

Abstract
Cellular metabolism is directly or indirectly associated with various cellular processes by producing a variety of metabolites. Metabolic alterations may cause adverse effects on cell viability. However, some alterations potentiate the rescue of the malfunction of the cell system. Here, we found that the alteration of glucose metabolism suppressed genome instability caused by the impairment of chromatin structure. Deletion of the TDH2 gene, which encodes glyceraldehyde 3-phospho dehydrogenase and is essential for glycolysis/gluconeogenesis, partially suppressed DNA damage sensitivity due to chromatin structure, which was persistently acetylated histone H3 on lysine 56 in cells with deletions of both HST3 and HST4, encoding NAD+-dependent deacetylases. tdh2 deletion also restored the short replicative lifespan of cells with deletion of sir2, another NAD+-dependent deacetylase, by suppressing intrachromosomal recombination in rDNA repeats increased by the unacetylated histone H4 on lysine 16. tdh2 deletion also suppressed recombination between direct repeats in hst3∆ hst4∆ cells by suppressing the replication fork instability that leads to both DNA deletions among repeats. We focused on quinolinic acid (QUIN), a metabolic intermediate in the de novo nicotinamide adenine dinucleotide (NAD+) synthesis pathway, which accumulated in the tdh2 deletion cells and was a candidate metabolite to suppress DNA replication fork instability. Deletion of QPT1, quinolinate phosphoribosyl transferase, elevated intracellular QUIN levels and partially suppressed the DNA damage sensitivity of hst3∆ hst4∆ cells as well as tdh2∆ cells. qpt1 deletion restored the short replicative lifespan of sir2∆ cells by suppressing intrachromosomal recombination among rDNA repeats. In addition, qpt1 deletion could suppress replication fork slippage between direct repeats. These findings suggest a connection between glucose metabolism and genomic stability.
Subject terms: Genomic instability, DNA metabolism
Introduction
Genome instability is closely connected with both carcinogenesis and aging1–3. DNA damage is an alteration in the chemical structure of DNA. Common types of DNA damage include DNA base modifications, DNA inter- and intrastrand crosslinks, and DNA single- and double-strand breaks (SSBs and DSBs, respectively)4. Endogenous and exogenous sources of DNA damage lead to genomic instability (reviewed in5). Endogenous sources of DNA damage include reactive oxygen species (ROS) or some other products of DNA metabolism. Endogenous sources of DNA damage can lead to DNA base modifications and the formation of bulky adducts. Problems in DNA metabolism (e.g., DNA replication and chromosomal segregation) can lead to DNA breaks (SSBs and DSBs). Exogenous sources of DNA damage are external agents, including ionizing radiation, ultraviolet radiation and a variety of chemical agents. Chemical agents can have various effects on the DNA strand (e.g., DNA intercalation, DNA crosslinking, and DNA alkylation). Cis-elements of the DNA sequence affect replication fork stability. The replication fork can skip among DNA repeats (e.g., trinucleotide repeats, inverted repeats or direct repeats), resulting in deletions or mutations1,6–9. The proteins involved in DNA damage repair, DNA replication and the cell cycle checkpoint work cooperatively to maintain genome integrity to fix DNA lesions or to prevent DNA replication fork instability. Mutations of these proteins drastically cause the accumulation of mutations in the chromosome, which results in carcinogenesis and progeria2,3,9.
Chromatin is composed of DNA fiber and chromatin-binding proteins such as histones that package chromosomal DNA into nuclei. The condensed structure usually becomes an obstacle for the execution of nuclear activities on chromatin. Therefore, chromatin regulator proteins (e.g., histone modifiers and chromatin remodeling factors) create an environment that allows the replisome, transcriptional machinery and DNA damage repair machinery to perform on chromatin10,11. The canonical histones (H2A, H2B, H3 and H4) harbor various posttranslational modifications (such as acetylation, methylation, phosphorylation and ubiquitination)10. Histone acetyltransferase (HAT) and histone deacetylase (HDAC) acetylate and deacetylate lysine residues in histone proteins, respectively. HAT and HDAC cooperatively manage gene transcription12,13, chromatin remodeling14, and DNA damage repair15,16. Hst3 and Hst4, NAD+-dependent deacetylases in budding yeast17, mutually deacetylate histone H3 on lysine 56 (H3-K56)18–20. Histone H3-K56 is persistently acetylated in hst3∆ hst4∆ cells, which causes sensitivity to DNA damaging agents because of the loosened chromatin structure18,19. Because the deletion of the RTT109 gene, encoding the HAT responsible for histone H3-K56 acetylation, allows histone H3-K56 to be deacetylated and confers severe DNA damage sensitivity, proper control of both acetylation and deacetylation on histone H3-K56 in chromatin is needed to perform DNA repair21. Sir2 is another NAD+-dependent deacetylase, and one substrate of Sir2 is histone H4 on K1622. Sir2 is involved in gene silencing of genes inserted within ribosomal DNA (rDNA) repeats and at telomere loci and the silent mating type loci HML and HMR23. sir2 deletion causes persistence of histone H4 acetylation on K16 in chromatin, which elevates the frequencies of intrachromosomal recombination in rDNA repeats by replication fork slippage and generates extrachromosomal ribosomal DNA circles (ERCs)24,25. The accumulation of ERCs reduces the replicative lifespan in sir2∆ cells24,25. Thus, persistence of either H3-K56 acetylation on chromatin or H4 K16 acetylation on rDNA repeats leads to genome instability.
In this study, we found that the alteration of glucose metabolism suppressed the genome instability caused by aberrant chromatin structure. Deletion of the TDH2 gene, which encodes glyceraldehyde 3-phosphate dehydrogenase (GAPDH), a metabolic enzyme in glycolysis/gluconeogenesis, partially suppressed the DNA damage sensitivity of hst3∆ hst4∆, although the tdh2 gene deletion did not affect the H3-K56 acetylation in hst3∆ hst4∆ cells. tdh2 deletion could also restore the short replicative lifespan of sir2∆ cells. In addition, tdh2 deletion could suppress replication fork slippage between direct repeats in both wild-type and hst3∆ hst4∆ cells. Furthermore, we examined the role of quinolinic acid (QUIN), which is a metabolic intermediate in the de novo nicotinamide adenine dinucleotide (NAD+) synthesis pathway (or kynurenine pathway) and was accumulated in tdh2∆ cells, and tested whether QUIN could suppress replication fork instability as observed in tdh2∆ cells. The cells with deletion of QPT1, quinolinate phosphoribosyl transferase, which synthesizes nicotinate mononucleotide (NaMN) from QUIN, experienced elevated intracellular QUIN concentrations and partially suppressed DNA damage sensitivity in hst3∆ hst4∆ cells similarly as observed in tdh2∆ cells. The qpt1 deletion restored the short replicative lifespan of sir2∆ cells. Furthermore, the qpt1 deletion suppressed replication fork slippage between direct repeats more compared to wild-type cells. These findings suggest that metabolic alteration contributes to preventing the genomic instability caused by the impaired function of chromatin regulation, and QUIN may be a candidate to stabilize the DNA replication fork to prevent DNA damage.
Results
Deletion of the TDH2 gene partially suppresses the DNA damage sensitivity of hst3∆ hst4∆ cells
The TDH2 gene encodes one of the yeast GAPDH genes (TDH1/2/3) involved in both glycolysis and gluconeogenesis. A previous study reported that hst3∆ hst4∆ cells activate gluconeogenesis in the presence of glucose, and the tdh2 gene deletion represses gluconeogenesis in enhanced hst3∆ hst4∆ cells and restores slow growth26. Because the slow growth of hst3∆ hst4∆ cells is due to the frequent occurrence of DNA damage during cell cycle progression18,19, we tested whether the tdh2 gene deletion was able to suppress the DNA damage sensitivity of hst3∆ hst4∆ cells. This was examined by monitoring cell growth in YPD (Yeast extract-Polypeptone-Dextrose) solid medium supplemented with the following DNA damaging agents: methyl methanesulfonate (MMS), hydroxyurea (HU) and camptothecin (CPT). The alkylating agent MMS attaches alkyl groups to DNA bases, and HU inhibits ribonucleotide reductase to reduce intracellular deoxyribonucleotide levels. CPT inhibits Type I topoisomerases, thus DNA SSBs are not repaired, and then creates a DNA DSB after passing the replication fork. These agents promote replication collapse and eventually cause DNA DSBs, which are usually fixed by homologous recombination (HR)8,16,27. hst3∆ hst4∆ tdh2∆ cells grew in YPD solid medium amended with each DNA damaging agent better than hst3∆ hst4∆ cells; however, the growth was not restored to the levels of the wild-type and tdh2∆ cells (Fig. 1a and Fig. S1). Rad53 is a member of both the DNA damage and the intra-S phase checkpoint systems and harbors multiple phosphorylation sites phosphorylated by checkpoint activation in response to DNA lesions or replication fork arrests28–34. The extent of Rad53 phosphorylation depends on the intensity of DNA damage. We monitored Rad53 phosphorylation to examine whether deleting tdh2 represses the occurrence of DNA damage in hst3∆ hst4∆ cells. Multiple phosphorylated Rad53 was indicated in wild-type and tdh2∆ cells treated with MMS by the presence of slow-migrated bands in sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), although a single band of unphosphorylated Rad53 was detected in cells without MMS (Fig. 1b; lanes 1, 2, 5 and 6). Phosphorylated Rad53 bands were detected in hst3∆ hst4∆ and hst3∆ hst4∆ tdh2∆ cells treated with MMS, similar to the patterns of wild-type and tdh2∆ cells (Fig. 1b; lanes 2, 4, 6 and 8). Even in the absence of MMS, a smeared Rad53 band was detected in hst3∆ hst4∆ cells (Fig. 1b; lane 3), indicating that the chromatin acetylated at histone H3-K56 becomes fragile and induces DNA damage during nuclear activities18,19. Interestingly, the smeared Rad53 band was not detected in hst3∆ hst4∆ tdh2∆ cells without MMS treatment or in wild-type cells (Fig. 1b; lanes 1 and 7). Thus, tdh2 deletion suppresses the occurrence of DNA damage originating from the chromatin associated with persistently acetylated histone H3-K56 in hst3∆ hst4∆ cells. Next, we asked whether DNA damage suppression by tdh2 deletion was restricted to the chromatin acetylated histone H3-K56. Rtt109 functions as an acetyltransferase for histone H3-K5621. rtt109∆ cells, which did not acetylate histone H3-K56, showed greater sensitivity to MMS, CPT and HU than hst3∆ hst4∆ cells21 (Fig. 1c and Fig. S2). The DNA damage sensitivity of hst3∆ hst4∆ rtt109∆ cells was the same as that of rtt109∆ cells, in that H3-K56 in chromatin remained deacetylated in hst3∆ hst4∆ rtt109∆ (Fig. 1c and Fig. S2). Both rtt109∆ tdh2∆ and hst3∆ hst4∆ rtt109∆ tdh2∆ cells exhibited the same DNA damage sensitivities as rtt109∆ cells (Fig. 1c and Fig. S2), suggesting that tdh2 deletion does not suppress the occurrence of DNA damage caused by chromatin deacetylated histone H3-K56. Next, we asked whether tdh2 deletion might reduce the acetylation level of histone H3-K56 in hst3∆ hst4∆ cells and gain resistance to DNA damage sensitivity. Our previous analysis showed that the level of acetyl-CoA, which is used as a substrate to acetylate lysine residue on target proteins, including histones, was increased in hst3∆ hst4∆ tdh2∆ cells more than in wild-type, hst3∆ hst4∆ and tdh2∆ cells26. To exclude the possibility that another histone modifier reduced the acetylation level of histone H3-K56 to a level sufficient to promote DNA damage repair in hst3∆ hst4∆ tdh2∆ cells, we examined whether tdh2 deletion influenced the H3-K56 acetylation level in hst3∆ hst4∆ cells. The acetylation of histone H3-K56 is regulated in a cell cycle-dependent manner, and this acetylation appears from late G1 to early G2 phase16,35. When the G2/M phase was arrested by treatment with nocodazole, an inhibitor of microtubule polymerization, H3-K56 was deacetylated in both wild-type and tdh2∆ cells (Fig. 1d; lanes 1 and 3). In contrast, the acetylation level of histone H3-K56 remained the same between hst3∆ hst4∆ and hst3∆ hst4∆ tdh2∆ cells treated with nocodazole (Fig. 1d; lanes 2 and 4). Thus, tdh2 deletion does not affect the acetylation level in hst3∆ hst4∆ cells. Altogether, tdh2 deletion is able to suppress the genome instability caused by the aberrant chromatin structure constitutively acetylated histone H3 on K56.
Figure 1.
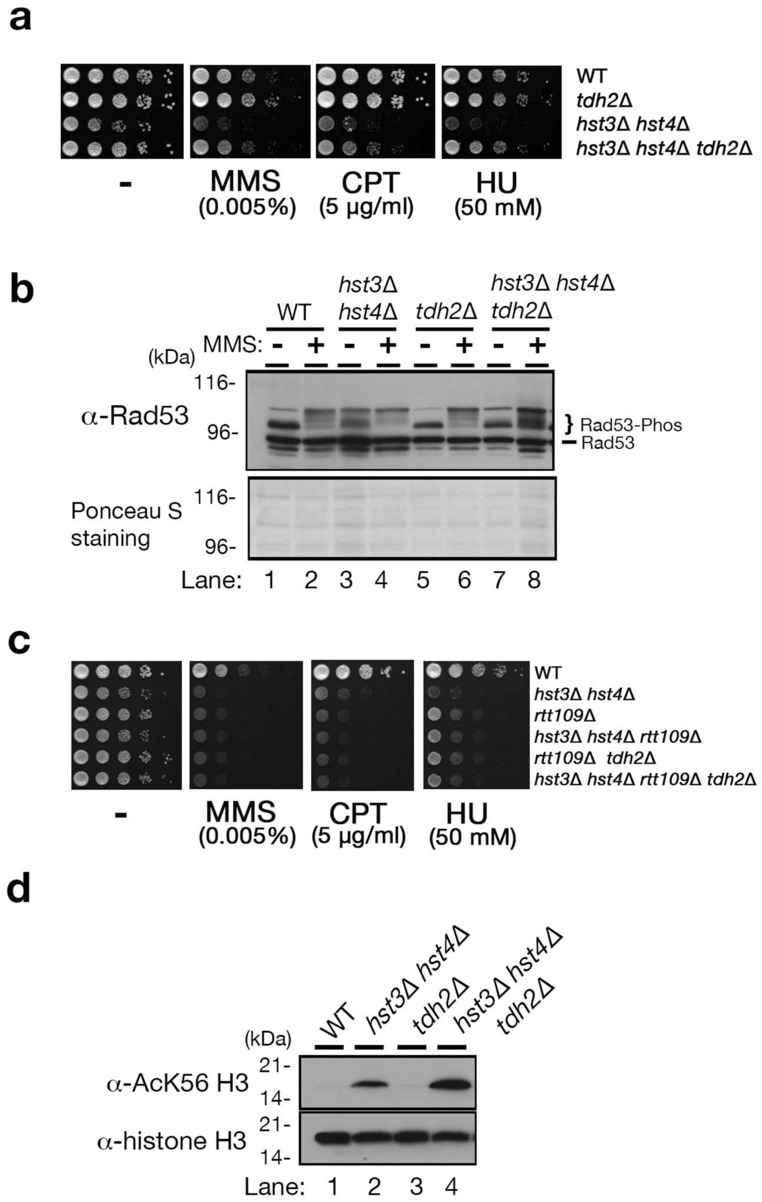
tdh2 gene deletion can suppress the DNA damage sensitivity of hst3∆ hst4∆ cells. (a,c) Plate assays monitoring the sensitivity of each DNA damaging agent (methyl methanesulfonate (MMS), hydroxyurea (HU) and camptothecin (CPT)). A ten-fold dilution of the cell suspension was sequentially diluted (from left to right) and then spotted on solid medium. WT: wild-type. (b) Rad53 phosphorylation was detected by western blotting using an anti-Rad53 antibody. Ponceau S staining indicates the total amount of protein on the membrane. (d) Acetylation level of histone H3-K56 on G2/M phase-arrested cells.
tdh2 deletion suppresses deletion between direct repeats (DRs) on the chromosome
Next, we tested whether tdh2 deletion could suppress the genome instability caused by chromatin acetylation other than H3-K56. The sir2 deletion increases the acetylation level of histone H4 on K16 at ribosomal DNA (rDNA) repeats and elevates the ratio of intrachromosomal recombination among rDNA repeats by replication fork slippage to bear ERCs25. Accumulation of ERCs reduces the replicative lifespan of sir2∆ cells36. Pedigree analysis monitors the replicative age of mother cells to count the number of divided daughter cells. The replicative lifespan of wild-type cells was approximately 22 divisions (50% viability), with a maximum of approximately 40 (Fig. 2a; wild-type). The lifespan of sir2∆ cells was significantly reduced to half the level (approximately 13 divisions (50% viability)) of wild-type cells (Fig. 2a; wild-type vs. sir2∆ (P = 4.457E-115)). tdh2∆ cells had an increased replicative lifespan compared to wild-type cells (Fig. 2a; wild-type vs. tdh2∆ (P = 0.017))26. sir2∆ tdh2∆ cells exhibited a significantly extended lifespan compared to the sir2∆ cells but did not reach the lifespan of the wild-type cells (Fig. 2a; sir2∆ tdh2∆ vs. sir2∆ (P = 3.676E-65), and sir2∆ tdh2∆ vs. wild-type (P = 0.018)). Fob1 functions as a replication fork barrier in rDNA repeats, which elevates the opportunities for intrachromosomal recombination among rDNA repeats37. The fob1 deletion restored the lifespan of sir2∆ cells such that the lifespan of sir2∆ fob1∆ cells was almost the same as that of wild-type (Fig. 2b; wild-type vs. sir2∆ fob1∆). The replicative lifespans were almost the same among wild-type, sir2∆ fob1∆ and sir2∆ fob1∆ tdh2∆ cells (Fig. 2b; wild-type vs. sir2∆ fob1∆ vs. sir2∆ fob1∆ tdh2∆), suggesting that tdh2 deletion suppresses the intrachromosomal recombination among rDNA repeats in the same manner as fob1 deletion. These data suggest that the tdh2 gene deletion can suppress replication fork slippage, causing intrachromosomal recombination among rDNA repeats in sir2∆ cells. To examine whether the genomic instability suppressed by tdh2 deletion is due to aberrant intrachromosomal recombination between direct repeats (DRs) in hst3∆ hst4∆ cells, we used a strain to monitor the ratio of the CaURA3 gene deletion by recombination between DRs due to replication fork slippage (Fig. 2c top). The CaURA3 deletion cells can grow in SC solid medium containing 5-floroorotic acid (5-FOA), a counterselecting agent for URA3 gene deletion. The frequency of CaURA3 gene deletion in hst3∆ hst4∆ tdh2∆ cells was significantly (P = 0.046) reduced to levels lower than those of hst3∆ hst4∆ cells (Fig. 2c; hst3∆ hst4∆ vs. hst3∆ hst4∆ tdh2∆). Furthermore, the frequency of CaURA3 gene deletion in tdh2∆ cells was significantly (P = 0.034) reduced to levels lower than those of wild-type cells (Fig. 2c; wild-type vs. tdh2∆). Thus, tdh2 deletion is involved in suppressing deletions among DRs due to replication fork slippage. Next, we examined whether tdh2 deletion supported the DNA repair machinery in preventing deletion by replication fork slippage. DNA repair machinery has a pivotal role in preventing genome rearrangement in DNA replication38. Sgs1, a RecQ family nucleolar DNA helicase, suppresses DNA replication-associated genome rearrangement39,40. sgs1∆ cells exhibited MMS and HU sensitivity, leading to DNA damage due to DNA replication fork stalling (Fig. S3; wild-type and sgs1∆). Because the HU and MMS sensitivities of tdh2∆ cells were almost the same as those of sgs1∆ cells (Fig. S3; sgs1∆ and sgs1∆ tdh2∆), the replication fork stability by tdh2 deletion depends on the DNA repair machinery. Thus, tdh2 deletion increases the replication fork stability together with the DNA repair machinery.
Figure 2.
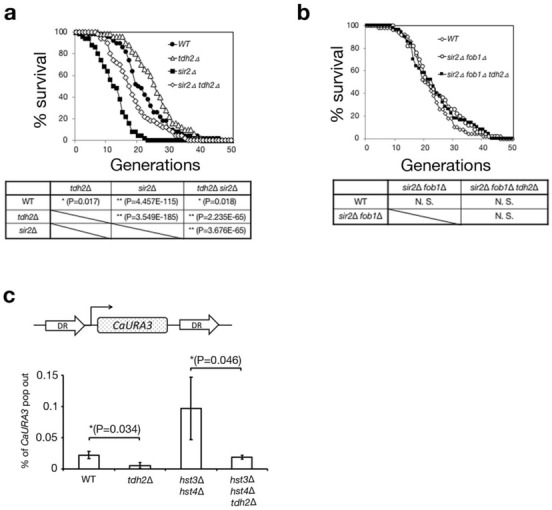
tdh2 gene deletion can suppress intrachromosomal recombination among repeats. (a,b) Pedigree analysis to count replicative lifespans among cells. *P < 0.05. **P < 0.01. N.S: not significant. Unpaired t-test (two-tailed). Over 50 cells/strain were used for analysis. (c) The frequencies of CaURA3 gene deletion among repeats. DRs: direct repeats. *P < 0.05. **P < 0.01. N.S: not significant. Unpaired t-test (two-tails). Error bars represent the standard deviation of three biological replicates.
Quinolinic acid (QUIN) is a metabolic candidate to suppress replication fork instability
We examined whether a metabolite increased in tdh2∆ cells might contribute to replication fork stability in both wild-type and hst3∆ hst4∆ cells. Using capillary electrophoresis time-of-flight mass spectrometry (CE-TOF/MS) analysis to compare the levels of cellular metabolites26, we focused on quinolinic acid (QUIN), which was significantly increased in tdh2∆ cells. QUIN is a metabolic intermediate in the de novo NAD+ synthetic pathway, or the kynurenine pathway (Fig. 3a). QPT1 encodes quinolinate phosphoribosyltransferase to converts QUIN to nicotinic acid mononucleotide (NaMN). We confirmed that the amount of intracellular QUIN significantly (P < 0.05) accumulated in both tdh2∆ and qpt1∆ cells more than wild-type cells (Fig. 3b; wild-type, tdh2∆ and qpt1∆). We used the qpt1 deletion strain to examine whether QUIN was able to suppress the DNA damage sensitivity of hst3∆ hst4∆ cells. hst3∆ hst4∆ qpt1∆ cells exhibited better tolerance to DNA damaging agents (MMS, HU and CPT) than hst3∆ hst4∆ cells. However, the tolerance was not recovered to the levels of the wild-type and tdh2∆ cells (Fig. 3c and Fig. S4; wild-type, qpt1∆, hst3∆ hst4∆ qpt1∆ and hst3∆ hst4∆). Furthermore, the sensitivity of hst3∆ hst4∆ tdh2∆ qpt1∆ cells was almost the same as that of hst3∆ hst4∆ tdh2∆ cells (Fig. 3c and Fig. S4; hst3∆ hst4∆ tdh2∆ and hst3∆ hst4∆ tdh2∆ qpt1∆). Thus, qpt1 deletion can partially restore the DNA damage sensitivity of hst3∆ hst4∆ cells in a Tdh2-dependent manner. Next, we examined whether qpt1 deletion was able to extend the lifespan of sir2∆ cells by pedigree analysis. As shown in Fig. 3d, sir2∆ qpt1∆ cells experienced significantly extended replicative lifespans compared to sir2∆ cells (sir2∆ qpt1∆ vs. sir2∆ (P = 8.324E-65)). Furthermore, the lifespan of sir2∆ tdh2∆ qpt1∆ cells was almost the same as that of sir2∆ tdh2∆ cells (Fig. 3e; sir2∆ tdh2∆ qpt1∆ vs. sir2∆ tdh2∆), suggesting that qpt1 deletion extends the lifespan of sir2∆ in a Tdh2-dependent manner. Next, we investigated whether the qpt1 deletion suppressed spontaneous URA3 gene deletion between DRs as well as the tdh2 deletion. The ratio of CaURA3 gene deletion in DRs was significantly (P = 0.042) reduced in qpt1∆ cells compared to wild-type cells (Fig. 3f). Altogether, these findings suggest that the elevated intracellular QUIN levels in the tdh2 deletion contribute to genome stability.
Figure 3.
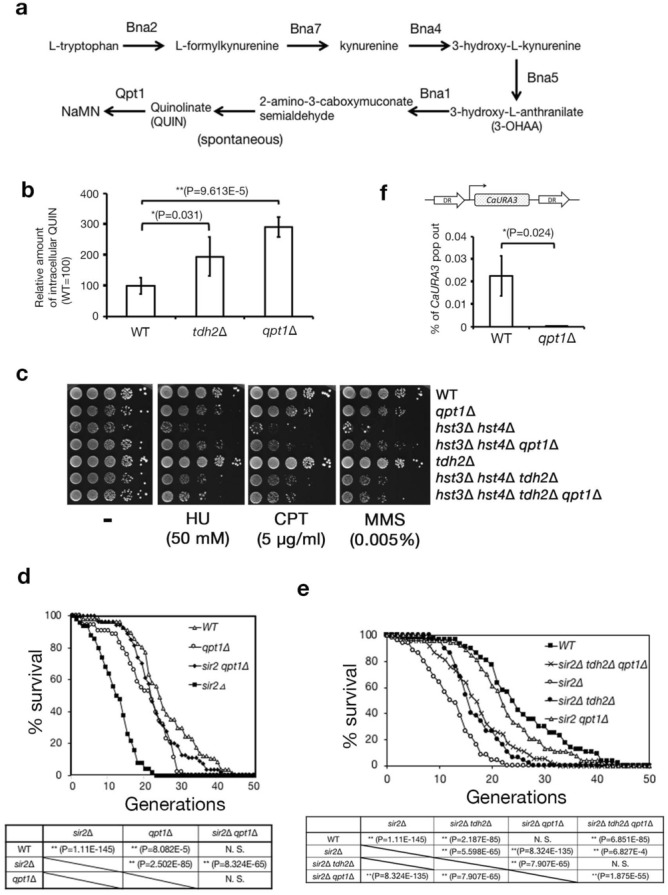
Quinolinic acid (QUIN) is a candidate metabolite that is increased in tdh2∆ cells to suppress replication fork slippage. (a) The metabolic pathway of the de novo NAD+ synthesis pathway (kynurenine pathway). NaMN: nicotinic acid mononucleotide. (b) Comparison of intracellular QUIN levels among strains. Relative amount of QUIN in each cell per wild-type cell (WT = 100). *P < 0.05. **P < 0.01. Unpaired t-test (two-tailed). Error bars represent the standard deviation of three biological replicates. (c) Plate assays monitoring the sensitivity of each DNA damaging agent. (d,e) Pedigree analysis to count replicative lifespans among cells. *P < 0.05. **P < 0.01. N.S: nonsignificant. Unpaired t-test (two-tails). Over 50 cells/strain were used for analysis. (f) The frequencies of the CaURA3 deletion among repeats. DRs: direct repeats. *P < 0.05. Unpaired t-test (two-tailed). Error bars represent the standard deviation of three biological replicates.
Intracellular NAD+, which is elevated in tdh2∆ cells, does not suppress the DNA damage sensitivity of hst3∆ hst4∆ cells
QUIN is utilized in the NAD+ salvage pathway to synthesize NAD+ (Fig. S5A). We hypothesized that QUIN accumulated to synthesize NAD+ de novo and that NAD+ functioned as an end product to suppress replication fork instability in tdh2∆ cells. To test this hypothesis, we examined whether tdh2 deletion elevated intracellular NAD+ levels. The cells were cultured, and the intracellular NAD+ concentration was measured by performing acid extractions of nucleotides over the course of cell growth in liquid cultures. The intracellular NAD+ concentration was almost the same between wild-type and tdh2∆ cells at first, although the concentration was severely decreased in cells lacking NPT1, the nicotinate phosphoribosyl transferase in the NAD+ salvage pathway (Fig. S5A and B)41. Intracellular NAD+ concentrations in both wild-type and npt1∆ cells steadily declined over the course of cell culture (Fig. S5B) because yeast cultures reached the end of the log phase and approached the diauxic shift, which was due to depletion of the limiting NAD+ precursor nicotinic acid from the growth medium41,42. In contrast, the NAD+ concentration remained constant in tdh2∆ cells over the course of cell culture (Fig. S5B), indicating that tdh2 deletion promotes NAD+ synthesis. In addition, tdh2 deletion slightly increased the intracellular NAD+ concentration in hst3∆ hst4∆ tdh2∆ cells, although the concentration declined in hst3∆ hst4∆ cells (Fig. S5C). Next, we examined whether de novo NAD+ synthesis induced by tdh2 deletion is necessary for the growth of hst3∆ hst4∆ cells. To monitor the contribution of tdh2 deletion to declining intracellular NAD+ levels in hst3∆ hst4∆ cells, we constructed npt1∆ hst3∆ hst4∆ and npt1∆ hst3∆ hst4∆ tdh2∆ strains containing the PHM286 plasmid harboring the HST3 and URA3 genes. Strains were streaked on SC solid medium supplemented with 5-FOA, a counterselecting agent, to induce loss of the URA3 plasmid. Although npt1∆ hst3∆ hst4∆ cells did not grow in SC medium with 5-FOA, npt1∆ hst3∆ hst4∆ tdh2∆ cells did grow (Fig. S5D). Thus, an increase in intracellular NAD+ levels by tdh2 deletion is necessary for the cell growth of hst3∆ hst4∆ cells. To reveal the mechanism that elevates intracellular NAD+ levels by tdh2 deletion, we compared the transcription levels of genes involved in NAD+ synthesis among strains. The BNA2 and BNA4 genes belong to the kynurenine pathway and provide QUIN for tryptophan (Fig. 3a), and TNA1 encodes the high affinity nicotinic acid plasma membrane permease responsible for the uptake of nicotinic acid (Fig. S5A). Although the expression levels of the BNA2 and BNA4 genes were almost the same between wild-type and tdh2∆ cells and the level of TNA1 was slightly increased in tdh2∆ cells compared with wild-type cells (Fig. S5E), the expression levels of these genes were significantly (P < 0.05) increased in hst3∆ hst4∆ tdh2∆ cells compared with hst3∆ hst4∆ cells (Fig. S5E and F). Thus, tdh2 deletion induces the gene expression involved in intracellular NAD+ synthesis in hst3∆ hst4∆ cells, which can improve the cell growth of hst3∆ hst4∆ cells.
Next, we examined whether elevated intracellular NAD+ levels suppressed the DNA damage sensitivity of hst3∆ hst4∆ cells. Isonicotinamide (INAM), an isostere of nicotinamide, raises intracellular NAD+ levels in budding yeast43. As shown in Fig. S6A, the intracellular NAD+ concentration was significantly (P < 0.05) elevated in the presence of INAM in both wild-type and hst3∆ hst4∆ cells. Treatment with INAM allowed both wild-type and hst3∆ hst4∆ cells to tolerate MMS (Fig. S6B). However, the addition of INAM did not suppress the sensitivities of hst3∆ hst4∆ cells to HU and CPT (Fig. S6B). These results indicate that elevated intracellular NAD+ levels do not contribute to the DNA damage sensitivity of hst3∆ hst4∆ cells. Altogether, these data suggest that NAD+ does not contribute to the suppression of replication fork instability in tdh2∆ cells.
Discussion
In this study, we elucidated that the deletion of TDH2 suppresses replication fork instability in the chromatin environment in which composed histones were acetylated. The acetylation of histone molecules loosens the chromatin structure by weakening the DNA-histone interaction. In particular, histone H3 on K56 is located within the histone core region and interacts with the DNA strand, and acetylation directly weakens the DNA-histone interaction16. The loosened chromatin structure becomes fragile, causing SSBs and DSBs during nuclear activities and exposes the naked DNA region to possible replication fork slippage at repeats. Both sir2∆ and hst3∆ hst4∆ cells exhibit high frequencies of loss of heterozygosity (LOH)20,44. LOH occurs in diploid cells that lose the chromosome arm or regenerate another chromosome arm instead of losing the arm by break-induced replication (BIR)4,45. In this study, tdh2 deletion suppresses recombination by replication fork slippage in both hst3∆ hst4∆ and sir2∆ cells. The frequency of recombination between DRs was reduced in tdh2∆ cells compared to wild-type cells (Fig. 2c), which contributes to the extension of the replicative lifespan of tdh2∆ cells (Fig. 2a)26.
ROS, hazardous byproducts of mitochondrial respiration, are well recognized as mediators of DNA damage46. DNA damage caused by ROS appears as an oxidized base, a sugar modification, a DNA or protein crosslink or a DNA strand break47–49. 8-Oxoguanine, a representative oxidized DNA adduct, works as a replication fork blocker50,51. The accumulation of DNA damage causes replication fork stalling, which provides an opportunity for HR between direct repeats on the DNA strand. DNA damage agents used in this study (MMS, HU and CPT) are involved in ROS production. MMS indirectly inhibits respiratory chain in mitochondria by caused mitochondrial DNA (mtDNA) damage, which induces ROS production in budding yeast52. CPT also inhibits DNA topoisomerase I in mitochondria to induce mtDNA damages, which inhibits the respiratory chain in mitochondrion to induce ROS production in mammals53. HU also induces ROS production in budding yeast54. ROS is usually scavenged by several cellular metabolites, NADPH, a reduced form of nicotinamide adenine dinucleotide phosphate (NADP), functions as a major scavenger of ROS and is provided from the pentose phosphate pathway (PPP) branched from glycolysis mainly. In addition of generating phosphopentoses and ribonucleotides, PPP plays a pivotal role to combat oxidative stress55. Our previous study showed that tdh2∆ and hst3∆ hst4∆ tdh2∆ cells accumulated NADP+, metabolic intermediates of PPP and various ribonucleotides26. The phenotype of tdh2∆ is similar to that of fbp1∆, essential gene in gluconeogenesis26. The combination with interruption of gluconeogenesis caused by fbp1∆ and triple sirtuin gene deletions (hst3∆ hst4∆ sir2∆) alters the metabolic flux from glycolysis to PPP and increase the ribonucleotide levels56. These suggests that tdh2∆ alters the metabolic flux from glycolysis to PPP, and increases NADPH level to scavenge ROS. Therefore, tdh2∆ brings the resistance for DNA damage agents (HU, MMS and CPT) in hst3∆ hst4∆ genetical background, and stabilizes replication fork to prevent homologous recombination between DRs.
In mammalian, QUIN has been shown to increase the production of free radicals, leading to oxidative stress, DNA damage, and increased poly(ADP-ribose) polymerase 1 (PARP-1) activity57,58 . However, moderate QUIN level can induce resistance to oxidative stress through increased NAD+ production59. NAD+ influences DNA repair and gene expression through its role as a substrate for PARP-1 in mammal cells. Although there is no PARP in budding yeast, another mechanism supports to allow QUIN to induce resistance for oxidative stress. It has been shown that QUIN chelates ferrous iron to generate ROS60. Iron–Sulfur (Fe–S) centers are metallic cofactors with electronic properties that are associated with proteins61. It is well known that numerous DNA-transacting proteins such as DNA replication machinery contains Fe–S centers54. Because Fe–S centers are sensitive to oxidative agents, a moderate QUIN level can activate the oxidative stress response, and leads to resistance for oxidative stress. In the future, approaches to repair or prevent DNA damage will focus more heavily on altering the metabolic state of cells.
Methods
Strains and media
The genotypes of the strains, plasmids and primers used in this study are listed in Table S1. The parental budding yeast strain used in the present study was BY4742 (MATα his3∆ leu2∆1 met15∆0 ura3∆0)62. Yeast cells were routinely grown at 30 °C in YPD (1% yeast extract, 2% peptone, 2% glucose) or appropriate synthetic complete (SC) medium63,64. If necessary, the media were solidified with 2% agar. A yeast strain harboring a single gene deletion was commercially available from the haploid yeast open reading frame deletion collection65 (GE Dharmacon, Lafayette, CO, USA). To construct a double or triple gene deletion strain, the different mating type single gene deletion haploid strains were crossed, and sporulation was subsequently induced. After dissection, the spores were germinated on YPD medium. The deletion of each gene was confirmed using either antibiotics or auxotrophic markers and checking for growth on agar plates containing antibiotics or SC agar plates without selective amino acids. We employed YPD media supplemented with the following antibiotics: G418 (Sigma-Aldrich, St. Louis, MO, USA) at a final concentration of 100 μg/ml for the kan gene, hygromycin B at a final concentration of 200 μg/ml for the hph gene and ClonNAT (Werner Bioagents, Germany) at a final concentration of 100 μg/ml for the nat gene. SC-histidine medium was employed to select the his5+ strain66,67. The strains with deletions in both HST3 and HST4 harbor the PHM286 URA3 plasmid, which contains the wild-type HST3 gene and prevents spontaneous DNA damage and genomic instability. These strains were counterselected for loss of the PHM286 plasmid by selecting colonies that grew in SC medium supplemented with 5-fluoroorotic acid (5-FOA) at a final concentration of 100 µg/ml prior to use in subsequent experiments. To construct the strains monitoring CaURA3 gene deletion frequencies, the plasmid PHM764 was digested with Bsu 36I and integrated into the TRP1 locus. The URA + strains were selected and confirmed for correct integration of the PHM764 plasmid by PCR.
A standard method was used for isolation of the yeast genomic DNA64. E. coli strain DH5α68 and standard media and methods were used for plasmid manipulations69. Plasmid DNA was isolated from E. coli using a QIAquick Spin Miniprep kit (Qiagen, Santa Clarita, CA, USA). DNA fragments from polymerase chain reaction (PCR) samples or agarose gels were isolated using the Wizard SV Gel and PCR Clean-up kit (Promega, Madison, WI, USA). Oligonucleotides were purchased from either Invitrogen (Invitrogen, Carlsbad, CA, USA) or FASMAC (FASMAC, Kanagawa, Japan).
Plasmid construction
DNA for plasmid construction was generated by PCR using the iProof High-Fidelity DNA polymerase (Bio-Rad, Hercules, CA, USA). The mix contained 10 µl of 5 × iProof buffer, 0.25 µl each of 100 µM PCR primer, 1 μl of 10 mM dNTP mix, 0.1 µg of template DNA and 0.5 µl of iProof Taq polymerase (final volume 50 μl). Reactions were run for 1 cycle of 10 s at 98 °C, 25 cycles of 10 s at 98 °C, 10 s at 55 °C, and 1 min/kb of desired product at 72 °C. These 25 cycles were followed by a 5-min extension at 72 °C. To construct plasmid PHM764, an ~ 2400 bp DNA fragment (Candida albicans URA3 gene flanking region with each 500 bp direct repeat) in pAG6170 was digested with Bam HI and EcoR V and then ligated into Bam HI/Hinc II-digested YIplac204 plasmid71.
DNA damage sensitivity test
Yeast strains were cultured in 5 ml of YPD liquid medium at 25 °C overnight and then adjusted to 5 × 106 cells/ml in 5 ml of YPD medium. Next, cell culture was continued for 3 h at 25 °C, and then cells were harvested and suspended in DIW at 5 × 107 cells/ml. Two hundred microliters of cell suspension (1 × 107 cells) was transferred to a 96-well plate and sequentially diluted tenfold to a concentration of 5 × 103 cells/ml. A small portion of the diluted cell suspension in each well was set on YPD medium containing each concentration of DNA damaging agent using a replica plater (Sigma-Aldrich, St. Louis, MO, USA). The plates were incubated at 25 °C for 3 to 5 days. YPD media was supplemented with the following DNA damaging agents: methyl methanesulfonate (MMS), hydroxyurea (HU) and camptothecin (CPT). The concentration of each agent is listed in figures.
Western blotting
Proteins were separated by 15% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose membranes (Amersham PROTORAN) (GE healthcare, Little Chalfont, Buckinghamshire, England). The protein level in each lane on a nitrocellulose membrane was adjusted equally and confirmed by staining with 0.1% Ponceau S solution (Sigma-Aldrich, St. Louis, MO, USA). Rabbit polyclonal anti-acetylated lysine 56 on the yeast histone H3 antibody (1:1000 dilution) was used16. For detection of phosphorylated species of the Rad53 protein, anti-Rad53 rabbit polyclonal antibody (the equivalent antibody is commercially available (Santa Cruz Biotechnology, Dallas, TX, USA)) was used.
Detection of Rad53 phosphorylation in response to DNA damage
Yeast strains were cultured in 5 ml of YPD liquid medium at 25 °C overnight and then adjusted to a concentration of 5 × 106 cells/ml in 5 ml of YPD medium containing MMS at a final concentration of 0.03%. Afterwards, cell culture was continued for 3 h at 25 °C, and then cells were harvested. The whole cell extraction method was described previously72. Multiple species of phosphorylated Rad53 and unphosphorylated Rad53 were separated by 7.5% SDS-PAGE and then detected by western blotting using an anti-Rad53 antibody.
Cell cycle arrest during G2/M phase
Yeast strains were cultured in 5 ml of YPD liquid medium at 25 °C overnight and then adjusted to a concentration of 5 × 106 cells/ml in 5 ml of YPD medium containing nocodazole at a final concentration of 10 µg/ml. Afterwards, cell culture was continued for 3 h at 25 °C, and then cells were harvested. The whole cell extraction method was previously described72.
Replicative lifespan assay (pedigree analysis)
The pedigree analysis procedure was described previously26. Typically, a minimum of 50 mother cells was counted for each strain tested. To compare the difference in replicative lifespans among strains statistically, we performed the unpaired t-test (two-tailed).
CaURA3 deletion assay
The yeast strains were streaked on SC-Ura plates to select URA3-positive strains prior to the assay. The URA3-positive colonies were inoculated in 5 ml of YPD liquid medium and cultured at 25 °C overnight. A small aliquot of culture (5 × 104 or 5 × 105 cells/strain) was plated in a YPD solid plate supplemented with 5-FOA at a final concentration of 100 µg/ml to select strains containing the ura3 gene deletion. The number of colonies was counted, and the frequencies of the CaURA3 gene deletion were calculated for plated cells. Three replicates were analyzed for each strain.
Synthetic lethality test
Yeast strains with or without the PHM286 plasmid (URA3) were grown on YPD medium at 25 °C overnight; then, the strains were streaked on SC solid medium supplemented with 5-FOA at a final concentration of 100 µg/ml at 25 °C for 3 days to counterselect for loss of the PHM286 plasmid.
RNA isolation and real time (RT)–PCR
Total RNA was isolated from budding yeast using the RNeasy Mini Kit (Qiagen, Santa Clarita, CA, USA). A relative comparison of the mRNA amount was performed using a One Step SYBR PrimeScript PLUS RT-PCR Kit (Takara-Bio, Kusatsu, Shiga, Japan). The mix contained 10 µl of 2 × One Step SYBR RT-PCR buffer 4, 1.2 µl of Takara Ex Taq HS Mix, 0.4 µl of PrimeScript PLUS RTase Mix, 0.8 µl of 10 µM PCR forward primer, 0.8 µl of 10 μM PCR reverse primer and 100 ng of total RNA (final volume 20 µl). Reactions were run for 1 cycle of 5 min at 42 °C, 1 cycle of 10 s at 95 °C, 40 cycles of 5 s at 95 °C, 1 cycle of 20 s at 55 °C, 1 cycle of 1 s at 95 °C, and 1 cycle of 15 s at 65 °C, followed by 1 s at 95 °C using either a Light Cycler 480 System II or Light Cycler Nano (Roche Life Science, Penzberg, Germany). The level of each mRNA was compared with the amount of ACT1 mRNA. PCR primers are listed in Table S1.
The measurement of NAD+ concentration and INAM treatment
The yeast strains were cultured in 250 ml of YPD liquid medium at 25 °C. During cell culture, the OD600 nm of a 1:10 cell dilution and the cell number were recorded, and 20 ml of culture was pelleted and washed with water. After harvesting, the cell pellet was stocked at − 80 °C until use. The preparation and quantification of intracellular NAD+ was described previously41. Over 10 independent cultures were routinely used to determine the NAD+ concentrations in duplicate.
For INAM treatment, the yeast strains (5 × 106 cells/ml at start) were cultured in 20 ml of YPD liquid medium supplemented with or without INAM at a final concentration of 25 mM at 25 °C until an OD600 of ~ 1.5 was reached and then harvested as described previously43. Cell pellets were employed to measure the intracellular NAD+ concentration.
Measurement of intracellular quinolinic acid (QUIN) concentration
For metabolite extraction, yeast cells (1 × 108 cells) were suspended in 50% methanol and immediately frozen in liquid nitrogen. Then, frozen samples were ground by a Multi Beads Shocker (Yasui Kikai, Osaka, Japan) then centrifuged at 13,000 × g for 10 min at 4 °C. The supernatant was mixed with an equal volume of chloroform, and the mixture was centrifuged again. The upper aqueous phase was transferred to a tube and evaporated using SpeedVac SPD 1010 (Thermo Fischer Scientific, Waltham, MA, USA). Levels of quinolinic acid (QUIN) in yeast cells were determined using an Agilent 6460 Triple Quad mass spectrometer coupled to an Agilent 1290 HPLC system with multiple reaction monitoring (MRM) mode. The MRM transition for QA was optimized as m/z 166 to 78. MS settings and chromatographic conditions were described previously73. The amount of QA was calculated by integrating the sum of the area using Mass Hunter Quantitative software (Agilent Technologies, Santa Clara, CA, USA).
Supplementary information
Acknowledgements
The authors would like to thank Rika Miyazaki, Mayumi Hachinohe, Midori Yamane, Daiki Akazawa, Kazuhiro Ohsawa, Mayumi Ohno, and Yuzu Terashita for supporting experiments.
Author contributions
H.M. designed experiments. M.H. and M.Y. carried out experiments. K.Y. and T.N. prepared Fig. 3b. H.M. prepared the manuscript. All authors checked the draft.
Funding
This work was supported by the Japan Society for the Promotion of Science ‘[15K14461 to H.M.]’. This work was also supported by NIG-JOINT (37A2020), Tohoku University-IDAC (2020, 2), Kobe University-BRC (2020, 201006) to H.M.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41598-020-78302-5.
References
- 1.Aguilera A, Garcia-Muse T. Causes of genome instability. Annu. Rev. Genet. 2013;47:1–32. doi: 10.1146/annurev-genet-111212-133232. [DOI] [PubMed] [Google Scholar]
- 2.Cha HJ, Yim H. The accumulation of DNA repair defects is the molecular origin of carcinogenesis. Tumour Biol. 2013;34:3293–3302. doi: 10.1007/s13277-013-1038-y. [DOI] [PubMed] [Google Scholar]
- 3.Basu AK. DNA damage, mutagenesis and cancer. Int. J. Mol. Sci. 2018 doi: 10.3390/ijms19040970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kramara J, Osia B, Malkova A. Break-induced replication: The where, the why, and the how. Trends Genet. 2018;34:518–531. doi: 10.1016/j.tig.2018.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA Repai and Mutagenesis. Washington, DC: ASM Press; 2005. [Google Scholar]
- 6.Cleary JD, Pearson CE. The contribution of cis-elements to disease-associated repeat instability: Clinical and experimental evidence. Cytogenet. Genome Res. 2003;100:25–55. doi: 10.1159/000072837. [DOI] [PubMed] [Google Scholar]
- 7.Pearson CE, Nichol Edamura K, Cleary JD. Repeat instability: Mechanisms of dynamic mutations. Nat. Rev. Genet. 2005;6:729–742. doi: 10.1038/nrg1689. [DOI] [PubMed] [Google Scholar]
- 8.Marians KJ. Lesion bypass and the reactivation of stalled replication forks. Annu. Rev. Biochem. 2018;87:217–238. doi: 10.1146/annurev-biochem-062917-011921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cotterill S. Diseases associated with mutation of replication and repair proteins. Adv. Exp. Med. Biol. 2018;1076:215–234. doi: 10.1007/978-981-13-0529-0_12. [DOI] [PubMed] [Google Scholar]
- 10.Margueron R, Reinberg D. Chromatin structure and the inheritance of epigenetic information. Nat. Rev. Genet. 2010;11:285–296. doi: 10.1038/nrg2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alabert C, Groth A. Chromatin replication and epigenome maintenance. Nat. Rev. Mol. Cell Biol. 2012;13:153–167. doi: 10.1038/nrm3288. [DOI] [PubMed] [Google Scholar]
- 12.Struhl K. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 1998;12:599–606. doi: 10.1101/gad.12.5.599. [DOI] [PubMed] [Google Scholar]
- 13.Kornberg RD, Lorch Y. Chromatin-modifying and -remodeling complexes. Curr. Opin. Genet Dev. 1999;9:148–151. doi: 10.1016/S0959-437X(99)80022-7. [DOI] [PubMed] [Google Scholar]
- 14.Zhu Q, Wani AA. Histone modifications: Crucial elements for damage response and chromatin restoration. J. Cell. Physiol. 2010;223:283–288. doi: 10.1002/jcp.22060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duan MR, Smerdon MJ. Histone H3 lysine 14 (H3K14) acetylation facilitates DNA repair in a positioned nucleosome by stabilizing the binding of the chromatin Remodeler RSC (remodels structure of chromatin) J. Biol. Chem. 2014;289:8353–8363. doi: 10.1074/jbc.M113.540732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Masumoto H, Hawke D, Kobayashi R, Verreault A. A role for cell-cycle-regulated histone H3 lysine 56 acetylation in the DNA damage response. Nature. 2005;436:294–298. doi: 10.1038/nature03714. [DOI] [PubMed] [Google Scholar]
- 17.Brachmann CB, et al. The SIR2 gene family, conserved from bacteria to humans, functions in silencing, cell cycle progression, and chromosome stability. Genes Dev. 1995;9:2888–2902. doi: 10.1101/gad.9.23.2888. [DOI] [PubMed] [Google Scholar]
- 18.Maas NL, Miller KM, DeFazio LG, Toczyski DP. Cell cycle and checkpoint regulation of histone H3 K56 acetylation by Hst3 and Hst4. Mol. Cell. 2006;23:109–119. doi: 10.1016/j.molcel.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 19.Celic I, et al. The sirtuins hst3 and Hst4p preserve genome integrity by controlling histone h3 lysine 56 deacetylation. Curr. Biol. 2006;16:1280–1289. doi: 10.1016/j.cub.2006.06.023. [DOI] [PubMed] [Google Scholar]
- 20.Hachinohe M, Hanaoka F, Masumoto H. Hst3 and Hst4 histone deacetylases regulate replicative lifespan by preventing genome instability in Saccharomyces cerevisiae. Genes Cells. 2011;16:467–477. doi: 10.1111/j.1365-2443.2011.01493.x. [DOI] [PubMed] [Google Scholar]
- 21.Han J, et al. Rtt109 acetylates histone H3 lysine 56 and functions in DNA replication. Science. 2007;315:653–655. doi: 10.1126/science.1133234. [DOI] [PubMed] [Google Scholar]
- 22.Sauve AA, Wolberger C, Schramm VL, Boeke JD. The biochemistry of sirtuins. Annu. Rev. Biochem. 2006;75:435–465. doi: 10.1146/annurev.biochem.74.082803.133500. [DOI] [PubMed] [Google Scholar]
- 23.Rusche LN, Kirchmaier AL, Rine J. The establishment, inheritance, and function of silenced chromatin in Saccharomyces cerevisiae. Annu. Rev. Biochem. 2003;72:481–516. doi: 10.1146/annurev.biochem.72.121801.161547. [DOI] [PubMed] [Google Scholar]
- 24.Gottlieb S, Esposito RE. A new role for a yeast transcriptional silencer gene, SIR2, in regulation of recombination in ribosomal DNA. Cell. 1989;56:771–776. doi: 10.1016/0092-8674(89)90681-8. [DOI] [PubMed] [Google Scholar]
- 25.Cesarini E, D'Alfonso A, Camilloni G. H4K16 acetylation affects recombination and ncRNA transcription at rDNA in Saccharomyces cerevisiae. Mol. Biol. Cell. 2012;23:2770–2781. doi: 10.1091/mbc.E12-02-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hachinohe M, et al. A reduction in age-enhanced gluconeogenesis extends lifespan. PLoS ONE. 2013;8:e54011. doi: 10.1371/journal.pone.0054011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Travesa A, et al. DNA replication stress differentially regulates G1/S genes via Rad53-dependent inactivation of Nrm1. EMBO J. 2012;31:1811–1822. doi: 10.1038/emboj.2012.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cimprich KA, Cortez D. ATR: An essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 2008;9:616–627. doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ciccia A, Elledge SJ. The DNA damage response: Making it safe to play with knives. Mol. Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marechal A, Zou L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 2013 doi: 10.1101/cshperspect.a012716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yeeles JT, Poli J, Marians KJ, Pasero P. Rescuing stalled or damaged replication forks. Cold Spring Harb. Perspect. Biol. 2013;5:a012815. doi: 10.1101/cshperspect.a012815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gan H, et al. Checkpoint kinase Rad53 couples leading- and lagging-strand DNA synthesis under replication stress. Mol. Cell. 2017;68:446–455. doi: 10.1016/j.molcel.2017.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ogi H, Wang CZ, Nakai W, Kawasaki Y, Masumoto H. The role of the Saccharomyces cerevisiae Cdc7-Dbf4 complex in the replication checkpoint. Gene. 2008;414:32–40. doi: 10.1016/j.gene.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 34.Chen ES, et al. Phospho-priming confers functionally relevant specificities for Rad53 kinase autophosphorylation. Biochemistry. 2017;56:5112–5124. doi: 10.1021/acs.biochem.7b00689. [DOI] [PubMed] [Google Scholar]
- 35.Xu F, Zhang K, Grunstein M. Acetylation in histone H3 globular domain regulates gene expression in yeast. Cell. 2005;121:375–385. doi: 10.1016/j.cell.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 36.Sinclair DA, Guarente L. Extrachromosomal rDNA circles—A cause of aging in yeast. Cell. 1997;91:1033–1042. doi: 10.1016/S0092-8674(00)80493-6. [DOI] [PubMed] [Google Scholar]
- 37.Kobayashi T, Horiuchi T. A yeast gene product, Fob1 protein, required for both replication fork blocking and recombinational hotspot activities. Genes Cells. 1996;1:465–474. doi: 10.1046/j.1365-2443.1996.d01-256.x. [DOI] [PubMed] [Google Scholar]
- 38.Lambert S, Carr AM. Impediments to replication fork movement: Stabilisation, reactivation and genome instability. Chromosoma. 2013;122:33–45. doi: 10.1007/s00412-013-0398-9. [DOI] [PubMed] [Google Scholar]
- 39.Larsen NB, et al. Stalled replication forks generate a distinct mutational signature in yeast. Proc Natl Acad Sci USA. 2017;114:9665–9670. doi: 10.1073/pnas.1706640114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chang EY, et al. RECQ-like helicases Sgs1 and BLM regulate R-loop-associated genome instability. J. Cell Biol. 2017;216:3991–4005. doi: 10.1083/jcb.201703168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Belenky P, et al. Nicotinamide riboside promotes Sir2 silencing and extends lifespan via Nrk and Urh1/Pnp1/Meu1 pathways to NAD+ Cell. 2007;129:473–484. doi: 10.1016/j.cell.2007.03.024. [DOI] [PubMed] [Google Scholar]
- 42.Sandmeier JJ, Celic I, Boeke JD, Smith JS. Telomeric and rDNA silencing in Saccharomyces cerevisiae are dependent on a nuclear NAD(+) salvage pathway. Genetics. 2002;160:877–889. doi: 10.1093/genetics/160.3.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McClure JM, Wierman MB, Maqani N, Smith JS. Isonicotinamide enhances Sir2 protein-mediated silencing and longevity in yeast by raising intracellular NAD+ concentration. J. Biol. Chem. 2012;287:20957–20966. doi: 10.1074/jbc.M112.367524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McMurray MA, Gottschling DE. An age-induced switch to a hyper-recombinational state. Science. 2003;301:1908–1911. doi: 10.1126/science.1087706. [DOI] [PubMed] [Google Scholar]
- 45.Sakofsky CJ, Malkova A. Break induced replication in eukaryotes: Mechanisms, functions, and consequences. Crit. Rev. Biochem. Mol. Biol. 2017;52:395–413. doi: 10.1080/10409238.2017.1314444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Srinivas US, Tan BWQ, Vellayappan BA, Jeyasekharan AD. ROS and the DNA damage response in cancer. Redox Biol. 2019;25:101084. doi: 10.1016/j.redox.2018.101084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grollman AP, Moriya M. Mutagenesis by 8-oxoguanine: An enemy within. Trends Genet. 1993;9:246–249. doi: 10.1016/0168-9525(93)90089-z. [DOI] [PubMed] [Google Scholar]
- 48.Marnett LJ. Oxyradicals and DNA damage. Carcinogenesis. 2000;21:361–370. doi: 10.1093/carcin/21.3.361. [DOI] [PubMed] [Google Scholar]
- 49.Wallace SS. Biological consequences of free radical-damaged DNA bases. Free Radic. Biol. Med. 2002;33:1–14. doi: 10.1016/s0891-5849(02)00827-4. [DOI] [PubMed] [Google Scholar]
- 50.Tolentino JH, Burke TJ, Mukhopadhyay S, McGregor WG, Basu AK. Inhibition of DNA replication fork progression and mutagenic potential of 1, N6-ethenoadenine and 8-oxoguanine in human cell extracts. Nucleic Acids Res. 2008;36:1300–1308. doi: 10.1093/nar/gkm1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Herman KN, Toffton S, McCulloch SD. Minimal detection of nuclear mutations in XP-V and normal cells treated with oxidative stress inducing agents. J. Biochem. Mol. Toxicol. 2014;28:568–577. doi: 10.1002/jbt.21599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kitanovic A, et al. Metabolic response to MMS-mediated DNA damage in Saccharomyces cerevisiae is dependent on the glucose concentration in the medium. FEMS Yeast Res. 2009;9:535–551. doi: 10.1111/j.1567-1364.2009.00505.x. [DOI] [PubMed] [Google Scholar]
- 53.Douarre C, et al. Mitochondrial topoisomerase I is critical for mitochondrial integrity and cellular energy metabolism. PLoS ONE. 2012;7:e41094. doi: 10.1371/journal.pone.0041094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang ME, et al. DNA replication inhibitor hydroxyurea alters Fe–S centers by producing reactive oxygen species in vivo. Sci. Rep. 2016;6:29361. doi: 10.1038/srep29361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Patra KC, Hay N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014;39:347–354. doi: 10.1016/j.tibs.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Masumoto H, Matsuyama S. The combination of NAD+-dependent deacetylase gene deletion and the interruption of gluconeogenesis causes increased glucose metabolism in budding yeast. PLoS ONE. 2018;13:e0194942. doi: 10.1371/journal.pone.0194942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Goda K, Kishimoto R, Shimizu S, Hamane Y, Ueda M. Quinolinic acid and active oxygens. Possible contribution of active Oxygens during cell death in the brain. Adv. Exp. Med. Biol. 1996;398:247–254. doi: 10.1007/978-1-4613-0381-7_38. [DOI] [PubMed] [Google Scholar]
- 58.Pérez-De La Cruz V, Carrillo-Mora P, Santamaría A. Quinolinic Acid, an endogenous molecule combining excitotoxicity, oxidative stress and other toxic mechanisms. Int. J. Tryptophan Res. 2012;5:1–8. doi: 10.4137/ijtr.s8158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bostian AC, Eoff RL. Aberrant kynurenine signaling modulates DNA replication stress factors and promotes genomic instability in gliomas. Chem. Res. Toxicol. 2016;29:1369–1380. doi: 10.1021/acs.chemrestox.6b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pláteník J, Stopka P, Vejrazka M, Stípek S. Quinolinic acid-iron(ii) complexes: Slow autoxidation, but enhanced hydroxyl radical production in the Fenton reaction. Free Radic. Res. 2001;34:445–459. doi: 10.1080/10715760100300391. [DOI] [PubMed] [Google Scholar]
- 61.Beinert H, Holm RH, Münck E. Iron–sulfur clusters: Nature's modular, multipurpose structures. Science. 1997;277:653–659. doi: 10.1126/science.277.5326.653. [DOI] [PubMed] [Google Scholar]
- 62.Brachmann CB, et al. Designer deletion strains derived from Saccharomyces cerevisiae S288C: A useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 1998;14:115–132. doi: 10.1002/(SICI)1097-0061(19980130)14:2<115::AID-YEA204>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 63.Sherman F. Getting started with yeast. Methods Enzymol. 2002;350:3–41. doi: 10.1016/S0076-6879(02)50954-X. [DOI] [PubMed] [Google Scholar]
- 64.Amberg DC, Burke FJ, Strathern JN. Methods in Yeast Genetics. 2005. New York: Cold Spring Harbor Laboratory Press; 2005. [Google Scholar]
- 65.Winzeler EA, et al. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science. 1999;285:901–906. doi: 10.1126/science.285.5429.901. [DOI] [PubMed] [Google Scholar]
- 66.Longtine MS, et al. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998;14:953–961. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 67.Goldstein AL, McCusker JH. Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast. 1999;15:1541–1553. doi: 10.1002/(SICI)1097-0061(199910)15:14<1541::AID-YEA476>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 68.Taylor RG, Walker DC, McInnes RR. E. coli host strains significantly affect the quality of small scale plasmid DNA preparations used for sequencing. Nucleic Acids Res. 1993;21:1677–1678. doi: 10.1093/nar/21.7.1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Struhl K. Current Protocols in Molecular Biology. New York: Wiley; 1995. [Google Scholar]
- 70.Goldstein AL, Pan X, McCusker JH. Heterologous URA3MX cassettes for gene replacement in Saccharomyces cerevisiae. Yeast. 1999;15:507–511. doi: 10.1002/(SICI)1097-0061(199904)15:6<507::AID-YEA369>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 71.Gietz RD, Sugino A. New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene. 1988;74:527–534. doi: 10.1016/0378-1119(88)90185-0. [DOI] [PubMed] [Google Scholar]
- 72.Kushnirov VV. Rapid and reliable protein extraction from yeast. Yeast. 2000;16:857–860. doi: 10.1002/1097-0061(20000630)16:9<857::AID-YEA561>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 73.Yaku K, Okabe K, Nakagawa T. Simultaneous measurement of NAD metabolome in aged mice tissue using liquid chromatography tandem-mass spectrometry. Biomed. Chromatogr. 2018;32:e4205. doi: 10.1002/bmc.4205. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.