

Abstract
Exomethylene acycloguanine nucleosides 4, 6 and its monophosphate derivatives 5, 7, and 8 have been synthesized. Mitsunobu-type coupling of 2-N-acetyl-6-O-diphenylcarbamoylguanine (11) with primary alcohols proceeded regioselectively to furnish the desired N9-substituted products in moderate yield. Evaluation of 4-8 for anti-HBV activity in HepG2 cells revealed that the phosphonate derivative 8 was found to exhibit moderated activity (EC50 value of 0.29 μM), but cytotoxicity (CC50 value of 39 μM) against the host cells was also observed.
Keywords: Nucleoside, acyclonucleoside, nucleoside phosphonate, pro-drug, hybrid, entecavir, adefovir, anti-HBV activity
Graphical Abstract

INTRODUCTION
Hepatitis B is one of the most prevalent viral diseases in the world and is known to be a major cause of chronic disease, leading to cirrhosis/hepatocellular carcinoma.[1] Among the most frequently used drugs for treatment of the disease[2] are the nucleoside analogue entecavir (1)[3a,b] and the nucleotide analogue adefovir (2)[4] (Figure 1). Entecavir 1 is especially considered as one of the best choices for chronic patients due to its lack of significant adverse effects.[5] Entecavir is structurally a carbocyclic analogue of 2′-deoxyguanosine. The exomethylene functionality at the 5′-position of 1 would appear to be an important pharmacophore for significant antiviral activity because the potency of carbocyclic dG that truncates the double bond is 10 times less than that of 1.[3a] Meanwhile, adefovir 2 is the phosphonate analogue of monophosphate of acycloadenine nucleoside. The feature of this class of nucleotide analogues is that the requisite first phosphorylation, which is a crucial step for the activation of biologically active nucleoside derivatives, has been bypassed.
FIGURE 1.
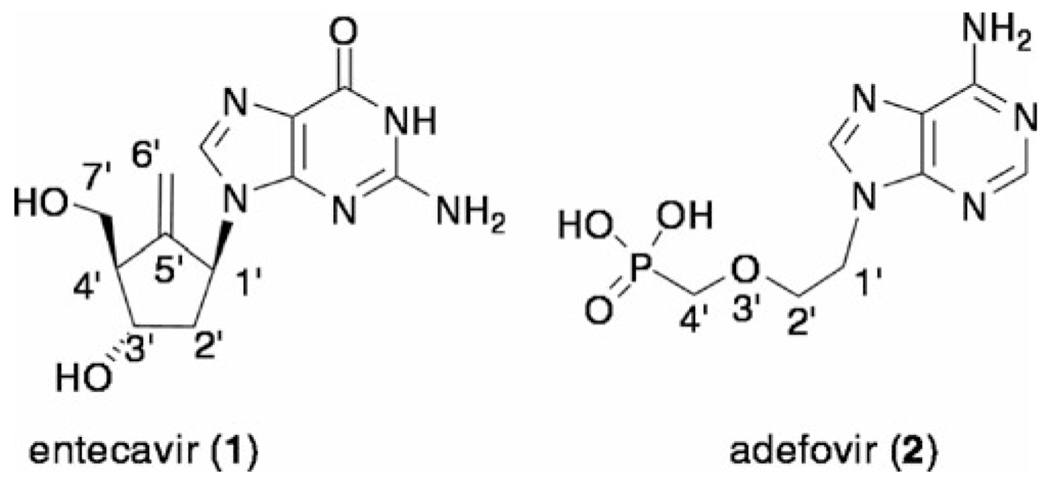
Structure of entecavir (1) and adefovir (2).
Further studies on the structure–activity relationship of these classes of nucleosides should increase our knowledge of structural requirements for developing novel antiviral agents for HBV, and will aid in the search for better anti-HBV agents. In this context, we have envisioned combining the above two structural features in one molecule and designed the exomethylene acyclic guanine nucleosides and its monophosphate derivatives as shown in Figure 2. The initial target molecules are exomethylene propyl-(4, MEP-G) and butyl-(6, MEB-G) guanine nucleosides. The number of constituted atoms (1′ to 4′-position) in the acyclic side chain of MEB-G 6 corresponds to the structure comprising C1′, C5′, C4′, and C7′ in entecavir 1, whereas MEP-G truncates one-carbon atom in acyclic moiety. l-ala-P-MEP-G 5 and l-ala-P-MEB-G 7 are the respective phosphoalaninate pro-drugs of the monophophates of 4 and 6. Moreover, the phosphonate analogue Piv-P-MEP-G 8 of 5 was also designed. The phosphonate 8 has a one-carbon elongated side chain (C1′ to C5′) compared with that of adefovir 2. Herein, we describe the results of the synthesis of 4-8 and evaluation of their anti-HBV activity.
FIGURE 2.

Structure of A-MEP (3) and the target molecules (4–8).
RESULTS AND DISCUSSION
Chemistry
Initially, synthesis of G-MEP (4) was carried out (Scheme 1). Synthesis of the adenine counterpart 3 (A-MEP) of the target molecule 4 has been reported.[6] Therefore, according to the procedure given in literature, 2-methylenepropane-1,3-diol (9) was utilized as a starting material. Compound 9 was converted into 2-O-(tert-butyldimethylsilyloxymethyl)prop-2-en-1-ol (10). The literature procedure for the coupling of adenine with acyclic moiety involved the mesylation of 10 followed by nucleophilic substitution of respective mesylate with nucleobase under basic reaction conditions. To reduce synthetic steps to the target G-MEP 4, Mitsunobu-type reaction of 10 with 2-N-acetyl-6-O-diphenylcarbamoylguanine (11)[7] was examined. Thus, when 10 was reacted with 11 in the presence of DIAD/Ph3P in THF at 70°C, the desired protected acyclopurine nucleoside 12 could be obtained in 53% yield. Removal of protecting groups in the base moiety was carried out by the treatment of 12 with ammonium hydroxide in methanol to give guanine derivative 13 in 88% isolated yield. In the Heteronuclear Multiple-Bond Correlation (HMBC) spectra of 13, the correlation between CH2-1′/C-4 and CH2-1′/C-8 was observed, by which 13 was assigned as N9-isomer. Compound 13 was converted to MEP-G 4 in 33% yield by treating with Bu4NF. Finally, 4 was transformed into the phosphoalaninate pro-drug 5 (16%) by reaction with methyl chlorophenylphosphoryl P→N-l-alaninate and N-mehtylimidazole in pyridine.[8]
SCHEME 1.

Synthesis of G-MEP (4) and its monophosphate pro-drug l-ala-P-MEP-G (5).
Next, synthesis of G-MEB (6) was performed (Scheme 2). Initially, 4-(tert-butyldiphenylsilyloxy)-2-methylenebutan-1-ol (16) was prepared from 14 in the following three steps: (1) silylation of 14, (2) epoxidation of the resulting silylated alkene, (3) β-elimination of the obtained epoxide 15 with diethylaluminium 2,2,6,6-tetramethylpiperidide.[9] When 16 was reacted with 11 under the above-mentioned reaction conditions, the desired N9-substituted 17 was obtained in 69% isolated yield as a single regio-isomer. Compound 17 was converted into 18 in 81% yield by ammonolysis in methanol, and its HMBC spectra revealed the correlation between CH2-1′/C-4 and CH2-1′/C-8. Desilylation of 18 gave G-MEB (6) in 60% yield. As described above for 4, MEB-G 6 was transformed into phosphoralaninate pro-drug 7 in 57% yield.
SCHEME 2.

Synthesis of G-MEB (6) and l-ala-P-MEB-G (7).
Finally, synthesis of the phosphonate analogue 8 of l-ala-P-MEB-G 7 was accomplished as illustrated in Scheme 3. Phosphonate alcohol 19 was prepared from 9 according to the published procedure.[6] Reaction of the alcohol 19 with 11 under the identical conditions for the synthesis of 17 gave the desired 20 in 62% isolated yield. Treatment of 20 with aqueous ammonia in methanol provided acycloguanine phosphonate derivative 21 in 54% yield, and at this stage, the regiochemistry was confirmed on the basis of HMBC spectrum, in which the same correlations as for 4 and 6 were observed. The phosphonate 21 was transformed into phosphonic acid (22) in 74% yield by treatment with TMSBr in CH2Cl2, and the resulting phosphonic acid 22 was converted into its pivaloyloxymethyl (POM) ester (8; Piv-P-MEP-G) in 10% yield.[10]
SCHEME 3.

Synthesis of Piv-P-MEP-G (8).
Biological Evaluation
Evaluation of anti-HBV activity of the novel acyclonucleosides synthesized in this study was conducted with HepG2 2.2.15 cells transfected with the HBV genome.[11] As shown in Table 1, G-MEP 4 and G-MEB 6 neither exhibit anti-HBV activity nor display any cytotoxicity toward HepG2 cells (entries 1 and 2). These results revealed that the conformationally rigid cyclopentane ring of entecavir is an essential structure for antiviral activity. Moreover, inactivity of phosphoalaninates 5 and 7 suggested that the initial phosphorylation step of 4 and 6 is not responsible for the lack of anti-HBV activity.
TABLE 1.
Anti-HBV activity and cytotoxicity of 4-8
| Compound | EC50 (μM) | CC50 (μM) (MT-4) |
|---|---|---|
| 4 | >1 | >100 |
| 5 | >1 | >100 |
| 6 | >1 | >100 |
| 7 | >1 | >100 |
| 8 | 0.29 | 39 |
| Entecavir | 0.00077 | NDa |
ND: not determined.
In contrast with the above results, the POM-protected phosphonate derivative 8 was found to possess antiviral activity with an EC50 of 0.29 μM, which is 1500 times less potent than that of entecavir. However, cytotoxicity toward HepG2 cells was also observed. With POM-protected prodrugs, it has been pointed out that the toxicity could be attributed to pivalic acid generated during the release of parent compounds.[10b] Therefore, we have evaluated the toxicity of pivalic acid in this assay system and found that the acid did not show any cytotoxicity up to 100 μM, which most likely ruled out this issue.
CONCLUSIONS
We have synthesized the novel exomethylene acycloguanine nucleosides MEP-G (4) and MEB-G (6). The respective monophosphate derivatives, that is, phosphoalaninate pro-drugs l-ala-P-MEP-5 and l-ala-P-MEB-G 7 of the monophosphate of 4 and 6 were also synthesized. Furthermore, the phosphonate analog Piv-P-MEP-G (8) of 7 was also evaluated for anti-HBV activity. Mitsunobu-type alkylation of 2-N-acetyl-6-O-diphenylcarbamoylguanine (11) with respective primary alcohols proceeded regioselectively at its N9-position and this synthetic strategy enabled us to shorten the synthetic route for the target molecules compared with the previously reported method. Evaluation of anti-HBV activity and its cytotoxicity toward HepG2 cells revealed that the rigidity of aglycon moiety is an important requirement for the inhibition of HBV replication. The phosphonate derivative 8 showed moderate activity with an EC50 of 0.29 μM, and the selectivity index (SI) was 137, although some cytotoxicity (CC50 of 39 μM) was observed.
EXPERIMENTAL
General
Melting points are uncorrected. Reagents and solvents were used without any further purification unless otherwise noted. Thin layer chromatography (TLC) was performed using precoated TLC plates (Merck, Silica gel 60 F254, 0.25 mm). 1H and 13C NMR spectra were recorded on a JEOL ECA 500 spectrometer operating at room temperature. Chemical shifts are reported in parts per million (δ) relative to the residual solvent peak. DSS served as internal standard for 13C NMR measurements in D2O. Mass spectra analysis was performed on a JEOL JMS-T100LP.
2-Acetyl -9-[2-(tert-butyldimethylsilyloxymethyl)allyl]-6-O-diphenylcarbamoyl (12)
To a solution of 10 (989 mg, 5.68 mmol), triphenylphosphine (1.49 g, 5.68 mmol) and 11 (2.10 g, 5.41 mmol) in THF (50 mL) was added DIAD (1.15 g, 5.68 mmol). The reaction mixture was heated to 70°C and stirred for 2 h. After cooling, the mixture was filtered through a celite pad. The filtrate was evaporated, and the residue was purified by silica gel column chromatography (Hexane-EtOAc 3:2 to 2:3, v/v) to provide 12 (1.71 g, 2.99 mmol, 53% yield) as a white foam. 1H NMR (500 MHz, CDCl3) δ 8.05 (brs, 1H), 7.95 (s, 1H), 7.43-7.25 (m, 10H), 5.23 (s, 1H), 4.96 (s, 1H), 4.80 (s, 2H), 4.10 (s, 2H), 2.56 (s, 3H), 0.89 (s, 9H), 0.05 (s, 6H); 13C NMR (126 MHz, CDCl3) δ 171.08, 156.09, 155.13, 152.13, 150.36, 144.18, 142.43, 141.69, 129.15, 127.08, 125.86, 120.52, 114.27, 64.08, 45.50, 25.77, 25.13, 18.22, −5.46. MS (ESI) m/z (M+Na)+ calcd. 595.2465, found 595.2485.
9-[2-(tert-Butyldimethylsilyloxymethyl)allyl]guanine (13)
To a solution of 12 (868 mg, 1.51 mmol) in methanol (6 mL) was added 28% ammonia solution (3 mL) and heated to 60°C in a sealed tube. After 2 h, the reaction mixture was concentrated and purified by silica gel column chromatography (CH2Cl2-MeOH 92:8 to 84:16, v/v) to provide 13 (446 mg, 1.33 mmol, 88% yield) as a white solid. 1H NMR (500 MHz, DMSO-d6) δ 10.58 (brs, 1H), 7.60 (s, 1H), 6.42 (brs, 2H), 5.12 (s, 1H), 4.65 (s, 1H), 4.54 (s, 2H), 4.11 (s, 2H), 0.86 (s, 9H), 0.03 (s, 6H); 13C NMR (126 MHz, DMSO-d6) δ 157.05, 153.80, 151.52, 144.56, 137.67, 116.57, 110.84, 63.69, 44.20, 25.96, 18.15, −5.31; MS (ESI) m/z (M+Na)+ calcd. 358.1675, found 358.1714.
9-[(Hydroxymethyl)allyl]guanine (4)
To a suspension of 13 (421 mg, 1.26 mmol) in THF (15 mL) was added 1 M TBAF in THF solution (3.14 mL, 3.14 mmol). After 2 h, the reaction mixture was concentrated. The residue was recrystalized from MeOH to provide 3 (91 mg, 0.41 mmol, 33% yield) as a white solid. 1H NMR (500 MHz, DMSO-d6) δ 10.57 (brs, 1H), 7.63 (s, 1H), 6.45 (brs, 2H), 5.07 (s, 1H), 5.05 (t, J = 5.4 Hz, 1H), 4.56 (s, 3H), 3.91(d, J = 5.7 Hz, 2H); 13C NMR (126 MHz, DMSO-d6) δ 157.01, 153.78, 151.51, 145.86, 137.82, 116.53, 110.25, 62.19, 44.44; MS (ESI) m/z (M+Na)+ calcd. 244.0810, found 244.0827.
Synthesis of l-ala-P-MEP-G (5)
To a solution of 4 (100 mg, 0.452 mmol) in pyridine (15 mL) was added a solution of methyl chlorophenylphosphoryl P→N-l-alaninate in THF (0.2 M, 11.3 mL, 2.26 mmol). After addition of N-methylimidazole (0.36 mL, 4.52 mmol), the mixture was stirred for 16 h. The solvents were evaporated and the residue was re-dissolved into AcOH (6 mL) and stirred at room temperature overnight. The solution was evaporated, and the residue was purified by silica gel column chromatography (EtOAc-MeOH 9:1 to 7:3, v/v) to provide 5 (33 mg, 0.072 mmol, 16%). 1H NMR (DMSO-d6, 500 MHz) δ 10.62 (brs, 1H), 7.63 and 7.61 (2s, 1H), 7.41-7.35 (m, 2H), 7.22-7.16 (m, 3H), 6.48 (brs, 2H), 6.16-6.05 (m, 1H), 5.22 and 5.18 (2s, 1H), 4.70 (s, 1H), 4.61 and 4.58 (2s, 2H), 4.54 (d, J = 6.3 Hz, 2H) and 4.49 (d, J = 5.7 Hz, 2H), 3.91-3.81 (m, 1H), 3.61 and 3.59 (2s, 3H), 1.26-1.19 (m, 3H); 13C NMR (DMSO-d6, 126 MHz) δ 173.98 and 173.90 (2d, J = 4.8 Hz), 157.06, 153.91, 151.56, 150.86 and 150.83, 140.63 and 140.53, 137.76, 129.91, 124.90, 120.50 and 120.47, 116.61, 114.00 and 113.74, 66.56 and 66.40 (2d, J = 4.8 Hz), 52.16, 50.03 and 49.90, 44.12 and 44.07, 19.87; MS (ESI) m/z (M+H)+ calcd. 463.1495; found 463.1540.
2-Acetylamino-9-[4-(tert-butyldiphenylsilyloxy)-2-methylenebutyl]-9H-purin-6-yl diphenylcarbamate (17)
To a solution of 16 (1.50 g, 4.40 mmol), triphenylphosphine (1.38 g, 5.28 mmol) and 11 (2.05 g, 5.28 mmol) in THF (35 mL) was added DIAD (1.07 g, 5.28 mmol) in THF (7 mL). The reaction mixture was heated to 70°C and stirred for 2 h. After cooling, the mixture was filtered through a celite pad. The filtrate was evaporated, and the residue was purified by silica gel column chromatography (Hexane-EtOAc 6:4 to 3:7, v/v) to provide compound 17 (2.17 g, 3.05 mmol, 69% yield) as a white foam. 1H NMR (CDCl3, 500 MHz) δ 7.92 (brs, 1H), 7.82 (s, 1H), 7.65-7.62 (m, 4H), 7.47-7.34 (m, 16H), 5.04 (s, 1H), 4.84 (s, 1H), 4.70 (s, 2H), 3.80 (t, J = 6.3 Hz, 2H), 2.50 (s, 3H), 2.24 (t, J = 6.3 Hz, 2H), 1.05 (s, 9H); 13C NMR (CDCl3, 126 MHz) δ 171.11, 156.12, 155.10, 152.16, 150.38, 143.98, 141.72, 140.85, 135.49, 133.31, 129.82, 129.17, 127.73, 125.92, 120.48, 115.42, 62.58, 48.48, 36.48, 26.82, 25.12, 19.14; MS (ESI) m/z (M+Na)+ calcd. 733.2935; found 733.2924.
2-Amino-9-[4-((tert-butyldiphenylsilyloxy)-2-methylenebutyl)-1H-purin-6(9H)-one (18)
A solution of compound 17 (1.00 g, 1.41 mmol) in 2 M NH3 (MeOH sol) (8 mL) was stirred at 70°C for 2 h. The solution was evaporated, and the residue was purified by silica gel column chromatography (CH2Cl2-MeOH 97:3 to 94:6, v/v) to provide 18 (540 mg, 1.14 mmol, 81% yield) as a white foam. 1H NMR (DMSO-d6, 500 MHz) δ 10.56 (brs, 1H), 7.64-7.58 (m, 4H), 7.54 (s, 1H), 7.50-7.41 (m, 6H), 6.39 (brs, 2H), 4.90 (s, 1H), 4.60 (s, 1H), 4.50 (s, 2H), 3.74 (t, J = 6.7 Hz, 2H), 2.26 (t, J = 6.7 Hz, 2H), 0.98 (s, 9H); 13C NMR (DMSO-d6, 126 MHz) δ 157.01, 153.78, 151.46, 142.15, 137.58, 135.22, 133.25, 130.08, 128.12, 116.56, 113.03, 63.23, 47.08, 36.51, 26.84, 18.94; MS (ESI) m/z (M+Na)+ calcd. 496.2145; found 496.2187.
2-Amino-9-(4-hydroxy-2-methylenebutyl)-1H-purin-6(9H)-one (6)
To a solution of 18 (437 mg, 0.923 mmol) in THF (35 mL) was added 1 M TBAF in THF (1.11 mL, 1.11 mmol). The mixture was stirred at room temperature for 1 h. The solvent was removed, and the residue was purified by silica gel column chromatography (CH2Cl2-MeOH 9:1 to 2:8, v/v) to provide 6 (130 mg, 0.55 mmol, 60% yield) as a white foam. 1H NMR (DMSO-d6, 500 MHz) δ 10.58 (brs, 1H), 7.61 (s, 1H), 6.45 (brs, 2H), 4.88 (s, 1H), 4.60 (t, J = 5.2 Hz, 1H), 4.53 (s, 2H), 4.50 (s, 1H), 3.52 (dt, J = 6.8, 5.2 Hz, 2H), 2.14 (t, J = 6.8 Hz, 2H); 13C NMR (DMSO-d6, 125 MHz) δ 157.08, 153.82, 151.54, 143.07, 137.85, 116.51, 111.97, 59.64, 47.15, 36.99; MS (ESI) m/z (M+Na)+ calcd. 258.0967; found 258.0966.
Synthesis of l-ala-P-MEB-G (7)
To a solution of 6 (50 mg, 0.213 mmol) in pyridine (7.5 mL) was added a solution of methyl chlorophenylphosphoryl P→N-L-alaninate in THF (0.2 M, 5.3 mL, 1.06 mmol). After addition of N-methylimidazole (0.17 mL, 2.13 mmol), the mixture was stirred for 16 h. The solvents were evaporated and the residue was re-dissolved into AcOH (6 mL) and stirred at room temperature overnight. The solution was evaporated, and the residue was purified by silica gel column chromatography (CH2Cl2-MeOH 97:3 to 91:9, v/v) to provide 7 (58 mg, 0.12 mmol, 57%). 1H NMR (DMSO-d6, 500 MHz) δ 10.59 (brs, 1H), 7.61 and 7.59 (2s, 1H), 7.44-7.28 (m, 2H), 7.25-7.08 (m, 3H), 6.45 (s, 2H), 6.09-5.91 (m, 1H), 4.95 and 4.93 (2s, 1H), 4.58 and 4.57 (2s, 1H), 4.55 and 4.53 (2s, 2H), 4.21-4.07 (m, 2H), 3.89-3.79 (m, 1H), 3.59 and 3.58 (2s, 3H), 2.39-2.30 (m, 2H), 1.26-1.18 (m, 3H); 13C NMR (DMSO-d6, 126 MHz) δ 174.02 and 173.90 (2d, J = 4.8 Hz), 157.06, 153.85, 151.55, 150.94, 141.38 and 141.30, 137.77, 129.82, 124.74, 120.41, 116.54, 113.07 and 113.01, 64.29 and 64.23 (2d, J = 4.8 Hz), 52.10, 50.04 and 49.87, 46.82 and 46.80, 34.02 and 33.98, 19.88; MS (ESI) m/z (M+H)+ calcd. 477.1651; found 477.1700.
2-Acetylamino-9-[(diisopropoxyphosphorylmethoxy)allyl]-6-diphenylcarbamoylpurine (20)
To a solution of (2-hydroxymethylallyloxymethyl)phosphonic acid diisopropyl ester 19 (2.13 g, 8.00 mmol), triphenylphosphine (2.73 g, 10.4 mmol) and 11 (3.73 g, 9.60 mmol) in THF (40 mL) was added DIAD (1.94 g, 9.60 mmol). The reaction mixture was heated to 70°C and stirred for 2 h. After cooling, the mixture was filtered through a celite pad. The filtrate was evaporated, and the residue was purified by silica gel column chromatography (EtOAc-MeOH 100:0 to 92:8, v/v) to provide 20 (3.14 g, 4.93 mmol, 62% yield) as a pale yellow foam. 1H NMR (500 MHz, CD3OD) δ 8.31 (s, 1H), 7.49-7.27 (m, 10H), 5.31 (s, 1H), 5.10 (s, 1H), 4.97 (s, 2H), 4.79-4.65 (m, 2H), 4.10 (s, 2H), 3.74 (d, J = 8.6 Hz, 2H), 2.31 (s, 3H), 1.36 (dd, J = 8.0, 6.3 Hz, 12H); 13C NMR (126 MHz, CD3OD) δ 172.63, 157.21, 156.76, 154.14, 152.58, 147.49, 143.55, 141.76, 130.61, 128.61, 121.78, 117.98, 72.24 (d, J = 12.0 Hz), 73.46 (d, J = 6.0 Hz), 65.53 (d, J = 167.9 Hz), 47.06, 25.06, 24.62 (d, J = 3.6 Hz), 24.58 (d, J = 4.8 Hz); MS (ESI) m/z (M+H)+ calcd. 637.2540, found 637.2569.
9-[(Diisopropoxyphosphorylmethoxy)allyl]guanine (21)
Compound 20 (361 mg, 0.567 mmol) was dissolved in ca. 9 M ammonia in methanol (5 mL) and heated to 70°C in a sealed tube. After 2 h, the reaction mixture was concentrated and purified by silica gel column chromatography (CH2Cl2-MeOH 95:5 to 88:12, v/v) to provide 21 (123 mg, 54% yield) as a white solid. 1H NMR (500 MHz, CDCl3) δ 11.81 (brs, 1H), 7.60 (s, 1H), 7.00 (brs, 2H), 5.22 (s, 1H), 4.86-4.79 (m, 3H), 4.62 (s, 2H), 4.12 (s, 2H), 3.78 (d, J = 9.7 Hz, 2H), 1.36 (dd, J = 8.0 and 6.3 Hz, 12H); 13C NMR (126 MHz, CDCl3) δ 158.40, 154.49, 151.16, 139.67, 137.52, 117.12, 116.53, 74.41 (d, J = 14.4 Hz), 71.85 (d, J = 6.0 Hz), 64.78 (d, J = 171.5 Hz), 44.69, 24.07 (d, J = 3.6 Hz), 24.03(d. J = 4.8 Hz); MS (ESI) m/z (M+Na)+ calcd. 422.1569, found 422.1581.
9-[2-(Hydroxymethyl)allyloxymethylphosphonic acid]guanine (22)
To a solution of 21 (475 mg, 1.19 mmol) in CH2Cl2 (10 mL) was added TMSBr (910 mg, 5.95 mmol). The reaction mixture was stirred at room temperature overnight. The reaction mixture was diluted with CH2Cl2 and extracted with water. The aqueous layer was lyophilized and the residue was purified by ODS column chromatography (H2O-MeOH 95:5 to 7:3, v/v) to provide 22 (278 mg, 74% yield) as a white solid. 1H NMR (500 MHz, D2O) δ 8.72 (s, 1H), 5.38 (s, 1H), 5.11 (s, 1H), 4.88 (s, 2H), 4.10 (s, 2H), 3.58 (d, J = 9.2 Hz, 2H); 13C NMR (126 MHz, D20) δ 158.52, 157.93, 152.92, 141.08, 140.82, 121.33, 111.85, 76.00 (d, J = 13.2 Hz), 68.34 (d, J = 158.4 Hz), 49.37; MS (ESI) m/z (M−H)− calcd. 314.0654 found 314.0690.
9-[Bis-O-(pivaloyoxymethyl)phosphorylmethoxy]allyl]guanine (8)
To a solution of 22 (101 mg, 0.320 mmol), triethylamine (179 μL, 1.28 mmol) and tetrabutylammonium bromide (103 mg, 0.320 mmol) in N-methylpyrrolidone (3 mL) was added chloromethyl pivalate (241 mg, 1.60 mmol). The reaction mixture was heated to 50°C and stirred for 20 h. The reaction was quenched with MeOH and directly applied to silica gel column chromatography (CH2Cl2-MeOH 98:2 to 9:1, v/v). Appropriate fractions were evaporated, triturated from acetonitrile/water = 1/1 to provide 8 (17.0 mg, 10% yield) as a white solid. 1H NMR (500 MHz, CD3OD) δ 7.72 (s, 1H), 5.74-5.69 (m, 4H), 5.26 (s, 1H), 4.94 (s, 1H), 4.71 (s, 2H), 4.11 (s, 2H), 3.93 (d, J = 8.6 Hz, 2H), 1.22 (s, 18H); 13C NMR (126 MHz, CD3OD) δ 178.42, 159.69, 155.64, 153.53, 142.19, 140.25, 117.75, 117.37, 83.52 (d, J = 6.0 Hz), 75.22 (d, J = 14.4 Hz), 64.81 (d, J = 166.74 Hz), 46.04, 40.03, 27.51; MS (ESI) m/z (M+Na)+ calcd. 566.1992 found 566.1963.
Cell Culture, Anti-HBV Assay, and Cytotoxicity Assay
HepG2 2.2.15 human hepatoblastoma cell line, which can stably produce HBV particles,[11] was kindly gifted from B. Korba of Georgetown University. The HepG2 2.2.15 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, USA) and MT-4 cells were grown in RPMI 1640-based culture medium, which supplemented with 10% (v/v) fetal calf serum (FCS; Gibco, USA) and 50 U/mL penicillin and 50 μg/mL streptomycin.
The HepG2 2.2.15 cells were plated at 1 × 105/mL in the presence or absence of various concentrations of a test compound in 96-well microtiter culture plates, then followed by incubation at 37°C for six days. After incubation, the concentration of HBV DNA in the supernatant was determined.
Cytotoxicity of a compound in MT-4 cells was also determined. Cells were plated in a 96-well plate at a density of 1 × 105/mL and cultured in the absence or the presence of various concentrations of a compound at 37°C for seven days. After 100 mL of the medium was removed from each well, 10 mL of 3-(4,5-dimetylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution (Nacalai Tesque, Kyoto, Japan) was added to each well in the plate, followed by incubation at 37°C for 2 h. After incubation to dissolve formazan crystals, 100 mL of acidified isopropanol containing 4% (v/v) Triton X-100 was added to each well, and the optical density was measured in a kinetic microplate reader (Vmax; Molecular Devices, Sunnyvale, CA).
Detection of HBV DNA by Real-Time Quantitative Polymerase Chain Reaction
Viral DNA in the supernatants was extracted using QIAamp MinElute Virus Spin Kit (Qiagen, Valencia, CA) according to the manufacture’s instruction. HBV DNA was quantified by real-time PCR relative to an external plasmid DNA standard on a Light Cycler instrument using LightCycler® FastStart DNA MasterPLUS SYBR Green I (Roche, Mannheim, Germany) and primers HBV-RT-F (5′-GAGTCTAGACTCGTGGTGGA-3′) and HBV-RT-R (5′-TGAGGCATAGGAGGAGGATG-3′), which amplified a 184-bp fragment in the RT region of the HBV genome. The PCR conditions used were an initial 3 min at 95° C, followed by 40 cycles of 95° C for 10 s, 55° C for 10 s, and 72°C for 10 s.
Acknowledgments
FUNDING
Financial support from a Health and Labor Sciences Research Grant [Practical Research on Hepatitis (Research on the innovative development and the practical application of new drugs for hepatitis B)] is gratefully acknowledged.
REFERENCES
- 1.White DO; Fenner FJ Medical Virology, 4th ed., Academic, New York, NY, 1994; Chapter 22. [Google Scholar]
- 2.Dienstag JL Drug therapy: Hepatitis B virus infection. N. Engl. J. Med 2008, 359, 1486–1500. [DOI] [PubMed] [Google Scholar]
- 3.(a). Innaimo SF; Seifer M; Bisacchi GS; Stabdring DN; Zahler R; Colonno RJ Identification of BMS-200475 as a potent and selective inhibitor of hepatitis B virus. Antimicrob. Agents Chemother 1997, 41, 1444–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yamanaka G; Wilson T; Innaimo S; Bisacchi GS; Egli P; Rinehart JK; Zahler R; Colonno RJ Metabolic studies on BMS-200475, a new antiviral compound active against hepatitis B virus. Antimicrob. Agents Chemother 1999, 43, 190–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De Clercq E Ten paths to the discovery of active nucleoside and nucleotide analogues. Nucleosides Nucleotides Nucleic Acids. 2012, 31, 339–352 and references cited therein. [DOI] [PubMed] [Google Scholar]
- 5.(a). Will H; Reiser W; Weimer T; Pfaff E; Büscher M; Sprengel R; Cattaneo R; Schaller H Replication strategy of human hepatitis B virus. J. Virol 1987, 61, 904–911. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Scott LJ; Kieting GM Entecavir. Drugs. 2009, 69, 1003–1033. [DOI] [PubMed] [Google Scholar]
- 6.Kim A; Hong H Synthesis and antiviral evaluation of novel exomethylene acyclic nucleosides and phosphonic acid nucleosides. Arch. Pharm. Chem. Life Sci 2005, 338, 528–533. [DOI] [PubMed] [Google Scholar]
- 7.Zou R; Robins MJ High-yield regioselective synthesis of 9-glycosyl guanine nucleosides and analogues via coupling with 2-N-acetyl-6-O-diphenylcarbamoylguanine. Can. J. Chem 1987, 65, 1436–1437. [Google Scholar]
- 8.Qiu Y-L; Ptak RG; Breitenbach JM; Lin J-S; Cheng Y-C; Drach JC; Kern ER Zemlicka, synthesis and antiviral activity of phosphoralaninate derivatives of methylenecyclopropane analogs of nucleosides. Antiviral Res. 1999, 43, 37–53. [DOI] [PubMed] [Google Scholar]
- 9.Weigant S; Brückner R Direct preparation of allylstannanes from allyl alcohols: Convenient synthesis of β-substituted allylstannanes and of stereo-defined δ-substituted allylstannanes. Synthesis. 1996, 475–482. [Google Scholar]
- 10.(a). Choi J-R; Cho D-G; Roh KY; Hwang J-T; Ahn S; Jang HS; Cho W-Y; Kim KW; Cho Y-G; Kim J; Kim Y-Z A novel class of phosphonate nucleosides. 9-[(1-phosphonomethoxycyclopropyl)methyl]guanine as a potent and selective anti-HBV agent. J. Med. Chem 2004, 47, 2864–2869. [DOI] [PubMed] [Google Scholar]; (b) Roux L; Priet S; Payrot N; Weck C; Fournier M; Zoulim F; Balzarini J; Canard B; Alvarez K Ester pro-drugs of acyclic nucleoside thiophosphonates compared to phosphonates: synthesis, antiviral activity and decomposition study. Eur. J. Med. Chem 2013, 63, 866–881. [DOI] [PubMed] [Google Scholar]
- 11.Sells MA; Chen ML; Acs G Production of hepatitis B virus particles in Hep G2 cells transfected with cloned hepatitis B virus DNA. PNAS. 1987, 84, 1005–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]