.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.0c03412.
Peptides and proteins, IRMPD of monoclonal antibody, detailed assignment of phosphopeptides fragments, formation and fragmentation of peptides carrying IP6, detailed fragmentation analysis of phospho-Bora, large protein complexes, IRMPD mass tag identification of a CRISPR–Cas protein, and IRMPD of the Mega-Dalton AaLS complex (PDF)
Author Contributions
The paper was written through contributions of all authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Wysocki V.; Jones C.; Galhena A.; Blackwell A. Surface-Induced Dissociation Shows Potential to Be More Informative Than Collision-Induced Dissociation for Structural Studies of Large Systems. J. Am. Soc. Mass Spectrom. 2008, 19, 903–913. 10.1016/j.jasms.2008.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macias L. A.; Santos I. C.; Brodbelt J. S. Ion Activation Methods for Peptides and Proteins. Anal. Chem. 2020, 92, 227–251. 10.1021/acs.analchem.9b04859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Boer M. A.; Greisch J.-F.; Tamara S.; Bondt A.; Heck A. J. R. Selectivity over coverage in de novo sequencing of IgGs. Chem. Sci. 2020, 10.1039/D0SC03438J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little D. P.; Speir J. P.; Senko M. W.; O’Connor P. B.; McLafferty F. W. Infrared Multiphoton Dissociation of Large Multiply Charged Ions for Biomolecule Sequencing. Anal. Chem. 1994, 66, 2809–2815. 10.1021/ac00090a004. [DOI] [PubMed] [Google Scholar]
- Rush M. J. P.; Riley N. M.; Westphall M. S.; Coon J. J. Top-Down Characterization of Proteins with Intact Disulfide Bonds Using Activated-Ion Electron Transfer Dissociation. Anal. Chem. 2018, 90, 8946–8953. 10.1021/acs.analchem.8b01113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganisl B.; Breuker K. Does Electron Capture Dissociation Cleave Protein Disulfide Bonds?. ChemistryOpen 2012, 1, 260–268. 10.1002/open.201200038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne A. H.; Glish G. L. Thermally Assisted Infrared Multiphoton Photodissociation in a Quadrupole Ion Trap. Anal. Chem. 2001, 73, 3542–3548. 10.1021/ac010245+. [DOI] [PubMed] [Google Scholar]
- Crowe M. C.; Brodbelt J. S. Infrared multiphoton dissociation (IRMPD) and collisionallyactivated dissociation of peptides in a quadrupole ion trap with selective IRMPD of phosphopeptides. J. Am. Soc. Mass Spectrom. 2004, 15, 1581–1592. 10.1016/j.jasms.2004.07.016. [DOI] [PubMed] [Google Scholar]
- Mikhailov V. A.; Liko I.; Mize T. H.; Bush M. F.; Benesch J. L. P.; Robinson C. V. Infrared Laser Activation of Soluble and Membrane Protein Assemblies in the Gas Phase. Anal. Chem. 2016, 88, 7060–7067. 10.1021/acs.analchem.6b00645. [DOI] [PubMed] [Google Scholar]
- Li H.; Nguyen H. H.; Ogorzalek Loo R. R.; Campuzano I. D. G.; Loo J. A. An integrated native mass spectrometry and top-down proteomics method that connects sequence to structure and function of macromolecular complexes. Nature Chem. 2018, 10, 139–148. 10.1038/nchem.2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flora J. W.; Muddiman D. C. Selective, Sensitive, and Rapid Phosphopeptide Identification in Enzymatic Digests Using ESI-FTICR-MS with Infrared Multiphoton Dissociation. Anal. Chem. 2001, 73, 3305–3311. 10.1021/ac010333u. [DOI] [PubMed] [Google Scholar]
- He Z.; Honeycutt C. W.; Xing B.; McDowell R. W.; Pellechia P. J.; Zhang T. Solid-State Fourier Transform Infrared and 31P Nuclear Magnetic Resonance Spectral Features of Phosphate Compounds. Soil Sci. 2007, 172, 501. 10.1097/SS.0b013e318053dba0. [DOI] [Google Scholar]
- Khalil F. L.; Brown T. L. Infrared Spectra of Adenosine Triphosphate Complexes in Deuterium Oxide Solution. J. Am. Chem. Soc. 1964, 86, 5113–5117. 10.1021/ja01077a014. [DOI] [Google Scholar]
- Siebert T.; Guchhait B.; Liu Y.; Costard R.; Elsaesser T. Anharmonic Backbone Vibrations in Ultrafast Processes at the DNA–Water Interface. J. Phys. Chem. B 2015, 119, 9670–9677. 10.1021/acs.jpcb.5b04499. [DOI] [PubMed] [Google Scholar]
- Scuderi D.; Bakker J. M.; Durand S.; Maitre P.; Sharma A.; Martens J. K.; Nicol E.; Clavaguéra C.; Ohanessian G. Structure of singly hydrated, protonated phospho-tyrosine. Int. J. Mass Spectrom. 2011, 308, 338–347. 10.1016/j.ijms.2011.08.031. [DOI] [Google Scholar]
- Correia C. F.; Balaj P. O.; Scuderi D.; Maitre P.; Ohanessian G. Vibrational Signatures of Protonated, Phosphorylated Amino Acids in the Gas Phase. J. Am. Chem. Soc. 2008, 130, 3359–3370. 10.1021/ja073868z. [DOI] [PubMed] [Google Scholar]
- Maitre P.; Scuderi D.; Corinti D.; Chiavarino B.; Crestoni M. E.; Fornarini S. Applications of Infrared Multiple Photon Dissociation (IRMPD) to the Detection of Posttranslational Modifications. Chem. Rev. 2020, 120, 3261–3295. 10.1021/acs.chemrev.9b00395. [DOI] [PubMed] [Google Scholar]
- Wu R. R.; He C. C.; Hamlow L. A.; Nei Y.-w.; Berden G.; Oomens J.; Rodgers M. T. N3 Protonation Induces Base Rotation of 2′-Deoxyadenosine-5′-monophosphate and Adenosine-5′-monophosphate. J. Phys. Chem. B 2016, 120, 4616–4624. 10.1021/acs.jpcb.6b04052. [DOI] [PubMed] [Google Scholar]
- Ligare M. R.; Rijs A. M.; Berden G.; Kabeláč M.; Nachtigallova D.; Oomens J.; de Vries M. S. Resonant Infrared Multiple Photon Dissociation Spectroscopy of Anionic Nucleotide Monophosphate Clusters. J. Phys. Chem. B 2015, 119, 7894–7901. 10.1021/acs.jpcb.5b02222. [DOI] [PubMed] [Google Scholar]
- van Outersterp R. E.; Martens J.; Berden G.; Steill J. D.; Oomens J.; Rijs A. M. Structural characterization of nucleotide 5′-triphosphates by infrared ion spectroscopy and theoretical studies. Phys. Chem. Chem. Phys. 2018, 20, 28319–28330. 10.1039/C8CP03314E. [DOI] [PubMed] [Google Scholar]
- Horn D. M.; Zubarev R. A.; McLafferty F. W. Automated de novo sequencing of proteins by tandem high-resolution mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 10313–10317. 10.1073/pnas.97.19.10313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Waterbeemd M.; Lössl P.; Gautier V.; Marino F.; Yamashita M.; Conti E.; Scholten A.; Heck A. J. R. Simultaneous Assessment of Kinetic, Site-Specific, and Structural Aspects of Enzymatic Protein Phosphorylation. Angew. Chem., Int. Ed. 2014, 53, 9660–9664. 10.1002/anie.201404637. [DOI] [PubMed] [Google Scholar]
- Greisch J.-F.; Tamara S.; Scheltema R. A.; Maxwell H. W. R.; Fagerlund R. D.; Fineran P. C.; Tetter S.; Hilvert D.; Heck A. J. R. Expanding the mass range for UVPD-based native top-down mass spectrometry. Chem. Sci. 2019, 10, 7163–7171. 10.1039/C9SC01857C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quaite-Randall E.; Joachimiak A.. Chaperonin Protocols; Schneider C., Ed.; Methods in Molecular Biology; Springer: New York: Totowa, NJ, 2000; pp 29–39. [Google Scholar]
- Voziyan P. A.; Fisher M. T. Chaperonin-assisted folding of glutamine synthetase under nonpermissive conditions: Off-pathway aggregation propensity does not determine the co-chaperonin requirement. Protein Sci. 2000, 9, 2405. 10.1110/ps.9.12.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Duijn E.; Bakkes P. J.; Heeren R. M. A.; van den Heuvel R. H. H.; van Heerikhuizen H.; van der Vies S. M.; Heck A. J. R. Monitoring macromolecular complexes involved in the chaperonin-assisted protein folding cycle by mass spectrometry. Nat. Methods 2005, 2, 371–376. 10.1038/nmeth753. [DOI] [PubMed] [Google Scholar]
- Sasaki E.; Hilvert D. Self-Assembly of Proteinaceous Multishell Structures Mediated by a Supercharged Protein. J. Phys. Chem. B 2016, 120, 6089–6095. 10.1021/acs.jpcb.6b02068. [DOI] [PubMed] [Google Scholar]
- Rosati S.; Rose R. J.; Thompson N. J.; van Duijn E.; Damoc E.; Denisov E.; Makarov A.; Heck A. J. R. Exploring an Orbitrap Analyzer for the Characterization of Intact Antibodies by Native Mass Spectrometry. Angew. Chem., Int. Ed. 2012, 51, 12992–12996. 10.1002/anie.201206745. [DOI] [PubMed] [Google Scholar]
- Fort K. L.; Dyachenko A.; Potel C. M.; Corradini E.; Marino F.; Barendregt A.; Makarov A. A.; Scheltema R. A.; Heck A. J. R. Implementation of Ultraviolet Photodissociation on a Benchtop Q Exactive Mass Spectrometer and Its Application to Phosphoproteomics. Anal. Chem. 2016, 88, 2303–2310. 10.1021/acs.analchem.5b04162. [DOI] [PubMed] [Google Scholar]
- Girod M.; Biarc J.; Enjalbert Q.; Salvador A.; Antoine R.; Dugourd P.; Lemoine J. Implementing visible 473 nm photodissociation in a Q-Exactive mass spectrometer: towards specific detection of cysteine-containing peptides. Analyst 2014, 139, 5523–5530. 10.1039/C4AN00956H. [DOI] [PubMed] [Google Scholar]
- Peters-Clarke T. M.; Schauer K. L.; Riley N. M.; Lodge J. M.; Westphall M. S.; Coon J. J. Optical Fiber-Enabled Photoactivation of Peptides and Proteins. Anal. Chem. 2020, 92, 12363–12370. 10.1021/acs.analchem.0c02087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasicek L. A.; Ledvina A. R.; Shaw J.; Griep-Raming J.; Westphall M. S.; Coon J. J.; Brodbelt J. S. Implementing Photodissociation in an Orbitrap Mass Spectrometer. J. Am. Soc. Mass Spectrom. 2011, 22, 1105–1108. 10.1007/s13361-011-0119-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein J.; Carvalho L.; Zaia J. Application of network smoothing to glycan LC-MS profiling. Bioinformatics 2018, 34, 3511–3518. 10.1093/bioinformatics/bty397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein J.; Heckendorf C.; Lukauskas S.. mobiusklein/msdeisotope: Release v0.0.10; Zenodo, 2019https://zenodo.org/record/3475687#.XnNv8HLTVPY.
- Park J.; Piehowski P. D.; Wilkins C.; Zhou M.; Mendoza J.; Fujimoto G. M.; Gibbons B. C.; Shaw J. B.; Shen Y.; Shukla A. K.; Moore R. J.; Liu T.; Petyuk V. A.; Tolić N.; Paša-Tolić L.; Smith R. D.; Payne S. H.; Kim S. Informed-Proteomics: open-source software package for top-down proteomics. Nat. Methods 2017, 14, 909–914. 10.1038/nmeth.4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colorado A.; Shen J. X.; Vartanian V. H.; Brodbelt J. Use of Infrared Multiphoton Photodissociation with SWIFT for Electrospray Ionization and Laser Desorption Applications in a Quadrupole Ion Trap Mass Spectrometer. Anal. Chem. 1996, 68, 4033–4043. 10.1021/ac9600565. [DOI] [PubMed] [Google Scholar]
- Boué S. M.; Stephenson J. L.; Yost R. A. Pulsed helium introduction into a quadrupole ion trap for reduced collisional quenching during infrared multiphoton dissociation of electrosprayed ions. Rapid Commun. Mass Spectrom. 2000, 14, 1391–1397. . [DOI] [PubMed] [Google Scholar]
- Tecklenburg R. E.; Miller M. N.; Russell D. H. Laser ion beam photodissociation studies of model amino acids and peptides. J. Am. Chem. Soc. 1989, 111, 1161–1171. 10.1021/ja00186a001. [DOI] [Google Scholar]
- Vasicek L.; Brodbelt J. S. Enhancement of Ultraviolet Photodissociation Efficiencies through Attachment of Aromatic Chromophores. Anal. Chem. 2010, 82, 9441–9446. 10.1021/ac102126s. [DOI] [PubMed] [Google Scholar]
- Cotham V. C.; Wine Y.; Brodbelt J. S. Selective 351 nm Photodissociation of Cysteine-Containing Peptides for Discrimination of Antigen-Binding Regions of IgG Fragments in Bottom-Up Liquid Chromatography–Tandem Mass Spectrometry Workflows. Anal. Chem. 2013, 85, 5577–5585. 10.1021/ac400851x. [DOI] [PubMed] [Google Scholar]
- Quick M. M.; Mehaffey M. R.; Johns R. W.; Parker W. R.; Brodbelt J. S. SITS Derivatization of Peptides to Enhance 266 nm Ultraviolet Photodissociation (UVPD). J. Am. Soc. Mass Spectrom. 2017, 28, 1462–1472. 10.1007/s13361-017-1650-y. [DOI] [PubMed] [Google Scholar]
- Vasicek L. A.; Wilson J. J.; Brodbelt J. S. Improved infrared multiphoton dissociation of peptides through N-terminal phosphonite derivatization. J. Am. Soc. Mass Spectrom. 2009, 20, 377–384. 10.1016/j.jasms.2008.10.016. [DOI] [PubMed] [Google Scholar]
- Wilson J. J.; Kirkovits G. J.; Sessler J. L.; Brodbelt J. S. Photodissociation of non-covalent peptide-crown ether complexes. J. Am. Soc. Mass Spectrom. 2008, 19, 257–260. 10.1016/j.jasms.2007.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancelot G.; Mayer R.; Hélène C. Models of interaction between nucleic acids and proteins Hydrogen bonding of arginine with nucleic acid bases, phosphate groups and carboxylic acids. Biochim. Biophys. Acta, Nucleic Acids Protein Synth. 1979, 564, 181–190. 10.1016/0005-2787(79)90217-X. [DOI] [PubMed] [Google Scholar]
- Braunewell K.-H.; Paul B.; Altarche-Xifro W.; Noack C.; Lange K.; Hofmann A. Interactions of Visinin-like Proteins with Phosphoinositides. Aust. J. Chem. 2010, 63, 350–356. 10.1071/CH09355. [DOI] [Google Scholar]
- Hirsch A. K. H.; Fischer F. R.; Diederich F. Phosphate Recognition in Structural Biology. Angew. Chem., Int. Ed. 2007, 46, 338–352. 10.1002/anie.200603420. [DOI] [PubMed] [Google Scholar]
- Parca L.; Gherardini P. F.; Helmer-Citterich M.; Ausiello G. Phosphate binding sites identification in protein structures. Nucleic Acids Res. 2011, 39, 1231–1242. 10.1093/nar/gkq987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki A.; Coppinger J. A.; Jang C.-Y.; Yates J. R.; Fang G. Bora and the Kinase Aurora A Cooperatively Activate the Kinase Plk1 and Control Mitotic Entry. Science 2008, 320, 1655–1658. 10.1126/science.1157425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutterer A.; Berdnik D.; Wirtz-Peitz F.; Žigman M.; Schleiffer A.; Knoblich J. A. Mitotic Activation of the Kinase Aurora-A Requires Its Binding Partner Bora. Dev. Cell 2006, 11, 147–157. 10.1016/j.devcel.2006.06.002. [DOI] [PubMed] [Google Scholar]
- Eckerdt F.; Maller J. L. Kicking off the polo game. Trends Biochem. Sci. 2008, 33, 511–513. 10.1016/j.tibs.2008.08.004. [DOI] [PubMed] [Google Scholar]
- Brunner A. M.; Lössl P.; Liu F.; Huguet R.; Mullen C.; Yamashita M.; Zabrouskov V.; Makarov A.; Altelaar A. F. M.; Heck A. J. R. Benchmarking Multiple Fragmentation Methods on an Orbitrap Fusion for Top-down Phospho-Proteoform Characterization. Anal. Chem. 2015, 87, 4152–4158. 10.1021/acs.analchem.5b00162. [DOI] [PubMed] [Google Scholar]
- Kettenbach A. N.; Schweppe D. K.; Faherty B. K.; Pechenick D.; Pletnev A. A.; Gerber S. A. Quantitative Phosphoproteomics Identifies Substrates and Functional Modules of Aurora and Polo-Like Kinase Activities in Mitotic Cells. Sci. Signaling 2011, 4, rs5. 10.1126/scisignal.2001497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson S. A.; McKenzie R. E.; Fagerlund R. D.; Kieper S. N.; Fineran P. C.; Brouns S. J. J. CRISPR-Cas: Adapting to change. Science 2017, 356, eaal5056 10.1126/science.aal5056. [DOI] [PubMed] [Google Scholar]
- Rouillon C.; Zhou M.; Zhang J.; Politis A.; Beilsten-Edmands V.; Cannone G.; Graham S.; Robinson C. V.; Spagnolo L.; White M. F. Structure of the CRISPR Interference Complex CSM Reveals Key Similarities with Cascade. Mol. Cell 2013, 52, 124–134. 10.1016/j.molcel.2013.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braig K.; Joachimiakt A.; Horwich A. L.; Sigler P. B. The crystal structure of the bacterial chaperonin GroEL at 2.8 A. Nature 1994, 371, 9. 10.1038/371578a0. [DOI] [PubMed] [Google Scholar]
- Rostom A. A.; Robinson C. V. Detection of the Intact GroEL Chaperonin Assembly by Mass Spectrometry. J. Am. Chem. Soc. 1999, 121, 4718–4719. 10.1021/ja990238r. [DOI] [Google Scholar]
- Sobott F.; Robinson C. V. Characterizing electrosprayed biomolecules using tandem-MS—the noncovalent GroEL chaperonin assembly. Int. J. Mass Spectrom. 2004, 236, 25–32. 10.1016/j.ijms.2004.05.010. [DOI] [Google Scholar]
- Rose R. J.; Damoc E.; Denisov E.; Makarov A.; Heck A. J. R. High-sensitivity Orbitrap mass analysis of intact macromolecular assemblies. Nat. Methods 2012, 9, 1084–1086. 10.1038/nmeth.2208. [DOI] [PubMed] [Google Scholar]
- Belov M. E.; Damoc E.; Denisov E.; Compton P. D.; Horning S.; Makarov A. A.; Kelleher N. L. From Protein Complexes to Subunit Backbone Fragments: A Multi-stage Approach to Native Mass Spectrometry. Anal. Chem. 2013, 85, 11163–11173. 10.1021/ac4029328. [DOI] [PubMed] [Google Scholar]
- Li H.; Nguyen H. H.; Ogorzalek Loo R. R.; Campuzano I. D. G.; Loo J. A. An integrated native mass spectrometry and top-down proteomics method that connects sequence to structure and function of macromolecular complexes. Nature Chem. 2018, 10, 139–148. 10.1038/nchem.2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Waterbeemd M.; Fort K. L.; Boll D.; Reinhardt-Szyba M.; Routh A.; Makarov A.; Heck A. J. R. High-fidelity mass analysis unveils heterogeneity in intact ribosomal particles. Nat. Methods 2017, 14, 283–286. 10.1038/nmeth.4147. [DOI] [PubMed] [Google Scholar]
- Quack M. Infrared laser chemistry and the dynamics of molecular multiphoton excitation. Infrared Phys. 1989, 29, 441–466. 10.1016/0020-0891(89)90087-0. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.