

Abstract
Necroptosis is programmed necrosis triggered by death receptor signaling. We investigated whether necroptosis contributes to neuronal damage and functional impairment in a model of retinal ischemia. Methods: Sprague-Dawley rats were subjected to raised intraocular pressure for 45 min and received intravitreal injections of the specific necroptosis inhibitor, Nec-1, its inactive analogue (Nec-1i) or vehicle. Seven days after ischemia, ERGs were performed and then the eyes were enucleated for histological analysis. In other animals, retinas were subjected to propodium iodide, TUNEL staining or Western Blotting and probed with anti-LC-3 antibody. Results: Retinal ischemia resulted in selective neuronal degeneration of the inner layers. Pretreatment with Nec-1 led to significant preservation in thickness and histoarchitecture of the inner retina and functional improvement compared with vehicle-treated controls. Pretreatment with Nec-1i did not provide histological or functional protection. Post-treatment with Nec-1 also significantly attenuated the ERG b-wave reduction compared with ischemic vehicle controls. Nec-1 had no effect on the number of caspase or TUNEL-labelled cells in the ischemic retina but did inhibit the induction of LC-3 II and reduced the number of PI-labelled cells after ischemia. Conclusion: Necroptosis is an important mode of neuronal cell death and involves autophagy in a model of retinal ischemia.
Keywords: necrosis, neuroprotection, ischemia, retina
Necroptosis is a recently discovered, caspase independent, regulated cell death that has the morphological features of necrosis (early membrane and organelle swelling followed by cell lysis) and is activated by death receptor signaling. A network of molecular signaling has recently been discovered in this type of cell death and is separate from signaling pathways mediating apoptosis (Hitomi et al., 2008). In some cell types, for example, TNFα induces apoptosis but, in the presence of caspase inhibitors, activates necroptosis instead (Degterev et al., 2005). Using an in vitro cell death assay involving death receptor activation, a chemical library screen has yielded a novel class of compounds called necrostatins that specifically inhibit necroptotic cell death but do not protect against caspase-mediated apoptosis or oxidative stress induced necrosis. Medicinal chemistry optimization yielded the potent and metabolically more stable necroptosis inhibitor, Necrostatin-1 (Nec-1) (Teng et al., 2005). This compound has been shown to inhibit RIP1 kinase phosphorylation that may be a key early signaling event in necroptosis (Degterev et al., 2008). The potential biological relevance of necroptosis has recently been shown in several disease models. For example, Nec-1 dose dependently reduces infarct volume in two mouse models of middle cerebral artery occlusion (MCAo) (Degterev et al., 2005; Savitz, 2007), suggesting that necroptosis may be an important mode of cell death in ischemic stroke.
It remains unknown, however, if necroptosis contributes to other types of neurological injury and if necrostatins inhibit necroptosis in vivo, directly protects neurons, and improves functional outcome after ischemia. We addressed these questions by examining the effects of Nec-1 in a model of global retinal-ischemia reperfusion injury. TNFα is a death inducing ligand that mediates ischemic neuronal injury and emerging evidence suggests that it plays a pivotal role in other models of neuronal death such as retinal ischemia (Fontaine et al., 2002; Nakazawa et al., 2006; Berger et al., 2008), a condition which shares many of the same pathophysiological mechanisms that occur in global cerebral ischemia and is a model of acute glaucoma, hypertensive retinopathy, and diabetic retinopathy. Given the central role of death-receptor signaling in cerebral and retinal ischemia, we tested the hypothesis that necroptosis contributes to cell death after retinal ischemia-reperfusion injury. We first determined whether Nec-1 attenuates retinal ischemic damage and then addressed whether Nec-1 reduces in vivo early plasmamembrane breakdown, a morphological hallmark of necroptosis, and autophagy identified through LC-II induction, which is a secondary marker of necroptosis (Degterev et al., 2005). We then investigated whether Nec-1 directly protects neurons in the inner retinal layers, which are selectively susceptible to global ischemia in this model. Finally, we studied the electrophysiological effects of Nec-1 on the ischemic retina to determine if inhibition of necroptosis leads to objective functional improvement after neuronal ischemic injury.
METHODS
For all studies, investigators were blinded to treatment during surgery, data acquisition, and analysis.
Administration of Necrostatins, Related Compounds, and Caspase Inhibitors
Necrostatin-1 (Nec-1; 5-(7-chloro-1H-indol-3-ylmethyl)-3-methylimidazolidine-2,4-dione), vehicle (DMSO), or an inactive analogue of necrostatin-1 analog (Nec-1i; 5-(7-chloro-1H-indol-3-ylmethyl)imidazolidine-2,4-dione) (2 μl each) was administered into the vitreous of the injured eye before or at various times after ischemia (Nec-1 and Nec-1i available from Sigma). For all experiments, a 4 mM solution of necrostatin-1 or its inactive analogue was used. In separate experiments, the pan-caspase, inhibitor, Z-VAD-FMK was administered right before ischemia.
Retinal Ischemia
All animals were treated in accordance with the Association for Research in Vision and Ophthalmology (ARVO) Statement for the Use of Animals in Ophthalmic and Vision Research. Male Sprague-Dawley rats weighing 150–175 g (Charles River) were subjected to high intraocular pressure (HIOP). Briefly, after deep anesthesia with an intramuscular injection of ketamine (30–40 mg/kg) and xylazine (2.5 mg/kg) in rats, the anterior chamber of the right eye was cannulated using a 27-gauge needle and attached via silastic tubing to an infusion of sterile 0.9% saline. Under direct vision, the needle tip was placed within the anterior chamber and the corneal puncture site sealed with cyanoacrylate cement. Intraocular pressure (IOP) was raised to 120 mmHg by elevating the saline container, in order to exceed systemic arterial blood pressure, for a duration of 45 min. Animals were kept normothermic at 36.7 ± 0.5°C with a rectal probe and heating pad during the procedure. Whitening of the iris and loss of the red reflex of the retina confirmed retinal ischemia. Following completion of the target period of ischemia, the needle was withdrawn and the intraocular pressure normalized. The contralateral eye of each animal served as a non-ischemic control. One drop of gentamicin ophthalmic solution was applied topically to the right eye before and after cannulating the anterior chamber. The animals were then euthanized at various times and their eyes were enucleated for morphologic and immunohistochemical studies.
Assessment of Histopathological Damage
The right (experimental) and left (untouched control) globes were enucleated 1 week after ischemia and were fixed in paraformaldehyde and embedded in paraffin. Enucleated globes were then sectioned in the vertical meridian and the inferior portion of the eye wall (retina, choroid and sclera). 5 μm-thick sections were stained with hematoxylin and eosin and the retinal histoarchitecture was evaluated by light microscopy. The thickness of the retinal layers was determined as follows: (1) outer limiting membrane (OLM) to inner limiting membrane (ILM); (2) outer nuclear layer (ONL); (3) outer plexiform layer (OPL); (4) inner nuclear layer (INL); (5) inner plexiform layer (IPL) to ILM. Averages for these measurements taken in four adjacent areas within 1 mm of the optic nerve were calculated. Selection of the same topographic area of retina for all of these measurements in the same eyes is implemented in order to reduce regional anatomic variations. Additionally, manual cell counts were performed over a length of 200 μm in the inferior peripapillary region (Singh et al., 2001; Junk et al., 2002).
Assessment of Neuronal Function with Electroretinograms
Procedures used in our laboratories have been described in detail previously (Roth et al., 1998; Li and Roth 1999; Lin and Roth 1999). Rats were dark adapted overnight, and briefly anesthetized with an intramuscular injection of ketamine (30 mg/kg) and an intramuscular injection of xylazine (2.5 mg/kg). Pupils were dilated with 1% tropicamide (Alcon, Humacao, Puerto Rico) and cyclomydril (0.2% cyclopentolate HCI and 0.1% phenylephrine HCl, Alcon, Fort Worth, TX). Animals were kept at 37 ± 0.5°C with a rectal probe and heating pad during the procedure until completely recovered from anesthesia. A platinum electrode was placed on the topically anesthetized cornea. Teca electrolyte electrode gel (Teca Corporation, Pleasantville, NY) was used as a conducting medium for the corneal electrode. A reference electrode was placed by the ipsilateral mastoid and a ground electrode was placed close to the midline of the cephalad dorsum. Light stimuli were generated by a Ganzfield xenon flash tube light source (ERG-jet, Nicolet Biomedical Inc., Madison, WI) with a 0.75 Log-flash intensity (cd-s/m2). Full field white light stroboscopic flashes lasting 10 msec were presented at a distance of 15 cm and a rate of 1.0/sec. Neuroelectric signals were impedance matched through a unity gain preamplifier and further differentially amplified with appropriate band-pass settings. To improve the signal-to-noise ratio, ERG responses elicited by identical stimuli were averaged on-line by a computer. Amplified signals (200 msec analysis time, 1–1500 Hz bandwidth) were stored and analyzed. ERG studies were performed in both the injured and contralateral control eyes before ischemia (baseline), immediately following reperfusion, and at 7 days after ischemia, which is immediately prior to animal sacrifice. ERG analysis consisted of determining both the latency to the b-wave peak from the stimulus onset and amplitude from the trough of the a-wave to the peak of the b-wave. The effects of treatment upon the a-wave and b-wave were assessed by dividing the amplitude measured in the experimental eye by the corresponding value of the control eye. All amplitudes were normalized to baseline values and expressed as a percent of baseline.
Administration of Propidium Iodide and Detection of PI-Positive Cells
Propidium iodide (PI; 1.0 mg/ml, Sigma, St. Louis, MO) was diluted in ddH2O 2:1. Two microliters were administered 20 min prior to sacrifice by intravitral injection. Rats were euthanized at 6 hr after retinal ischemia. Eyes were frozen in nitrogen vapor, and cryostat sections (12 μm) were placed on poly-L-lysine slides and then stored at −80°C. For detection of PI-labeled cells, retinal sections were fixed in 100% ethanol for 10 min at room temperature and photographed using a Nikon Eclipse T300 fluorescence microscope (Tokyo, Japan) fitted with excitation/emission filters 568/585 for PI.
Caspase-3 Immunohistochemistry
Eyes were enucleated 24 hr after ischemia and were fixed in 4% paraformaldehyde for 2 hr. After removing the anterior segment of the globe, the eye cups were further fixed in paraformaldehyde overnight then transferred into a 25% sucrose solution for an additional 24 hr. The eyes were then embedded in OCT compound with 2-methylbutane and dry ice. Twelvemicron-thick cryosections were prepared, fixed in cold methanol for 15 min, rinsed in 1 × PBS for 5 min, and incubated with 10% goat serum for 45 min at room temperature. Incubation with the primary antibody (BD Biosciences Pharmingen rabbit anti active caspase-3 polyclonal Ab, 1:1000) was performed overnight at 4°C. Sections were then washed with 1×PBS three times and incubated with either fluorescein- or rhodamine-conjugated secondary antibody (anti rabbit IgG; 1:100) at room temperature for 45 min. Sections were mounted with Antifade and were analyzed by fluorescent microscopy. Corresponding negative controls were prepared by substitution of the primary antibody with 10% normal goat serum in PBS.
TUNEL
This technique was performed using the In Situ Cell Death Detection Kit (Roche Applied Science) according to the specifications and directions of the manufacturer. Briefly, 12 μm cryosections were incubated in methanol at −20°C for 10 min, washed in 1×PBS for 5 min, and then incubated in a dUTP/Tdt mixture (Roche Applied Science) at 37°C for 1 hr followed by three rinses in 1×PBS and mounted with anti-fade. Corresponding negative (without terminal transferase) and positive control (DNase-I-treated) sections were also prepared. The sections were analyzed under fluorescent microscopy.
Western Blotting
After euthanasia and enucleation, retinas harvested at the same time points for immunohistochemistry were rapidly dissected, frozen in liquid nitrogen, and subsequently crushed using a tissue pulverizer (Beckman) chilled by dry ice. These retinas were solubilized in 9 M urea, 4% Nonidet P-40, and 2% 2-mercaptoethanol, at pH 9.5. Protease inhibitor cocktail (P8340, Sigma) consisting of 4-(2-aminoethyl) benzenesulfonyl fluoride, pepstatin A, E-64, bestatin, leupeptin, and aprotinin, was added to inhibit protease activity. Samples were centrifuged for 10 min at approximately 14,000 × g. The supernatant was used for SDS-PAGE and the pellet discarded. Protein concentration was determined with a modified Bradford assay using a kit purchased from Biorad (Hercules, CA). Equal amounts of retinal protein per lane (40 μg) were diluted with SDS sample buffer and loaded onto gels for SDS-PAGE (4%–20% or 16%; Invitrogen). Proteins were electroblotted to polyvinylidene difluoride (PVDF) membranes (Immobilon-P; Millipore, Bedford, MA) and the efficiency of transfer was confirmed by staining the membrane with Ponceau S red (Sigma). Non-specific binding was blocked with 5% nonfat dry milk in Tween-Tris-buffered saline (TTBS). Then membranes were incubated overnight at 4°C with antibodies against LC-3 (1:500, MBL, Japan) prepared in 5% non-fat dry milk solution in TTBS. Anti-rabbit HRP-conjugated secondary antibody was applied at 1:20000 (anti-rabbit Ab, Jackson). Negative controls were performed without primary antibody. Chemiluminescence was developed with a kit (Super Signal West Pico; Pierce, Rockford, IL). Protein bands were digitally imaged with a commercial system (CCDBIO 16SC Imaging System; Hitachi Genetic Systems/MiraiBio, Alameda, CA) and quantified by densitometry (Gene Snap and Gene Tools software; Hitachi). Equal protein loading was checked by Ponceau S red staining of gels, by non-specific bands, and by immunoblotting with anti-ERK antibody (rabbit polyclonal anti-ERK (Santa Cruz), 1:2000).
Statistical Analyses
Data are expressed as mean ± SEM. Cell count data were analyzed by the rank sum test. Retinal thickness were analysed by ANOVA. ERG data were analyzed with a student’s t-test. For all comparisons, P < 0.05 was regarded as statistically significant.
RESULTS
Necrostatin-1 Reduces Retinal Tissue Damage and Neuronal Cell Death After Ischemia
We hypothesized that Nec-1, an inhibitor of TNF-mediated caspase-independent cell death, would reduce injury after retinal ischemia. To address this question, we examined the effects of Nec-1 on retinal histopathology at 7 days following ischemia. Retinal ischemia for 45 min causes selective neuronal degeneration in the inner layers and reduction in retinal thickness compared with untreated controls (Fig. 2A,B). Pretreatment with an intravitreal injection of Nec-1 at the time of and 2 hr after ischemia led to significant preservation in thickness and histoarchitecture of the inner retina compared with vehicle-treated controls (n = 6 per group, P < 0.05) (Fig. 2A,B). Pretreatment with an inactive analogue of Nec-1 (Nec-1i), previously shown to lack necroptosis inhibitory activity, abolished this protective effect (Fig. 2B). We then quantified neuronal death in the selectively vulnerable inner retina. Average neuronal cell counts in the inner retina were reduced by 25% in the vehicle-treated, ischemic group but were nearly 100% in the Nec-1 treated, ischemic animals (n = 6, P < 0.05) (Fig. 2C,D). Nec-1i, however, did not protect neurons compared with vehicle controls (Fig. 2C,D); thus, Nec-1 confers significant protection of neurons under ischemic conditions.
Fig. 2.

Necrostatin-1 reduces retinal injury and protects neurons in the inner retinal layers A, B: Following 45 min of retinal ischemia of the right eye, animals treated with Nec-1 led to significant preservation in thickness and histoarchitecture of the inner retina compared with vehicle-treated controls or inactive-Nec-1 treated animals (n = 6 per group, P < 0.05). A: Representative photomicrographs of the different treatment groups. B: Percent retinal thickness of the inner retina normalized to the untouched contralateral eye. C, D: The bar graphs represent the percentage of neurons normalized to the non-ischemic control. The control group shows a 25% decrease in neurons. In the INL (C), there was significant neuronal protection when the eyes were treated with Nec-1 as compared to the inactive Nec-1 analogue or the DMSO control group (n = 6, *P < 0.05 compared with vehicle control). In the GCL (D), there was a trend towards a decrease in the number of cells that had undergone cell death in the Nec-1 treated group (n = 6, p = 0.0621 compared to vehicle control). [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Necrostatin-1 Reduces Acute Plasmalemma Permeability in Injured Cells after Retinal Ischemia
Since an early hallmark of necroptosis is plasma membrane permeability, we assessed the effects of Nec-1 on loss of plasma membrane integrity using propidum iodide uptake in the GCL and INL. Compared with vehicle-treated retinas, Nec-1 led to a significant reduction in PI-labeled cells at 6 hr after ischemia (P < 0.05; Fig. 3A–C). Because plasma membrane permeability may also occur late in apoptosis, we assessed whether Nec-1 inhibits two classic markers of apoptosis at 1 day after ischemia: caspase expression and TUNEL staining. Caspase-3 expression was examined by immunohistochemistry in the retina at 24 hr after ischemia in animals pretreated with Nec-1 or its vehicle. At 24 hr after ischemia, caspase-3 stained cells were observed in the GCL and INL of vehicle-treated animals. Pretreatment with Nec-1 did not change the number of caspase-3 labeled cells in either layer compared with vehicle controls (Fig. 4A). At 24 hr after ischemia, TUNEL positive cells were noted in the vehicle-treated group in the GCL and INL. Pretreatment with Nec-1 did not change the number of TUNEL positive cells in these layers compared with vehicle treated controls (Fig. 4B).
Fig. 3.

Pre-treatment with Nec-1 reduces propidium iodide (PI)-positive cells at 6 hr after retinal ischemia in the injured inner nuclear and ganglion cell layer. A: Representative photomicrographs showing reduced numbers of PI-positive cells in the INL and GCL after retinal ischemia in Nec-1 and vehicle-treated rats. Magnification ×40. B, C: Quantitation of PI-positive cells in injured INL (B) and GCL (C). *P < 0.05 vs. vehicle treated animals (n = 6). Left eye is untouched, non-ischemic control.
Fig. 4.

There is no effect of Nec-1 on caspase-3 (A) or TUNEL (B) stained cells after retinal ischemia. Rats were pretreated with 4 mM Nec-1 or DMSO by an intravitreal injection at the time of and 2 hr after ischemia. TUNEL or caspase-3 staining was then performed after 24 hr of reperfusion. Cell counting in the GCL is shown. N = 4 per group.
Necrostatin-1 Inhibits Autophagy After Retinal Ischemia
To further examine specificity of Nec-1, we tested whether Nec-1 inhibited autophagy, which is a caspase-independent process activated during necroptosis, and is directly associated with the conversion of the microtubule associated protein, LC-3 (I) to LC-3 (II). In vehicle treated animals, there is robust induction of LC-3 (II) at 8 hr after ischemia (Fig. 5). Nec-1 inhibits this induction while Nec-1i has no effect on LC-3 (II) compared with vehicle-treated controls (Fig. 5). Western blot densitometric analysis of LC-3 (II) protein levels demonstrated a statistically significant decrease in the Nec-1-treated ischemic retina compared with Nec-1i-treated ischemic retina (230% ± 48 vs. 567% ± 86, P < 0.05; n = 4, as a percentage of normal, non-ischemic eye).
Fig. 5.
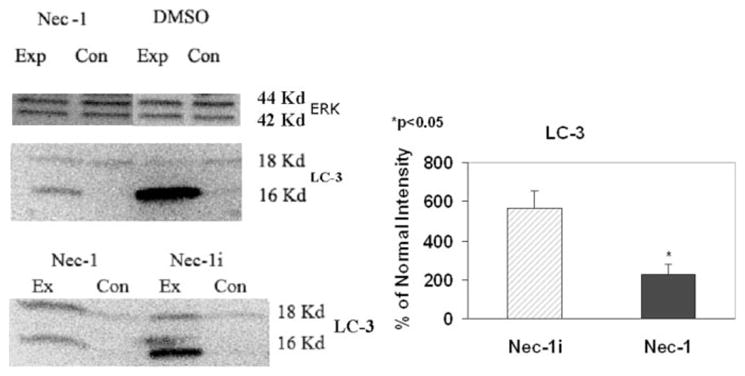
Nec-1 inhibits autophagy after retinal ischemic injury. Rats were pretreated with an intravitreal injection at the time of and 2 hr after ischemia with DMSO, Nec-1, or its inactive analogue (Nec-1i). After 8 hr of reperfusion, the eyes were enucleated, sectioned and exposed to an antibody against LC-3. A: Representative blots of 4 samples. The results show in the DMSO group two bands: autophagy is characterized by the induction of LC3-II (16kD: lower band) from LC3-I (18kd: upper band). Nec-1 partially inhibited the induction of LC-3 (II). However, there was no inhibition when Nec-1 was substituted for Nec-1i. Equal loading was confirmed by ERK protein. B: Densitometry revealed a significant reduction in the 16kd LC-3 (II) in the Nec-1 treated samples compared with the Nec-1i treated samples (P < 0.05; N = 4). Ex = experimental group; Con = untouched control eye.
Necrostatin-1 Reduces Functional Impairment after Retinal Ischemia
While the above data suggest that Nec-1 reduces neuronal cell death after retinal ischemic injury through caspase-independent mechanisms, a crucial question is whether inhibition of necroptosis is functionally significant. We addressed this question by assessing the effects of Nec-1 using electroretinography. Representative images 7 days after ischemia are shown in Figures 6A and 6B of animals pre-treated with Nec-1, Nec-1i, or vehicle. The mean ERG b-wave, a measure of inner retinal function, at 7 days after retinal ischemia was reduced to 13% of baseline in the vehicle treated ischemic animals, which is consistent with the level of impairment seen in untreated ischemic retinas. The administration of Nec-1 led to a significant preservation of the ERG b-wave (27% of baseline) in the ischemic group (Fig. 6C). However, Nec-1i had no protective effect as shown in Figure 6A. We then addressed the question whether Nec-1 is neuroprotective when administered at time points after ischemia. In post-treatment paradigms, Nec-1 also significantly attenuated the ERG b-wave reduction when given 2 and 4 hr after ischemia compared with vehicle controls (P < 0.05, n = 6, Fig. 6D).
Fig. 6.

Nec-1 leads to functional protection in both pre and post-treatment paradigms. A, B: Representative ERGs of animals pre-treated with Nec-1 or vehicle or Nec-1i. C is the baseline ERG prior to ischemia. S and E were pretreated with an intravitreal injection at the time of and 2 hr post ischemia. S was treated with Nec-1 (4 mM) while E was treated with an inactive analogue of Nec-1 (Nec-1i). C: The mean ERG b-wave at 7 days after retinal ischemia was reduced to 13% of baseline in the vehicle treated group while the administration of Nec-1 led to a significant preservation of the ERG b-wave; 27% of baseline (*P < 0.05, n = 8). D: Animals were treated at 2 and 4 hr after retinal ischemia and ERG b-wave was significantly higher compared to the vehicle-controlled eyes (*P < 0.05, n = 8).
DISCUSSION
The present study supports the existence of a novel form of caspase-independent programmed necrosis involving the activation of autophagy in retinal ischemia. Nec-1 attenuated retinal degeneration, preserved retinal thickness, rescued neurons in the inner retinal layers, and reduced functional impairment after ischemia. These results together with prior work (Degterev et al., 2005) provide strong evidence that necroptosis is a significant cell death pathway in conditions involving neuronal injury.
Several lines of evidence suggest that necroptosis is an important mode of cell death in retinal injury. First, necroptosis is a pathway of regulated necrotic cell death triggered by death receptor ligands in the presence of broad caspase inhibition (Degterev et al., 2005). Retinal ganglion cells exposed to such stimuli, for example, as TNFα in culture are not rescued from death by co-treatment with caspase inhibitors and, instead, follow an alternative pathway of PI positive, caspase-independent cell death (Tezel and Yang, 2004). These data suggest that retinal ganglion cells express alternative pathways of non-apoptotic cell death and prompted us to directly examine whether necroptosis may contribute to retinal ischemic injury. Our results support this hypothesis as Nec-1, a specific inhibitor of necroptosis, reduced post-ischemic lesion size in the retina.
Multiple lines of evidence support the possibility that Nec-1 targets necroptotic cells in vivo, consistent with its mechanism of action in vitro. Nec-1 does not alter the activation of caspase-3 or TUNEL staining in the ischemic retina or in the ischemic brain (Degterev et al., 2005), further supporting that Nec-1 does not target apoptotic or caspase-mediated pathways. However, it should be acknowledged that caspase and TUNEL are not absolute markers of apoptosis. Furthermore, Nec-1 inhibits the induction of LC-3 (II), a marker of autophagy, which is known to be directly activated in necroptosis (Degterev et al., 2005). Nec-1i, a closely related analog of Nec-1 lacking anti-necroptotic activity in vitro, displayed no cytoprotective effect in vivo and did not attenuate the formation of LC-3-II. These data closely parallel the activity profile of Nec-1 in necroptotic cells and suggest that in vivo Nec-1 targets a caspase-independent necroptotic pathway involving autophagy. Finally, Nec-1 reduces the acute loss of plasma membrane integrity at an early time point after ischemia. The loss of plasma membrane integrity at 6 hr after ischemia argues for a mechanism involving necrosis because plasma membrane permeability typically occurs late in the course of apoptosis and there was no evidence for an effect of Nec-1 on caspase expression at 24 hr, a time point of maximal caspase expression in this model. However, it is important to acknowledge that PI staining merely refers to plasma membrane integrity and is not specific for necroptosis per se.
The signaling pathways underlying necroptosis and its association with autophagy remain to be explored. An in vitro study found that Nec-1 reduces oxidative cell death in the hippocampal cell line, HT-22. AIF translocation in that study was inhibited by Nec-1 (Xu et al., 2007), suggesting a potential role for AIF in necroptosis. Holler et al. (2000) have found that RIP kinase is an important early event in the death signaling of Jurkat cells exposed to concurrent treatment of TNFα and Z-VAD. Furthermore, detailed analysis of Nec-1’s mechanism of action have demonstrated that Nec-1 is a specific inhibitor of RIP1 kinase (Degterev et al., 2008), and a series of genes that may act downstream and/or serve as regulators of RIP1 in necroptosis have recently been described (Hitomi et al., 2008). Our analysis suggests that RIP1 kinase may be a promising new target for future therapies against conditions involving neuronal ischemia.
Inhibition of necroptosis may be a new strategy for the development of cytoprotective agents in acute neurological disorders. We show for the first time that Nec-1 protects neurons with objective functional electrophysiological data on ERG. Dysfunction of the inner retinal layer is involved in the ERG-b wave abnormalities observed in this model (Audo et al., 2008). Such functional data may be a more predictive sensitive measure of clinical outcome than histology, which traditionally has much more been relied upon in neuroprotection studies. This reliance on histological outcome may be one reason for the disconnect between laboratory and clinical studies on neuroprotective agents. Protection in this study was even seen when the drug was administered at 2 and 4 after ischemia, a time window which is clinically relevant and suggests that the kinetics of necroptosis may be slower than other types of non-specific necrosis. Nec-1 can also be administered in a clinically relevant manner by intravitreal injection whereas prior studies have only shown protection with intracerebro-ventricular injection in brain ischemia.
In summary, necroptosis may be an important, emerging mode of cell death in retinal ischemia. The therapeutic effects demonstrated with Nec-1 support its use as a novel neuroprotective agent that targets necrotic cell death mechanisms. It is important to identify different types of cell death mechanisms in order to better understand the molecular pathophysiology of acute neural injuries. Future studies examining the effects of necrostatins in combination with inhibitors of other cell death pathways are therefore warranted. Such combination approaches may offer therapeutic benefit for the treatment of acute neurological disorders, including retinal ischemia. It remains to be determined if necroptosis and apoptosis are mutually exclusive cell death pathways in neurons or if they are both activated in the same cell.
Fig. 1.
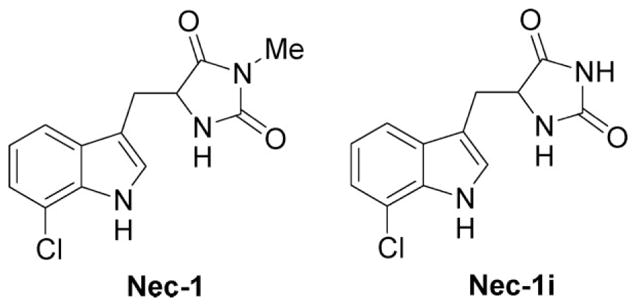
Chemical structures of Nec-1 and its inactive analogue, Nec-1i.
Acknowledgments
Contract grant sponsor: AHA; Contract grant number: 0475008N (to S.I.S.); Contract grant sponsor: Howard Hughes Medical Institute (to S.I.S.); Contract grant sponsor: NIH EY; Contract grant number: 10343 (to S.R.).
References
- Audo I, Robson AG, Holder GE, Moore AT. The negative ERG: clinical phenotypes and disease mechanisms of inner retinal dysfunction. Surv Ophthalmol. 2008;53:16–40. doi: 10.1016/j.survophthal.2007.10.010. [DOI] [PubMed] [Google Scholar]
- Berger S, Savitz SI, Nijhawan S, Singh M, David J, Rosenbaum PS, Rosenbaum DM. Deleterious role of TNF-alpha in retinal ischemia-reperfusion injury. Invest Ophthalmol Vis Sci. 2008;49:3605–3610. doi: 10.1167/iovs.07-0817. [DOI] [PubMed] [Google Scholar]
- Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, Hedrick SM, Gerber SA, Lugovskoy A, Yuan J. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4:313–321. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontaine V, Mohand-Said S, Hanoteau N, Fuchs C, Pfizenmaier K, Eisel U. Neurodegenerative and neuroprotective effects of tumor Necrosis factor (TNF) in retinal ischemia: opposite roles of TNF receptor 1 and TNF receptor 2. J Neurosci. 2002;22:RC216. doi: 10.1523/JNEUROSCI.22-07-j0001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitomi J, Christofferson DE, Ng A, Yao J, Degterev A, Xavier RJ, Yuan J. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell. 2008;135:1311–1323. doi: 10.1016/j.cell.2008.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, Bodmer JL, Schneider P, Seed B, Tschopp J. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol. 2000;1:489–495. doi: 10.1038/82732. [DOI] [PubMed] [Google Scholar]
- Junk AK, Mammis A, Savitz SI, Singh M, Roth S, Malhotra S, Rosenbaum PS, Cerami A, Brines M, Rosenbaum DM. Erythropoietin administration protects retinal neurons from acute ischemia-reperfusion injury. Proc Natl Acad Sci USA. 2002;99:10659–10664. doi: 10.1073/pnas.152321399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Roth S. Retinal ischemic preconditioning in the rat: requirement for adenosine and repetitive induction. Invest Ophthalmol Vis Sci. 1999;40:1200–1216. [PubMed] [Google Scholar]
- Lin J, Roth S. Ischemic preconditioning attenuates hypoperfusion after retinal ischemia in rats. Invest Ophthalmol Vis Sci. 1999;40:2925–2931. [PubMed] [Google Scholar]
- Nakazawa T, Nakazawa C, Matsubara A, Noda K, Hisatomi T, She H, Michaud N, Hafezi-Moghadam A, Miller JW, Benowitz LI. Tumor necrosis factor-alpha mediates oligodendrocyte death and delayed retinal ganglion cell loss in a mouse model of glaucoma. J Neurosci. 2006;26:12633–12641. doi: 10.1523/JNEUROSCI.2801-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth S, Li B, Rosenbaum PS, Gupta H, Goldstein IM, Maxwell KM, Gidday JM. Preconditioning provides complete protection against retinal ischemic injury in rats. Invest Ophthalmol Vis Sci. 1998;39:777–785. [PubMed] [Google Scholar]
- Savitz SI, Degterev A, Yuan J, Moskowitz MA. Inhibition of necroptosis is neuroprotective in permanent focal cerebral ischemia. Stroke. 2007;38:453. [Google Scholar]
- Singh M, Savitz SI, Hoque R, Gupta G, Roth S, Rosenbaum PS, Rosenbaum DM. Cell-specific caspase expression by different neuronal phenotypes in transient retinal ischemia. J Neurochem. 2001;77:466–475. doi: 10.1046/j.1471-4159.2001.00258.x. [DOI] [PubMed] [Google Scholar]
- Teng X, Degterev A, Jagtap P, Xing X, Choi S, Denu R, Yuan J, Cuny GD. Structure-activity relationship study of novel necroptosis inhibitors. Bioorg Med Chem Lett. 2005;15:5039–5044. doi: 10.1016/j.bmcl.2005.07.077. [DOI] [PubMed] [Google Scholar]
- Tezel G, Yang X. Caspase-independent component of retinal ganglion cell death, in vitro. Invest Ophthalmol Vis Sci. 2004;45:4049–4059. doi: 10.1167/iovs.04-0490. [DOI] [PubMed] [Google Scholar]
- Xu X, Chua CC, Kong J, Kostrzewa RM, Kumaraguru U, Hamdy RC, Chua BH. Necrostatin-1 protects against glutamate-induced glutathione depletion and caspase-independent cell death in HT-22 cells. J Neurochem. 2007;103:2004–2014. doi: 10.1111/j.1471-4159.2007.04884.x. [DOI] [PubMed] [Google Scholar]