

Abstract
Rheumatoid arthritis (RA) is a complex autoimmune disease with an etiology that is not yet well understood, disproportionally affects women and also varies in incidence and prevalence by population. The presence of anti-citrullinated protein antibodies (ACPA) is a highly specific biomarker for the diagnosis of clinically apparent RA. ACPA are also present in the serum for an average of 3–5 years prior to the onset of RA during an asymptomatic period characterized by mucosal inflammation and local ACPA production at these sites. We hypothesized that systemic complement activation products might be generated during the pre-clinical initiation of RA and/or provide a second hit that promotes subsequent arthritis development in the joints. In addition, we evaluated which demographic and genetic features and environmental exposures could influence the complement activation process. We analyzed plasma from healthy subjects, subjects at-risk for the development of RA based on serum ACPA positivity in absence of inflammatory arthritis (IA), and ACPA positive RA subjects by Multiplex Assay and ELISA for eighteen complement system components, factors and activation products belonging to the classical, lectin and alternative pathways. By using regression models, associations between complement proteins and various demographic, genetic, and environmental factors previously found to be associated with RA, including sex, smoking, shared epitope, and oral contraceptive use, were examined. We found no evidence of systemic complement activation in ACPA positive subjects without IA, but in contrast found evidence of systemic involvement of the both classical and alternative pathways during the stage of the disease where classified RA is present, (i.e. during joint inflammation and damage). With regard to the demographic, genetic, and environmental variables, females who reported current or past oral contraceptive use and subjects with current tobacco exposure demonstrated alterations of the alternative pathway of complement. Furthermore, RA subjects with established disease who have a body mass index categorized as obese demonstrated higher levels of C2 compared to RA subjects who are not considered obese. In sum, the complement system may be involved in the pathogenesis of RA, with only localized mucosal effects during the preclinical period in those at-risk for RA but in the joint as well as systemically in those who have developed clinically apparent arthritis.
Keywords: Complement system, arthritis, ACPA, inflammation, RF, risk factors
1. Introduction
Rheumatoid arthritis (RA) is a complex autoimmune disease which presents clinically as a debilitating inflammatory autoimmune disease of the joints that affects approximately 0.24% of the world population (Cross et al., 2014). The mortality rate in patients with RA is higher than the general population (Widdifield et al., 2018), and the serious morbidity of this condition impacts the public health care system substantially (Helmick et al., 2008). Regardless of the origin of this disease, the primary target tissue is the synovium, resulting in synovitis and pannus formation in the joints. In addition, the disease process in RA can affect other organs of the body, one of the primary examples being the lung.
The exact etiology of RA and roles of the tightly associated anti-citrullinated protein antibodies (ACPA) in the pathogenesis of RA are not completely understood. While ACPAs may not be present in all incident cases of RA, their presence in serum and/or in sputum in individuals before the development of clinically apparent joint disease has provided important information regarding the initiation of this disease in humans (Demoruelle et al., 2013; Willis et al., 2013).
With regard to the pathogenic potential of ACPA, RA patients who are ACPA positive demonstrate worse clinical disease outcomes as compared to ACPA negative patients (Kroot et al., 2000; Meyer et al., 2003). In addition, murine ACPA as well as human IgG monoclonal antibodies derived from RA patients can enhance arthritis in mice (Kuhn et al., 2006; Petkova et al., 2006), providing evidence that ACPA have the potential to be pathogenic. The decrease in rheumatoid factors (RF) and anti-CCP levels with clinical improvement following anti-CD20 treatment in RA patients (Cambridge et al., 2003) further strengthens the argument that APCA might be centrally involved in disease pathogenesis.
The presence of ACPA in serum many years prior to the clinical signs of disease (Deane et al., 2009; Nielen et al., 2004; Rantapaa-Dahlqvist et al., 2003) indicates that the development of autoantibodies to altered-self alone is not sufficient to trigger synovitis in RA. Rather, a “second hit” has been postulated to be required for localization of the pathogenic immune response to the joint (Banda et al., 2018; Firestein and McInnes, 2017). Furthermore, it has been shown in mice that tolerance is induced in ACPA-specific B cells in vivo, and ACPA production requires additional crucial steps beyond the generation of neoantigens by citrullination to develop arthritis (Yamada et al., 2018). This study challenges the central dogma that ACPA alone are not pathogenic in human RA and directly support a hypothesis that a second hit, perhaps provided by complement activation, is necessary to precipitate disease in the joints.
The complement system is an effector arm of the immune system that could in principle be causing tissue damage in an ACPA positive environment. Many studies have demonstrated that complement activation is necessary to develop experimental antibody-mediated arthritis [reviewed in (Holers and Banda, 2018)]. Therefore, understanding the role of the complement system as an effector mechanism and also an in-depth analysis of various complement components in peripheral blood during the initiation (preclinical) and perpetuation (clinically apparent arthritis) stages of human RA could be highly informative.
Many risk factors have been associated with susceptibility for the development of RA. These include: twin, sibling and family relationships (Aho et al., 1986; Silman et al., 1993); HLA susceptibility genes including an epitope in the third hypervariable region of the HLA-DR, also commonly called shared epitope (SE) (de Vries et al., 2002); cigarette smoking (Liao et al., 2009; Pedersen et al., 2007); bacterial infections or expansions, such as Porphyromonas gingivalis and Prevotella copri (Bartold et al., 2005; de Pablo et al., 2008; Marotte et al., 2006; Mercado et al., 2001; Mikuls et al., 2009; Rosenstein et al., 2004; Scher et al., 2013); and viral infections, such as Epstein-Barr virus (EBV) and Human T-lymphotropic virus type I (HTLV-I) (Eguchi et al., 1996; Ferrell et al., 1981; La Cava et al., 1997). Additional environmental exposures such as obesity (Crowson et al., 2013; Lu et al., 2014), and oral contraceptive use (Pedersen et al., 2006) may also impact the risk of developing RA.
Polymorphisms in complement genes and loci have been shown to influence risk in many diseases (Harris et al., 2012). Furthermore, small changes in the activities of polymorphic variants when inherited in some combinations dramatically change complement activity and also impact risk (Harris et al., 2012; Heurich et al., 2011). Additionally, TNF receptor-associated factor (TRAF) 1 and complement C5 highly polymorphic region on chromosome 9 has been linked to RA (Kurreeman et al., 2007). The C2 gene is also located in the HLA Class III region on short arm of chromosome 6. C1q and C2 are the key component of the CP. It has been shown that genetic variants of C1q predispose to RA (Trouw et al., 2013). In that study single nucleotide polymorphisms (SNPs) in and around the C1q gene in a Dutch RA population were correlated with C1q levels and were suggested to be a risk for RA development. In another study, it was concluded that, although both age-related macular degeneration (AMD) and RA are characterized by activation of the alternative pathway (AP), SNPs in the AP inhibitor complement factor H that predispose strongly to AMD do not confer substantial risk to RA (Trouw et al., 2011).
None of these prior studies have evaluated the role of activation of the complement system or variation in the complement proteins as a risk factor or modifier during the transition of pre-clinical to the clinical stage in RA patients. However, many mouse models of RA have shown that mice deficient in certain complement components such as C5 do not develop arthritis (Wang et al., 2000), and treatment of arthritic mice with anti-C5 inhibitory antibody (Wang et al., 1995) or with GalNac-conjugated C5 duplex (Anna Borodovsky, 2014) or GalNAc-conjugated MASP-3 duplex prevents arthritis (Banda et al., 2018). We and others have performed numerous studies demonstrating that genetic and pharmacologic modulation of complement function decreases disease progression in mouse models of RA. Nonetheless, pharmacological manipulation of complement has been less successful for the treatment of human RA. While a number of small molecule complement inhibitors and inhibitory antibodies have been developed and work well in vitro, they have failed in RA clinical trials. The reasons for this are unknown. For example, the anti-C5 monoclonal antibody BB5.1, which is a neutralizing antibody, is capable of decreasing or inhibiting arthritis in the collagen-induced arthritis and anti-collagen antibody-induced arthritis models, but clinical effects of C5 inhibition were modest. GalNAc-C5siRNA duplexes have not been tested in clinical trials for human RA but very effective in ameliorating arthritis in mice (Anna Borodovsky, 2014). Complete removal of components of the C5–C5aR axis via gene disruption is capable of profoundly affecting the course of disease, and a small molecule inhibitor; PMX53, resulted in a dose dependent block of C5a mediated activation; however, RA clinical trials were again less successful. Similarly, a humanized anti-C5 antibody showed excellent efficacy when used to treat paroxysmal nocturnal haemoglobinuria (Hill et al., 2005), however, its use in a phase IIb (unpublished) trial for the treatment of RA was unsuccessful (discussed in (Vergunst et al., 2007). Similarly murine anti-factor B inhibitory antibody reduces AP complement activation in vitro but was totally ineffective in mouse models of arthritis (Banda, NK unpublished data). Avacopan (CCX168) is being developed for inflammatory and autoimmune diseases, it blocks the activity of C5a (http://www.chemocentryx.com). Furthermore, the complement system as a part of innate immunity protects the body from infections, which might itself be a contributing risk factor due to over activation in the pathogenesis of RA.
The complement system is activated by three different pathways which share a common terminal pathway. The classical pathway (CP) of the complement includes C1q, C1r and C1s complement components. The lectin pathway (LP) includes complement components mannan-binding lectin (MBL), ficolins (FCNs) and collectins (CLs), along with three MBL-associated serum proteases (MASPs). The classical and lectin pathway share C2 and C4 complement components in assembly of the C3 convertase. The AP includes C3, factor B (FB), factor D (FD) and Properdin (P) complement components. The terminal pathway includes C5 and C5b-C9, also designated the membrane attack complex (MAC).
One study has shown that ACPA activate the complement system in vitro via the CP and the AP but not by the LP in RA (Trouw et al., 2009). In this study ACPA from all 60 patients activated the complement system. This important observation leads to the suggestion that complement activation can play very important roles in the pathogenesis of RA. Additionally, it has been shown that citrullination locally in the joints can increase inflammation through direct targeting by ACPA (Kuhn et al., 2006; Sokolove et al., 2011). Furthermore, IgM RF and IgA RF amplify complement activation mediated by ACPA-containing immune complexes (IC) (Anquetil et al., 2015). These findings suggest that ACPA-IC, incorporating IgM or IgA RF, participate in the triggering of the inflammation-promoting activation of complement cascades occurring in RA joints.
The question addressed here is whether there may be a change in the levels or balance of systemic complement proteins during early stages of the disease in those without clinical symptoms, i.e. individuals at-risk for future RA, as compared to patients with clinically diagnosed RA. In addition, differences in the complement protein levels in subjects at-risk for, or with classified and clinically active RA, might associate with various environmental risk factors to modify the steady state systemic inflammation and affect not only at-risk subjects but also those with sustained systemic inflammation and local joint damage. The associations and relationships among at-risk subjects, RA patients and complement proteins has not been explored in-depth. An abundance of serum complement protein or activation in subjects at-risk for RA could switch the balance between tolerance and autoimmunity due to their effector function and also due to epitope spreading.
2. Material and methods
2.1. Subjects
Subjects were recruited from the Studies of the Etiology of Rheumatoid Arthritis (SERA) cohort, described previously (Deane et al., 2009; Kolfenbach et al., 2009). SERA was designed to study the development of future RA through recruitment and longitudinal follow-up of individuals at-risk for future RA as well as subjects with classified RA and healthy controls. For the current study, we selected 18 at-risk subjects with the highest anti-CCP3 titers, assuming these subjects were statistically closest to RA development (Ford et al., 2018). We then randomly matched anti-CCP3 positive RA subjects and control subjects who were negative for RA-related autoantibodies to the previously selected 18 at-risk subjects by sex and age within ±10 years. All subjects were selected from a larger group of subjects who had plasma available for complement testing.
2.2. RA subjects
Sixteen subjects with classified RA by 1987 ACR criteria determined by chart review were included in the study. An additional 16 RA subjects were selected after initial analyses and analyzed separately to confirm associations. The additional 16 RA subjects were matched to the initial study groups by sex and age within ±10 years. These additional RA subjects were selected so that there were 8 RA subjects with BMI <30 and 8 RA subjects with BMI ≥30. All subjects were serum anti-CCP3 positive at the selected visit.
2.3. At-risk subjects
Eighteen subjects who were considered at-risk for future RA based on testing serum anti-CCP3 positive prior to study entry were included in the current study. Subjects were recruited from Colorado-based rheumatology clinics (27.8%) or Colorado-based health fairs (72.2%) and tested positive for ACPA. All of the at-risk subjects tested anti-CCP3 positive again at the selected study visit and were without current or past inflammatory arthritis.
Control subjects
Seventeen healthy control subjects were recruited through local advertisement. An additional 17 healthy control subjects were recruited from Colorado-based health fairs. Healthy control subjects recruited from health-fairs participated in a free rheumatoid arthritis screening where they had their serum tested for anti-CCP3, and the results of the test were negative. All healthy control subjects were seronegative for RA-related autoantibodies and without current or past inflammatory arthritis at the selected study visit.
2.5. Study visits
During the initial (baseline) visit and follow-up visits, subjects completed questionnaires relating to basic demographics, current and past environmental exposures including disease and illness, and a family history of disease and illness. Serum from blood draw was tested for ACPA using a commercially available enzyme-linked immunosorbent assay (ELISA); anti-CCP3 (Inova Diagnostics). Serum positivity was based on the manufacturer’s recommended cutoff level of ≥20 units. Blood for anti-CCP3 testing was drawn into clinical grade serum separation tubes (Fisher Scientific BD VacutainerTM)) and allowed to clot for 15 min and then centrifuged at 3000 rpm for 10 min. Serum was aliquoted into 2 ml graduated tubes and stored at −80 degrees within 1 hour of blood draw until the sample was tested. Blood for complement testing was drawn into clinical grade K2 EDTA vacutainer tubes (Fisher Scientific BD Vacutainer™) and centrifuged at 3000 rpm for 10 minutes within 15 minutes of collection. Plasma was aliquoted into 2 ml graduated tubes and stored at −80 degrees within 1 hour of blood draw until the sample was tested. Serum and plasma processing and storage was identical across the selected study groups. Although there are many new autoantibodies which are a common characteristic feature of rheumatic autoimmune diseases, many do not play a major pathogenic role. Nonetheless, some of them are extremely useful biomarkers and are being used to diagnose RA and these include ACPA, RF, anti-CarP, anti-PAD and RA33. In this study, we have used anti-CCP3 ACPA levels to confirm RA positivity, as this biomarker has been included in American College of Rheumatology (ACR) and European League for Rheumatoid arthritis (EULAR) criteria for RA classification (Aletaha et al., 2010)
2.6. Human leukocyte antigen shared epitope genotyping
DNA was extracted from buffy coat samples on each subject and was tested for the presence of specific alleles of the human leukocyte antigen (HLA) that encode a shared epitope (SE) associated with RA e.g. HLA-DR4 and HLA-DR1 alleles, as described previously (Kolfenbach et al., 2009). A subject was considered SE positive if one or more allele contained the following SE subtypes: DRB1*0401, 0404, 0405, 0408, 0409, 0410, and 0413; DRB1*0101, 0102; DRB1*1001.
2.7. Complement factor measurements
Complement analysis was performed in plasma that had not been previously thawed by a combination of multiplex and single assay methods. For the multiplex analysis the human complement bead-based xMAP technology (Luminex Corp, Northbrook IL) and commercially available kits (EMD Millipore, Milliplex Map, Burlington, MA) were used to measure thirteen complement proteins, spanning all three activation arms and the terminal pathway of complement. Measurements were made on a MagPix Luminex instrument. The Millipore Panel #1 was used to measure C2, C4b, C5a, C9, FD, MBL and Factor I (FI). Panel #2 was used to measure C1q, C3, C3b & iC3b, C4, FB, Factor H (FH), and Properdin (P). In addition, the complement activation markers Bb, C3a and the soluble terminal complement complex, sC5b-9, were measured by ELISA (Quidel Corp, San Diego CA). All testing methods had been optimized and validated, and for these studies were performed, within Exsera BioLabs, a College of American Pathologists (CAP) and Clinical Laboratory Improvement Amendments (CLIA) certified laboratory.
All analysis was performed in duplicate with the resulting mean values reported. For the multiplex Luminex data, the mean fluorescent intensity was the raw value and for the ELISA analysis the raw values was optical density. Standard curves and a four parameter parametric curve fit were utilized to calculate the absolute quality in ng/mL or μg/mL, as appropriate. Three quality controls (QC) were included in each run, including at least one laboratory developed and characterized QC. The QCs were monitored for performance, and for all testing in the study the values returned were within the required parameter, demonstrating assay performance. Human reference ranges for the analytes tested have been determined within Exsera by the measurement of greater than 100 normal individual. The normal individual pool was gender and racially inclusive.
2.8. Demographic, genetic, and environmental factors
Demographic information and environmental exposures were self-reported on study questionnaires and completed at the time of the study visit and blood draw. Demographic and environmental exposures that have previously been shown to be associated with RA were examined (Deane et al., 2017). Specifically, the association between complement levels and the following demographics, genetic, and environmental exposures were analyzed; sex (female/male), shared epitope (SE: positive/negative), body mass index (BMI: calculated as weight (kg)/height (m2)) as a dichotomous variable (obese: BMI≥30/non-obese: BMI<30), ever smoker (yes/no), and current smoker (yes/no). In addition, ever use of oral contraceptive (yes/no) was examined in female subjects.
2.9. Statistical analysis
For the current study, one study visit per subject was selected based on the inclusion and matching criteria and for each study group mentioned previously. Characteristics of the subjects were compared by study group and summarized in Table 1. Differences by study group were analyzed excluding the RA replication subjects but, we include these subjects in table 1 for reference. For continuous variables, the Kruskal-Wallis for group comparisons was used. For dichotomous variables, a chi-square test was used. Where cell size was less than 5, a Fisher’s exact test was performed. Unadjusted complement levels were compared across all three study groups using the Kruskal-Wallis test followed by post hoc Dunn’s test when the Kruskal-Wallis P-value ≤0.10 (Figure 1 and Figure 2). Complement levels were also compared across all three study groups after model adjustment for sex, race/ethnicity, and age in multivariable generalized linear regression models (Table 2).
Table 1.
Characteristics of study subjects by study group
| Control | At-Risk | RA | RA-replication | P-value | |
|---|---|---|---|---|---|
| N | 34 | 18 | 16 | 16 | |
| Age: mean (SD) | 55.4 (13.0) | 53.6 (13.9) | 55.4 (14.3) | 55.1 (13.0) | 0.90 |
| Sex: % female | 79.4 | 77.8 | 81.3 | 81.3 | 1.00 |
| RaceEthnicity: %NHW | 88.2 | 72.2 | 56.3 | 68.8 | 0.04 |
| Shared Epitope: %positive | 41.2 | 66.7 | 68.8 | 43.8 | 0.09 |
| BMI: =30 | 20.6 | 33.3 | 31.3 | 50.0 | 0.22 |
| Ever smoker: % yes | 38.2 | 27.8 | 50.0 | 56.3 | 0.41 |
| Current smoker: % yes | 0.0 | 11.1 | 18.8 | 18.8 | 0.02 |
| Female Specific | |||||
| N | 27 | 14 | 13 | 13 | |
| OC Use Ever: % yes | 77.8 | 76.9 | 84.6 | 84.6 | 1.00 |
Figure 1.
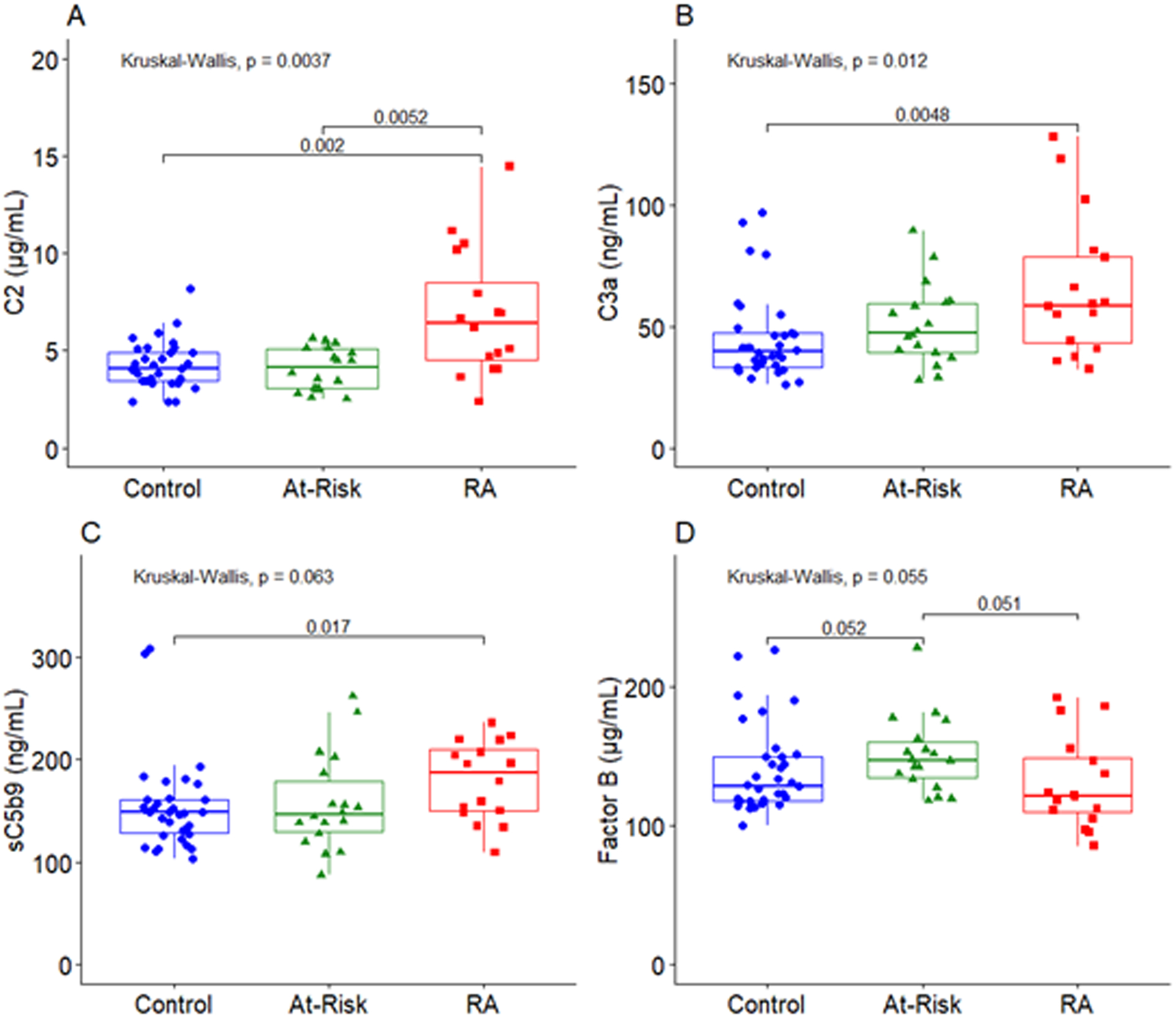
Complement levels differ significantly across RA patients, at-risk subjects, and healthy controls. A: Levels of complement component C2 were significantly higher in RA patients compared to both at-risk and control subjects. B: Levels of complement component C3a were significantly higher in RA patients compared to control subjects. C: Levels of sC5b9 were significantly higher in RA patients compared to controls subjects. D: Levels of Factor B were marginally higher in at-risk subjects compared to both RA patients and controls subjects. The middle line indicates the median value with the upper and lower hinges corresponding to the first and third quartiles respectively.
Figure 2.
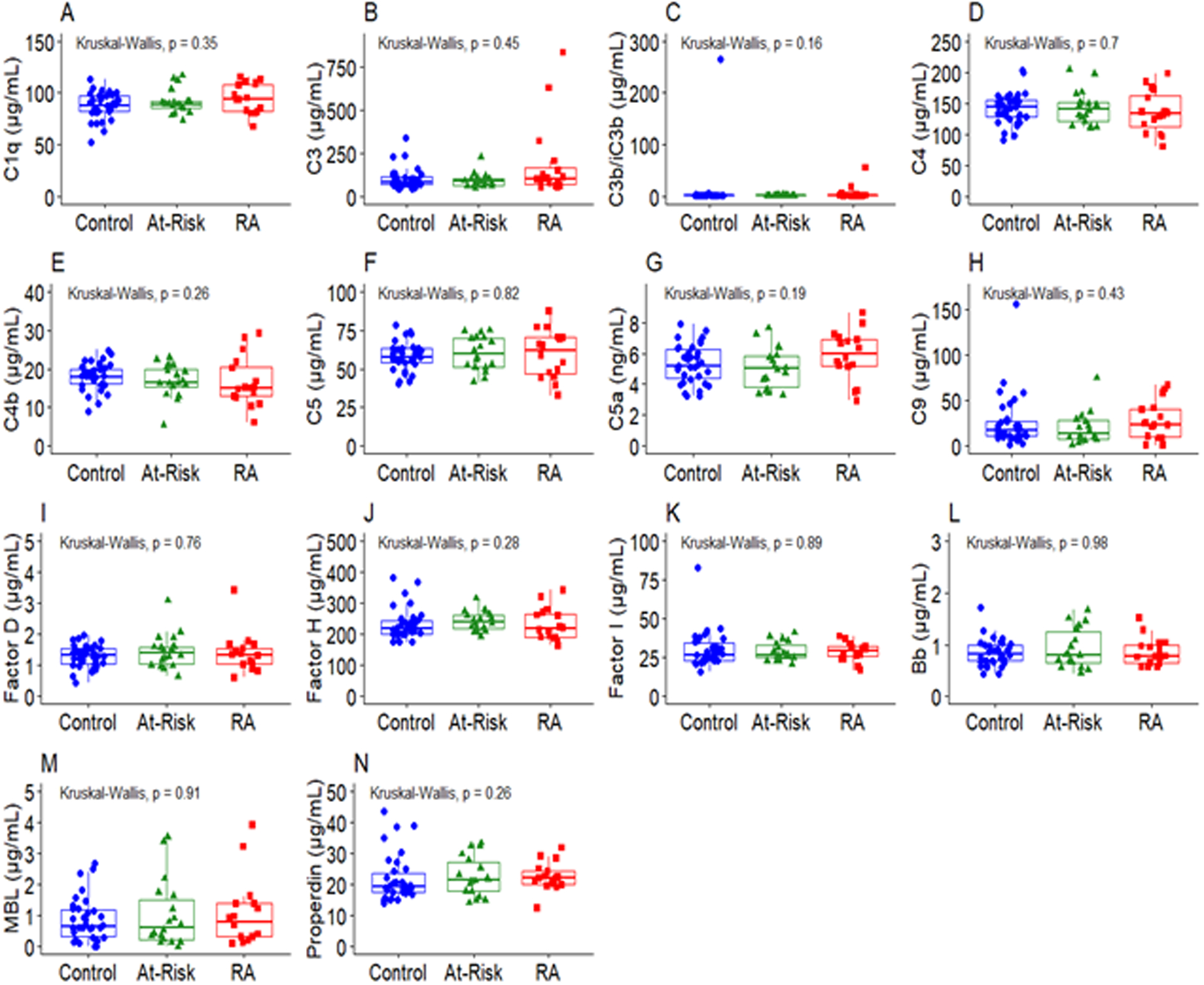
Complement whose levels did not differ among RA patients, at-risk subjects, and control subjects. The middle line indicates the median value with the upper and lower hinges corresponding to the first and third quartiles respectively. P-value from Kruskal-Wallis test shown on plots.
Table 2.
Associations between complement and selected study group after adjusting for sex, race ethnicity, and age
| Complement | Adjusted Modela | Complement | Adjusted Modela | ||||
|---|---|---|---|---|---|---|---|
| LS-Mean | 95% CI | p-value | LS-Mean | 95% CI | p-value | ||
| C1q (μg/mL) | C5b-9 (ng/mL) | ||||||
| Control | 88.74 | 83.22–94.63 | Ref | Control | 158.53 | 141.81–177.23 | Ref |
| At-Risk | 92.07 | 85.58–99.06 | 0.39 | At-Risk | 161.87 | 142.81–183.48 | 0.77 |
| RA | 95.08 | 88.07–102.64 | 0.13 | RA | 183.55 | 161.12–209.10 | 0.06 |
| C2 (μg/mL) | C9 (μg/mL) | ||||||
| Control | 4.25 | 3.70–4.88 | Ref. | Control | 30.76 | 20.99–45.08 | Ref |
| At-Risk | 4.09 | 3.48–4.79 | 0.67 | At-Risk | 23.66 | 14.96–37.42 | 0.28 |
| RA | 6.80 | 5.74–8.06 | <0.01 | RA | 35.45 | 22.36–56.19 | 0.62 |
| RA v. At-Riskb | <0.01 | ||||||
| C3 (μg/mL) | Bb (ng/mL) | ||||||
| Control | 107.94 | 83.14–140.14 | Ref | Control | 0.85 | 0.74–0.98 | Ref |
| At-Risk | 99.61 | 73-36-135.26 | 0.63 | At-Risk | 0.92 | 0.78–1.09 | 0.36 |
| RA | 174.73 | 128.78–237.07 | 0.01 | RA | 0.86 | 0.472–1.02 | 0.93 |
| RA v. At-Riskb | 0.01 | ||||||
| C3a (ng/mL) | Factor B (μg/mL) | ||||||
| Control | 42.74 | 36.52–50.01 | Ref | Control | 140.31 | 127.96–153.85 | Ref |
| At-Risk | 49.53 | 41.41–59.24 | 0.15 | At-Risk | 150.03 | 135.39–166.25 | 0.27 |
| RA | 65.59 | 54.66–78.69 | <0.01 | RA | 127.37 | 114.22–142.03 | 0.14 |
| RA v. At-Riskb | 0.02 | RA v. At-Riskb | 0.02 | ||||
| C3b/iC3bc (μg/mL) | Factor D (μg/mL) | ||||||
| Control | 1.38 | 0.93–2.05 | Ref | Control | 1.24 | 1.08–1.42 | Ref |
| At-Risk | 1.29 | 0.82–2.03 | 0.77 | At-Risk | 1.40 | 1.19–1.65 | 0.18 |
| RA | 4.11 | 2.57–6.58 | <0.01 | RA | 1.36 | 1.15–1.60 | 0.36 |
| C4 (μg/mL) | Factor H (μg/mL) | ||||||
| Control | 141.23 | 129-85-153.61 | Ref | Control | 229.73 | 211.78–249.19 | Ref |
| At-Risk | 140.81 | 128.15–154.72 | 0.96 | At-Risk | 239.22 | 218.47–261.94 | 0.45 |
| RA | 129.95 | 117.73–143.45 | 0.16 | RA | 223.51 | 203.11–245.96 | 0.63 |
| C4b (μg/mL) | Factor Ic (μg/mL) | ||||||
| Control | 18.11 | 16.02–20.49 | Ref | Control | 26.94 | 24.55–29.56 | Ref |
| At-Risk | 16.61 | 14.47–19.08 | 0.28 | At-Risk | 27.86 | 25.10–30.92 | 0.58 |
| RA | 15.92 | 13.74–18.46 | 0.14 | RA | 27.17 | 24.38–30.29 | 0.89 |
| C5 (μg/mL) | MBL (μg/mL) | ||||||
| Control | 56.79 | 52.13–61.88 | Ref | Control | 1.29 | 0.86–1.92 | Ref |
| At-Risk | 58.88 | 53.37–64.97 | 0.52 | At-Risk | 1.44 | 0.91–2.30 | 0.66 |
| RA | 57.82 | 52.17–64.08 | 0.77 | RA | 1.24 | 0.81–1.92 | 0.91 |
| C5a (ng/mL) | Properdin (μg/mL) | ||||||
| Control | 5.26 | 4.69–5.90 | Ref | Control | 22.21 | 19.65–25.10 | Ref |
| At-Risk | 5.01 | 4.41–5.69 | 0.51 | At-Risk | 22.71 | 19.83–26.00 | 0.78 |
| RA | 5.74 | 5.02–6.55 | 0.28 | RA | 21.99 | 19.09–25.33 | 0.91 |
Multivariable generalized linear model adjusted for sex, race ethnicity, and age
RA v. At-Risk p-value shown when significant P<0.05
1 observation with Cook’s D > 1 removed from model
Generalized linear models were also used to examine the association between complement levels and demographic, genetic, and environmental exposures in order to explore effects of the environment on plasma complement levels. Associations between the independent genetic and environmental exposures and the various complement proteins as the continuous dependent variable were analyzed using multivariable generalized linear regression models adjusting for selected study group (control, at-risk, RA), race [Non-Hispanic white (NHW), other], and the matching factors sex (female/male) and age (continuous years) to control for any additional bias from the matching process (Pearce, 2016). Any observation with a complement value below the limit of detection was set to one-half the limit of detection for inclusion in regression models. Sensitivity analysis comparing models excluding complement values below the limit of detection and including imputed values did not change statistical inference. In addition, any data point with a Cook’s distance value > 1 from regression model was used to identify and remove any influential data points for final regression analysis. A p-value for exposure variable less than 0.05 was considered significant. For these analyses, we also assessed effect modification to determine if the association between the environmental exposure and complement differed by selected study group. A p-value less than 0.05 for the interaction term was used to determine whether to stratify results by study group. A false discovery rate (FDR) adjustment was made using the Benjamini and Hochberg adaptive step-down Bonferroni method to account for multiple comparisons (Glickman et al., 2014). We present both the original and FDR-adjusted p-values in Table 3. After initial analyses, we selected an additional 16 RA subjects to replicate the association between the BMI and complement C2. Analyses were performed in SAS version 9.4 (Cary, NC) and R 3.5.4 (www.R-project.org).
Table 3.
Associations between complement and the environment
| Exposure | Complement Protein | P-valuea (FDR P-value)b | Description of association |
|---|---|---|---|
| Female sex | C4 C9 Factor I MBL |
0.02 (0.14) 0.02 (0.11) 0.01 (0.06) 0.03 (0.18) |
Female subjects had higher levels of C4 and Factor I and lower levels of C9 and MBL compared to male subjects. |
| Shared Epitope | C3a | 0.05 (0.15) | SE positive subjects had marginally higher levels of C3a compared to SE negative subjects. |
| BMI |
C2 C4 C4b C5 Factor B Factor I |
<0.01 (0.04) <0.01 (0.01) 0.01 (0.04) 0.01 (0.07) 0.01 (0.05) 0.01 (0.04) |
BMI ≥30 is associated with higher levels of C2, C4. C4b. C5, Factor B. Factor I. and Properdin compared to those with BMI <30. |
| Study Group*C2 | 0.03c | The association between C2 and BMI differs significantly by study group. | |
| Ever Smoker | Factor D | 0.02 (0.11) | Subjects who ever smoked cigarettes had lower levels of Factor D compared to subjects who never smoked cigarettes. |
| Current Smoker | C3 C3b/iC3b Factor H |
0.01 (0.05) <0.01 (0.01) 0.04 (0.16) |
Subjects who report current smoking had lower levels of C3 and C3b/iC3b and higher levels of Factor H compared to subj ects who do not currently smoke cisarettes. |
| Oral Contraceptive Use | Properdin | 0.03 (0.12) | Female subjects who had ever used an oral contraceptive had lower levels of Properdin compared to female subjects who had never used and oral contraceptive. |
P-value from generalized linear regression models adjusted for selected group, age. race and sex.
P-value adjusted by Benjamini and Hochberg adaptive step-down Bonferroni method
P-Value for significant interaction term
3. Results
3.1. Subjects
Characteristics of the subjects are presented in Table 1 by study group. RA subjects were more likely to report race other than NHW compared to both the at-risk and control subjects. RA subjects were also more likely to be current smokers. No other significant differences were noted by study group. Characteristics of the RA replication subjects are also presented in Table 1. Characteristics of the additional RA subjects did not significantly differ by selected BMI categorization (non-obese/obese).
3.2. Complement factor levels differ across RA, at-risk, and healthy control subjects
No substantial differences were noted in plasma levels of complement factors when comparing healthy controls to at-risk subjects, suggesting the lack of systemic inflammation in these subjects to the level required for complement activation. In contrast, unadjusted levels of C2, C3a, and sC5b9 were significantly higher in RA subjects compared to control subjects (PC2=<0.01, PC3a=<0.01, PsC5b9=0.02). Levels of the classical pathway protein C2 were also significantly higher in RA subjects compared to at-risk subjects (P=0.01). Levels of FB were marginally higher in at-risk subjects compared to both RA subjects and healthy controls (P=0.05) (Figure 1). Complement levels that did not significantly differ by study group are presented in Figure 2.
In addition to the unadjusted differences in complement levels presented in Figure 1, there were complement levels that differed significantly by study group in models adjusted for race, sex, and age (Table 2). RA subjects demonstrated higher levels of C2 and C3a compared to the control subjects (C2 and C3a PRA v. Control=<0.01). Levels of C2, C3, and C3a were also higher in the RA subjects compared to the at-risk subjects, in addition to the previously presented difference in the RA subjects compared to control subjects for C3a (Figure 1; C2 PRA v. At-Risk=<0.01; C3 PRA v. At-Risk=0.01; C3a PRA v. At-Risk=0.02). And, the levels of FB were higher in at-risk subjects compared to RA subjects (P=0.02).
3.3. Demographic, Genetic, and Environmental factors are associated with complement protein levels
Environmental exposures and demographic and genetic factors were associated with various complement proteins after model adjustment for selected study group, race/ethnicity, sex, and age (Table 3). Female subjects, regardless of study group, had higher levels of C4 and Factor I and lower levels of C9 and MBL compared to male subjects. However, after FDR adjustment these associations were no longer significant. Subjects who are SE positive had higher levels of C3a compared to SE negative subjects. Again, after FDR adjustment this association was no longer significant.
Increasing BMI was associated with numerous complement proteins where BMI categorized as obese (≥30) was associated with increased levels of complement factors C2, C4, C4b, C5, FB, and FI. Notably, after adjustment for multiple comparisons, the association between BMI and complement C2, C4, C4b, and Factor I remained significant. The association between C2 and BMI significantly differed by study group (p-value for interaction term = 0.03). In order to better understand the association we stratified results by study group. Among subjects with RA, those who had a BMI categorized as obese (≥30) had higher levels of C2 compared to non-obese RA subjects (p=0.01). This association was not seen among the control and at-risk groups (Figure 3). To replicate the significant association between C2 and BMI among the RA subjects, we selected an additional 16 RA subjects and repeated the analysis and a similar association was seen. RA subjects who had a BMI categorized as obese (≥30) had higher levels of C2 compared to non-obese RA subjects (p<0.01). To note, one subject who had BMI ≥30 also had a high value for C2 which appears as an outlying data point in figure 3. This observation was not considered an influential data point in the adjusted regression model (Cook’s D < 1). However, after removal of this observation the association was still significant (p=0.03). And, when combining all RA subjects, those with obese BMI had higher levels of C2 compared to non-obese RA subjects (p<0.01). The median C2 value for all RA subjects with obese BMI was 10 μg/mL (range 3.8–35.0) compared to a median value of 5 μg/mL (range 1.6–11.3) among the non-obese RA subjects.
Figure 3.
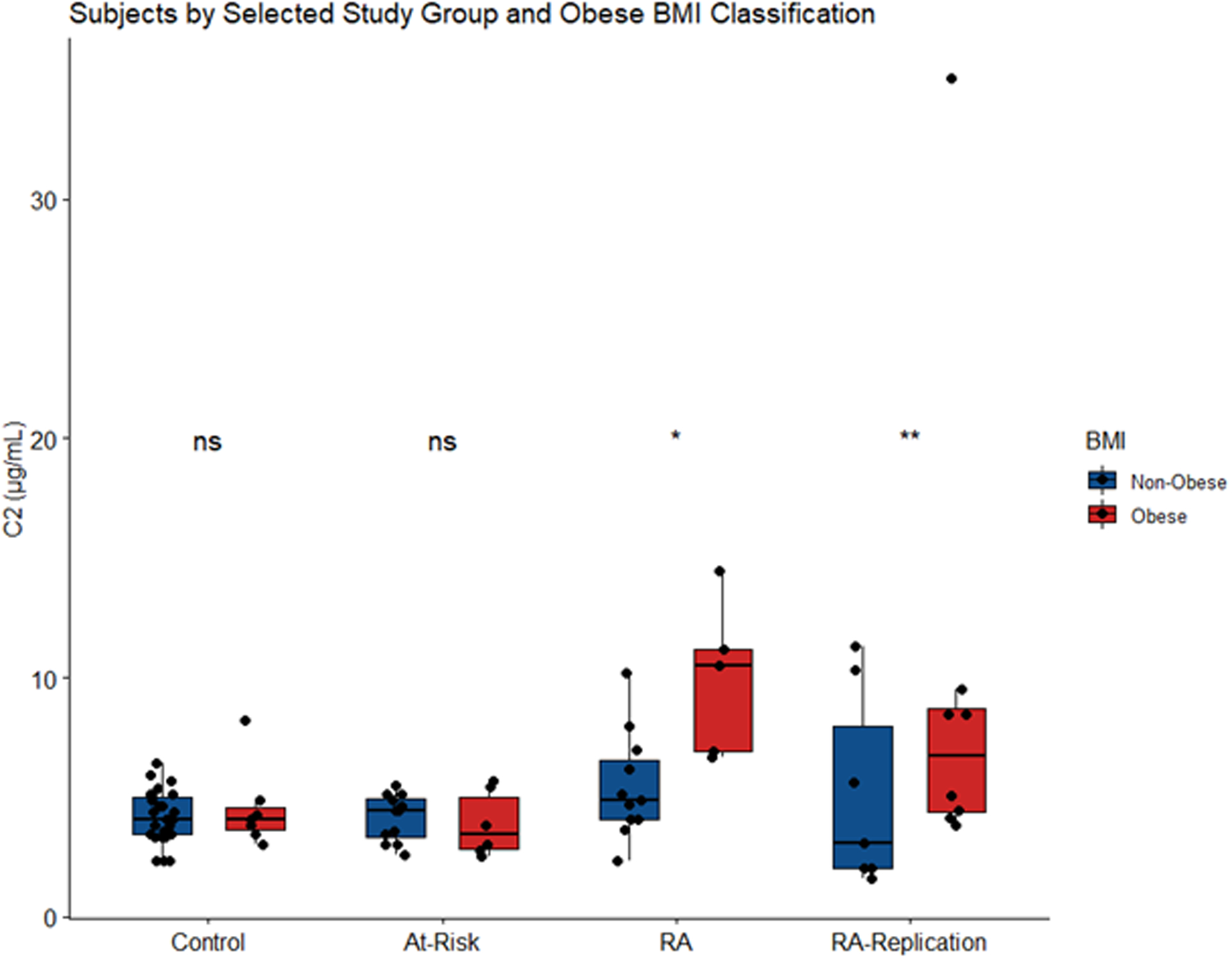
In regression models adjusting for age, sex, and race, complement C2 levels were significantly higher in RA subjects with BMI ≥30 compared to those RA subjects who had a BMI <30 (p=0.01). A similar association was not seen in the control (p=0.19) or at-risk subjects (p=0.48). P-value for interaction = 0.03. In a replication analysis of RA subjects, a significant association was seen (p<0.01). The middle line indicates the median value with the upper and lower hinges corresponding to the first and third quartiles respectively (ns ≥0.05, *P < 0.05, **P < 0.01).
Subjects who report ever smoking more than 100 cigarettes in their lifetime had lower levels of FD compared to never smokers. After adjustment for multiple comparisons this association was no longer significant. Those who were current smokers had lower levels of C3 and C3b/iC3b (one observation removed) and higher levels FH compared to subjects who did not currently smoke cigarettes. The association between C3 and current smoking remained marginally significant after multiple comparison adjustment (p=0.05). And, the association between current smoking and C3b/iC3b remained significant after multiple comparisons adjustment and removing one influential data point (p=0.01).
Finally, among female subjects those who report ever taking an oral contraceptive had lower levels of Properdin compared to females who never used an oral contraceptive. The association was not significant after FDR adjustment.
4. Discussion
In this study, differences in the absolute levels of complement proteins present in the plasma of at-risk, RA, and healthy control subjects along with associations between various RA risk factors and complement have been found. The data analyzed by both unadjusted and adjusted regression models support three main conclusions. First, the localized inflammation that has been identified in subjects at-risk for RA, which is characterized by the presence of autoantibodies and activated neutrophils, does not manifest itself as substantial increases in systemic complement activation fragments. Second, the measurements of various complement proteins in plasma suggest that there is the potential for enhanced CP activation, as manifest by increased C2 levels, in subjects with RA. In addition, the presence of elevated C3a and sC5b-9 levels, which could derive from the CP, AP and/or LP, in RA subjects is consistent with a potential role for C3 and/or C5-derived pro-inflammatory products in the pathogenesis of disease at this point. And third, the finding of elevated complement factors in association with elevated BMI suggests that these factors may play a role in the increased risk for development of RA with a higher BMI (Feng et al., 2016). In addition, although several of the additional epidemiologic factors studied do not provide a direct relationship between increased/decreased risk of RA, both the presence of the SE and oral contraceptive use demonstrate changes in complement factors that are consistent with increased (SE and C3a) and decreased (oral contraceptives and properdin) levels. In contrast, current use and a history of smoking demonstrate changes that are variable relative to the increased risk of development of RA.
Overall higher levels of C2, C3a and sC5b-9 in the plasma of RA subjects compared to healthy controls suggests that the classical pathway might be particularly prominent in the development of clinical arthritis, which is consistent with the concept that the CP plays an important role in mediating inflammation and injury through ACPA and RF. ACPA and anti-CarP autoantibodies have been reported to present many years before the onset of RA and there is a strong association with the onset of RA. Furthermore CP activation is antigen-antibody-dependent resulting from an adaptive immune response during ongoing inflammation in RA subjects, potentially leading to activation of the terminal pathway and generation of C5b-9.
Here we also show that in at-risk subjects, especially when studied in adjusted analyses (Table 2), there was a moderately significant increase in the levels of FB, a protein of the AP, compared with healthy subjects with no signs of disease or with RA subjects. The higher levels of FB in at-risk subjects suggests the potential for AP activation at this very early stage of the disease, perhaps acting at sites of mucosal inflammation such as lung and gut. To follow up this finding will require additional study of samples from these particular sites, which are of interest as they are characterized by active neutrophil NET formation (Demoruelle et al., 2018). As another potential mechanism, AP activation could be due EBV infection in at-risk RA subjects. Notably, both purified EBV gp350 and EBV in human serum have been shown to activated the AP (Mold et al., 1988a, b), and EBV regulates and processes C3 (Mold et al., 1988a). We have not yet tested EBV load and anti-EBV antibodies in the plasma of at-risk RA subjects. Supporting evidence that the AP is important in RA pathogenesis is found in mouse models of RA which have shown that the AP is sufficient and necessary to precipitate disease in mice (Banda et al., 2006).
RA is 2.6 times more prevalent in females than in males, which is a trait that is commonly found in other autoimmune diseases. One of the most intriguing questions is why women are more affected by RA than men. Although not significant, we found that in female subjects there may be activation of the CP and AP marked by slightly higher levels of C3b/iC3b, a split product of C3 and Factor I, compared to male subjects (data not shown). Factor I, along with co-factor, Factor H, mediates the cleavage and inactivation of C3b into iC3b. The increased levels of Factor I indicate that counter regulatory mechanisms to control CP and AP may be operative in order to control inflammation in females, who are generally more susceptible to arthritis. Furthermore the analysis of female specific exposures including oral contraceptive use provided some insight into how hormonal changes may be involved in complement activation. In the current study, females who report ever using an oral contraceptive have lower levels of Properdin, a component and a positive regulation of the AP, that can promote consumption by stabilizing C3 and C5 convertases. Lower activation of AP including lower Properdin levels in females has also been shown in a recent study indicating sex-specific changes in the complement activity healthy Caucasian population (Gaya da Costa et al., 2018). That study also demonstrated in the healthy population there were no significant differences in CP or CP components between males and females, but there were higher levels of terminal components (Gaya da Costa et al., 2018). Therefore, the significant changes in the AP activity we see in females with RA may be related to an oral contraceptive use. In contrast, we have also observed in all combined unadjusted groups, (i.e. in healthy controls, at-risk subjects and RA subjects), that females had higher levels of C4 and C9 (data not shown). The reason for this discrepancy is unknown, but in their study the use of oral contraceptives was not examined. In a large population-based case-control Swedish Epidemiological Investigation of RA study, it was shown that women who ever use oral contraceptive has a significantly decreased risk of developing ACPA positive RA (Orellana et al., 2017). If so then our data with lower AP activation due to lower level Properdin in women with oral contraceptive use will be consistent with those findings. Mechanistically Properdin stabilizes AP convertase, and lower levels of Properdin mean complement Factor H and Factor I can destabilize the AP convertase at physiological concentrations more efficiently in females using oral contraceptives. The role of AP has been well documented in our previous arthritis-related studies (Banda et al., 2006). Overall, natural sex-specific differences in the levels of complement components do exist, consistent with the aforementioned study findings (Gaya da Costa et al., 2018).
Obese BMI is associated with higher levels of C2, C4, C4b, C5, FB, and FI (Table 3). In addition, there is a significant difference in the association between C2 and BMI among the RA subjects compared to the control and at-risk subjects (Figure 3). Interestingly, those with established disease who have a BMI categorized as obese have higher levels of C2 compared to RA subjects who are not considered obese. BMI is normally used to quantify the amount of fat, muscle and bone, and how complement proteins are associated with BMI is not known. However, these data are consistent with our previously published study regarding the role of adipocytes (fat), which predominately generates complement FD (a.k.a. adipsin), a rate limiting factor of the AP in the pathogenic activation of the AP in the arthritic joints in concert with fibroblast like-synoviocytes (Arend et al., 2013). Complement C2 protein predominately generated by the liver but it is also synthesized by human monocyte and macrophages (Lappin and Whaley, 1989), and macrophages are resident cells in the human adipose tissue embedded in the human synovium (Banda NK unpublished data) and these macrophages could locally contribute to the pathogenesis of RA in obese subjects. C2 is a component common to both the CP and LP. Synergistic CP and LP complement activation can happen under acute inflammatory conditions such as in RA patients. Normally high levels of complement components are either due to infection or due to inflammation and low levels could be due to inherited complement deficiencies such as in lupus. But higher C2 levels in RA subjects who have BMI categorized as obese might be related to inflammation. Individuals at the high end of the complotype spectrum are more prone to chronic inflammation (Harris et al., 2012).
Lymphocytes isolated from the synovial membrane of RA patients in contrast to the PMBCs were able to stimulate C2 synthesis without exposure to mitogens or antigens (Lappin and Whaley, 1989), indicating hyper activation may be associated with a direct role of CP locally in the joint damage in RA patients. Furthermore, mouse models of arthritis have shown that C3 or C5 deficiency ameliorate arthritis (Banda et al., 2006; Hietala et al., 2002; Ji et al., 2002) indicating the crucial role of complement in the pathogenesis of RA. In addition, complement proteins are not only critical in an activated immune system but also in bone growth and bone homeostasis during physiological development (Modinger et al., 2018). It has been shown bone-cell development, bone-cell metabolism, bone-forming osteoblast and bone-reabsorbing osteoclasts are dependent or critically influenced by complement proteins (Modinger et al., 2018).
We found that current smokers have a lower levels of C3 and C3b/iC3b, but higher levels of Factor H compared with subjects who don’t currently smoke cigarettes (Table 3). The presence of higher levels of Factor H indicate that overall there is a less spontaneous activation of the AP systemically in current smokers, which could be a complement-mediated counter-regulatory defense mechanism. This is consistent with the traditional role of Factor H to protect tissue injury due to complement-mediated damage in lungs. The affinity of Factor H is 10-fold higher for C3b on host cells and other non-activators. It might be a natural mechanism to control more C3b deposits on the mucosal surface such as lungs, which are directly affected by smoking or cigarette smoke contents. There is also possibility that Factor H might be recognizing a specific marker on the surface of lungs of smokers to avoid AP activation to proceed followed by inhibition of inflammation at the mucosal sites, which have been suspected for a while to triggering arthritis. Here we also don’t rule out the role of other soluble or membrane bound complement regulatory proteins on the surface of lungs in smokers leading to a lower levels of C3b/iC3b in plasma.
One of the major limitations of current study is that we have not measured complement proteins in the synovium fluid or synovial tissue of healthy controls, subjects at-risk for RA, and RA subjects, due to the non-availability of the samples. Many complement proteins and their cleaved products have been reported in the synovial fluid of RA patients such as C1q, C4, C3a, C5a, C5b-9 [reviewed in (Modinger et al., 2018)]. These types of case-control samples and studies could have provided a better understanding of the pathogenesis from preclinical to clinical status in at-risk subjects to RA patients. We have also not measured C-reactive protein (CRP), an acute phase reactant protein, as an inflammatory marker for it is non-specific inflammatory marker and its concentration varies a lot with multiple factors, gender and ethnicity (Khera et al., 2005). Another limitation of the current study is the small sample size. Future studies should include larger sample size to better assess how environmental exposures effect the relationship between complement activation and disease stage.
We conclude that the complement system is activated systemically in subjects with RA and may play an important role in RA pathogenesis in the transition from an at-risk state to clinically apparent disease. Importantly, the complement system is an increasingly studied component in disease processes, and there are opportunities to alter complement activation and effector pathways with inhibitors at different stages of the disease, (i.e. in subjects at-risk and subjects with established disease), when initial changes occur in the synovial membrane. For example, earliest joint changes in RA occur in the synovial membrane, leading to synovitis (Capitanescu et al., 2011). The synovium is the main site for inflammatory process and if untreated it will likely lead to adjacent cartilage and bone destruction. It has been postulated that at very early stage an acute inflammatory change occur but if the process continues it is replaced by chronic inflammation localized in the synovium (Hitchon and El-Gabalawy, 2011). As such, further studies elucidating the relationship between the complement system and disease progression are warranted.
Highlights.
The complement system, in an ACPA-containing environment, may play an important role in driving the pathogenesis of rheumatoid arthritis from the pre-clinical to clinical stage.
Subjects with active RA and ongoing joint inflammation and damage showed signs of systemic complement activation, but subjects at risk for RA did not.
In RA subjects, the alternative pathway of the complement system may be affected by tobacco exposure and, in females, oral contraceptive use.
RA subjects with established disease who have a body mass index categorized as obese have higher levels of C2 compared to RA subjects who are not considered obese.
Acknowledgements
We thank the healthy donors and the RA and at-risk subjects at the University of Colorado AMC and 9 Health Fair for contributing to this research by donating their blood. Serum and plasma samples were analyzed by the Exsera BioLabs at the University of Colorado Denver.
Supported by National Institutes of Health (NIH) grants U01 AI101981 to VMH (PI), R01 AR51749 to VMH (PI) and NKB (Co-I), and Rheumatology Research Foundation investigator grant to VMH. Additional support was from an investigator-initiated grant from Abbvie to KDD, NIH R56 AI103023 to KDD, the Walter S. and Lucienne Driskill Foundation to KDD, and a Rheumatology Research Foundation investigator grant to KDD.
Abbreviations used in this manuscript:
- RF
Rheumatoid factor
- ACPA
anti-citrullinated protein antibodies
- HLA
human leukocyte antigen
- SE
shared epitope
- BMI
body mass index
- FB
Factor B
- FD
Factor D
- FH
Factor H
- FI
Factor I
- P
Properdin
References
- Aho K, Koskenvuo M, Tuominen J, Kaprio J, 1986. Occurrence of rheumatoid arthritis in a nationwide series of twins. J Rheumatol 13, 899–902. [PubMed] [Google Scholar]
- Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO 3rd, Birnbaum NS, Burmester GR, Bykerk VP, Cohen MD, Combe B, Costenbader KH, Dougados M, Emery P, Ferraccioli G, Hazes JM, Hobbs K, Huizinga TW, Kavanaugh A, Kay J, Kvien TK, Laing T, Mease P, Menard HA, Moreland LW, Naden RL, Pincus T, Smolen JS, Stanislawska-Biernat E, Symmons D, Tak PP, Upchurch KS, Vencovsky J, Wolfe F, Hawker G, 2010. 2010 rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Ann Rheum Dis 69, 1580–1588. [DOI] [PubMed] [Google Scholar]
- Anna Borodovsky KY, Andrew Sprague, Nirmal K Banda V Michael Holers, Akshay Vaishnaw, Martin Maier, Rajeev Kallanthottathil, Klaus Charisse, Satya Kuchimanchi, Muthiah Manoharan, David J, Salant, Kevin Fitzgerald, Rachel Meyers, and Benny Sorensen, 2014. Aln-CC5, an Investigational RNAi Therapeutic Targeting C5 for Complement Inhibition. Blood 124, 1606. [Google Scholar]
- Anquetil F, Clavel C, Offer G, Serre G, Sebbag M, 2015. IgM and IgA rheumatoid factors purified from rheumatoid arthritis sera boost the Fc receptor- and complement-dependent effector functions of the disease-specific anti-citrullinated protein autoantibodies. Journal of Immunology 194, 3664–3674. [DOI] [PubMed] [Google Scholar]
- Arend WP, Mehta G, Antonioli AH, Takahashi M, Takahashi K, Stahl GL, Holers VM, Banda NK, 2013. Roles of adipocytes and fibroblasts in activation of the alternative pathway of complement in inflammatory arthritis in mice. Journal of Immunology 190, 6423–6433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banda NK, Desai D, Scheinman RI, Pihl R, Sekine H, Fujita T, Sharma V, Hansen AG, Garred P, Thiel S, Borodovsky A, Holers VM, 2018. Targeting of Liver Mannan-Binding Lectin-Associated Serine Protease-3 with RNA Interference Ameliorates Disease in a Mouse Model of Rheumatoid Arthritis. Immunohorizons 2, 274–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banda NK, Thurman JM, Kraus D, Wood A, Carroll MC, Arend WP, Holers VM, 2006. Alternative complement pathway activation is essential for inflammation and joint destruction in the passive transfer model of collagen-induced arthritis. J Immunol 177, 1904–1912. [DOI] [PubMed] [Google Scholar]
- Bartold PM, Marshall RI, Haynes DR, 2005. Periodontitis and rheumatoid arthritis: a review. J Periodontol 76, 2066–2074. [DOI] [PubMed] [Google Scholar]
- Cambridge G, Leandro MJ, Edwards JC, Ehrenstein MR, Salden M, Bodman-Smith M, Webster AD, 2003. Serologic changes following B lymphocyte depletion therapy for rheumatoid arthritis. Arthritis Rheum 48, 2146–2154. [DOI] [PubMed] [Google Scholar]
- Capitanescu B, Simionescu C, Margaritescu C, Stepan A, Ciurea R, 2011. Clinical and morphological aspects of sinovitis in early rheumatoid arthritis. Curr Health Sci J 37, 17–20. [PMC free article] [PubMed] [Google Scholar]
- Cross M, Smith E, Hoy D, Carmona L, Wolfe F, Vos T, Williams B, Gabriel S, Lassere M, Johns N, Buchbinder R, Woolf A, March L, 2014. The global burden of rheumatoid arthritis: estimates from the global burden of disease 2010 study. Annals of the rheumatic diseases 73, 1316–1322. [DOI] [PubMed] [Google Scholar]
- Crowson CS, Matteson EL, Davis JM 3rd, Gabriel SE, 2013. Contribution of obesity to the rise in incidence of rheumatoid arthritis. Arthritis Care Res (Hoboken) 65, 71–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Pablo P, Dietrich T, McAlindon TE, 2008. Association of periodontal disease and tooth loss with rheumatoid arthritis in the US population. J Rheumatol 35, 70–76. [PubMed] [Google Scholar]
- de Vries N, Tijssen H, van Riel PL, van de Putte LB, 2002. Reshaping the shared epitope hypothesis: HLA-associated risk for rheumatoid arthritis is encoded by amino acid substitutions at positions 67–74 of the HLA-DRB1 molecule. Arthritis Rheum 46, 921–928. [DOI] [PubMed] [Google Scholar]
- Deane KD, Demoruelle MK, Kelmenson LB, Kuhn KA, Norris JM, Holers VM, 2017. Genetic and environmental risk factors for rheumatoid arthritis. Best Pract Res Clin Rheumatol 31, 3–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deane KD, Striebich CC, Goldstein BL, Derber LA, Parish MC, Feser ML, Hamburger EM, Brake S, Belz C, Goddard J, Norris JM, Karlson EW, Holers VM, 2009. Identification of undiagnosed inflammatory arthritis in a community health fair screen. Arthritis Rheum 61, 1642–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demoruelle MK, Bowers E, Lahey LJ, Sokolove J, Purmalek M, Seto NL, Weisman MH, Norris JM, Kaplan MJ, Holers VM, Robinson WH, Deane KD, 2018. Antibody Responses to Citrullinated and Noncitrullinated Antigens in the Sputum of Subjects With Rheumatoid Arthritis and Subjects at Risk for Development of Rheumatoid Arthritis. Arthritis Rheumatol 70, 516–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demoruelle MK, Parish MC, Derber LA, Kolfenbach JR, Hughes-Austin JM, Weisman MH, Gilliland W, Edison JD, Buckner JH, Mikuls TR, O’Dell JR, Keating RM, Gregersen PK, Norris JM, Holers VM, Deane KD, 2013. Performance of anti-cyclic citrullinated Peptide assays differs in subjects at increased risk of rheumatoid arthritis and subjects with established disease. Arthritis Rheum 65, 2243–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguchi K, Origuchi T, Takashima H, Iwata K, Katamine S, Nagataki S, 1996. High seroprevalence of anti-HTLV-I antibody in rheumatoid arthritis. Arthritis Rheum 39, 463–466. [DOI] [PubMed] [Google Scholar]
- Feng J, Chen Q, Yu F, Wang Z, Chen S, Jin Z, Cai Q, Liu Y, He J, 2016. Body Mass Index and Risk of Rheumatoid Arthritis: A Meta-Analysis of Observational Studies. Medicine (Baltimore) 95, e2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrell PB, Aitcheson CT, Pearson GR, Tan EM, 1981. Seroepidemiological study of relationships between Epstein-Barr virus and rheumatoid arthritis. J Clin Invest 67, 681–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firestein GS, McInnes IB, 2017. Immunopathogenesis of Rheumatoid Arthritis. Immunity 46, 183–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford JA, Liu X, Marshall AA, Zaccardelli A, Prado MG, Wiyarand C, Lu B, Karlson EW, Schur PH, Deane KD, Sparks JA, 2018. Impact of Cyclic Citrullinated Peptide Antibody Level on Progression to Rheumatoid Arthritis in Clinically Tested CCP-Positive Patients Without RA. Arthritis Care Res (Hoboken). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaya da Costa M, Poppelaars F, van Kooten C, Mollnes TE, Tedesco F, Wurzner R, Trouw LA, Truedsson L, Daha MR, Roos A, Seelen MA, 2018. Age and Sex-Associated Changes of Complement Activity and Complement Levels in a Healthy Caucasian Population. Front Immunol 9, 2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glickman ME, Rao SR, Schultz MR, 2014. False discovery rate control is a recommended alternative to Bonferroni-type adjustments in health studies. J Clin Epidemiol 67, 850–857. [DOI] [PubMed] [Google Scholar]
- Harris CL, Heurich M, Rodriguez de Cordoba S, Morgan BP, 2012. The complotype: dictating risk for inflammation and infection. Trends Immunol 33, 513–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmick CG, Felson DT, Lawrence RC, Gabriel S, Hirsch R, Kwoh CK, Liang MH, Kremers HM, Mayes MD, Merkel PA, Pillemer SR, Reveille JD, Stone JH, 2008. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part I. Arthritis and rheumatism 58, 15–25. [DOI] [PubMed] [Google Scholar]
- Heurich M, Martinez-Barricarte R, Francis NJ, Roberts DL, Rodriguez de Cordoba S, Morgan BP, Harris CL, 2011. Common polymorphisms in C3, factor B, and factor H collaborate to determine systemic complement activity and disease risk. Proc Natl Acad Sci U S A 108, 8761–8766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hietala MA, Jonsson IM, Tarkowski A, Kleinau S, Pekna M, 2002. Complement deficiency ameliorates collagen-induced arthritis in mice. Journal of Immunology 169, 454–459. [DOI] [PubMed] [Google Scholar]
- Hill A, Hillmen P, Richards SJ, Elebute D, Marsh JC, Chan J, Mojcik CF, Rother RP, 2005. Sustained response and long-term safety of eculizumab in paroxysmal nocturnal hemoglobinuria. Blood 106, 2559–2565. [DOI] [PubMed] [Google Scholar]
- Hitchon CA, El-Gabalawy HS, 2011. The synovium in rheumatoid arthritis. Open Rheumatol J 5, 107–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holers VM, Banda NK, 2018. Complement in the Initiation and Evolution of Rheumatoid Arthritis. Front Immunol 9, 1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji H, Ohmura K, Mahmood U, Lee DM, Hofhuis FM, Boackle SA, Takahashi K, Holers VM, Walport M, Gerard C, Ezekowitz A, Carroll MC, Brenner M, Weissleder R, Verbeek JS, Duchatelle V, Degott C, Benoist C, Mathis D, 2002. Arthritis critically dependent on innate immune system players. Immunity 16, 157–168. [DOI] [PubMed] [Google Scholar]
- Khera A, McGuire DK, Murphy SA, Stanek HG, Das SR, Vongpatanasin W, Wians FH Jr., Grundy SM, de Lemos JA, 2005. Race and gender differences in C-reactive protein levels. J Am Coll Cardiol 46, 464–469. [DOI] [PubMed] [Google Scholar]
- Kolfenbach JR, Deane KD, Derber LA, O’Donnell C, Weisman MH, Buckner JH, Gersuk VH, Wei S, Mikuls TR, O’Dell J, Gregersen PK, Keating RM, Norris JM, Holers VM, 2009. A prospective approach to investigating the natural history of preclinical rheumatoid arthritis (RA) using first-degree relatives of probands with RA. Arthritis Rheum 61, 1735–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroot EJ, de Jong BA, van Leeuwen MA, Swinkels H, van den Hoogen FH, van’t Hof M, van de Putte LB, van Rijswijk MH, van Venrooij WJ, van Riel PL, 2000. The prognostic value of anti-cyclic citrullinated peptide antibody in patients with recent-onset rheumatoid arthritis. Arthritis Rheum 43, 1831–1835. [DOI] [PubMed] [Google Scholar]
- Kuhn KA, Kulik L, Tomooka B, Braschler KJ, Arend WP, Robinson WH, Holers VM, 2006. Antibodies against citrullinated proteins enhance tissue injury in experimental autoimmune arthritis. The Journal of clinical investigation 116, 961–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurreeman FA, Padyukov L, Marques RB, Schrodi SJ, Seddighzadeh M, Stoeken-Rijsbergen G, van der Helm-van Mil AH, Allaart CF, Verduyn W, Houwing-Duistermaat J, Alfredsson L, Begovich AB, Klareskog L, Huizinga TW, Toes RE, 2007. A candidate gene approach identifies the TRAF1/C5 region as a risk factor for rheumatoid arthritis. PLoS Med 4, e278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Cava A, Nelson JL, Ollier WE, MacGregor A, Keystone EC, Thorne JC, Scavulli JF, Berry CC, Carson DA, Albani S, 1997. Genetic bias in immune responses to a cassette shared by different microorganisms in patients with rheumatoid arthritis. J Clin Invest 100, 658–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappin D, Whaley K, 1989. Modulation of monocyte complement synthesis by lymphocytes and lymphocyte-conditioned media. Clin Exp Immunol 76, 86–91. [PMC free article] [PubMed] [Google Scholar]
- Liao KP, Alfredsson L, Karlson EW, 2009. Environmental influences on risk for rheumatoid arthritis. Curr Opin Rheumatol 21, 279–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B, Hiraki LT, Sparks JA, Malspeis S, Chen CY, Awosogba JA, Arkema EV, Costenbader KH, Karlson EW, 2014. Being overweight or obese and risk of developing rheumatoid arthritis among women: a prospective cohort study. Ann Rheum Dis 73, 1914–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marotte H, Farge P, Gaudin P, Alexandre C, Mougin B, Miossec P, 2006. The association between periodontal disease and joint destruction in rheumatoid arthritis extends the link between the HLA-DR shared epitope and severity of bone destruction. Ann Rheum Dis 65, 905–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercado FB, Marshall RI, Klestov AC, Bartold PM, 2001. Relationship between rheumatoid arthritis and periodontitis. J Periodontol 72, 779–787. [DOI] [PubMed] [Google Scholar]
- Meyer O, Labarre C, Dougados M, Goupille P, Cantagrel A, Dubois A, Nicaise-Roland P, Sibilia J, Combe B, 2003. Anticitrullinated protein/peptide antibody assays in early rheumatoid arthritis for predicting five year radiographic damage. Ann Rheum Dis 62, 120–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikuls TR, Payne JB, Reinhardt RA, Thiele GM, Maziarz E, Cannella AC, Holers VM, Kuhn KA, O’Dell JR, 2009. Antibody responses to Porphyromonas gingivalis (P. gingivalis) in subjects with rheumatoid arthritis and periodontitis. Int Immunopharmacol 9, 38–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modinger Y, Loffler B, Huber-Lang M, Ignatius A, 2018. Complement involvement in bone homeostasis and bone disorders. Semin Immunol 37, 53–65. [DOI] [PubMed] [Google Scholar]
- Mold C, Bradt BM, Nemerow GR, Cooper NR, 1988a. Activation of the alternative complement pathway by EBV and the viral envelope glycoprotein, gp350. J Immunol 140, 3867–3874. [PubMed] [Google Scholar]
- Mold C, Bradt BM, Nemerow GR, Cooper NR, 1988b. Epstein-Barr virus regulates activation and processing of the third component of complement. J Exp Med 168, 949–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielen MM, van Schaardenburg D, Reesink HW, van de Stadt RJ, van der Horst-Bruinsma IE, de Koning MH, Habibuw MR, Vandenbroucke JP, Dijkmans BA, 2004. Specific autoantibodies precede the symptoms of rheumatoid arthritis: a study of serial measurements in blood donors. Arthritis Rheum 50, 380–386. [DOI] [PubMed] [Google Scholar]
- Orellana C, Saevarsdottir S, Klareskog L, Karlson EW, Alfredsson L, Bengtsson C, 2017. Oral contraceptives, breastfeeding and the risk of developing rheumatoid arthritis: results from the Swedish EIRA study. Ann Rheum Dis 76, 1845–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce N, 2016. Analysis of matched case-control studies. BMJ 352, i969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen M, Jacobsen S, Garred P, Madsen HO, Klarlund M, Svejgaard A, Pedersen BV, Wohlfahrt J, Frisch M, 2007. Strong combined gene-environment effects in anti-cyclic citrullinated peptide-positive rheumatoid arthritis: a nationwide case-control study in Denmark. Arthritis Rheum 56, 1446–1453. [DOI] [PubMed] [Google Scholar]
- Pedersen M, Jacobsen S, Klarlund M, Pedersen BV, Wiik A, Wohlfahrt J, Frisch M, 2006. Environmental risk factors differ between rheumatoid arthritis with and without autoantibodies against cyclic citrullinated peptides. Arthritis Res Ther 8, R133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkova SB, Konstantinov KN, Sproule TJ, Lyons BL, Awwami MA, Roopenian DC, 2006. Human antibodies induce arthritis in mice deficient in the low-affinity inhibitory IgG receptor Fc gamma RIIB. J Exp Med 203, 275–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rantapaa-Dahlqvist S, de Jong BA, Berglin E, Hallmans G, Wadell G, Stenlund H, Sundin U, van Venrooij WJ, 2003. Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheum 48, 2741–2749. [DOI] [PubMed] [Google Scholar]
- Rosenstein ED, Greenwald RA, Kushner LJ, Weissmann G, 2004. Hypothesis: the humoral immune response to oral bacteria provides a stimulus for the development of rheumatoid arthritis. Inflammation 28, 311–318. [DOI] [PubMed] [Google Scholar]
- Scher JU, Sczesnak A, Longman RS, Segata N, Ubeda C, Bielski C, Rostron T, Cerundolo V, Pamer EG, Abramson SB, Huttenhower C, Littman DR, 2013. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. Elife 2, e01202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silman AJ, MacGregor AJ, Thomson W, Holligan S, Carthy D, Farhan A, Ollier WE, 1993. Twin concordance rates for rheumatoid arthritis: results from a nationwide study. Br J Rheumatol 32, 903–907. [DOI] [PubMed] [Google Scholar]
- Sokolove J, Zhao X, Chandra PE, Robinson WH, 2011. Immune complexes containing citrullinated fibrinogen costimulate macrophages via Toll-like receptor 4 and Fcgamma receptor. Arthritis and rheumatism 63, 53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trouw LA, Bohringer S, Daha NA, Stahl EA, Raychaudhuri S, Kurreeman FA, Stoeken-Rijsbergen G, Houwing-Duistermaat JJ, Huizinga TW, Toes RE, 2011. The major risk alleles of age-related macular degeneration (AMD) in CFH do not play a major role in rheumatoid arthritis (RA). Clin Exp Immunol 166, 333–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trouw LA, Daha N, Kurreeman FA, Bohringer S, Goulielmos GN, Westra HJ, Zhernakova A, Franke L, Stahl EA, Levarht EW, Stoeken-Rijsbergen G, Verduijn W, Roos A, Li Y, Houwing-Duistermaat JJ, Huizinga TW, Toes RE, 2013. Genetic variants in the region of the C1q genes are associated with rheumatoid arthritis. Clin Exp Immunol 173, 76–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trouw LA, Haisma EM, Levarht EW, van der Woude D, Ioan-Facsinay A, Daha MR, Huizinga TW, Toes RE, 2009. Anti-cyclic citrullinated peptide antibodies from rheumatoid arthritis patients activate complement via both the classical and alternative pathways. Arthritis and rheumatism 60, 1923–1931. [DOI] [PubMed] [Google Scholar]
- Vergunst CE, Gerlag DM, Dinant H, Schulz L, Vinkenoog M, Smeets TJ, Sanders ME, Reedquist KA, Tak PP, 2007. Blocking the receptor for C5a in patients with rheumatoid arthritis does not reduce synovial inflammation. Rheumatology (Oxford) 46, 1773–1778. [DOI] [PubMed] [Google Scholar]
- Wang Y, Kristan J, Hao L, Lenkoski CS, Shen Y, Matis LA, 2000. A role for complement in antibody-mediated inflammation: C5-deficient DBA/1 mice are resistant to collagen-induced arthritis. Journal of Immunology 164, 4340–4347. [DOI] [PubMed] [Google Scholar]
- Wang Y, Rollins SA, Madri JA, Matis LA, 1995. Anti-C5 monoclonal antibody therapy prevents collagen-induced arthritis and ameliorates established disease. Proceedings of the National Academy of Sciences of the United States of America 92, 8955–8959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widdifield J, Paterson JM, Huang A, Bernatsky S, 2018. Causes of Death in Rheumatoid Arthritis: How Do They Compare to the General Population? Arthritis Care Res (Hoboken) 70, 1748–1755. [DOI] [PubMed] [Google Scholar]
- Willis VC, Demoruelle MK, Derber LA, Chartier-Logan CJ, Parish MC, Pedraza IF, Weisman MH, Norris JM, Holers VM, Deane KD, 2013. Sputum autoantibodies in patients with established rheumatoid arthritis and subjects at risk of future clinically apparent disease. Arthritis Rheum 65, 2545–2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada H, Ozawa T, Kishi H, Okada S, Nakashima Y, Muraguchi A, Yoshikai Y, 2018. Cutting Edge: B Cells Expressing Cyclic Citrullinated Peptide-Specific Antigen Receptor Are Tolerized in Normal Conditions. J Immunol. [DOI] [PubMed] [Google Scholar]