

Abstract
Interferon-β has therapeutic efficacy in Multiple Sclerosis by reducing disease exacerbations and delaying relapses. Previous studies have suggested that the effects of type I IFN in Experimental Autoimmune Encephalomyelitis (EAE) in mice were targeted to myeloid cells. We used mice with a conditional deletion(cKO) of the type I IFN receptor (IFNAR) in T regulatory (Treg) cells to dissect the role of IFN signaling on Tregs. cKO mice developed severe EAE with an earlier onset than control mice. Although Treg cells from cKO mice were more activated, the activation status and effector cytokine production of CD4+Foxp3− T cells in the draining lymph nodes (dLN) was similar in WT and cKO mice during the priming phase. Production of chemokines (CCL8, CCL9, CCL22) by CD4+Foxp3− T cells and LN resident cells from cKO mice was suppressed. Suppression of chemokine production was accompanied by a substantial reduction of myeloid derived suppressor cells (MDSCs) in the dLN of cKO mice, while generation of MDSCs and recruitment to peripheral organs was comparable. This study demonstrates that signaling by type I IFNs in Tregs reduces their capacity to suppress chemokine production, with resultant alteration of the entire microenvironment of draining lymph nodes leading to enhancement of MDSC homing, and beneficial effects on disease outcome.
Keywords: Regulatory T cells, Myeloid Derived Suppressor Cells, Type 1 IFN Receptor-α, Experimental Autoimmune Encephalomyelitis, Chemokines
1. Introduction
Type I interferons (IFNs), while providing a first line of defense against viruses and microorganisms [1], can have either beneficial or detrimental effects depending on context [2–5]. While all type I IFNs signal through the IFN alpha receptor (IFNAR), the IFNAR is ubiquitously expressed [6–9], rendering the exact target cell of type I IFNs difficult to dissect. Multiple sclerosis (MS) patients are treated with IFNβ to reduce clinical symptoms [10]. Multiple effects have been claimed for the therapeutic effects of IFNβ in MS including inhibition of the disruption of the blood brain barrier [11], increase in nerve growth factor secretion [12], alteration of the expression adhesion molecules on endothelial cells and leukocytes [13–15], and reduction of osteopontin and IL-17 production by CD4+ T cells [16]. Other studies have shown that IFNβ switches the immune phenotype of reactive cells from pro-inflammatory to an anti-inflammatory phenotype [17, 18] and differentially modulates the expression of genes belonging to various immune related pathways [19, 20]. However, precise analysis of the exact target cell of IFN in MS is clouded by the ubiquitous expression of IFNAR.
A previous study in experimental autoimmune encephalomyelitis (EAE), an animal model of MS, using mouse strains deficient in B cells, T cells or myeloid cells concluded IFNAR signaling on myeloid cells is critical for prevention of severe EAE [21]. However, this study failed to address the potential role of IFNAR signaling on T regulatory cells (Treg) as they only analyzed mice with a deletion of the IFNAR on all T cells including Treg cells. We have recently shown that IFNAR signaling is critical for the homeostasis of Treg and their suppressive function under stress conditions [22]. In addition, conditional deletion of IFNAR on Treg resulted in enhancement of their suppressive function and delayed viral clearance in a model of chronic lymphocytic choriomeningitis virus (LCMV) infection as well as an enhancement of growth of a transplantable tumor [23]. Here, we demonstrate that mice with a conditional deletion of the IFNAR on Treg develop more severe EAE. The enhancement of disease was secondary to an altered microenvironment in the draining LN (dLN) due to enhanced Treg suppression of chemokine production, and the resultant failure of the migration of myeloid derived suppressor cells (MDSCs) to the site of priming.
2. Methods
2.1. Mice
C57BL/6, LysM-Cre, 2D2 and Foxp3YFP-Cre mice were obtained from The Jackson Laboratories (Bar Harbor, ME). IFNARfl/fl mice were kindly provided by Ulrich Kalinke (Paul-Ehrlich Institut, Langen, Germany) and crossed to Foxp3YFP-Cre to generate Treg lineage specific IFNAR deficient mice. All the mice used in the experiments were 10–12 weeks of age, bred in-house and age matched. The Animal Care and Use Committee of the National Institute of Allergy and Infectious Diseases approved all experiments.
2.2. Induction of EAE and Clinical evaluation
As described in earlier paper [24].
2.3. Flow cytometry and sorting
Cell surface staining was done using following antibodies (from BD Biosciences or Thermo Fisher Scientific unless otherwise specified): anti-CD4 (RM4–5), −CD8 (53–6.7), −CD19 (1D3), −CD90.2 (53–2.1), −CD25(7D4), −CD44(IM7), −CD45.1(A20), −CD45.2(104), −CD62l (MEL-14), −CD69(H1.2F3), −CD16/CD32 (2.4G2), −CD11b (M1/70), −Ly6G (1A8), −Ly6C (HK1.4), −IL–17a (eBio17B7), −Foxp3 (FJK-16s), −Lag-3 (eBioC9B7W), −CTLA-4 (UC10–4B9), −OX-40 (OX-86), −CCL8(Invitrogen), −CD31 (Biolegend clone 390), −IFN-γ (Biolegend clone XMG1.2). For endothelial cell staining, tissues were incubated in 0.2mg/ml Collagenase P (Roche), 0.8 mg/ml Dispase I (Sigma-Worthington) and RPMI for 20 min at 37 °C before disruption.
2.4. CNS isolation
Tissue was dissected and processed using Neural Tissue Dissociation Kit (P) (Miltenyi Biotec) according to manufacturer’s instructions.
2.5. Edu staining
Click-iT Plus Edu flow cytometry assay kit (Thermo Fisher Scientific) was used according to manufacturer’s instructions. Mice were given Edu (200mg/kg) on d 0 and d2 and euthanized at d 3 to quantitate Edu labeled populations in bone marrow and spleen.
2.6. ELISA
CD4+Foxp3YFP−, CD8+, CD19+ and CD4−CD8−CD19−CD31+ cells were stimulated for 8h with eBioscience™ cell stimulation cocktail (500X) (Thermo Fisher Scientific). Cell supernatants were assayed for chemokines using Quantibody Mouse Chemokine Array (Ray Biotech) according to manufacturer’s instructions.
2.7. Chemokine Array
FACS sorted CD4+Foxp3YFP− or CD4+Foxp3YFP+ cells were subjected to RT2Profiler™ PCR array for Mouse Chemokines and Receptors (Qiagen) according to manufacturer’s instructions.
2.8. RNA-Seq
Raw RNA-Seq data was processed using Pipeliner, which is an in-house pipeline (https://github.com/CCBR/Pipeliner). Adapter sequences were trimmed with cutadapt (version 1.18). Trimmed reads were aligned to the mouse GRCm38.p6 reference genome and GENCODE release 18 reference transcriptome using STAR v2.5.3 run in 2-pass mode [25]. In total, 13.8–17.9 million read pairs were mapped to the reference transcriptome among the 12 samples. RSEM v1.3.0 [26] was used to compute the expected counts which were further converted to counts per million (cpm) by iDEP [27]. Genes that were expressed with at least 1 cpm in a minimum of three among the six samples (within each sample group) were included in the differential expression testing by DESeq2 [28] within iDEP. 13367 genes passed this threshold in the Treg sample set, whereas 12818 genes passed this threshold in the CD4+Foxp3− sample set. False discovery rate (FDR) cut-off value of 0.05 was used to detect differentially expressed genes and for downstream analysis. Heatmaps, volcano plots, and barplots were generated using gplots (v3.0.1.1), EnhancedVolcano (v1.0.1), and ggpubr (0.2.2) packages in R, respectively. Upstream regulator and canonical pathway enrichment analyses were performed using Ingenuity Pathway Analysis (IPA, Qiagen, Redwood City, CA, USA).
2.9. Statistical analysis
Flow cytometry data were analysed using Flow jo software version 9.9.6 (Flow Jo LLC, Ashland, OR). Graphs were prepared by GraphPad Prism software version 7.0 (GraphPad Software, Inc. La Jolla, CA). Statistical analysis was done through unpaired two-tailed Student’s t-test. All data are presented as Mean±SEM. Values were considered statistically significant when p < 0.05.
3. Results
3.1. Mice with a conditional deletion of the IFNAR in Tregs develop enhanced EAE
To specifically address the role of type I IFN signaling in Treg, we immunized IFNARfl/flxFoxp3YFP-Cre mice (cKO) [23] with MOG35–65 in CFA and injected the animals with Pertussis toxin at day 0 and day 2. cKO mice had a slightly earlier onset and more severe disease (Fig. 1A) as compared to WT mice. A higher percentage of CD4+ T cells were detected in the CNS tissue of cKO mice during the peak phase of disease (Fig. 1B) and a higher percentage of these cells secreted IL-17A, but not IFN-γ (Fig. 1C). We also examined the dLN during the priming phase, but overall the numbers, phenotype, and cytokine production of conventional CD4+Foxp3− T cells and Treg did not differ between WT and cKO mice except for a slight increase in percentage of activated CD4+CD44hiFoxp3− T cells and an increase in activated Ly-6Clo Treg [29] in the cKO mice (Supplementary Fig. 1A–F). Interestingly, an increase in the percentage of activated Foxp3+Ly-6Clo Treg was also observed in the spleen consistent with a systemic effect of the lack of IFNAR expression on Treg. Expression of other membrane molecules including CD25, CD103, LAG-3, CTLA-4, CD69, OX-40 and TIGIT on Tregs showed no major differences between WT and cKO mice (data not shown). Additionally, T cells from 2D2 (expressing a MOG-specific TCR) transferred to WT or cKO mice showed no difference as compared to endogenous CD4+ T cells in cytokine production and the expression of activation antigens (Supplementary Fig. 1G–I).
Fig. 1.
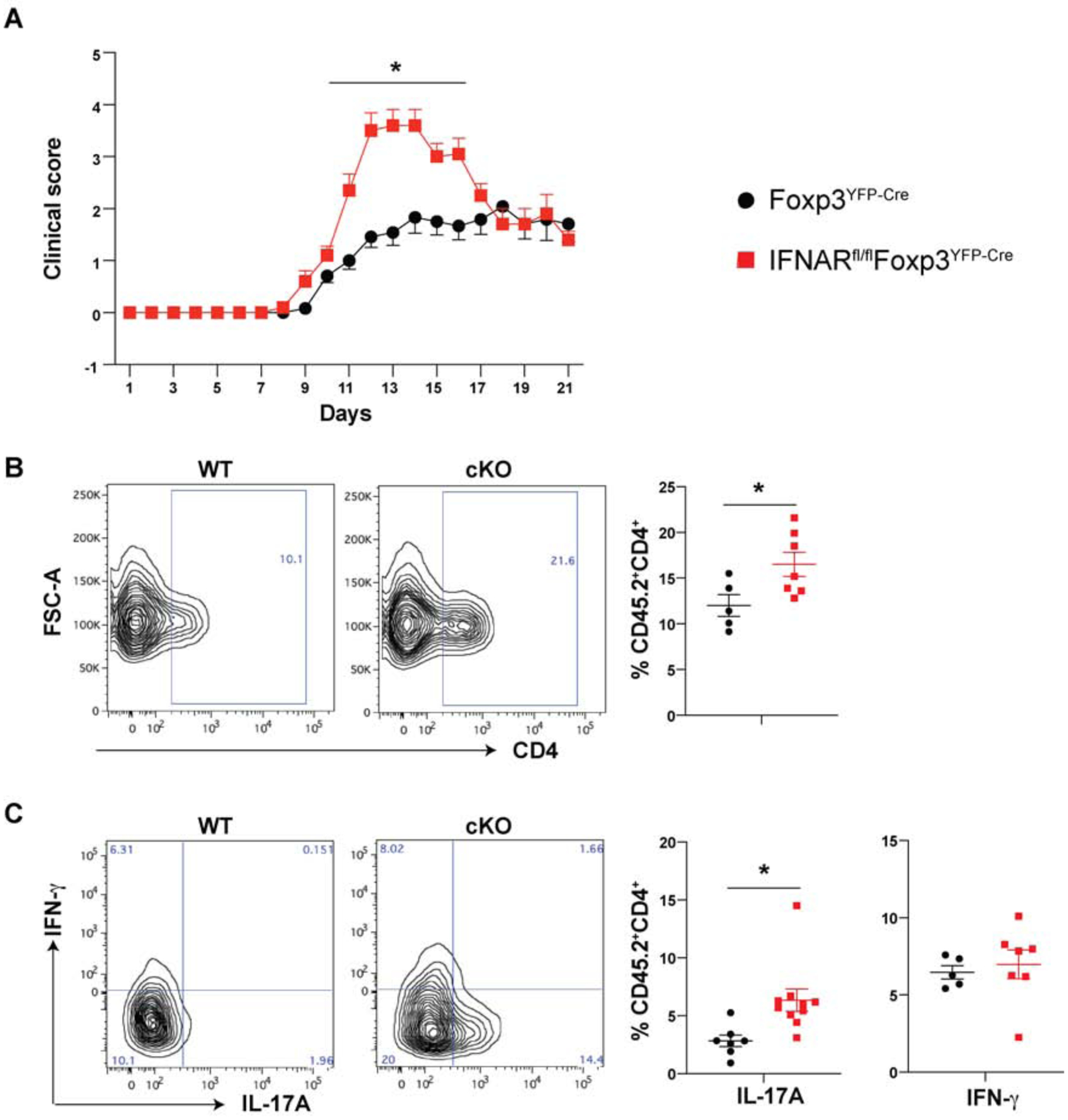
Lack of IFNAR signaling on Tregs exacerbates EAE disease pathology
(A) Mice were immunized with MOG-CFA and disease scored as described in Methods. Each data point represents mean of at least 10 mice, mean± SE, *p < 0.05.
(B) Proportions of CD4+ T cells infiltrating the CNS (spinal cord and brain) tissue on d 14 after induction of EAE.
(C) IL-17A and IFN-γ production by CD4+ T cells in CNS tissue on d 14 after EAE induction. Data cumulative of three independent experiments represent mean± SE, N≥5 unless indicated, *p < 0.05.
As we had previously shown that under stress conditions signaling via type I IFN was needed for Treg viability [22], we examined both Treg viability and the stability of Foxp3 expression. We sorted Treg from WT and cKO mice from dLN on the basis of YFP expression and parked these activated Tregs into naïve congenic CD45.1 mice. After 18 d, splenocytes from the recipient mice were analyzed for the presence of parked Tregs as well as Foxp3 expression. Overall, the numbers of donor WT and cKO Tregs detected were similar and they also retained high levels of Foxp3 expression (Supplementary Fig. 1J–K).
3.2. IFNAR signaling on Treg cells modulates chemokine production by CD4+Foxp3− T cells
To further probe for differences between Treg from cKO mice and WT mice, we extensively phenotyped Treg from unimmunized mice, but did not detect any significant differences between WT and cKO Treg (Supplementary Fig. 2A). Similarly, RNA-Seq analysis of WT and cKO Treg from unimmunized mice did not reveal any major differences in their transcriptome (Supplementary Fig. 2B). Next, we sorted Foxp3+ cells from the dLN of cKO or WT mice on d 4 post induction of EAE and subjected them to bulk RNA sequencing. RNA-Seq analysis revealed that out of 93 differentially regulated genes that passed the filter of FDR (false discovery rate) adjusted p-value ≤ 0.05, 72 genes were down-regulated and 21 genes were up-regulated in cKO Tregs as compared to WT Tregs (Fig. 2A–B). Not surprisingly, multiple IRFs (IRF1, IRF7, IRF8 and IRF9) were down-regulated in cKO Tregs as compared to WT Tregs. Upstream regulator analysis with these differentially expressed genes revealed various IRFs, particularly STAT1 and IRF1were inactivated in cKO Tregs (Fig. 2C). In addition, multiple chemokines (CCL8, CCL5, CCL12, CCL22 and CXCL10) were down-regulated in Treg in the absence of Type I IFN signaling (Fig. 2B).
Fig. 2.
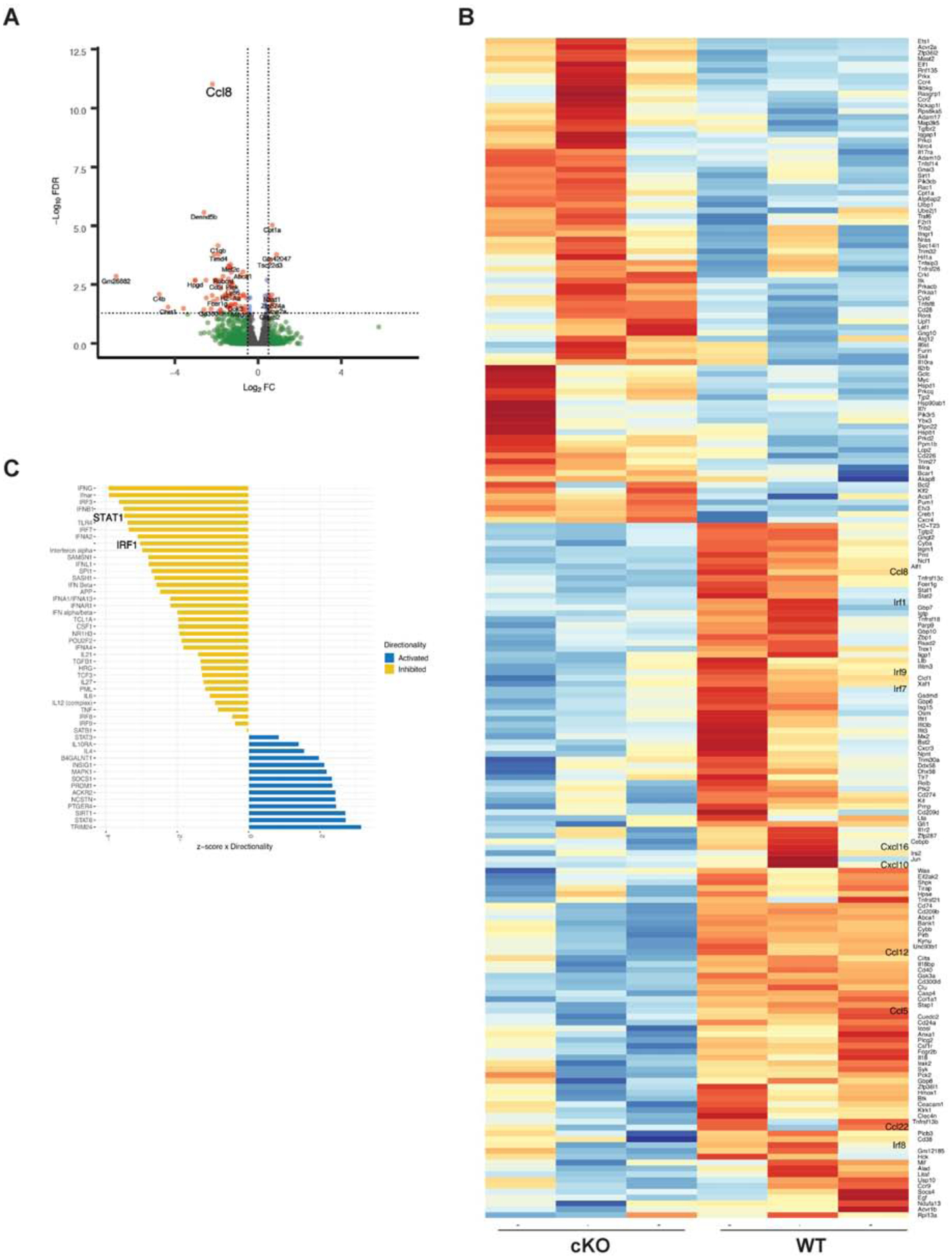
IFNAR signaling modulates chemokine and IRF gene expression on CD4+Foxp3+ T cells Foxp3YFP-Cre or IFNARfl/fl Foxp3YFP-Cre mice were immunized with MOG-CFA. dLN were isolated on d 4, CD4+Foxp3+ cells were purified by FACS, and subjected to RNA-Seq analysis
(A) Volcano plots generated for differential gene expression in CD4+Foxp3+ cells.
(B) Heat map analysis of chemokine/cytokine related genes in CD4+Foxp3+ cells from WT and cKO mice. Colors indicate up-regulation (red) or down-regulation (blue). (C) Predicted activated or inhibited transcription factors using up-stream regulator analysis of differentially regulated genes obtained after RNA-Seq analysis of d 4 immunized mice. Molecules that were predicted to be inhibited in cKO mice are shown in yellow, while the blue bars show molecules that are predicted to be activated in Tregs of cKO mice. (N=3 mice).
We also performed an RNA-Seq analysis of CD4+Foxp3− cells from d 4 dLN of cKO and WT mice. Out of 263 differentially regulated genes that passed the filter of FDR adjusted p-value ≤ 0.05, 139 genes were down-regulated and 124 genes were up-regulated in CD4+Foxp3− T cells from cKO mice as compared to that of WT mice (Fig. 3A–B). Of note, one of the most significantly down-regulated genes in the CD4+Foxp3− T cells from cKO mice was CCL8. To validate this data at the protein level, we induced EAE in cKO and WT mice and stained cells from dLN at day 5 for the intracellular expression of CCL8 (Fig. 3C). CD4+ T cells from dLN of naïve WT and cKO mice had an identical MFI for CCL8. However, immunization with MOG increased CCL8 expression in dLN from WT mice by more than 2-fold, but CD4+ T cells from the cKO failed to up-regulate CCL8 expression and the level of CCL8 remained at the resting level. The failure to up-regulate CCL8 in the dLN of cKO mice was not restricted to CD4+Foxp3− T lymphocytes as non-T cells also failed to up-regulate CCL8 expression (Fig. 3C). The heat map that visualizes the expression levels of differentially regulated genes associated with cytokine/chemokine-related gene ontology (GO) terms indicated that CD4+Foxp3− T cells from cKO mice had an altered capacity to produce multiple chemokines in addition to CCL8 as compared to WT mice. CCL8 and CCL22 were most prominently down-regulated in CD4+ T cells from cKO mice (Fig. 3B). These results were confirmed in a quantitative chemokine ligand ELISA assay on cells from day 5 dLN which again demonstrated that not only CD4+ T cells, but CD8+ T cells, B cells and endothelial cells were defective in CCL9 secretion (Fig. 3D). We examined chemokine receptor expression on CD4+Foxp3− T lymphocytes as well as Tregs from d3 draining lymph nodes of cKO and WT mice using the chemokine pcr array but did not find any differences among any of the groups (data not shown).
Fig. 3.
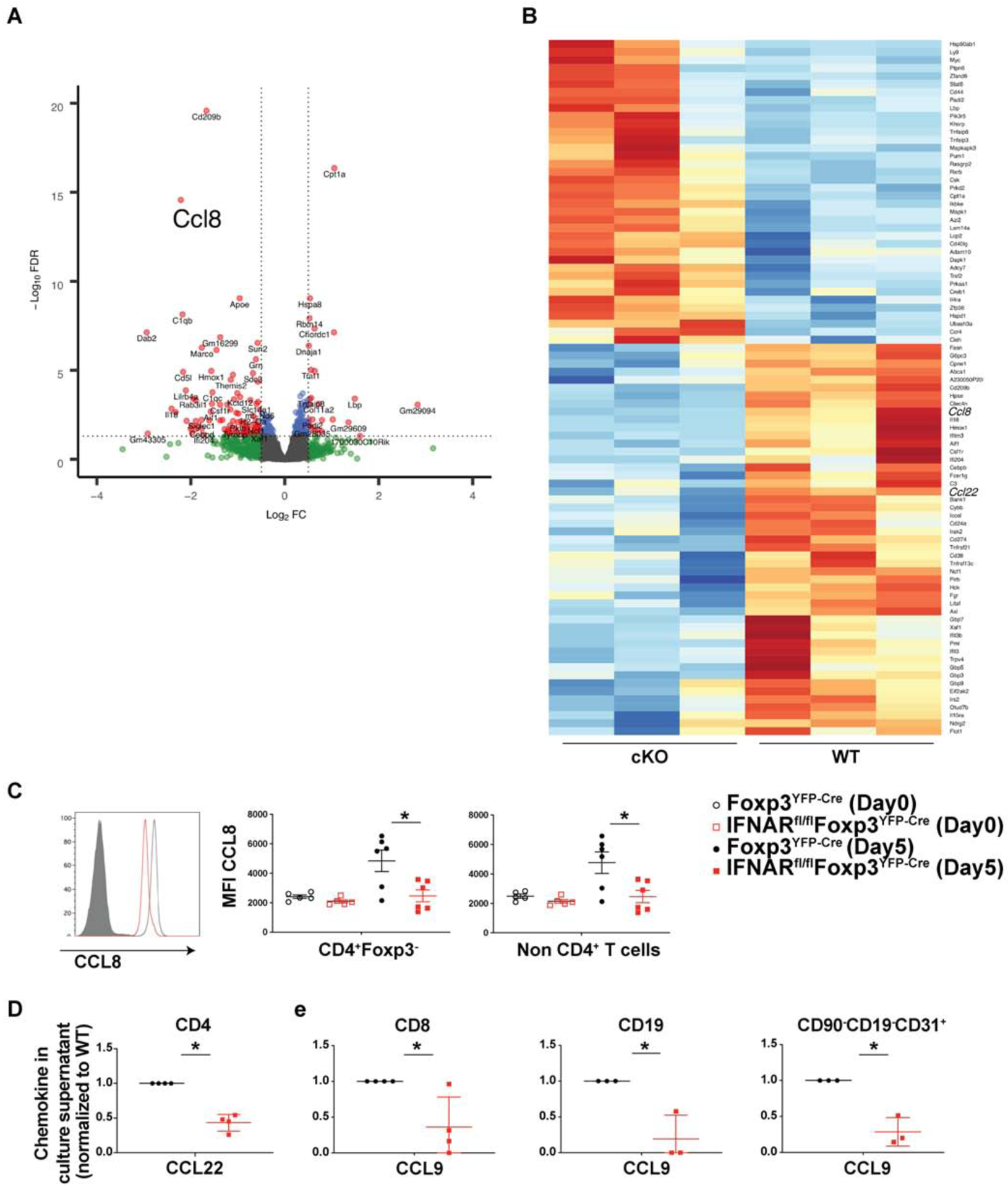
IFNAR signaling on Tregs modulates chemokine secretion by CD4+Foxp3− T cells dLN were isolated on d 4 from Foxp3YFP-Cre or IFNARfl/fl Foxp3YFP-Cre mice immunized with MOG-CFA, CD4+Foxp3− cells were purified by FACS, and subjected to RNA-Seq analysis (N=3 mice). (A) Volcano plots generated for differential gene expression in CD4+Foxp3− cells.
(B) Heat map analysis of chemokine/cytokine related genes in CD4+Foxp3− cells from WT and cKO mice. Colors indicate up-regulation (red) or down-regulation (blue).
(C) dLN cells of WT and cKO mice from naïve or from mice d 5 post immunization were stained for intra-cellular CCL8. Mean Fluorescence Intensity (MFI) plotted on CD4+Foxp3− cells, isotype and conjugate control (grey filled histograms), cKO mice (red) and WT mice (black). N=5, mean± SE, representative of two independent experiments.
(D–E) The indicated cell types were FACS sorted from dLN on d 4 post immunization and stimulated with PMA-Ionomycin without protein transport inhibitor for 4 h. Cell supernatants were assayed for chemokines by ELISA according to manufacturer’s recommendation. Amount of chemokine normalized to WT to remove inter-experimental variability. N≥4, mean± SE, cumulative of independent experiments.
3.3. cKO mice have a defect in the migration of MDSCs to the dLN during priming
The defect in the production of chemokines in the priming environment prompted us to examine in more detail the phenotype of cells present in the dLN early during the course of the inflammatory response. We initially focused our efforts on the analysis of CD11b+ cells in the dLN. Surprisingly, we observed a gradual infiltration of CD11b+ cells in the dLN of WT mice during the first seven days of priming, but a complete absence of these cells in the cKO mice during the same time interval. By day 9 after priming the numbers of CD11b+ cells were similar in the WT and cKO mice (Fig. 4A).
Fig. 4.
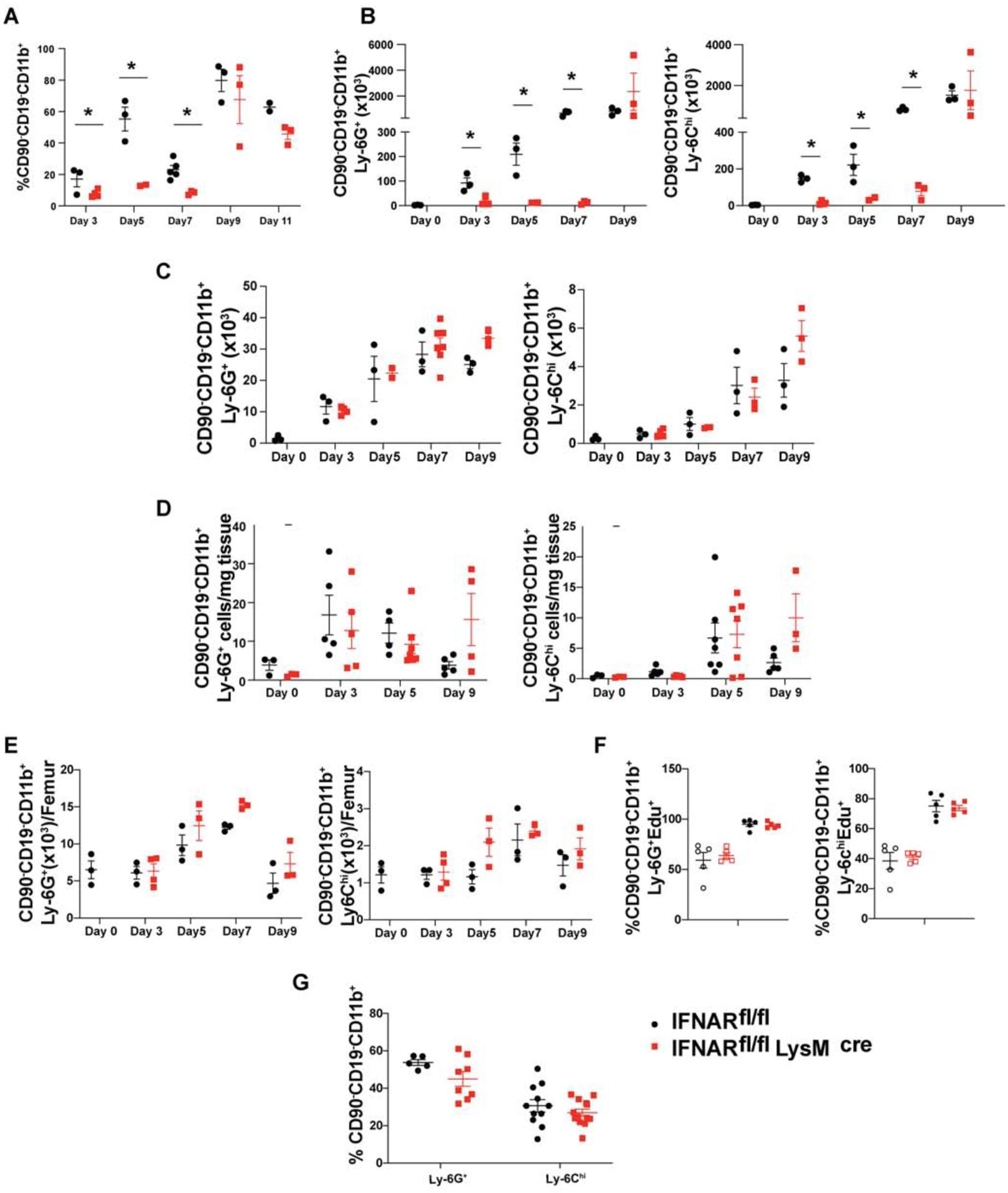
Lack of IFNAR signaling on Tregs leads to defective recruitment of MDSCs to dLN during priming
EAE was induced in Foxp3YFP-Cre (black spheres) or IFNARfl/fl Foxp3YFP-Cre (red cubes) mice and (A) Percentages of CD90−/CD19−CD11b+ cells in draining lymph nodes and absolute number of G-MDSCs (CD90−/CD19−CD11b+Ly6G+Ly6C−) and M-MDSCs (CD90−/CD19−CD11b+Ly6G−Ly6Chi) cells in (B) draining lymph nodes, (C) spleen, (D) CNS (brain and spinal cord) tissue and (E) bone marrow were measured at the indicated time points. N=3, mean± SE, representative of two independent experiments.
(F) Foxp3YFP-Cre or IFNARfl/fl Foxp3YFP-Cre mice were injected Edu (200mg/kg) i.p at d 0 and d 2, immunized with MOG-CFA (filled black circle=WT, filled red square=cKO) or left unimmunized (open black circle=WT, open red square=cKO), and euthanized on d 3. The incorporation of Edu in G-MDSCs and M-MDSCs from bone marrow was determined (N=5, mean± SE, representative of two independent experiments).
(G) IFNARfl/fl or IFNARfl/fl LysMCre mice were immunized with MOG-CFA and the percentages of G-MDSCs and M-MDSCs in dLN were quantitated on d 7 (N≥5, mean± SE, cumulative of two independent experiments.
The most likely candidates for the CD11b+ cells population in the dLN of the WT mice are myeloid derived suppressor cells (MDSCs), a heterogenous pool of cells consisting of progenitors and immature stages of myeloid cells expressing CD11b and Gr1 on their cell surface [30, 31]. MDSCs have been divided into two subsets based on surface markers and function as either granulocytic CD11b+Ly6G+Ly6Clow (G-MDSC) or monocytic CD11b+Ly6G−Ly6Chi (M-MDSC). Under steady state, they make up 1–2% of the spleen and 20–30% of bone marrow, lack any immune-suppressive properties, and quickly differentiate into their mature counterparts [32, 33]. However, during inflammation, they quickly expand, retain their immature phenotype, and acquire immunosuppressive functions [30, 32, 33]. Upon careful examination of the CD11b+ cells, we detected an increase of both G-MDSCs and M-MDSCs as early as d 3 to d 7 post priming in WT, but not in cKO mice (Fig. 4B). The increase was dependent on antigen-specific T cell stimulation as it occurred in mice immunized with MOG-IFA and was independent of the use of pertussis toxin in the priming protocol (Supplementary Fig. 3). Surprisingly, the defect in migration of the MDSCs was specific to the site of priming, as the numbers of MDSCs in spleen (Fig. 4C), CNS (brain and spinal cord) (Fig. 4D), and bone marrow (Fig. 4E) at various time points after priming were similar in WT and cKO mice.
To determine if the lack of MDSCs was secondary to a failure of generation in bone marrow, we injected cKO and WT mice with Edu (5-ethynyl-2’-deoxyuridine) with or without EAE induction. In the absence of priming, approximately 50% of G-MDSCs and 40% of M-MDSCs were labeled (Fig. 4F). The proportion of labeled cells increased to more than 95% for G-MDSCs and 80% for M-MDSCs in response to EAE induction indicating a major increase in MDSC turnover as a result of inflammation. However, cKO and WT mice had equivalent proportions of Edu labelled cells in bone marrow under both basal and priming conditions ruling out differential generation of MDSCs as a reason for the absence of MDSCs in the dLN of cKO mice. To determine that the failure of recruitment of MDSCs to dLN was specific to downstream effects of IFNAR signaling in Tregs, we induced EAE in IFNARfl/flxLysMCre mice and observed no difference in the proportion of MDSCs in dLN (Fig. 4G).
3.4. Mice with a deletion of myeloid cells develop very severe EAE
MDSCs have been shown to both suppress and enhance EAE under different experimental conditions. It seemed likely that MDSCs were suppressive in this model as disease severity was less in WT compared to cKO mice. To further define the role of myeloid cells during the early stages of priming, we injected WT mice with a single dose of either a myeloid cell depleting antibody (Ly-6G clone RB6–8C5) or control antibody (rat IgG2b) at the time of EAE induction. Treatment with the depleting antibody resulted in a 50% systemic reduction of myeloid cells with minimal change in other populations (Fig. 5A–B). By d 7, the myeloid compartment was completely reconstituted with newly generated cells from the bone marrow. Interestingly, the transient systemic reduction of myeloid cells during the priming phase of disease resulted in 50% mortality and severe exacerbation of disease severity (Fig. 5C–D). Concomitantly, myeloid cell depleted mice had higher proportions of activated CD4+ T cells and a higher percentage of IFN-γ producers in spleen (Fig. 5E–F). This result is consistent with the role of MDSCs as a suppressor population that helps control overt activation of responding CD4+ T cells during the priming phase of disease.
Fig. 5.
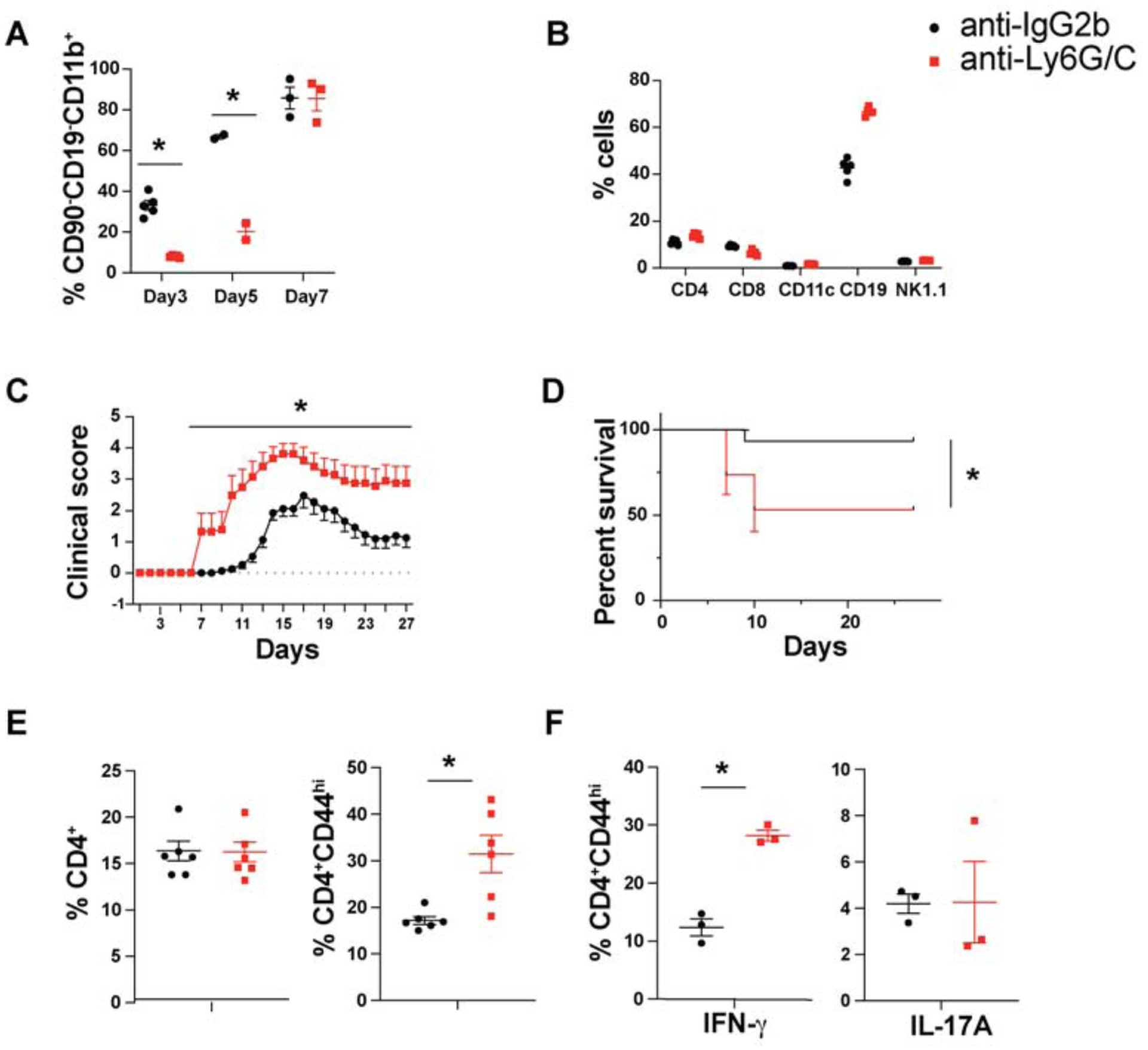
Transient depletion of myeloid cells during the priming phase of EAE exacerbates the disease
C57BL/6 mice were injected with either anti-IgG2b or anti-Ly6G/C (RB6–8C5; 100μg/mouse) at the time of EAE induction. (A) The percentages of CD90−CD19−CD11b+ cells in spleen at the indicated time points (B) and percentages of immune cell subpopulations at d 3 in spleen (N≥3, mean± SE). (C) EAE disease course (D) Mortality (N=15, cumulative of independent experiments, mean± SE, *p < 0.05). CD4+ T cells from spleen at d 7 post EAE induction were analyzed for (E) CD44 expression (N=6, cumulative of two independent experiments, mean± SE) and (F) for IL-17A and IFN-γ cytokine staining. (N=3, mean± SE, representative of two independent experiments).
4. Discussion
This study extends previous observations and provides a mechanism by which type I IFNs play a critical suppressive function in development of EAE. In contrast to a previous study [21] which demonstrated an important role of type I IFN signaling on myeloid cells, we observed marked enhancement of the severity of EAE when the IFNAR was specifically deleted from Treg. The deletion of IFNAR on Treg resulted in a complex change in the microenvironment of the dLN during the early phase (d 4–8) of T cell priming marked by the absence of MDSCs. As we had observed in LCMV infection [23], signaling by type I IFN attenuates Treg mediated suppression. In the absence of type I IFN signaling on Treg early during the course of T cell priming, Treg suppression is enhanced with decrease in the production of several chemokines which play critical roles in the migration of MDSCs to the dLN where they play a role in modulating effector T cell activation. Modulation of MDSC movement depended on type I IFN signaling on Treg, was specific to the priming site, and was independent of IFN signaling on MDSCs. The suppression of chemokine production observed in the absence of IFNAR signaling on Treg most prominently involved CCL8, CCL9, and CCL22; which can bind to CCR1, CCR3, CCR4, CCR5, and CCR8, all implicated in the recruitment of myeloid cells to sites of inflammation [34, 35].
Myeloid cells migrate in response to temporal and spatial chemokine gradients [36], a capability that defines their response during steady state or inflammation [37]. The role of MDSCs in EAE remains controversial and some studies have shown that MDSCs play a role in perpetuation of disease while others demonstrate an important role in disease resolution. Ioannou [38] have shown that adoptive transfer of G-MDSCs from MOG/CFA immunized mice decreased EAE severity when transferred 4 and 7 d after antigenic challenge. G-MDSCs in this context acted at the level of the target tissue with reduced inflammation and demyelination in the spinal cord. Treatment of mice at disease onset with IFN-β increased the proportion of MDSCs in spleen with a correlative amelioration of EAE clinical score [39]. In contrast, our studies demonstrated equivalent recruitment of MDSCs in cKO mice to spleen and CNS tissue, but a defect in homing only to the dLN. Although we could not specifically deplete MDSCs, we observed that a reduction of all granulocytes during the first 5 days after priming markedly enhanced disease pathology which is consistent with our observations that the presence of MDSCs during the initial stages of priming plays an important regulatory role in the development of EAE.
While numerous studies have focused on Treg mediated suppression of cytokine production by CD4+ T cells [40], the capacity of Treg to suppress chemokine production has not been extensively studied. Lund [41] demonstrated a benefit of Treg suppression of chemokine production in that the enhanced chemokine production in dLN following the global depletion of Treg diverted T effector cells from the vaginal mucosa during the course of herpes simplex virus infection with resultant spread of the infection to the CNS and death of the animals. While we did not observe a major effect on cytokine production by CD4+ T cells in the dLN, the selective suppression of chemokine production is consistent with previous studies [42, 43] which demonstrated that under conditions of strong antigen stimulation the capacity of Treg to inhibit cytokine production was abrogated, but their capacity to suppress chemokine production was maintained.
The potential molecular mechanisms used by type I IFNs to down-regulate suppressor function remain unclear. The ability of Treg to modulate chemokine production by a variety of cell types in the dLN including Foxp3− T cells, DCs, and other cell types supports the concept that Treg suppression is a much more complex process than just preventing the priming and expansion of antigen-specific Foxp3− T cells. The ability of Treg to control the migration of a critical suppressor population, MDSCs, to the draining LN indicates that Treg suppression involves the modulation of the entire lymph node microenvironment. We did not detect any enhancement of IL-10 or TGF-β production by cKO Treg. A recent study has demonstrated that type I IFNs rapidly induce IRF1 in hepatocytes with the subsequent induction of a pro-inflammatory cascade including chemokine production [44]. This study is consistent with our RNA-seq studies which demonstrated downregulation of various IRFs and STATs particularly IRF1 and STAT1 in cKO Tregs isolated on d4 after priming. Thus, activation of the IRF1 pathway in Treg following stimulation with type I IFNs is likely to be the mechanism for attenuation of Treg suppression by type I IFNsMore detailed studies of the Treg proteome and metabolome may be indicted to resolve this issue, but are technically challenging given the frequency of Tregs in the dLN. It also remains unclear how our results directly relate to the therapeutic effects of type I IFN in chronic MS. Our studies solely address the issue of the role of Tregs during the priming phase of EAE. Nevertheless, as epitope spreading occurs in MS, it remains possible that type I IFN treatment plays a role in modulating this process during the evolution of chronic disease.
5. Conclusions
Tregs respond to type I IFN secreted by dendritic cells during the very early stages of antigen priming and shape the response of CD4+ T cells by changing the microenvironment of the priming site resulting in the recruitment of other suppressor populations, such as MDSC. Taken together, these findings offer new insights into the complex process of suppression by Treg which are not restricted to a single aspect of T lymphocyte function, but involves the entire tissue priming microenvironment. The modulation of chemokine production by multiple cell types represents a powerful mechanism whereby Treg can play a critical “tuning” function to control the overall severity of disease.
Supplementary Material
Highlights.
Type I IFN signaling modulates IRF, STAT1 and chemokine gene expression on Treg
IFNAR on Treg modulates chemokine secretion by priming tissue resident cells
Lack of IFNAR leads to defective recruitment of MDSCs to priming site
MDSCs play suppressive role during priming phase of EAE
Acknowledgements
Authors would like to thank and acknowledge Ke Wang, Flow Cytometry Section, NIAID for assistance in cell sorting and D. Glass for genotyping of mice.
This work is supported by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Intitutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Statement:
All of the authors concur with this submission. The manuscript has not been submitted elsewhere. The authors report no conflicts of interest.
References
- [1].Stetson DB, Medzhitov R. Type I interferons in host defense. Immunity, 2006;25:373–81. [DOI] [PubMed] [Google Scholar]
- [2].Boxx GM, Cheng G. The Roles of Type I Interferon in Bacterial Infection. Cell Host Microbe, 2016;19:760–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Murira A, Lamarre A. Type-I Interferon Responses: From Friend to Foe in the Battle against Chronic Viral Infection. Front Immunol, 2016;7:609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Soper A, Kimura I, Nagaoka S, Konno Y, Yamamoto K, Koyanagi Y et al. Type I Interferon Responses by HIV-1 Infection: Association with Disease Progression and Control. Front Immunol, 2017;8:1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Blank T, Prinz M. Type I interferon pathway in CNS homeostasis and neurological disorders. Glia, 2017;65:1397–406. [DOI] [PubMed] [Google Scholar]
- [6].Floris S, Ruuls SR, Wierinckx A, van der Pol SM, Dopp E, van der Meide PH et al. Interferon-beta directly influences monocyte infiltration into the central nervous system. J Neuroimmunol, 2002;127:69–79. [DOI] [PubMed] [Google Scholar]
- [7].Heine S, Ebnet J, Maysami S, Stangel M. Effects of interferon-beta on oligodendroglial cells. J Neuroimmunol, 2006;177:173–80. [DOI] [PubMed] [Google Scholar]
- [8].Okada K, Kuroda E, Yoshida Y, Yamashita U, Suzumura A, Tsuji S. Effects of interferon-beta on the cytokine production of astrocytes. J Neuroimmunol, 2005;159:48–54. [DOI] [PubMed] [Google Scholar]
- [9].Pogue SL, Preston BT, Stalder J, Bebbington CR, Cardarelli PM. The receptor for type I IFNs is highly expressed on peripheral blood B cells and monocytes and mediates a distinct profile of differentiation and activation of these cells. J Interferon Cytokine Res, 2004;24:131–9. [DOI] [PubMed] [Google Scholar]
- [10].Panitch HS, Hirsch RL, Schindler J, Johnson KP. Treatment of multiple sclerosis with gamma interferon: exacerbations associated with activation of the immune system. Neurology, 1987;37:1097–102. [DOI] [PubMed] [Google Scholar]
- [11].Stone LA, Frank JA, Albert PS, Bash C, Smith ME, Maloni H et al. The effect of interferon-beta on blood-brain barrier disruptions demonstrated by contrast-enhanced magnetic resonance imaging in relapsing-remitting multiple sclerosis. Ann Neurol, 1995;37:611–9. [DOI] [PubMed] [Google Scholar]
- [12].Biernacki K, Antel JP, Blain M, Narayanan S, Arnold DL, Prat A. Interferon beta promotes nerve growth factor secretion early in the course of multiple sclerosis. Arch Neurol, 2005;62:563–8. [DOI] [PubMed] [Google Scholar]
- [13].Corsini E, Gelati M, Dufour A, Massa G, Nespolo A, Ciusani E et al. Effects of beta-IFN-1b treatment in MS patients on adhesion between PBMNCs, HUVECs and MS-HBECs: an in vivo and in vitro study. J Neuroimmunol, 1997;79:76–83. [DOI] [PubMed] [Google Scholar]
- [14].Muraro PA, Liberati L, Bonanni L, Pantalone A, Caporale CM, Iarlori C et al. Decreased integrin gene expression in patients with MS responding to interferon-beta treatment. J Neuroimmunol, 2004;150:123–31. [DOI] [PubMed] [Google Scholar]
- [15].Chabot S, Williams G, Yong VW. Microglial production of TNF-alpha is induced by activated T lymphocytes. Involvement of VLA-4 and inhibition by interferonbeta-1b. J Clin Invest, 1997;100:604–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Chen M, Chen G, Nie H, Zhang X, Niu X, Zang YC et al. Regulatory effects of IFN-beta on production of osteopontin and IL-17 by CD4+ T Cells in MS. Eur J Immunol, 2009;39:2525–36. [DOI] [PubMed] [Google Scholar]
- [17].Kozovska ME, Hong J, Zang YC, Li S, Rivera VM, Killian JM et al. Interferon beta induces T-helper 2 immune deviation in MS. Neurology, 1999;53:1692–7. [DOI] [PubMed] [Google Scholar]
- [18].Mirandola SR, Hallal DE, Farias AS, Oliveira EC, Brandao CO, Ruocco HH et al. Interferon-beta modifies the peripheral blood cell cytokine secretion in patients with multiple sclerosis. Int Immunopharmacol, 2009;9:824–30. [DOI] [PubMed] [Google Scholar]
- [19].Hecker M, Goertsches RH, Fatum C, Koczan D, Thiesen HJ, Guthke R et al. Network analysis of transcriptional regulation in response to intramuscular interferon-beta-1a multiple sclerosis treatment. Pharmacogenomics J, 2012;12:134–46. [DOI] [PubMed] [Google Scholar]
- [20].Sellebjerg F, Datta P, Larsen J, Rieneck K, Alsing I, Oturai A et al. Gene expression analysis of interferon-beta treatment in multiple sclerosis. Mult Scler, 2008;14:615–21. [DOI] [PubMed] [Google Scholar]
- [21].Prinz M, Schmidt H, Mildner A, Knobeloch KP, Hanisch UK, Raasch J et al. Distinct and nonredundant in vivo functions of IFNAR on myeloid cells limit autoimmunity in the central nervous system. Immunity, 2008;28:675–86. [DOI] [PubMed] [Google Scholar]
- [22].Metidji A, Rieder SA, Glass DD, Cremer I, Punkosdy GA, Shevach EM. IFN-alpha/beta receptor signaling promotes regulatory T cell development and function under stress conditions. J Immunol, 2015;194:4265–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Gangaplara A, Martens C, Dahlstrom E, Metidji A, Gokhale AS, Glass DD et al. Type I interferon signaling attenuates regulatory T cell function in viral infection and in the tumor microenvironment. PLoS Pathog, 2018;14:e1006985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Rieder SA, Metidji A, Glass DD, Thornton AM, Ikeda T, Morgan BA et al. Eos Is Redundant for Regulatory T Cell Function but Plays an Important Role in IL-2 and Th17 Production by CD4+ Conventional T Cells. J Immunol, 2015;195:553–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics, 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics, 2011;12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ge SX, Son EW, Yao R. iDEP: an integrated web application for differential expression and pathway analysis of RNA-Seq data. BMC Bioinformatics, 2018;19:534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol, 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lee JY, Kim J, Yi J, Kim D, Kim HO, Han D et al. Phenotypic and Functional Changes of Peripheral Ly6C(+) T Regulatory Cells Driven by Conventional Effector T Cells. Front Immunol, 2018;9:437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Bronte V, Zanovello P. Regulation of immune responses by L-arginine metabolism. Nat Rev Immunol, 2005;5:641–54. [DOI] [PubMed] [Google Scholar]
- [31].Rodriguez PC, Ernstoff MS, Hernandez C, Atkins M, Zabaleta J, Sierra R et al. Arginase I-producing myeloid-derived suppressor cells in renal cell carcinoma are a subpopulation of activated granulocytes. Cancer Res, 2009;69:1553–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol, 2009;9:162–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol, 2008;181:5791–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Klementowicz JE, Mahne AE, Spence A, Nguyen V, Satpathy AT, Murphy KM et al. Cutting Edge: Origins, Recruitment, and Regulation of CD11c(+) Cells in Inflamed Islets of Autoimmune Diabetes Mice. J Immunol, 2017;199:27–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Sokol CL, Camire RB, Jones MC, Luster AD. The Chemokine Receptor CCR8 Promotes the Migration of Dendritic Cells into the Lymph Node Parenchyma to Initiate the Allergic Immune Response. Immunity, 2018;49:449–63 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Petrie Aronin CE, Zhao YM, Yoon JS, Morgan NY, Prustel T, Germain RN et al. Migrating Myeloid Cells Sense Temporal Dynamics of Chemoattractant Concentrations. Immunity, 2017;47:862–74 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Delano MJ, Kelly-Scumpia KM, Thayer TC, Winfield RD, Scumpia PO, Cuenca AG et al. Neutrophil mobilization from the bone marrow during polymicrobial sepsis is dependent on CXCL12 signaling. J Immunol, 2011;187:911–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Ioannou M, Alissafi T, Lazaridis I, Deraos G, Matsoukas J, Gravanis A et al. Crucial role of granulocytic myeloid-derived suppressor cells in the regulation of central nervous system autoimmune disease. J Immunol, 2012;188:1136–46. [DOI] [PubMed] [Google Scholar]
- [39].Melero-Jerez C, Suardiaz M, Lebron-Galan R, Marin-Banasco C, Oliver-Martos B, Machin-Diaz I et al. The presence and suppressive activity of myeloid-derived suppressor cells are potentiated after interferon-beta treatment in a murine model of multiple sclerosis. Neurobiol Dis, 2019;127:13–31. [DOI] [PubMed] [Google Scholar]
- [40].Shevach EM. Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity, 2009;30:636–45. [DOI] [PubMed] [Google Scholar]
- [41].Lund JM, Hsing L, Pham TT, Rudensky AY. Coordination of early protective immunity to viral infection by regulatory T cells. Science, 2008;320:1220–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Dal Secco V, Soldani C, Debrat C, Asperti-Boursin F, Donnadieu E, Viola A et al. Tunable chemokine production by antigen presenting dendritic cells in response to changes in regulatory T cell frequency in mouse reactive lymph nodes. PLoS One, 2009;4:e7696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Morlacchi S, Dal Secco V, Soldani C, Glaichenhaus N, Viola A, Sarukhan A. Regulatory T cells target chemokine secretion by dendritic cells independently of their capacity to regulate T cell proliferation. J Immunol, 2011;186:6807–14. [DOI] [PubMed] [Google Scholar]
- [44].Forero A, Ozarkar S, Li H, Lee CH, Hemann EA, Nadjsombati MS et al. Differential Activation of the Transcription Factor IRF1 Underlies the Distinct Immune Responses Elicited by Type I and Type III Interferons. Immunity, 2019;51:451–64 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Lercher A, Bhattacharya A, Popa AM, Caldera M, Schlapansky MF, Baazim H et al. Type I Interferon Signaling Disrupts the Hepatic Urea Cycle and Alters Systemic Metabolism to Suppress T Cell Function. Immunity, 2019;51:1074–87 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.