

Abstract
PURPOSE
Epichaperome network maintenance is vital to survival of tumors that express it. PU-H71 is an epichaperome inhibitor that binds to the ATP-binding site of HSP90 and has demonstrated antitumor activity in breast cancer xenograft models and clinical safety in patients. PU–positron emission tomography (PET) is a theragnostic imaging tool that allows visualization of the epichaperome target. In this phase Ib trial, we present safety and tolerability for PU-H71 plus nab-paclitaxel in HER2-negative patients with metastatic breast cancer (MBC) and the utility of PU-PET as a noninvasive predictive biomarker.
METHODS
We performed a 3 + 3 dose-escalation study with escalating PU-H71 doses and standard nab-paclitaxel. The primary objective was to establish safety and determine maximum tolerated dose (MTD)/recommended phase 2 dose. Secondary objectives were to assess pharmacokinetics and clinical efficacy. Patients could enroll in a companion PU-PET protocol to measure epichaperome expression before treatment initiation to allow exploratory correlation with treatment benefit.
RESULTS
Of the 12 patients enrolled, dose-limiting toxicity occurred in one patient (G3 neutropenic fever) at dose level 1; MTD of PU-H71 was 300 mg/m2 plus nab-paclitaxel 260 mg/m2 administered every 3 weeks. Common toxicities included diarrhea, fatigue, peripheral neuropathy, and nausea. PU-H71 systemic exposure was not altered by nab-paclitaxel administration. Two of 12 patients had partial response (overall response rate, 17%) and the clinical benefit rate was 42% (5 of 12). Time to progression was associated with baseline epichaperome positivity and PU-H71 peak standard uptake value (SUV), with more durable disease control observed with high epichaperome levels.
CONCLUSION
The combination of PU-H71 and nab-paclitaxel was well tolerated, with evidence of clinical activity. More durable disease control without progression was observed in patients with high baseline epichaperome expression. A phase II trial of this combination with PU-PET as a companion diagnostic for patient selection is currently planned.
INTRODUCTION
In healthy cells under physiologic conditions, normal cellular function is maintained by the coordinated action of a complex protein machinery that consists of molecular chaperones, cochaperones, and protein-folding enzymes, collectively called the chaperome.1,2 Although the chaperome acts in protein folding and degradation, stressors may resculpt the chaperome and its function.3 Stresses associated with oncogenic transformation may increase connectivity among chaperome members, which in turn remodels, proteome-wide, the activity of oncogenic protein pathways. This proteome remodeling is accomplished by increasing both the interaction strength and number of interactions among the participant proteins, which is not necessarily accompanied by a change in their expression levels. Termed “epichaperomes,” these chaperome pools therefore act not as folders but rather as multimolecular scaffolds that pathologically remodel proteome-wide cellular processes.3-5
CONTEXT
Key Objective
To test the safety and tolerability for PU-H71 (epichaperome inhibitor) plus nab-paclitaxel in patients with HER2-negative metastatic breast cancer and the utility of PU–positron emission tomography (PET) as a noninvasive predictive biomarker.
Knowledge Generated
PU-H71 combined with nab-paclitaxel was safe; the frequency of most adverse events was in line with single-agent nab-paclitaxel, with the exception of GI adverse effects. Maximum tolerated dose of PU-H71 was 300 mg/m2 plus nab-paclitaxel 260 mg/m2 administered every 3 weeks. Four out of 12 patients achieved partial response as best overall response (all triple-negative breast cancer), with two confirmed partial responses. Time to progression was associated with baseline epichaperome positivity measured by PU-PET.
Relevance
PU-H71 plus nab-paclitaxel is safe. Promising activity was found in this small phase Ib trial. PU-PET worked as a predictor biomarker of treatment benefit. The study illustrates the power of precision medicine approaches beyond panel sequencing in tailoring effective therapy approaches.
Maintenance of the epichaperome network is vital to survival of tumors that express it, and cancers with this altered protein–protein interaction network configuration become susceptible to drugs that target critical epichaperome components such as heat shock protein 90 (HSP90), HSC70, HSP-organizing protein, and others.4,6
Targeting HSP90, which resides in the epichaperome, is an attractive anticancer strategy, and several HSP90 inhibitors (HSP90i) have been tested in phase I and II clinical trials.7-15 However, no agent has been approved for use in clinical practice, and HSP90i development has been challenging because of a variety of issues, including limited understanding and insights of heterogeneity of the target and plasma pharmacokinetics (PK), lack of selectivity for the epichaperome over abundant HSP90 pools, toxicity, and lack of predictive biomarkers of response and/or resistance.
PU-H71 is an epichaperome inhibitor that binds to the ATP-binding site of HSP90 and kinetically selects for HSP90 integrated into the epichaperome.4,16 It has demonstrated antitumor activity in xenograft models, including breast cancer models.17 Single-agent clinical safety has been established in patients with solid and hematologic tumors.18 In vitro, the sensitivity of a tumor cell to PU-H71 is directly proportional to the content of HSP90 integrated in the epichaperome but is independent of the levels of chaperome members, HSP90 client proteins, antiapoptotic proteins, and genetic alterations.4 Therefore, strategies that enhance the stress in tumor cells and consequently increase the integration of HSP90 into the epichaperome, such as cytotoxic therapies including nab-paclitaxel, may be synergistic with PU-H71.19 Each agent administered alone or together has modest antitumor effects, whereas the sequential administration of nab-paclitaxel followed by PU-H71 eradicated xenografted tumors, resulting in cures in mice. This formed the rationale for conducting this phase Ib trial to establish the safety and determine the maximum tolerated dose and/or recommended phase 2 dose (RP2D) of PU-H71 plus nab-paclitaxel in patients with HER2-negative metastatic breast cancer (MBC). Secondary objectives include assessment of PK and clinical activity of this combination.
The parallel development of a drug-specific pharmacometric/companion diagnostic assay is essential in optimizing clinical development of targeted therapies and an unmet need for epichaperome-targeted agents.4,20 Our group has developed a positron-emitting form of PU-H71, radiolabeled iodine-124 ([124I]-PU-H71), which enables visualization of the PU-H71 uptake in the epichaperome target and facilitates noninvasive real-time tumor pharmacometric measurements.4,18,20,21 PU-positron emission tomography (PET) informs baseline epichaperome expression in individual tumors,4 allowing the development of treatment and predictive biomarkers in parallel. We explored the potential of PU-PET imaging of tumors at baseline as a predictive biomarker of clinical benefit to PU-H71 plus nab-paclitaxel.
METHODS
Study Design and Patient Selection
We conducted a phase Ib, open-label, classic 3 + 3 dose-escalation trial of PU-H71 plus nab-paclitaxel in patients with HER2-negative MBC (ClinicalTrials.gov identifier: NCT03166085). Participants gave informed consent before entering the study, which was approved by the institutional research ethics board of Memorial Sloan Kettering Cancer Center (MSKCC).
Eligibility criteria included patients age ≥ 18 years with histologically confirmed (ASCO/College of American Pathologists version 2013) HER2-negative MBC. Patients with estrogen receptor–positive (ER+) MBC were required to experience disease progression on or be intolerant of at least one prior endocrine therapy. All patients were required to receive at least one cytotoxic therapy for metastatic disease and experience disease progression on their most recent therapy. Additional eligibility criteria included good performance status (Eastern Cooperative Oncology Group performance status < 2), adequate end-organ function, and life expectancy ≥ 3 months as assessed by the investigator.
The key exclusion criteria were symptomatic brain or CNS metastases, any active cancer treatment within 2 weeks of study treatment, prior treatment with nab-paclitaxel, and peripheral neuropathy of grade ≥ 2 per National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE), v4.0 at the time of or within 3 weeks of study therapy. Other exclusion criteria are detailed in the protocol.
Study Treatment
Extrapolating from the phase I monotherapy trial of PU-H71 and on the basis of tumor pharmacometric measurements that indicated a dose > 180 mg/m2 as target engaging,20 PU-H71 was administered intravenously over 1 hour, at a starting dose of 225 mg/m2, with plans to test another dose of 300 mg/m2. Nab-paclitaxel was administered intravenously over 30 minutes at a standard dose of 260 mg/m2 on day 1 of a 21-day cycle per the US Food and Drug Administration (FDA)–approved label for MBC. Both drugs were administered on the same day, sequentially, with nab-paclitaxel first, followed by PU-H71 approximately 6 hours later (±1 hour), as guided by the preclinical data.19 Therapy was continued until progression, severe or unexpected toxicity, patient withdrawal, more than two dose reductions for either PU-H71 or nab-paclitaxel, or death.
Toxicity Assessment and Dose Reduction
Patients were examined and assessed for toxicities during and before each cycle. Toxicity was graded according to NCI CTCAE v4.0. Patients were evaluated for dose-limiting toxicity (DLT) during cycle 1. DLT was defined as any grade ≥ 4 nonhematologic adverse event (AE), any grade 3 nonhematologic AE not improving to baseline or grade ≤ 1 by day 14 (despite adequate supportive care/toxicity management), grade 4 neutropenia lasting ≥ 7 days, or febrile neutropenia (more broad criteria for DLT are described in the Appendix).
Nab-paclitaxel or PU-H71 dose reductions were not permitted during the DLT period. For dosing beyond cycle 1, nab-paclitaxel was held if patients experienced any other grade 3 or 4 toxicity believed to be related to nab-paclitaxel until symptoms resolved to grade 1/baseline grade. Two dose reductions for nab-paclitaxel and/or for PU-H71 were permitted, as described in the Appendix.
PK Assessment
Blood samples for plasma PU-H71 PK were drawn during cycles 1 and 2 on days 1 and 2. Detailed sample collection and storage are provided in the Appendix. Plasma concentration profiles of PU-H71 and its metabolites as well as PK parameters including area under the curve (AUC), pre-dose trough concentration, time to obtain maximum concentration, clearance, terminal half-life, and volume of distribution were calculated. The areas under the plasma concentration–time curves after each single dose (AUC0-all) were calculated using the log-linear trapezoidal method.
Assessment of Treatment Response
Patients were evaluated for tumor response using computed tomography (CT) of the chest/abdomen/pelvis (CT-CAP) with contrast and a bone scan or fluorodeoxyglucose (FDG) PET scan every two cycles (±7 days) from start of therapy according to RECIST v.1.1 criteria.22 The best overall response (BOR) was defined as the best response recorded from the start of treatment until progression or withdrawal from the study. Clinical benefit rate (CBR) was defined as the proportion of patients with complete response (CR), partial response (PR), or stable disease (SD) lasting for at least 24 weeks. Time to progression (TTP) was defined as the time from randomization to disease progression or treatment discontinuation, whichever occurred first.23
PU-PET Measurement and Clinical Benefit Correlation
Patients had the option to enroll in a companion phase I diagnostic PU-PET protocol (ClinicalTrials.gov identifier: NCT01269593) to radiologically assess epichaperome expression before initiating treatment with PU-H71 plus nab-paclitaxel in the phase Ib therapy trial. For PU-PET, patients received a single microdose (nontherapeutic microdose) of up to 407 MBq of [124I]-PU-H71 intravenously and underwent imaging at 3-4 hours and 20-24 hours after infusion using a single PET/CT scanner. CT scans for attenuation correction and anatomic coregistration were performed before [124I]-PU-H71 tracer injection. Interpretation of PU-PET was done by an experienced MSKCC nuclear medicine physician (M.P.D.), using dedicated PET/CT analysis software.
Coadministration of [124I]-PU-H71 with therapeutic doses of PU-H71 enabled measurement of [124I]-PU-H71 peak standard uptake volume (SUVpeak) for the selected lesions per patient and allowed calculation of tumor-specific drug concentration (moles) of PU-H71 achieved on a per-lesion basis, as previously described by our group.20 A Mann-Whitney (Wilcoxon) test for unpaired data yields was done to evaluate the correlation between baseline tumor PU-H71 tracer concentrations (SUVpeak) and PU-H71 tumor-specific molar concentration with TTP. More specific details about PU-PET technique and data analysis and interpretation are included in the Appendix.
Statistical analyses were performed using GraphPad Prism (version 7). In each group of data, estimate variation was considered and is indicated in each figure as SEM. P values for unpaired comparisons between two groups with comparable variance were calculated by two-tailed Student’s t test. Pearson’s tests were used to identify correlations among variables.
RESULTS
Patients and Treatment
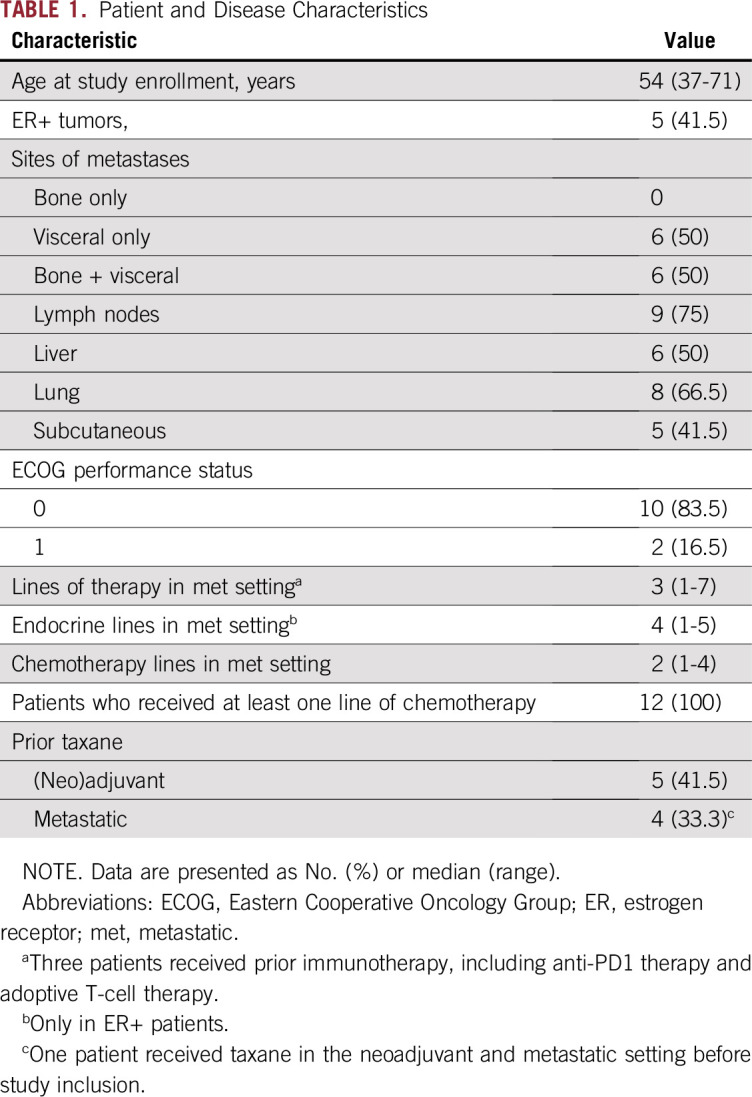
Twelve patients were enrolled: six at 225 mg/m2 of PU-H71 and another six at 300 mg/m2. Patients characteristics are described in Table 1. Median age was 54 years (range, 37-71 years), five patients were ER+, and the other seven had triple-negative breast cancer (TNBC). Median Eastern Cooperative Oncology Group performance status was 0 (range, 0-1). All patients had visceral disease. The median lines of therapy in the metastatic setting was three (range, one to seven). Eight patients had received prior taxane, including four in the metastatic setting.
TABLE 1.
Patient and Disease Characteristics
Overall Safety
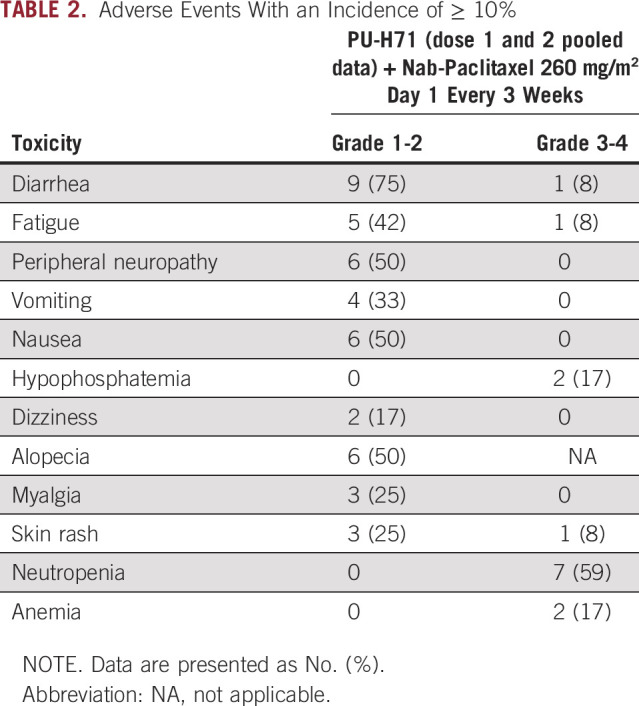
All patients were included in the safety analysis. One patient in cohort 1 developed a DLT of febrile neutropenia. After dose reduction of nab-paclitaxel and with growth factor support starting cycle 2, this patient continued treatment without any further neutropenia. No other DLTs were observed in cohort 2. Hence, the RP2D of PU-H71 was defined as 300 mg/m2 with 260 mg/m2 of nab-paclitaxel every 3 weeks.
Overall, the regimen was well tolerated. The most common nonhematological AEs were grade 1/2, as described in Table 2. The frequency of most AEs was similar to single agent nab-paclitaxel, with the exception of GI AEs, which were noted at higher rates with the combination.24 No patients required PU-H71 dose reductions. Four patients required nab-paclitaxel dose reduction, including one patient who discontinued therapy because of worsening peripheral neuropathy. There were no treatment-related deaths in the study.
TABLE 2.
Adverse Events With an Incidence of ≥ 10%
Plasma PK
Plasma PK evaluations were carried out in all patients to evaluate the effect of nab-paclitaxel on PU-H71 absorption and the PK variability according to PU-H71 dose. Cmax increased by approximately 111%, with a 56% increase in the AUCall mean values after the 300 mg/m2 dose compared with the 225 mg/m2 dose. In addition, harmonic mean apparent terminal elimination half-life values were 5.7 and 5.9 hours for the 225- and 300-mg/m2 dose groups, respectively, suggesting similar PK between the two groups (Fig 1). The systemic exposure to PU-H71 was not altered by coadministration with nab-paclitaxel, as evidenced by similar plasma PK profiles for both single-agent PU-H7120 (data extracted from NCT01393509) and sequential nab-paclitaxel PU-H71 therapy (Appendix).
FIG 1.

Concentration of PU‐H71 (ng/mL) during the first 24 hours after infusion. Plasma pharmacokinetics of PU‐H71: Mean concentration‐time profiles after single doses of 225 or 300 mg/m2 PU‐H71 after cycles 1 and 2 combined are shown. Green circles represent PU‐H71 dose level 1 (225 mg/m2) and orange circles represents PU‐H71 dose level 2 (300 mg/m2), both administered at 6 hours after the nab‐paclitaxel infusion.
Antitumor Activity
Of the 12 patients enrolled, the BOR of PR was noted in four of 12 (33%) patients, all with TNBC. The confirmed overall response rate (ORR) was two of 12 (17%), with one PR at each dose level. Both patients with confirmed PR received prior taxane, including one in the metastatic setting. This latter patient who had received five previous therapies for metastatic disease had a durable PR that lasted 30 weeks. At the time of the data cut-off of February 21, 2019, clinical benefit was seen in five of 12 (42%) patients. Specifically, CBR in TNBC was four of seven (57%) and that for ER+/HER2-negative breast cancer was one of five (20%). TTP was 18.6 weeks (95% CI, 11 to 28.7 weeks), with one patient remaining in the study with SD.
PU-PET as a Predictor Biomarker of Response
Nine of the 12 patients (three with ER+ and six with TNBC) underwent PU-PET at baseline to measure epichaperome expression (Fig 2). Each patient had up to two lesions selected on PU-PET as target lesions (total, 17 target lesions for nine patients) for response assessment by CT-CAP and FDG PET. Of the 17 lesions, three were bone metastases not measurable on CT and assessed by FDG PET only.
FIG 2.

Patient flowchart. CT, computed tomography; ER+, estrogen receptor positive; FDG PET, fluorodeoxyglucose positron emission tomography; HER2-negative, human epidermal growth factor receptor negative; TNBC, triple-negative breast cancer; TTP, time to progression.
TTP was associated with baseline epichaperome positivity and PU-H71 SUVpeak, with more durable disease control without progression (> 12 weeks) observed among patients with tumors with high epichaperome levels (Fig 3A, P = .0018).
FIG 3.
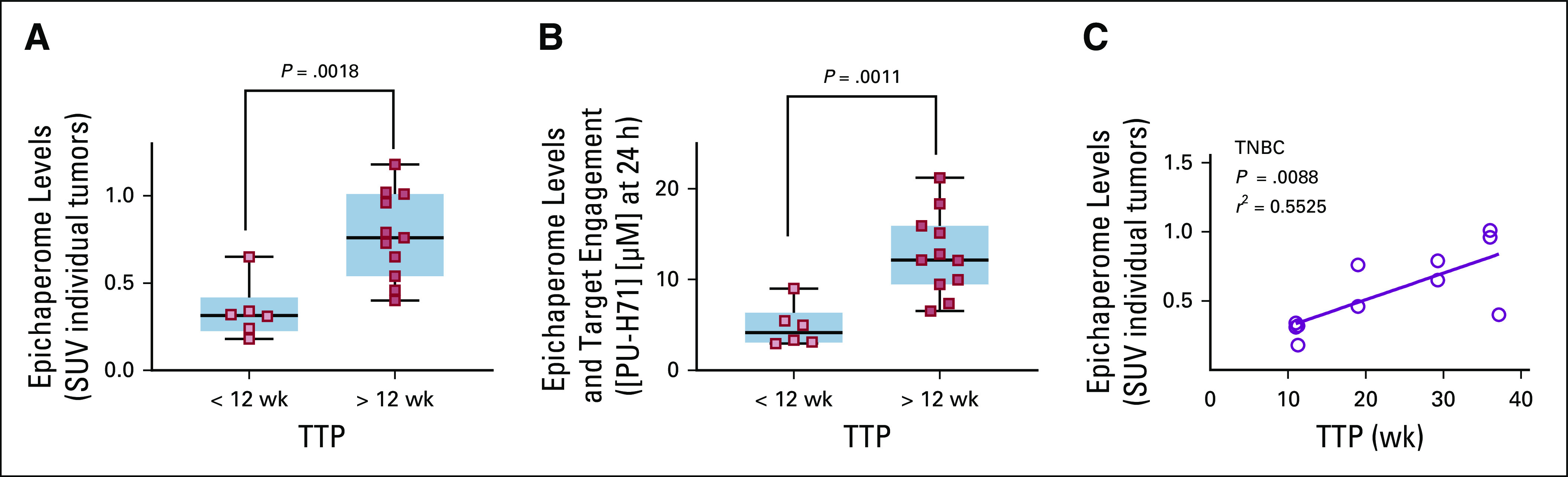
Tumor epichaperome positivity predicts durability of disease control without progression. (A) All but one of the nine patients contributed with two measurable lesions. PU–positron emission tomography measured baseline epichaperome levels (peak standard uptake value [SUV]) in individual tumors (three patients contributed with six lesions [time to progression (TTP) < 12 weeks] and six patients contributed with 11 lesions [TTP > 12 weeks]). (B) Tumor-specific molar concentration that is determined to be achieved by the therapeutic PU‐H71 dose administered and correlation with TTP. (C) Correlative analysis between epichaperome levels measured as in A and durability of response on the PU‐H71/nab-paclitaxel therapy for patients with triple-negative breast cancer (TNBC).
In epichaperome-positive tumors, the molar PU-H71 concentrations measured by PU-PET were more likely to be 5 µM and higher (6.5-21 µM) than in epichaperome-negative tumors, and this also correlated with TTP (Fig 3B, P = .0011). This estimation of PU-H71 molar concentration was irrespective of the administered dose of PU-H71 of 225 or 300 mg/m2, confirming that both doses delivered effective target-engaging concentrations. We have previously reported that an intravenous dose of > 180 mg/m2 of PU-H71 is needed to deliver an intratumoral concentration of at least 5 µM measured at 24 hours after PU-H71 injection and is associated with effective epichaperome engagement.20 These data also support the observation that nab-paclitaxel does not interfere with the tumor uptake of PU-H71.
Epichaperome positivity was observed in both ER+ and TNBC lesions, but the low number of ER+ patients imaged on PU-PET precludes meaningful assessments in this subgroup. For patients with TNBC, we observed a significant correlation between TTP and epichaperome positivity measured by SUVpeak (Fig 3C; P = .0088, r2 = 0.55, Pearson’s coefficient). Four of the six patients with TNBC were epichaperome positive and remained in the study without progression for >3 months (4.7, 7.3, 8.6, and > 13 months).
The importance of selectivity for the epichaperome by PU-PET is further illustrated in Figure 4. These two patients treated at 225 mg/m2 of PU-H71 underwent FDG PET with dedicated CT CAP and PU-PET imaging at baseline and subsequently had another FDG PET with dedicated CT CAP for response evaluation post PU-H71 plus nab-paclitaxel therapy. As shown, the patient with epichaperome-positive tumor on PU-PET at baseline achieved PR (duration of response of 34 weeks). This 53-year-old female with metastatic TNBC had epichaperome-positive lesions in thoracic metastases (Fig 4A), which were also detected on FDG PET (Fig 4B). FDG PET/CT post treatment with PU-H71 and nab-paclitaxel demonstrated PR disease (Fig 4C). In contrast, a 72-year-old patient who had MBC with thoracic metastases that were detectable by FDG PET (Fig 4E) but showed no epichaperome positivity on PU-PET (Fig 4D) had progression at 8 weeks, as demonstrated on FDG PET/CT (Fig 4F).
FIG 4.

Illustrative treatment evolution according to baseline PU–positron emission tomography (PET) result and fluorodeoxyglucose (FDG) PET. (A-C) Patient with positive baseline PU-PET and response to treatment. (A) Baseline PU-PET and (B) companion computed tomography (CT) image and FDG PET recorded at baseline (ie, before PU-H71–nab-paclitaxel therapy). (C) Post-treatment CT and FDG PET. (D-F) Patient with negative baseline PU-PET and response to treatment. (D) Baseline PU-PET and (E) companion CT image and FDG PET recorded at baseline (ie, before PU-H71–nab-paclitaxel therapy). (F) Post-treatment CT and FDG PET. Representative cross-sectional images are shown for each monitored tumor, each at the same transaxial plane. PU-PET images were obtained at 24 hours after intravenous infusion of a microdose of [124I]-PU-H71. The arrows and arrow heads point at distinct location of metastases for an individual patient. (*) Aorto-cardiac blood pool. Scale bars, PET window display intensity scales, with upper and lower SUV thresholds.
DISCUSSION
Although targeting genomic alterations has been on the forefront of drug development, our group has highlighted the importance of targeting the epichaperome using PU-H71. Preclinically, PU-H71–sensitive tumors (high epichaperome expressors) were characterized by highly interconnected or hyperconnected HSP90 and HSP70 chaperome machineries and represent approximately 10% of solid tumors or hematologic malignancies, and this frequency of high epichaperome expression can be increased up to 60% with a system stressor like nab-paclitaxel.4 This advanced understanding of how these chaperome networks form, the way they are regulated, and how they might be co-opted and augmented with stressors such as nab-paclitaxel provides us an opportunity to improve our delivery and the potential to achieve greater success with these agents than has been seen in prior clinical trials of HSP90i.
To our knowledge, this is the first clinical trial combining PU-H71 with nab-paclitaxel for treating patients with HER2-negative MBC. Previous trials of HSP90i have largely been conducted in patients with HER2-positive MBC.7,10,25-27 Consistent with our preclinical experience and the known toxicity profiles of both agents, the combination of nab-paclitaxel and PU-H71 was well tolerated, with only one DLT of febrile neutropenia in 12 treated patients.
Despite a heavily pretreated population, PU-H71 and nab-paclitaxel demonstrated an ORR of 17% and a 24-week CBR of 42%. Of note, responses were observed in patients with TNBC despite prior taxane therapy for an ORR of two of seven (29%) for this subset. Median TTP was 18.6 weeks (95% CI, 11 to 28.7 weeks). Using a more permissive CBR, as used in other phase I MBC trials, defined as CR + PR + SD ≥ 16 weeks, the 16-week CBR would be eight of 12 (67%). To place our results in context, the ORR in TNBC in the second-line metastatic setting is reported in the range of 6%-15%, and the median progression-free survival (PFS) is < 3 months.28,29 Hence the activity seen in this small phase Ib study is promising.
Recently, two phase III trials showed statistically significant benefit in PFS for patients with metastatic TNBC who received checkpoint inhibitor to PD1/PDL1 axis combined with chemotherapy in the first-line metastatic setting. The combination of nab-paclitaxel plus atezolizumab, tested in IMpassion130 (ClinicalTrials.gov Identifier: NCT02425891), has received accelerated approval by the FDA.30 This benefit and accelerated approval are restricted to patients with PD-L1–positive tumors, which is approximately 40% of the population in IMpassion130 (sp142 assay). Unfortunately, for the remaining larger proportion of patients with PD-L1–negative metastatic TNBC, there is a continued need for promising therapies.
Our group has developed PU-PET, a unique companion theragnostic assay to measure epichaperome expression and understand real-time tumor pharmacometric measurements.20 In this trial, we demonstrated that tumor epichaperome positivity at baseline serves as a predictive biomarker of benefit and that tumors with high tumor epichaperome expression have prolonged disease control from treatment with PU-H71 and nab-paclitaxel.
In conclusion, the combination of PU-H71 with nab-paclitaxel was well tolerated, with an RP2D of PU-H71 at 300 mg/m2. Based on the clinical activity, this combination warrants additional study, particularly in TNBC. PU-PET has the potential to serve as a theragnostic platform for precision medicine targeting of the epichaperome and serve as predictive biomarker of benefit to PU-H71 combination therapy. Future directions for PU-H71 development include a phase II trial of the PU-H71 with nab-paclitaxel in TNBC PD-L1–negative tumors in the first- or second-line metastatic setting and continued evaluation of PU-PET as a correlative imaging biomarker. An important issue for future trials is to define the optimal interval between nab-paclitaxel and PU-H71 infusions and determine how this interval could be affected with the use of an oral formulation of PU-H71, which is currently in clinical evaluation.
Appendix
Nab-Paclitaxel and PU-H71 Dose Adjustment
Nab-paclitaxel or PU-H71 dose reductions were not permitted during the DLT period. For dosing beyond cycle 1, nab-paclitaxel was held if patients experienced any other grade 3 or 4 toxicity thought to be related to nab-paclitaxel until symptoms resolved to grade 1/baseline grade. Two dose reductions for nab-paclitaxel (220 mg/m2 and 180 mg/m2) were permitted. Similarly, two dose reductions for PU-H71 (a decrease by 50 mg/m2 and 100 mg/m2 from the baseline dose level, respectively) were permitted.
Blood Sample Collection and Storage for Pharmacokinetics Assessment
Blood samples for plasma PU-H71 PK were drawn during cycles 1 and 2, on days 1 and 2, at the following time points relative to the start of the PU-H71 infusion: 5 minutes prior to infusion; immediately after stopping PU-H71 infusion; +30 minutes; +1.5 hour; +18-24 hours after stopping infusion of PU-H71, respectively. Each sample was collected in K2 ethylenediaminetetraacetic acid (EDTA) lavender top tubes and transferred into two polypropylene tubes (0.5 ml each). Tubes were stored at minus 70°C or lower prior to sending for analysis. Plasma concentration profiles of PU-H71 and its metabolites as well as PK parameters including area under the curve (AUC), pre-dose trough concentration; time to obtain maximum concentration, clearance, terminal half-life, and volume of distribution were calculated. The areas under the plasma concentration-time curves after each single dose (AUC0-all) were calculated using the log-linear trapezoidal method.
PU-PET Measurement and Clinical Benefit Correlation
Patients received a single microdose (<100 micrograms, non-therapeutic dose) of up to 407 megabecquerel (MBq) of [124I]-PU-H71 intravenously and underwent imaging at 3-4 hours and 20-24 hours after infusion using a single PET/CT scanner (Discovery DSTE, GE Healthcare Integrated IT Solutions, Barrington, IL). CT scans for attenuation correction and anatomic co-registration were performed before [124I]-PU-H71 tracer injection. PET data were reconstructed using a standard ordered subset expected maximization iterative algorithm. Emission data were corrected for scatter, attenuation, and decay. Interpretation of PU-PET was done by an experienced MSKCC nuclear medicine physician (M.D.), using dedicated PET/CT analysis software (AW Centricity Imaging-PACS/AW Suite, GE Healthcare Integrated IT Solutions). The PET appearance of tumors was dichotomized into categories of high (termed positive) versus low PU-H71-avidity (termed negative). High avidity was defined by tumor to blood pool tracer concentration ratios > 1 at both scan time points, where ratios ≤ 1 at either time point were considered low avidity. [124I]-PU-H71 tracer concentrations in tumors and non-tumor tissues were measured from reconstructed PET images from each time point. [124I]-PU-H71 tracer amounts were quantified in terms of standard uptake value (SUV). SUV was calculated as (PET-measured activity per cc of tissue-of-interest) divided by (total activity injected ÷ total grams of body mass). SUVpeak represents the average voxel SUV values within a 1cm3 spherical region of interest that includes the highest SUV voxel. For each patient, we calculated the tumor-specific molar concentrations that would be achieved by the therapeutic PU-H71 dose to be administered, predicted by tumor SUV measurements from the pretreatment tracer microdose [124I]-PU-H71 PET scans, using the following formalism:
Where [PU-H71tumor]t is the PET-derived intratumoral PU-H71 concentration (μM) ; PU-H71dose is the single therapeutic dose (mg) of nonradioactive PU-H71 administered for that patient each cycle; [Atumor]t is the tumor tracer concentration (in percentage of injected tracer-dose per cc tissue) measured by PET (divided by 100% to convert to decimal form); 0.8 (mL/g) is a typical tumor water space ; 512 is the molecular weight of PU-H71 ; and factor 1×106 converts the molar metric prefix to μM. PUH71 tracer was absent from blood pool by the 24-hour time point, thus no correction was needed for tumor blood pool activity at that time point.
Dose-Limiting Toxicity
The dose-limiting toxicity (DLT) observation period starts at Day 1 of Cycle 1, up to and including the assessments prior to drug administration on cycle 2, day1. Dose-limiting toxicity is defined as any of the following events:
• Any grade ≥ 4 nonhematologic adverse event.
• Any grade 3 nonhematologic adverse event not improving to baseline or grade ≤ 1 by day 14, despite adequate supportive care/toxicity management, unless listed below
• Any grade ≥ 3 elevation of liver enzymes of any duration
• Grade 4 thrombocytopenia
• Any grade 3 thrombocytopenia that has not recovered to grade ≤ 2 by day 7
• Grade 4 neutropenia lasting ≥ 7 days
• Febrile neutropenia
• Any treatment-related toxicity prompting a dose reduction of PU-H71 or nab-paclitaxel during the DLT observation period
• Grade ≥ 3 non-hematologic adverse event that is not due to disease progression or another clearly identifiable cause with the following exceptions: alopecia of any grade; grade 3 diarrhea that responds to therapy; grade 3 nausea or vomiting in the absence of premedication that responds to therapy.
- • Grade > 3 laboratory toxicities that are thought by the investigator to be clinically insignificant or related to an underlying condition will be discussed with the principle investigator and may not be considered DLT.
FIG A1.
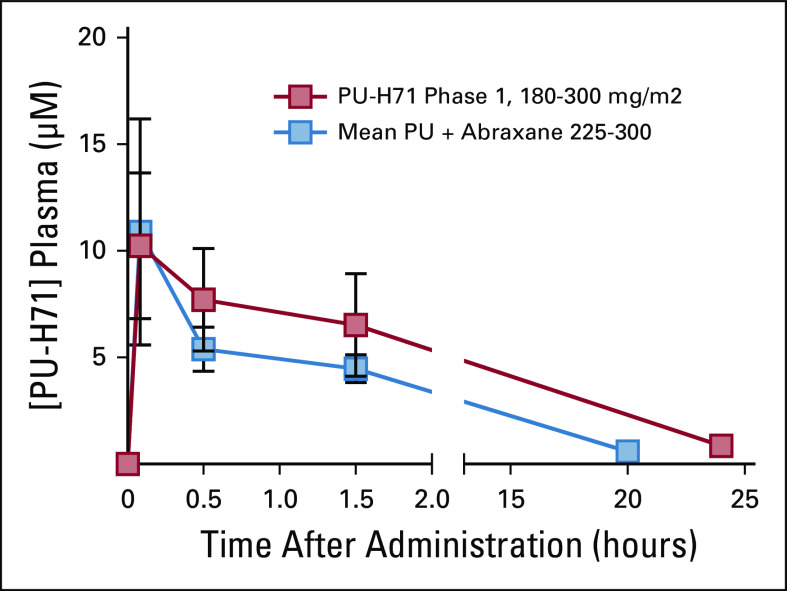 Plasma pharmacokinetics of PU-H71 +/- nab-paclitaxel. Concentration-time profiles after single doses of 225 or 300 mg/m2 PU-H71 on the PU-H71-nab-paclitaxel therapy (blue squares, n = 12) and after 180 through 300 mg/m2 (n = 11) on the single agent PU-H71, dose escalation therapy (NCT01393509, red squares). Values are mean ± SD.
Plasma pharmacokinetics of PU-H71 +/- nab-paclitaxel. Concentration-time profiles after single doses of 225 or 300 mg/m2 PU-H71 on the PU-H71-nab-paclitaxel therapy (blue squares, n = 12) and after 180 through 300 mg/m2 (n = 11) on the single agent PU-H71, dose escalation therapy (NCT01393509, red squares). Values are mean ± SD.

PRIOR PRESENTATION
Presented at the San Antonio Breast Cancer Symposium, San Antonio, TX, December 4-8, 2018.
SUPPORT
Supported by Mr. William H. Goodwin and Mrs. Alice Goodwin and the Commonwealth Foundation for Cancer Research and the Experimental Therapeutics Center of the Memorial Sloan Kettering Cancer Center, Grants No. R01 CA172546 and P30 CA008748 (NCI Core Facility Grant).
EQUAL CONTRIBUTION
K.L.J. and C.H.d.A. contributed equally.
M.P.D. and S.M. contributed equally.
AUTHOR CONTRIBUTIONS
Conception and design: Komal L. Jhaveri, Rui Wang, Nagavarakishore Pillarsetty, Gabriela Chiosis, Mark P. Dunphy, Shanu Modi
Administrative support: Weining Ma, Susan Duggan, Sarhe Khoshi, Nathan Kadija
Provision of study material or patients: Komal L. Jhaveri, Monica Fornier, Jacqueline F. Bromberg
Collection and assembly of data: Komal L. Jhaveri, Carlos H. dos Anjos, Tony Taldone, Elizabeth Comen, Monica Fornier, Jacqueline F. Bromberg, Weining Ma, Anna Rodina, Susan Duggan, Nathan Kadija, Mark P. Dunphy, Shanu Modi
Data analysis and interpretation: Komal L. Jhaveri, Carlos H. dos Anjos, Sujata Patil, Sarhe Khoshi, Gabriela Chiosis, Mark P. Dunphy, Shanu Modi
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Komal L. Jhaveri
Consulting or Advisory Role: Novartis, Pfizer, Spectrum Pharmaceuticals, AstraZeneca, Taiho Pharmaceutical, Jounce Therapeutics, ADC Therapeutics, Synthon, Intellisphere, Bristol Myers Squibb, Genentech, AbbVie, Eli Lilly
Research Funding: Novartis (Inst), Genentech (Inst), Debio Pharmaceuticals (Inst), ADC Therapeutics (Inst), Pfizer (Inst), Novita Pharmaceuticals (Inst), Clovis Oncology (Inst), Eli Lilly (Inst), Zymeworks (Inst), Immunomedics (Inst), Puma Biotechnology (Inst)
Travel, Accommodations, Expenses: Taiho Pharmaceutical, Jounce Therapeutics, Pfizer, AstraZeneca, Intellisphere
Carlos H. dos Anjos
Consulting or Advisory Role: MSD Oncology
Tony Taldone
Consulting or Advisory Role: Samus Therapeutics
Patents, Royalties, Other Intellectual Property: I am an inventor on patents covering associated composition of matter.
Elizabeth Comen
Employment: Survivornet
Stock and Other Ownership Interests: Survivornet.com
Honoraria: Navigant Consulting, Kantar Health, Grey Global Group, ClearView Healthcare Partners, Decision Resources, Gerson Lehrman Group, Pfizer
Consulting or Advisory Role: Bristol Myers Squibb, Pfizer, Genentech, HERON, Novartis
Research Funding: Roche
Travel, Accommodations, Expenses: Pfizer, Novartis
Monica Fornier
Honoraria: Eisai, Roche/Genentech
Consulting or Advisory Role: Eisai
Research Funding: Roche/Genentech
Nagavarakishore Pillarsetty
Stock and Other Ownership Interests: Amedisys, Madrigal Pharmaceuticals, Teva, Verastem, Ampio Pharmaceuticals, Merrimack, Celldex, ZIOPHARM Oncology, Immunogen, Bayer, OncoMed, Lexicon, ION Pharma, Viking Therapeutics
Patents, Royalties, Other Intellectual Property: N. Pillarsetty is coinventor on intellectual property on Hsp90/epichaperome targeting agents that were developed at MSKCC and is currently licensed to Samus Therapeutics. N. Pillarsetty has received financial royalties for his IP contributions that are licensed to Samus Therapeutics.
Sue Duggan
Employment: Samus Therapeutics
Gabriela Chiosis
Stock and Other Ownership Interests: Samus Therapeutics
Patents, Royalties, Other Intellectual Property: Samus Therapeutics; hold patents, have patents pending, receive royalties
Mark P. Dunphy
Patents, Royalties, Other Intellectual Property: Memorial Sloan Kettering Cancer Center holds the intellectual rights to PU-H71 and [124I]-PU-H71 (PCT/US06/03676, PCT/US2012/045861), which have been licensed to Samus Therapeutics. Dr. Dunphy has contributed to the patents and has received financial royalties for his IP contributions.
Shanu Modi
Honoraria: Novartis
Consulting or Advisory Role: Daiichi Sankyo, Macrogenics, AstraZeneca
Speakers' Bureau: Genentech, Daiichi Sankyo, AstraZeneca, Seattle Genetics
Research Funding: Roche/Genentech, Novartis, Seattle Genetics, Synta, Daiichi Sankyo
Travel, Accommodations, Expenses: Genentech, Daiichi Sankyo
No other potential conflicts of interest were reported.
REFERENCES
- 1.Hadizadeh Esfahani A, Sverchkova A, Saez-Rodriguez J, et al. A systematic atlas of chaperome deregulation topologies across the human cancer landscape. PLOS Comput Biol. 2018;14:e1005890. doi: 10.1371/journal.pcbi.1005890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brehme M, Voisine C, Rolland T, et al. A chaperome subnetwork safeguards proteostasis in aging and neurodegenerative disease. Cell Rep. 2014;9:1135–1150. doi: 10.1016/j.celrep.2014.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Joshi S, Wang T, Araujo TLS, et al. Adapting to stress - chaperome networks in cancer. Nat Rev Cancer. 2018;18:562–575. doi: 10.1038/s41568-018-0020-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rodina A, Wang T, Yan P, et al. The epichaperome is an integrated chaperome network that facilitates tumour survival. Nature. 2016;538:397–401. doi: 10.1038/nature19807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang T, Rodina A, Dunphy MP, et al. Chaperome heterogeneity and its implications for cancer study and treatment. J Biol Chem. 2019;294:2162–2179. doi: 10.1074/jbc.REV118.002811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kourtis N, Lazaris C, Hockemeyer K, et al. Oncogenic hijacking of the stress response machinery in T cell acute lymphoblastic leukemia. Nat Med. 2018;24:1157–1166. doi: 10.1038/s41591-018-0105-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jhaveri K, Wang R, Teplinsky E, et al. A phase I trial of ganetespib in combination with paclitaxel and trastuzumab in patients with human epidermal growth factor receptor-2 (HER2)-positive metastatic breast cancer. Breast Cancer Res. 2017;19:89. doi: 10.1186/s13058-017-0879-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jhaveri K, Chandarlapaty S, Lake D, et al. A phase II open-label study of ganetespib, a novel heat shock protein 90 inhibitor for patients with metastatic breast cancer. Clin Breast Cancer. 2014;14:154–160. doi: 10.1016/j.clbc.2013.12.012. [DOI] [PubMed] [Google Scholar]
- 9.Jhaveri K, Miller K, Rosen L, et al. A phase I dose-escalation trial of trastuzumab and alvespimycin hydrochloride (KOS-1022; 17 DMAG) in the treatment of advanced solid tumors. Clin Cancer Res. 2012;18:5090–5098. doi: 10.1158/1078-0432.CCR-11-3200. [DOI] [PubMed] [Google Scholar]
- 10.Modi S, Stopeck A, Linden H, et al. HSP90 inhibition is effective in breast cancer: A phase II trial of tanespimycin (17-AAG) plus trastuzumab in patients with HER2-positive metastatic breast cancer progressing on trastuzumab. Clin Cancer Res. 2011;17:5132–5139. doi: 10.1158/1078-0432.CCR-11-0072. [DOI] [PubMed] [Google Scholar]
- 11.Jhaveri K, Ochiana SO, Dunphy MP, et al. Heat shock protein 90 inhibitors in the treatment of cancer: Current status and future directions. Expert Opin Investig Drugs. 2014;23:611–628. doi: 10.1517/13543784.2014.902442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu W, Neckers L. Targeting the molecular chaperone heat shock protein 90 provides a multifaceted effect on diverse cell signaling pathways of cancer cells. Clin Cancer Res. 2007;13:1625–1629. doi: 10.1158/1078-0432.CCR-06-2966. [DOI] [PubMed] [Google Scholar]
- 13.Jhaveri K, Modi S. HSP90 inhibitors for cancer therapy and overcoming drug resistance. Adv Pharmacol. 2012;65:471–517. doi: 10.1016/B978-0-12-397927-8.00015-4. [DOI] [PubMed] [Google Scholar]
- 14.Jhaveri K, Taldone T, Modi S, et al. Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochim Biophys Acta. 2012;1823:742–755. doi: 10.1016/j.bbamcr.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Workman P, Clarke PA, Al-Lazikani B. Blocking the survival of the nastiest by HSP90 inhibition. Oncotarget. 2016;7:3658–3661. doi: 10.18632/oncotarget.6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Taldone T, Wang T, Rodina A, et al. A chemical biology approach to the chaperome in cancer-HSP90 and beyond. Cold Spring Harb Perspect Biol. 2020;12:a034116. doi: 10.1101/cshperspect.a034116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Caldas-Lopes E, Cerchietti L, Ahn JH, et al. Hsp90 inhibitor PU-H71, a multimodal inhibitor of malignancy, induces complete responses in triple-negative breast cancer models. Proc Natl Acad Sci USA. 2009;106:8368–8373. doi: 10.1073/pnas.0903392106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gerecitano J, Modi S, Rampal R, et al: Phase I trial of the HSP-90 inhibitor PU-H71. J Clin Oncol 33, 2015 (suppl 15; abstr 2537) [Google Scholar]
- 19. Chiosis G, Taldone T, Shrestha L, et al: Rational combination therapy for the treatment of cancer. US patent application PCT/US2016/055594, October 4, 2018.
- 20. doi: 10.1016/j.ccell.2019.09.007. Pillarsetty N, Jhaveri K, Taldone T, et al: Paradigms for precision medicine in epichaperome cancer therapy. Cancer Cell 36:559-573.e7, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dunphy MPS, Pressl C, Pillarsetty N, et al. First-in-human trial of epichaperome-targeted PET in patients with cancer. Clin Cancer Res. 2020;26:5178–5187. doi: 10.1158/1078-0432.CCR-19-3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 23.Johnson JR, Williams G, Pazdur R. End points and United States Food and Drug Administration approval of oncology drugs. J Clin Oncol. 2003;21:1404–1411. doi: 10.1200/JCO.2003.08.072. [DOI] [PubMed] [Google Scholar]
- 24.Gradishar WJ, Tjulandin S, Davidson N, et al. Phase III trial of nanoparticle albumin-bound paclitaxel compared with polyethylated castor oil-based paclitaxel in women with breast cancer. J Clin Oncol. 2005;23:7794–7803. doi: 10.1200/JCO.2005.04.937. [DOI] [PubMed] [Google Scholar]
- 25.Basso AD, Solit DB, Munster PN, et al. Ansamycin antibiotics inhibit Akt activation and cyclin D expression in breast cancer cells that overexpress HER2. Oncogene. 2002;21:1159–1166. doi: 10.1038/sj.onc.1205184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Modi S, Saura C, Henderson C, et al. A multicenter trial evaluating retaspimycin HCL (IPI-504) plus trastuzumab in patients with advanced or metastatic HER2-positive breast cancer. Breast Cancer Res Treat. 2013;139:107–113. doi: 10.1007/s10549-013-2510-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Modi S, Stopeck AT, Gordon MS, et al. Combination of trastuzumab and tanespimycin (17-AAG, KOS-953) is safe and active in trastuzumab-refractory HER-2 overexpressing breast cancer: A phase I dose-escalation study. J Clin Oncol. 2007;25:5410–5417. doi: 10.1200/JCO.2007.11.7960. [DOI] [PubMed] [Google Scholar]
- 28.Perez EA, Patel T, Moreno-Aspitia A. Efficacy of ixabepilone in ER/PR/HER2-negative (triple-negative) breast cancer. Breast Cancer Res Treat. 2010;121:261–271. doi: 10.1007/s10549-010-0824-0. [DOI] [PubMed] [Google Scholar]
- 29.Pivot X, Marmé F, Koenigsberg R, et al. Pooled analyses of eribulin in metastatic breast cancer patients with at least one prior chemotherapy. Ann Oncol. 2016;27:1525–1531. doi: 10.1093/annonc/mdw203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schmid P, Adams S, Rugo HS, et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N Engl J Med. 2018;379:2108–2121. doi: 10.1056/NEJMoa1809615. [DOI] [PubMed] [Google Scholar]