

Abstract
Nearly fifty protein families have been identified that inhibit CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats)-Cas mediated adaptive immune systems. Here, we analyze the available anti-CRISPR (Acr) structures and describe common themes and unique mechanisms of stoichiometric and enzymatic suppressors of CRISPR-Cas. Stoichiometric inhibitors sterically block interactions with DNA or prevent conformational changes that recruit or activate Cas nucleases, whereas enzymatic inhibitors covalently modify Cas proteins or cleave the CRISPR RNA. Here, we discuss some of the trade-offs associated with each of these strategies and highlight mechanistic insights revealed by atomic-resolution structures of Acrs.
Keywords: CRISPR-Cas, Anti-CRISPR, Bacteriophage, Cas9, Immunity
INTRODUCTION
Clustered Regularly Interspaced Short Palindromic Repeats (CRISPRs) and associated cas genes are essential components of diverse adaptive immune systems that defend bacteria and archaea from infection by foreign genetic elements. These immune systems are partitioned into two classes that have evolved independently but have been exchanged horizontally across taxa (39; 42). Class1 systems are divided into three types (I, III, IV) and 18 subtypes (A, B, C, etc.), but all class 1 systems consist of a multi-subunit RNA-guided surveillance complex (42; 43). Similarly, class 2 systems are divided into three types (II, V, VI) and 26 subtypes, but all class 2 systems consist of a single-protein effector that is guided by a CRISPR RNA (crRNA) (42; 43). Despite the phylogenetic and functional diversity of these systems, they all seem to participate in defense.
Considerable effort has been dedicated to understanding how Cas proteins integrate fragments of foreign DNA at one end of the CRISPR locus, and how CRISPR DNA is transcribed and processed into short crRNAs that guide Cas nucleases to the DNA or RNA of invading parasites (Figure 1). Progress in this field has been frenetic and numerous reviews dedicated to mechanisms of CRISPR adaptation (1; 31; 44; 69), crRNA biogenesis (13; 14) and interference (26; 36; 53) are available. As sophisticated and diverse as these immune systems are, phages and other genetic parasites have evolved mechanisms to neutralize these immune systems. Originally discovered in 2013, anti-CRISPRs (Acr) appear to mirror the diversity of the CRISPR systems themselves (7; 8; 48). Much like CRISPR systems, anti-CRISPRs have attracted considerable attention and several reviews have recently been published that address Acr function, evolution, and methods of discovery (9; 28; 49; 66). Here we complement existing reviews by first introducing recent work on the expression and regulation of Acrs. The implications of acr regulation are discussed in the context of CRISPR-Cas expression, before we move on to a discussion focused on the structures of Acr proteins and what they have taught us about the vulnerabilities of CRISPR RNA-guided defense systems.
Figure 1. CRISPR-Cas adaptive immunity and structurally determined anti-CRISPRs (Acrs).
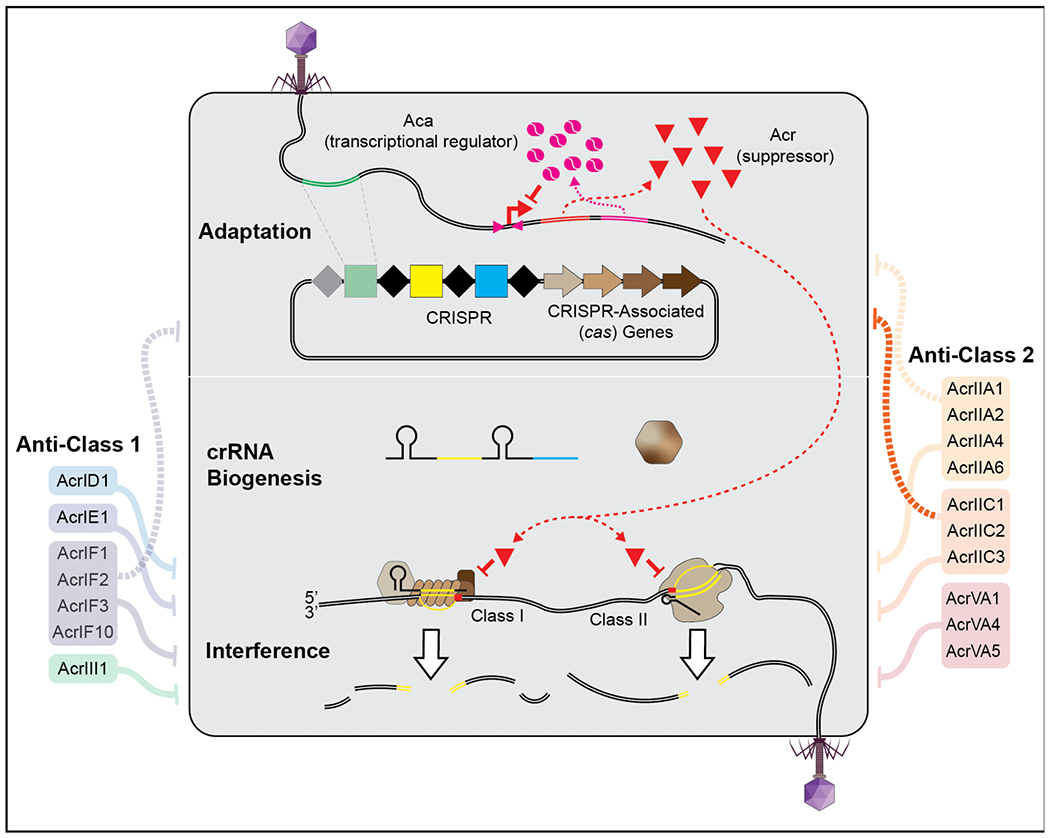
Homodimeric Acr-associated (Aca) proteins (pink) regulate Acr (red triangles) expression by binding to an upstream inverted repeat. Most suppressors of class 1 (left) and 2 (right) immune systems target the crRNA-guided surveillance and block DNA binding or nuclease activation.
Timing is everything
How do crRNA-guided surveillance complexes find complementary targets in a crowded intracellular environment, and on a time scale that affords protection from an invading virus that will (in some cases) program the cell for lysis (i.e., death) in the first few minutes after infection? The mechanism(s) that explain effective surveillance are complicated, but we know that detection of a complementary DNA target does not initially rely on unwinding the dsDNA duplex, which would be slow and energetically expensive (60; 68). Instead, detection of invading dsDNA (crRNA-guided detection of RNA relies on alternative mechanisms), starts with the identification of a short-duplexed sequence motif called a PAM (protospacer adjacent motif) (45; 62; 68). PAM binding is thought to destabilize the duplex and thereby facilitate crRNA-guided strand invasion (2; 58; 68). If the adjacent sequence is not complementary, then the interaction is ephemeral, and the search continues (64; 74). In contrast, a PAM with an adjacent complementary sequence triggers a conformational change that activates the nuclease and prompts target destruction (30; 46; 56; 71; 73; 82; 84). Collectively, this is an efficient process and some crRNA-guided surveillance complexes (e.g. Cas9 and Cascade) are predicted to find their targets in less than a minute (16; 33; 74).
But if crRNA-guided immune surveillance is rapid and efficient, then how do phages escape detection and elimination? DNA mutations and modifications play an important role in phage escape (11; 17; 61; 75), but here we focus on the delivery of immune suppressors. To effectively suppress the immune system, phages must quickly produce or deliver Acrs before the genome is identified and destroyed by crRNA-guided immune complexes (10; 40). It is conceivable (maybe even probable) that some phages package and inject Acr proteins along with their genomes. In fact, Stone et al recently determined the structure of a phage decoration protein that is a structural homolog of AcrIIC1 (70). Although this structural similarity raises the intriguing possibility that proteins associated with the virion could serve as readymade CRISPR-Cas antidotes, recent evidence now indicates that acrs are among the first genes to be transcribed and translated during an infection (Figure 1) (5; 65).
To date, nearly fifty families of unique Acrs (some containing just a single known homolog) have been experimentally demonstrated to suppress type I, II, III or V CRISPR immunity (Figure 1 and 2) (6). These proteins are small (52-333 amino acids) and diverse, sharing little to no sequence similarity with other proteins (8; 48). The small size and diverse sequences make it difficult to identify new Acrs using standard homology-based search methods. However, Pawluk et al. first identified a conserved gene with a helix-turn-helix (HTH) motif that is encoded downstream of known anti-CRISPR genes, but is absent in related phages lacking anti-CRISPRs (50). These anti-CRISPR associated (aca) genes have become effective new tools that serve as genetic landmarks for locating new Acrs, but until recently their functions have gone unreported. Stanley et al. recently demonstrate that acr and aca genes are immediately transcribed as a single RNA from the upstream promoter (i.e., polycistronic) (65). Collectively, this work, and a paper from Birkholz et al., now show that Aca proteins are homodimers that repress expression of the operon by binding to inverted repeats in the promoter (Figure 1) (5; 65). This results in a temporally controlled negative feedback loop that helps explain how phages deliver an early dose of Acrs, without the detrimental effects of runaway gene expression that occur in the absence of the repressor (5; 65). However, since Acr delivery appears to require transcription and translation (as opposed to delivery of the proteins directly) there is an intrinsic delay, and this delay provides an initial advantage to a previously “vaccinated” cell containing bespoke crRNA-guided complexes targeting that phage (10; 40). Borges et al. and Landsberger et al. recently demonstrated that infections of cells containing a CRISPR system targeting that phage are typically cleared, but each infection delivers small doses of Acr proteins that temporarily immunocompromise the cell. Thus, at high viral titers, Acrs accumulate to a critical intracellular threshold that eventually overwhelms the immune system (10; 40). Remarkably, the intracellular threshold necessary for immunosuppression differs between Acrs and correlates with the Acr binding affinity (i.e., weak binders require higher Acr concentrations and vice versa) (10; 40).
Figure 2. Anti-CRISPR (Acr) and Anti-CRISPR associated (aca) genes.
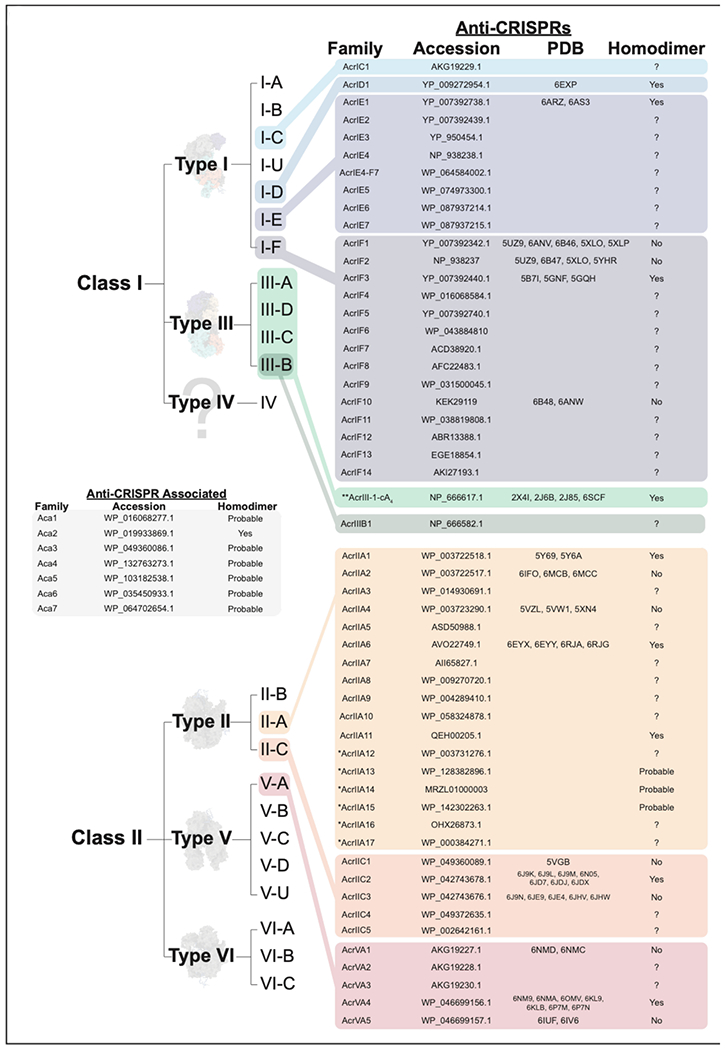
Acr proteins that have been experimentally demonstrated to prevent CRISPR interference, with representative protein accession numbers, PDB identifiers, and propensity to form homodimers. Proteins with HTH-domains, but not confirmed to form homodimers are listed as ‘Probable’. *currently under peer-review. **Acr with suggested naming convention that includes the secondary messenger.
While the role of Acrs in blocking interference in previously vaccinated cells has been well established, very little work has been done to understand how Acrs impact adaptation or crRNA biogenesis (Figure 1). With the exception of a single paper, indicating that Acrs inhibit new sequence acquisition in type I-F systems (76), no work has been published on the role of Acrs in processes upstream of interference. This is especially surprising since Cas9 plays a critical role in both interference and new sequence adaptation (25; 80). We anticipate that Acr proteins play an underappreciated role in limiting the efficiency of new sequence integration and that efforts to interrogate this aspect of the biology will reveal new insights into the CRISPR-anti-CRISPR dynamic.
Stoichiometric Inhibitors of CRISPR Defense
Anti-CRISPR Proteins that Masquerade as dsDNA
Parasites routinely use molecular mimicry to evade host immune responses (27; 54). Since many of the CRISPR-Cas systems target double-stranded DNA (dsDNA) (types I, II, and V), it is not surprising that phages have evolved a diverse repertoire of Acr proteins that serve as dsDNA decoys, which intercept the immune systems and prevent detection of invading DNA (15; 18; 23; 32; 41; 52; 63; 83). However, if dsDNA mimicry is a shared mechanism for diverse Acrs, then why does each Acr only interact with a specific surveillance complex? To address this question, we compared Acr structures to idealized B-form DNA and to the distorted DNA bound by each of the corresponding surveillance complexes (Figure 3A and D). These comparisons reveal a negative charge distribution on Acrs that more closely resembles the pattern of phosphates on DNA bound by the surveillance complex than idealized B-form (Figure 3B–C and E–F). In addition to the pseudo-helical presentation of negatively charged residues, we found that Acrs disguised as (bound) dsDNA are also often adorned with additional structural features (Figure 3E–F). These ancillary structural “decorations” often obscure the helical charge distribution characteristic of dsDNA, but they are expected to play important roles in affinity and specificity of Acr target selection. Collectively, the shape and charge distribution of a specific Acr, as well as the additional structural adornments that are unique to each Acr, may help explain why each DNA mimic exclusively targets the surveillance complex of a specific subtype.
Figure 3. Anti-CRISPRs more closely mimic bound dsDNA than idealized B-DNA.
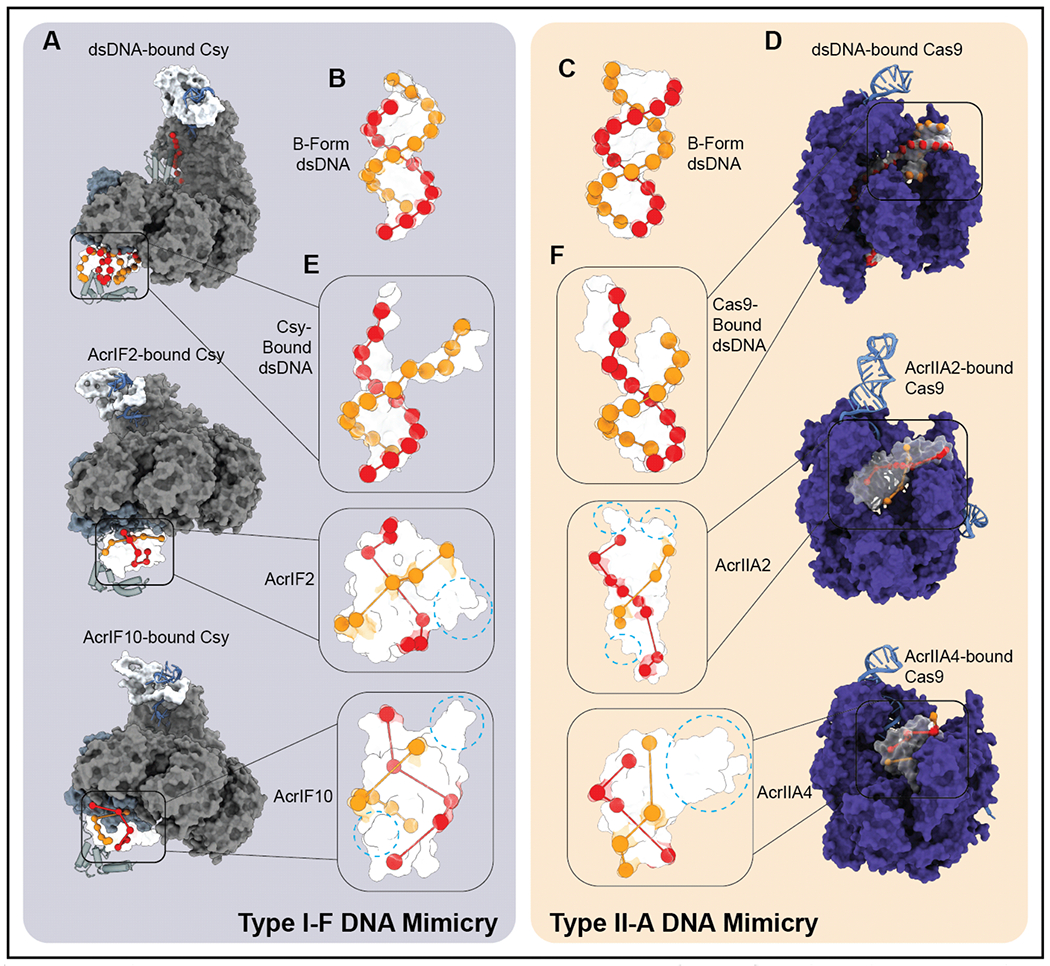
A) Surface representation of Csy bound to dsDNA, AcrIF2, and AcrIF10 (PDB: 6NE0, 5UZ9, 6B48). B-C) Surface representation of modeled B-form DNA with orbs highlighting phosphate groups of DNA backbone. D) Surface representation of Cas9 bound to dsDNA, AcrIIA2, and IIA4 (PDB: 4UN3, 6MCB, 5VW1). E-F) Surface representation of DNA bound by surveillance complex with orbs highlighting phosphate groups of DNA backbone (top) and surface representations of bound anti-CRISPRs with orbs highlighting pseudo-helical arrangement of acidic residues and auxiliary structural features (dashed blue circles in bottom two insets).
Acr-mediated Dimerization
Anti-CRISPRs that target diverse immune subtypes often form homodimers (22; 29; 34; 37; 38; 51; 72; 77; 78; 85; 86). This strategy may increase the effective size of an otherwise small Acr, which may in turn increase the affinity and specificity of target interactions. Moreover, homodimeric-Acrs often dimerize their corresponding immune system targets (e.g., AcrIIA6 and AcrVA4), which may balance the costs of making more Acrs upon infection (i.e., two Acrs neutralize two Cas). AcrIIA6 forms a stable homodimer that recognizes one molecule of Cas9 through a series of high-affinity interactions contributed by each Acr subunit (Figure 4A) (22; 72; 86). Residues that produce interactions on one face of the homodimer are symmetrically presented on the opposing face and these residues are free to engage with an additional molecule of Cas9. Thus, one homodimer neutralizes two molecules of Cas9, and formation of the homodimeric Acr simultaneously increases the affinity for both molecules of the Cas9 target.
Figure 4. Acrs that oligomerize Cas protein targets.
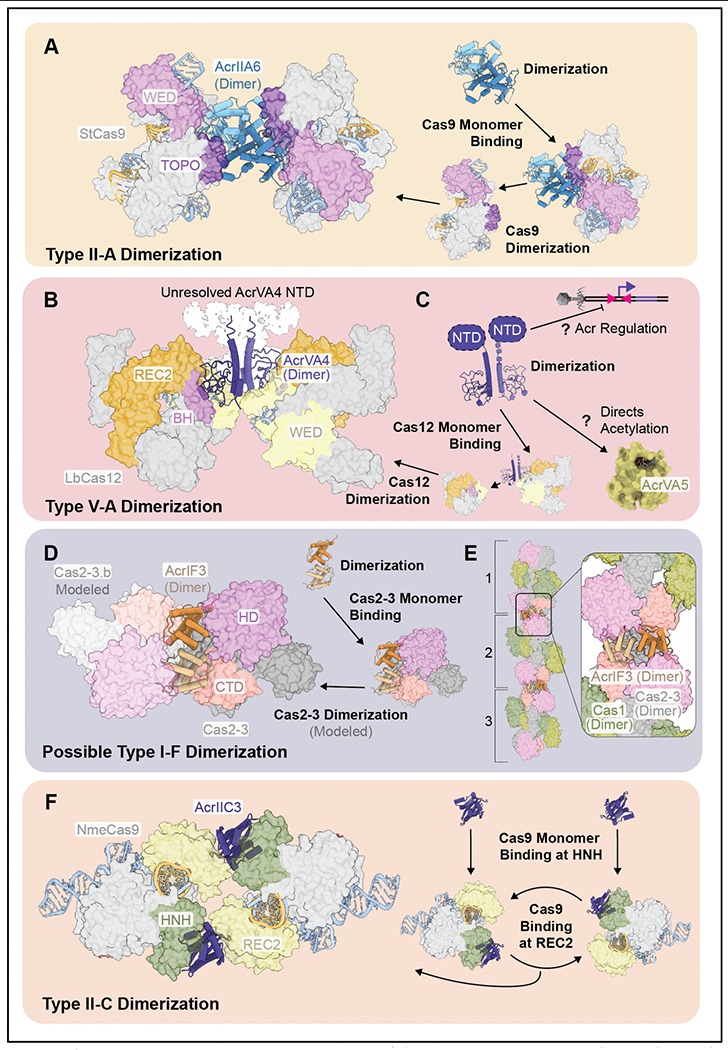
A-B) AcrIIA6 and AcrVA4 dimerize StCas9 and LbCas12, respectively. Unresolved density in the as cryo-EM reconstruction of AcrVA4 is shown in white (PDB: 6RJA, 6JE4, EMDB: 9398). C) AcrVA4 homodimer formation may facilitate acr regulation and/or guide AcrVA5 to the appropriate target (PDB: 6JE4, 6IUF). D) Model for AcrIF3 mediated oligomerization of the Cas1-2/3 integration complex of the type I-F immune system. The AcrIF3 dimer interacts with the HD nuclease domain and carboxy terminal domain (CTD) of Cas2/3 (PDB: 5B7I). E) Proposed model for AcrIF3-mediated dimerization would enable bidirectional oligomerization of Cas1-2/3 heterohexamers, three of which are illustrated. Modeled Cas1-2/3 super complex joined by AcrIF3 (PDB: 5B7I, 3GOD, EMDB: 8558). F) Two AcrIIC3 molecule join two molecules of NmeCas9. Domains of contact colored (PDB: 6NM9).
The benefits of homodimer formation are less clear in the case of AcrVA4. In contrast to the coordinated binding interactions by AcrIIA6, one subunit of an AcrVA4 dimer forms the majority of contacts with a single molecule of Cas12 (Figure 4B) (37; 51; 85). Theoretically, a monomer of AcrVA4 would be just as effective as the dimer. Consistent with this idea, the flexible N-terminal domain (NTD) of AcrVA4 (unresolved in Cryo-EM structures) was shown to be required for Acr dimerization but dispensable for Cas12 inhibition (37). However, additional selective pressures may be at play. Dimerization may be critical for regulating expression of acrVA4. Since the anti-CRISPR is not flanked by a known aca gene but does occur immediately downstream of two inverted repeats (IRs) (79), AcrVA4 may be regulated through an aca-like mechanism. While the role of the AcrVA4 NTD remains unknown, work recently deposited on Bioarchive (BioRxiv) and not yet peer reviewed, shows that the NTD regions of some Acrs (e.g., AcrIIA1) are not required for immune system inhibition, but rather function as Aca-type regulators of acr expression (47). In fact, the NTD of AcrIIA1 is also required for dimerization (34), again suggesting that while an Acr may form a dimer, this is not always necessary for Acr function. The NTD of AcrVA4 may play a similar role in regulating expression of the Acr, and this function may be dependent on dimerization of the suppressor. An alternate possibility for the biological role of AcrVA4 dimerization is that two molecules of the anti-CRISPR may be needed for interactions with AcrVA5. These Acrs cooccur in phage genomes and have been reported to form a complex (79), however this assembly dissociates upon AcrVA4-LbCas12 binding (85). Since AcrVA5 is an enzymatic inhibitor with little substrate specificity (see below), then its association with AcrVA4 may impart target specificity on an otherwise non-specific enzyme.
Unlike Acrs that dimerize the crRNA-guided surveillance complex, AcrIIC2 forms a homodimer but fails to dimerize Cas9, suggesting two molecules of the suppressor are needed for each Cas target (72; 86). The AcrIIC2 dimer binds to apo-Cas9 (i.e., sans RNA) and prevents crRNA (or sgRNA) guide-loading, which may increase proteolytic degradation of newly expressed Cas9 proteins (72; 86). This makes AcrIIC2 one of the more confounding and potentially interesting Acrs, since this mechanism implies that it is unable to prevent interference by Cas9 that is already loaded with a guide. This Acr would have limited utility in a cell with preformed Cas9s carrying crRNAs targeting that phage. However, since Cas9 must associate with the crRNA to fulfill its role in new sequence acquisition (25), then it is possible that AcrIIC2 serves as a suppressor that inhibits both interference and Cas9-dependent integration of foreign DNA into the CRISPR (72).
Based on the emerging theme of homodimeric Acrs that trigger dimerization of their targets, we wondered if other Acr dimers might be capable of achieving a similar function. AcrIF3, forms a homodimer that binds the type I-F transacting nuclease/helicase (Cas2/3) (56; 57; 77; 78). Structures of the AcrIF3 homodimer bound to Cas2/3 reveal a series of contacts made by each AcrIF3 molecule that are arranged along one face of the homodimer (77; 78). To determine if AcrIF3 might be capable of dimerizing Cas2/3, we modeled an additional molecule of Cas2/3 onto the crystal structure of dimeric AcrIF3 bound to Cas2/3 (Figure 4C). In this model, there are no major structural impediments that preclude AcrIF3-mediated dimerization of Cas2/3, and the majority of residues on the solvent exposed binding face of AcrIF3 are available to interact with a second molecule of Cas2/3. This suggests that AcrIF3 may tether two molecules of the helicase-nuclease to prevent interference. AcrIF3 has also been shown to block adaptation (76). The mechanism for blocking adaption has not been well-established but Rollins et al. previously shown that AcrIF3 binds to Cas2/3 alone and to Cas2/3 in complex with Cas 1 (57). In most type I systems, Cas2 and Cas3 are separate proteins involved in adaptation and interference, respectively. However, in I-F systems, these proteins are fused into a single polypeptide that forms a large (~375 kDa) heterohexameric complex with Cas1 (Cas14-Cas2/32) (Figure 4D) (21; 55; 57). This suggests that AcrIF3 might be a more efficient immune suppressor than has previously been appreciated and its ability to block adaptation and interference, might be complemented by the ability to trigger oligomerization of the Cas1-2/3 complex.
Why Two Acrs?
Unlike the homodimeric Acrs that benefit from larger binding surfaces, and in many cases dimerize their targets, a few Acrs function as monomers that bind a single surveillance complex at multiple sites (e.g., AcrIF1 and AcrIIC3). The advantage of this ‘strength in numbers’ strategy is unclear, since multiple Acrs binding a single Cas protein will increase the anti-CRISPR concentration needed to saturate targets and slow down the process of immune suppression. For example, AcrIFl blocks access to the crRNA-guide by binding to two different sites on the Csy complex (15; 23; 52), but it is unknown if binding at both sites is necessary to prevent crRNA-guided target engagement. After PAM-binding, the crRNA is expected to directionally unwind the DNA duplex, starting at the PAM proximal end of the guide. Mismatches nearest the PAM (referred to as the seed region of the crRNA) prevent target binding, indicating that hybridization within the seed region of crRNA is critical to target recognition (12; 81). One of the two AcrIF1 molecules that bind the Csy complex block access to the crRNA seed. However, it is unclear if this binding site is sufficient to impede target binding or if a second molecule of AcrIF1 must also bind further up the crRNA backbone to prevent target hybridization.
The advantages of multiple binding sites is more obvious for AcrIIC3, which binds to both the recognition lobe (REC2) and the HNH-nuclease domain of Cas9 (35; 71; 86). In fact, AcrIIC3-mediated dimerization of Cas9 is necessary for suppression, as a single molecule of AcrIIC3 is unable to prevent Cas9 target cleavage (86). Recently published structures reveal that two molecules of AcrIIC3 bridge oppositely oriented molecules of Cas9, creating a ring-shaped structure (Figure 4E) (71). AcrIIC3 prevents activation of the Cas9 HNH-nuclease (71), but it is unclear why one Acr protein is unable to block this conformational rearrangement. To understand the mechanism of AcrIIC3, we superimposed a molecule of the Acr onto structures of Cas9 in multiple conformational states. This analysis reveals that Cas9 conformational changes induced by target binding and nuclease activation disrupt contacts with AcrIIC3, and these conformational changes must dislodge the anti-CRISPR in the absence of additional stabilizing contacts with a second molecule of Cas9. This indicates that assembly of the ring-shaped heterotetramer provides additional contacts to both molecules of AcrIIC3, allowing each suppressor to pin the HNH domains of the two Cas9s in inactive conformations. Collectively, this suggests a model where two AcrIIC3-Cas9 heterodimers bind one another to neutralize each surveillance complex (Figure 4E), and that a monomer of AcrIIC3 is insufficient to inhibit one molecule of Cas9 but two Acrs collectively inhibit two surveillance complexes.
Enzymatic Suppressors of CRISPR Immunity
Covalent Modification of the Surveillance Complex
In contrast to Acrs with steric and allosteric mechanisms of immunosuppression, AcrVA1 and AcrVA5 are enzymes that covalently modify the Cas12-crRNA complex, making them potent suppressors even at sub-stoichiometric concentrations (19; 38; 85). This may be especially beneficial for type V suppressors, since Cas12 is capable of multi-turnover targeting (67). AcrVA1 is an endoribonuclease that cleaves between the fifth and sixth position of the crRNA (Figure 5A and B), while AcrVA5 is a GCN5-related N-acetyltransferase (GNAT) that acetylates lysines (Figure 5C and D) (19; 38; 85). Initially, AcrVA5 was thought to target acetylation to a specific PAM sensing residue (i.e., K635), but Dong et al. detected widespread acetylation of Cas12, which may indicate that AcrVA5 is more promiscuous than previously appreciated (Figure 5C). The biochemical promiscuity of AcrVA5 is consistent with the lack of an N-terminal specificity domain that is found in other GNAT homologs (Figure 5D) (59). However, untargeted acetylation seems dangerous and ineffective. The previously reported formation of a complex between AcrVA5 and AcrVA4 (85), suggests that AcrVA4 may direct the acetyltransferase activity of AcrVA5 to the appropriate (i.e., Cas12) substrate.
Figure 5. Enzymatic Acrs that inhibit type V-A CRISPR-Cas systems.
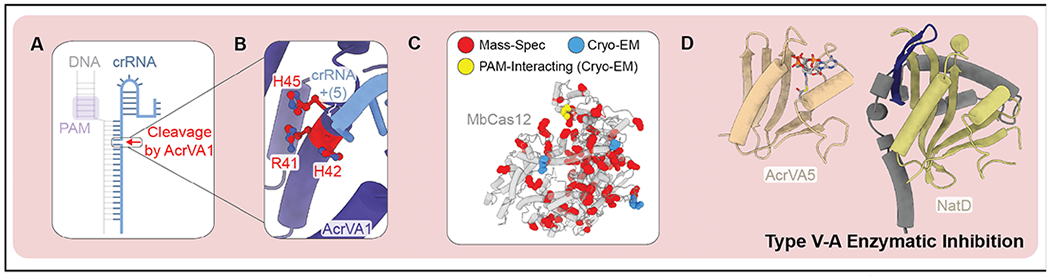
A) AcrVA1 is an endoribonuclease that cleaves between the fifth and sixth position of the crRNA-guide. B) Active site residues of AcrVA1 (red) responsible for crRNA cleavage (PDB: 6NMD). C) Residues of MbCas12 acetylated by AcrVA5 according to mass spectrometry (red) or cryo-EM and mass spec (blue and yellow) (PDB: 6IV6). D) Comparison of AcrVA5 (tan) and closest structural homolog (NatD from Homo sapien) (RMSD = 0.82 Å for 36 equivalently positioned C-alpha atoms). Structural features of NatD shared with AcrVA5 colored in olive. Two NatD β-strands that determine acceptor substrate specificity (dark blue) and additional N-terminal structural features (grey) (PDB: 6IUF, 4U9W).
The Future of Anti-CRISPR
Gazing into the anti-CRISPR crystal ball, it seems that only one thing is certain. The discovery of new anti-CRISPRs will continue to provide new insights about Acr mechanisms, while simultaneously revealing mechanistic vulnerabilities of the immune systems that they inhibit. To date, we are aware of fourteen stoichiometric inhibitors, which either block DNA binding (PAM recognition or hybridization to the guide) or allow binding but prevent nuclease activation. DNA mimics appear to be a common theme in the type I, II, and V systems and we anticipate the discovery of Acrs that adopt similar strategies for RNA targeting systems (type III and type VI). Protein mimics, like AcrIF3 (which mimics the helical bundle of Cas8f), appear to be effective suppressors, and we expect that protein mimics will be identified with increasing frequency. Similarly, we anticipate that enzymatic inhibitors are currently underrepresented, and we expect that multi-turnover enzymes that post-translationally modify Cas proteins, or post-transcriptionally modify CRISPR RNAs will continue to reveal the versatility of these inhibitors. As an example of this versatility, an unreviewed paper that is currently available on the bioRxiv, reports the identification of a widespread enzymatic inhibitor (i.e., AcrIII-1) that rapidly degrades cyclic tetra-adenylate (cA4), which is produced by type III CRISPR systems after binding viral RNA and has been shown to activate nucleases necessary for defense (3). In addition to its novel activity, this enzyme relies on a novel fold that assembles into a homodimer which specifically recognizes the symmetry of cA4. Though it may be a coincidence, we find the importance of symmetry imposed by homodimers to be remarkable. This includes homodimeric Acrs that dimerize the Cas proteins they target, homodimeric Acas that recognize the symmetry of inverted DNA repeats in the promoter, and now homodimeric Acrs that recognize the symmetry of second messengers.
Given the prevalence of CRISPR systems in archaeal genomes (~85%) (40), we expect that archaeal viruses will deploy the most diverse, abundant and innovative Acr-based solutions for subverting CRISPR-based immunity and that these Acrs will in turn drive immunological innovation. Aside from AcrIII-1, only two other archaeal anti-CRISPR proteins have described to date (i.e., AcrID1 and AcrIIIB1) (4; 24). Remarkably, SIRV2 contains up to 12 copies of AcrID1 (24). This observation suggests that duplicated Acrs may be involved in dosage compensations, in a way that is conceptually similar to the expansions of K3L suppressors in poxviruses (“DNA accordions”) (20). Alternatively, or perhaps in addition to, it is possible that each of these paralogs have nuanced, non-redundant functions.
In our opinion, AcrIII-1 represents an emerging ‘class’ of anti-CRISPR that transcend traditional subtype boundaries because of their unique mechanisms of action. Given the lack of reliance on a specific protein-protein interaction, this anti-CRISPR is presumed to be capable of inhibiting any type III system that relies on this messenger. Not restricted to inhibition of a specific subtype, AcrIII-1 inhibits systems that signal with cA4, but not other secondary messengers (i.e., cA6) (3). At a biological level, this suggests that like Cas proteins, second messengers are also in conflict, and we expect that Acrs will mirror the diversity of the messengers themselves. At a practical level, Acrs that target second messengers, rather than a subtype specific protein, may defy the existing Acr naming scheme, which typically includes a letter to designate a specific subtype rather than a second messenger that might be present in many different subtypes. For this new generation of Acrs, we suggest a naming scheme that incorporates the specific messenger (e.g., AcrIII-1-cA4).
The work on AcrIII-1 highlights the biological innovation that occurs in response to the selective pressures imposed by viruses and encourages us to think beyond the stochiometric or enzymatic proteins that bind directly to Cas proteins or CRISPR RNAs. We anticipate that viruses will also use non-coding RNAs or peptide-based inhibitors that will serve as decoys or molecular jamming devices that block or redirect the immune systems. Understanding how viruses evade detection and how hosts overcome immune suppression will continue to be a transformative area of discovery that will benefit from a holistic approach, that includes scientists with diverse expertise and backgrounds.
ACKNOWLEDGMENTS
We are grateful to friends and colleagues for thoughtful discussions and critical feedback. This work was supported by the National Institutes of Health (1R35GM134867) to BW. CRISPR-Cas work in the Bondy-Denomy Lab is supported by the UCSF Program for Breakthrough Biomedical Research funded in part by the Sandler Foundation, an NIH Director’s Early Independence Award DP5-OD021344, and R01GM127489.
Works Cited
- 1.Amitai G, Sorek R. 2016. CRISPR–Cas adaptation: insights into the mechanism of action. Nat Rev Microbiol 14:67. [DOI] [PubMed] [Google Scholar]
- 2.Anders C, Niewoehner O, Duerst A, Jinek M. 2014. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature 513:569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Athukoralage JS, McMahon S, Zhang C, Grüschow S, Graham S, et al. 2019. A viral ring nuclease anti-CRISPR subverts type III CRISPR immunity. BioRxiv:778746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bhoobalan-Chitty Y, Johansen TB, Di Cianni N, Peng X. 2019. Inhibition of Type III CRISPR-Cas Immunity by an Archaeal Virus-Encoded Anti-CRISPR Protein. Cell 179:448–58. e11 [DOI] [PubMed] [Google Scholar]
- 5.Birkholz N, Fagerlund RD, Smith LM, Jackson SA, Fineran PC. 2019. The autoregulator Aca2 mediates anti-CRISPR repression. Nucleic Acids Res 47:9658–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bondy-Denomy J, Davidson AR, Doudna JA, Fineran PC, Maxwell KL, et al. 2018. A unified resource for tracking anti-CRISPR names. The CRISPR journal 1:304–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bondy-Denomy J, Garcia B, Strum S, Du M, Rollins MF, et al. 2015. Multiple mechanisms for CRISPR-Cas inhibition by anti-CRISPR proteins. Nature 526:136–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bondy-Denomy J, Pawluk A, Maxwell KL, Davidson AR. 2013. Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature 493:429–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Borges AL, Davidson AR, Bondy-Denomy J. 2017. The Discovery, Mechanisms, and Evolutionary Impact of Anti-CRISPRs. Annu Rev Virol 4:37–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Borges AL, Zhang JY, Rollins MF, Osuna BA, Wiedenheft B, Bondy-Denomy J. 2018. Bacteriophage Cooperation Suppresses CRISPR-Cas3 and Cas9 Immunity. Cell 174:917–25.e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bryson AL, Hwang Y, Sherrill-Mix S, Wu GD, Lewis JD, et al. 2015. Covalent Modification of Bacteriophage T4 DNA Inhibits CRISPR-Cas9. MBio 6:e00648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cady KC, Bondy-Denomy J, Heussler GE, Davidson AR, O’Toole GA. 2012. The CRISPR/Cas adaptive immune system of Pseudomonas aeruginosa mediates resistance to naturally occurring and engineered phages. J Bacteriol 194:5728–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Charpentier E, Richter H, van der Oost J, White MF. 2015. Biogenesis pathways of RNA guides in archaeal and bacterial CRISPR-Cas adaptive immunity. FEMS microbiology reviews 39:428–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Charpentier E, van der Oost J, White MF. 2013. crRNA biogenesis In CRISPR-Cas Systems: 115–44: Springer; Number of 115-44 pp. [Google Scholar]
- 15.Chowdhury S, Carter J, Rollins MF, Golden SM, Jackson RN, et al. 2017. Structure Reveals Mechanisms of Viral Suppressors that Intercept a CRISPR RNA-Guided Surveillance Complex. Cell 169:47–57 e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dagdas YS, Chen JS, Sternberg SH, Doudna JA, Yildiz A. 2017. A conformational checkpoint between DNA binding and cleavage by CRISPR-Cas9. Science advances 3:eaao0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deveau H, Barrangou R, Garneau JE, Labonte J, Fremaux C, et al. 2008. Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J Bacteriol 190:1390–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dong, Guo M, Wang S, Zhu Y, Wang S, et al. 2017. Structural basis of CRISPR-SpyCas9 inhibition by an anti-CRISPR protein. Nature 546:436–9 [DOI] [PubMed] [Google Scholar]
- 19.Dong L, Guan X, Li N, Zhang F, Zhu Y, et al. 2019. An anti-CRISPR protein disables type V Cas12a by acetylation. Nat Struct Mol Biol 26:308–14 [DOI] [PubMed] [Google Scholar]
- 20.Elde NC, Child SJ, Eickbush MT, Kitzman JO, Rogers KS, et al. 2012. Poxviruses deploy genomic accordions to adapt rapidly against host antiviral defenses. Cell 150:831–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fagerlund RD, Wilkinson ME, Klykov O, Barendregt A, Pearce FG, et al. 2017. Spacer capture and integration by a type I-F Cas1-Cas2-3 CRISPR adaptation complex. Proc Natl Acad Sci U S A 114:E5122–E8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fuchsbauer O, Swuec P, Zimberger C, Amigues B, Levesque S, et al. 2019. Cas9 Allosteric Inhibition by the Anti-CRISPR Protein AcrIIA6. Mol Cell [DOI] [PubMed] [Google Scholar]
- 23.Guo TW, Bartesaghi A, Yang H, Falconieri V, Rao P, et al. 2017. Cryo-EM Structures Reveal Mechanism and Inhibition of DNA Targeting by a CRISPR-Cas Surveillance Complex. Cell 171:414–26 e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.He F, Bhoobalan-Chitty Y, Van LB, Kjeldsen AL, Dedola M, et al. 2018. Anti-CRISPR proteins encoded by archaeal lytic viruses inhibit subtype ID immunity. Nat Microbiol 3:461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heler R, Samai P, Modell JW, Weiner C, Goldberg GW, et al. 2015. Cas9 specifies functional viral targets during CRISPR-Cas adaptation. Nature 519:199–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hille F, Richter H, Wong SP, Bratovič M, Ressel S, Charpentier E. 2018. The biology of CRISPR-Cas: backward and forward. Cell 172:1239–59 [DOI] [PubMed] [Google Scholar]
- 27.Hurford A, Day T. 2013. Immune evasion and the evolution of molecular mimicry in parasites. Evolution 67:2889–904 [DOI] [PubMed] [Google Scholar]
- 28.Hwang S, Maxwell KL. 2019. Meet the Anti-CRISPRs: Widespread Protein Inhibitors of CRISPR-Cas Systems. CRISPR J 2:23–30 [DOI] [PubMed] [Google Scholar]
- 29.Hynes AP, Rousseau GM, Agudelo D, Goulet A, Amigues B, et al. 2018. Widespread anti-CRISPR proteins in virulent bacteriophages inhibit a range of Cas9 proteins. Nat Commun 9:2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jackson RN, van Erp PB, Sternberg SH, Wiedenheft B. 2017. Conformational regulation of CRISPR-associated nucleases. Current opinion in microbiology 37:110–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jackson SA, McKenzie RE, Fagerlund RD, Kieper SN, Fineran PC, Brouns SJ. 2017. CRISPR-Cas: Adapting to change. Science 356 [DOI] [PubMed] [Google Scholar]
- 32.Jiang F, Liu J-J, Osuna BA, Xu M, Berry JD, et al. 2019. Temperature-responsive competitive inhibition of CRISPR-Cas9. Mol Cell 73:601–10. e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jones DL, Leroy P, Unoson C, Fange D, Ćurić V, et al. 2017. Kinetics of dCas9 target search in Escherichia coli. Science 357:1420–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ka D, An SY, Suh J-Y, Bae E. 2017. Crystal structure of an anti-CRISPR protein, AcrIIA1. Nucleic Acids Res 46:485–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim Y, Lee SJ, Yoon HJ, Kim NK, Lee BJ, Suh JY. 2019. Anti-CRISPR AcrIIC3 discriminates between Cas9 orthologs via targeting the variable surface of the HNH nuclease domain. The FEBS journal [DOI] [PubMed] [Google Scholar]
- 36.Klompe SE, Sternberg SH. 2018. Harnessing “A Billion Years of Experimentation”: the ongoing exploration and exploitation of CRISPR-Cas immune systems. The CRISPR journal 1:141–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Knott GJ, Cress BF, Liu JJ, Thornton BW, Lew RJ, et al. 2019. Structural basis for AcrVA4 inhibition of specific CRISPR-Cas12a. Elife 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Knott GJ, Thornton BW, Lobba MJ, Liu JJ, Al-Shayeb B, et al. 2019. Broad-spectrum enzymatic inhibition of CRISPR-Cas12a. Nat Struct Mol Biol 26:315–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koonin EV, Makarova KS, Wolf YI, Krupovic M. 2019. Evolutionary entanglement of mobile genetic elements and host defence systems: guns for hire. Nat Rev Genet:1–13 [DOI] [PubMed] [Google Scholar]
- 40.Landsberger M, Gandon S, Meaden S, Rollie C, Chevallereau A, et al. 2018. Anti-CRISPR Phages Cooperate to Overcome CRISPR-Cas Immunity. Cell 174:908–16.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu L, Yin M, Wang M, Wang Y. 2019. Phage AcrIIA2 DNA Mimicry: Structural Basis of the CRISPR and Anti-CRISPR Arms Race. Mol Cell 73:611–20 e3 [DOI] [PubMed] [Google Scholar]
- 42.Makarova KS, Aravind L, Wolf YI, Koonin EV. 2011. Unification of Cas protein families and a simple scenario for the origin and evolution of CRISPR-Cas systems. Biology direct 6:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Makarova KS, Wolf YI, Iranzo J, Shmakov SA, Alkhnbashi OS, et al. 2019. Evolutionary classification of CRISPR-Cas systems: a burst of class 2 and derived variants. Nat Rev Microbiol [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McGinn J, Marraffini LA. 2019. Molecular mechanisms of CRISPR-Cas spacer acquisition. Nat Rev Microbiol 17:7–12 [DOI] [PubMed] [Google Scholar]
- 45.Mojica FJ, Díez-Villaseñor C, García-Martínez J, Almendros C. 2009. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology 155:733–40 [DOI] [PubMed] [Google Scholar]
- 46.Nishimasu H, Ran FA, Hsu PD, Konermann S, Shehata SI, et al. 2014. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 156:935–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Osuna BA, Karambelkar S, Mahendra C, Christie KA, Garcia B, et al. 2019. Listeria phages induce Cas9 degradation to protect lysogenic genomes. BioRxiv:787200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pawluk A, Bondy-Denomy J, Cheung VH, Maxwell KL, Davidson AR. 2014. A new group of phage anti-CRISPR genes inhibits the type I-E CRISPR-Cas system of Pseudomonas aeruginosa. MBio 5:e00896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pawluk A, Davidson AR, Maxwell KL. 2018. Anti-CRISPR: discovery, mechanism and function. Nat Rev Microbiol 16:12. [DOI] [PubMed] [Google Scholar]
- 50.Pawluk A, Staals RH, Taylor C, Watson BN, Saha S, et al. 2016. Inactivation of CRISPR-Cas systems by anti-CRISPR proteins in diverse bacterial species. Nat Microbiol 1:16085. [DOI] [PubMed] [Google Scholar]
- 51.Peng R, Li Z, Xu Y, He S, Peng Q, et al. 2019. Structural insight into multistage inhibition of CRISPR-Cas12a by AcrVA4. Proceedings of the National Academy of Sciences 116:18928–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Peng R, Xu Y, Zhu T, Li N, Qi J, et al. 2017. Alternate binding modes of anti-CRISPR viral suppressors AcrF1/2 to Csy surveillance complex revealed by cryo-EM structures. Cell Res 27:853–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Plagens A, Richter H, Charpentier E, Randau L. 2015. DNA and RNA interference mechanisms by CRISPR-Cas surveillance complexes. FEMS Microbiol Rev 39:442–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Putnam CD, Tainer JA. 2005. Protein mimicry of DNA and pathway regulation. DNA Repair (Amst) 4:1410–20 [DOI] [PubMed] [Google Scholar]
- 55.Richter C, Gristwood T, Clulow JS, Fineran PC. 2012. In vivo protein interactions and complex formation in the Pectobacterium atrosepticum subtype IF CRISPR/Cas System. PloS one 7:e49549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rollins MF, Chowdhury S, Carter J, Golden SM, Miettinen HM, et al. 2019. Structure Reveals a Mechanism of CRISPR-RNA-Guided Nuclease Recruitment and Anti-CRISPR Viral Mimicry. Mol Cell 74:132–42 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rollins MF, Chowdhury S, Carter J, Golden SM, Wilkinson RA, et al. 2017. Cas1 and the Csy complex are opposing regulators of Cas2/3 nuclease activity. Proc Natl Acad Sci U S A 114:E5113–E21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rollins MF, Schuman JT, Paulus K, Bukhari HS, Wiedenheft B. 2015. Mechanism of foreign DNA recognition by a CRISPR RNA-guided surveillance complex from Pseudomonas aeruginosa. Nucleic Acids Res 43:2216–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Salah Ud-Din AI, Tikhomirova A, Roujeinikova A. 2016. Structure and Functional Diversity of GCN5-Related N-Acetyltransferases (GNAT). Int J Mol Sci 17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Santiago-Frangos A, Wiegand T, Wiedenheft B. 2019. Cas9 slide-and-seek for phage defense and genome engineering. EMBO J 38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Semenova E, Jore MM, Datsenko KA, Semenova A, Westra ER, et al. 2011. Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RNA is governed by a seed sequence. Proc Natl Acad Sci U S A 108:10098–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shibata M, Nishimasu H, Kodera N, Hirano S, Ando T, et al. 2017. Real-space and real-time dynamics of CRISPR-Cas9 visualized by high-speed atomic force microscopy. Nat Commun 8:1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shin J, Jiang F, Liu JJ, Bray NL, Rauch BJ, et al. 2017. Disabling Cas9 by an anti-CRISPR DNA mimic. Sci Adv 3:e1701620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Singh D, Sternberg SH, Fei J, Doudna JA, Ha T. 2016. Real-time observation of DNA recognition and rejection by the RNA-guided endonuclease Cas9. Nat Commun 7:12778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stanley SY, Borges AL, Chen K-H, Swaney DL, Krogan NJ, et al. 2019. Anti-CRISPR-Associated Proteins Are Crucial Repressors of Anti-CRISPR Transcription. Cell 178:1452–64. e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stanley SY, Maxwell KL. 2018. Phage-Encoded Anti-CRISPR Defenses. Annu Rev Genet 52:445–64 [DOI] [PubMed] [Google Scholar]
- 67.Stella S, Mesa P, Thomsen J, Paul B, Alcon P, et al. 2018. Conformational Activation Promotes CRISPR-Cas12a Catalysis and Resetting of the Endonuclease Activity. Cell 175:1856–71 e21 [DOI] [PubMed] [Google Scholar]
- 68.Sternberg SH, Redding S, Jinek M, Greene EC, Doudna JA. 2014. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 507:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sternberg SH, Richter H, Charpentier E, Qimron U. 2016. Adaptation in CRISPR-Cas systems. Mol Cell 61:797–808 [DOI] [PubMed] [Google Scholar]
- 70.Stone NP, Hilbert BJ, Hidalgo D, Halloran KT, Lee J, et al. 2018. A hyperthermophilic phage decoration protein suggests common evolutionary origin with herpesvirus triplex proteins and an anti-CRISPR protein. Structure 26:936–47. e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sun W, Yang J, Cheng Z, Amrani N, Liu C, et al. 2019. Structures of Neisseria meningitidis Cas9 Complexes in Catalytically Poised and Anti-CRISPR-Inhibited States. Mol Cell [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Thavalingam A, Cheng Z, Garcia B, Huang X, Shah M, et al. 2019. Inhibition of CRISPR-Cas9 ribonucleoprotein complex assembly by anti-CRISPR AcrIIC2. Nat Commun 10:2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.van Erp PBG, Patterson A, Kant R, Berry L, Golden SM, et al. 2018. Conformational Dynamics of DNA Binding and Cas3 Recruitment by the CRISPR RNA-Guided Cascade Complex. ACS Chem Biol 13:481–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vink JNA, Martens KJA, Vlot M, McKenzie RE, Almendros C, et al. 2019. Direct Visualization of Native CRISPR Target Search in Live Bacteria Reveals Cascade DNA Surveillance Mechanism. Mol Cell [DOI] [PubMed] [Google Scholar]
- 75.Vlot M, Houkes J, Lochs SJA, Swarts DC, Zheng P, et al. 2017. Bacteriophage DNA glucosylation impairs target DNA binding by type I and II but not by type V CRISPR-Cas effector complexes. Nucleic acids research 46:873–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vorontsova D, Datsenko KA, Medvedeva S, Bondy-Denomy J, Savitskaya EE, et al. 2015. Foreign DNA acquisition by the I-F CRISPR-Cas system requires all components of the interference machinery. Nucleic Acids Res 43:10848–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang J, Ma J, Cheng Z, Meng X, You L, et al. 2016. A CRISPR evolutionary arms race: structural insights into viral anti-CRISPR/Cas responses. Cell Res 26:1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang X, Yao D, Xu JG, Li AR, Xu J, et al. 2016. Structural basis of Cas3 inhibition by the bacteriophage protein AcrF3. Nat Struct Mol Biol 23:868–70 [DOI] [PubMed] [Google Scholar]
- 79.Watters KE, Fellmann C, Bai HB, Ren SM, Doudna JA. 2018. Systematic discovery of natural CRISPR-Cas12a inhibitors. Science 362:236–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wei Y, Terns RM, Terns MP. 2015. Cas9 function and host genome sampling in Type II-A CRISPR-Cas adaptation. Genes Dev 29:356–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wiedenheft B, van Duijn E, Bultema JB, Waghmare SP, Zhou K, et al. 2011. RNA-guided complex from a bacterial immune system enhances target recognition through seed sequence interactions. Proc Natl Acad Sci U S A 108:10092–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Xiao Y, Luo M, Dolan AE, Liao M, Ke A. 2018. Structure basis for RNA-guided DNA degradation by Cascade and Cas3. Science 361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yang H, Patel DJ. 2017. Inhibition Mechanism of an Anti-CRISPR Suppressor AcrIIA4 Targeting SpyCas9. Mol Cell 67:117–27 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yang M, Peng S, Sun R, Lin J, Wang N, Chen C. 2018. The Conformational Dynamics of Cas9 Governing DNA Cleavage Are Revealed by Single-Molecule FRET. Cell Rep 22:372–82 [DOI] [PubMed] [Google Scholar]
- 85.Zhang H, Li Z, Daczkowski CM, Gabel C, Mesecar AD, Chang L. 2019. Structural Basis for the Inhibition of CRISPR-Cas12a by Anti-CRISPR Proteins. Cell Host Microbe 25:815–26 e4 [DOI] [PubMed] [Google Scholar]
- 86.Zhu Y, Gao A, Zhan Q, Wang Y, Feng H, et al. 2019. Diverse Mechanisms of CRISPR-Cas9 Inhibition by Type IIC Anti-CRISPR Proteins. Mol Cell 74:296–309 e7 [DOI] [PMC free article] [PubMed] [Google Scholar]