

Abstract
PURPOSE
Limited data are available on the prevalence and clinical impact of Lynch syndrome (LS)–associated genomic variants in non-European ancestry populations. We identified and characterized individuals harboring LS-associated variants in the ancestrally diverse BioMe Biobank in New York City.
PATIENTS AND METHODS
Exome sequence data from 30,223 adult BioMe participants were evaluated for pathogenic, likely pathogenic, and predicted loss-of-function variants in MLH1, MSH2, MSH6, and PMS2. Survey and electronic health record data from variant-positive individuals were reviewed for personal and family cancer histories.
RESULTS
We identified 70 individuals (0.2%) harboring LS-associated variants in MLH1 (n = 12; 17%), MSH2 (n = 13; 19%), MSH6 (n = 16; 23%), and PMS2 (n = 29; 41%). The overall prevalence was 1 in 432, with higher prevalence among individuals of self-reported African ancestry (1 in 299) than among Hispanic/Latinx (1 in 654) or European (1 in 518) ancestries. Thirteen variant-positive individuals (19%) had a personal history, and 19 (27%) had a family history of an LS-related cancer. LS-related cancer rates were highest in individuals with MSH6 variants (31%) and lowest in those with PMS2 variants (7%). LS-associated variants were associated with increased risk of colorectal (odds ratio [OR], 5.0; P = .02) and endometrial (OR, 30.1; P = 8.5 × 10−9) cancers in BioMe. Only 2 variant-positive individuals (3%) had a documented diagnosis of LS.
CONCLUSION
We found a higher prevalence of LS-associated variants among individuals of African ancestry in New York City. Although cancer risk is significantly increased among variant-positive individuals, the majority do not harbor a clinical diagnosis of LS, suggesting underrecognition of this disease.
INTRODUCTION
People with genetic variants linked to hereditary cancers are at increased risk for adverse health outcomes and often escape clinical diagnosis.1,2 Lynch syndrome (LS) is an autosomal dominant cancer syndrome caused by pathogenic variants in the mismatch repair (MMR) genes (MLH1, MSH2, MSH6, and PMS2) and deletions in the 3′ end of the EPCAM gene. Although mainly associated with colorectal cancer (CRC) and endometrial cancer (EC), LS also increases lifetime risk for various other cancers, including those of the ovaries, stomach, small intestine, upper urologic tract, brain, biliary system, pancreas, and cutaneous sebaceous glands.3,4 Individuals with LS-associated variants are at increased risk of developing multiple malignancies during their lifetime and are more likely to develop cancer at an early age.
CONTEXT
Key Objective
We sought to evaluate the prevalence and clinical impact of Lynch syndrome (LS)–associated genomic variants in unselected and diverse patient populations.
Knowledge Generated
Expected pathogenic variants in mismatch repair (MMR) genes were identified in approximately 1 in 430 participants of an ancestrally diverse, electronic health record–linked biobank in New York City. The highest prevalence was seen in individuals of self-reported African ancestry (approximately 1 in 300). Variant-positive individuals had significantly increased risk of endometrial cancer and colorectal cancer but not of breast cancer. The vast majority of variant-positive individuals did not have a documented diagnosis of LS in their medical records, even when they had LS-related cancers, suggesting underrecognition of this syndrome.
Relevance
Genomic screening for LS-associated variants in MMR genes may be an effective tool to identify individuals with elevated cancer risk.
Early identification of individuals harboring LS-associated variants in MMR genes provides the opportunity toreduce cancer-related morbidity and mortality through specialized cancer screening, prophylactic surgical measures, and/or chemoprevention.5-9 In current clinical practice, patients typically obtain genetic testing for LS when they meet clinical diagnostic criteria, including the Amsterdam II criteria or revised Bethesda guidelines.10,11 These criteria rely on personal and family cancer history, age of cancer onset, and/or molecular tumor characteristics, with sensitivities as low as 25% and 50%, respectively.12 As such, certain at-risk individuals may be missed. Recently, universal screening of all CRCs and ECs using microsatellite instability (MSI) and/or immunohistochemistry (IHC) for MMR protein expression has been recommended.13-15 Although more effective than clinical diagnostic criteria, this method of testing still misses patients with LS who do not present with EC or CRC.14
The prevalence of LS has been estimated as 1 in 440, with the majority of cases resulting from pathogenic variants in MLH1 and MSH2.16,17 However, prevalence estimates are based on studies ascertaining predominantly patients with European ancestry (EA) who met clinical diagnostic criteria for LS or were diagnosed with CRC and/or EC.18-21 Additionally, clinical testing for MSH6 and PMS2 was developed after testing for MLH1 and MSH2, such that many earlier studies of prevalence did not account for these genes.22 More recent epidemiologic data suggest that LS may be more common than previously appreciated, with an estimated prevalence as high as 1 in 279.23 Improved understanding of the prevalence and phenotypic spectrum of MMR gene variants is needed, especially within non-EA populations. This study evaluated the prevalence and clinical impact of expected pathogenic MMR variants in an ancestrally diverse, unselected population biobank.
PATIENTS AND METHODS
Study Population
The BioMe Biobank is an electronic health record (EHR)–linked biobank with > 55,000 participants enrolled nonselectively from the Mount Sinai Health System in New York City. The study population consisted of 30,223 consented BioMe participants age 18 years or older (upon enrollment) and with exome sequence data available through a collaboration with the Regeneron Genetics Center. Details of the study population have previously been described.1 Self-reported ancestry of participants was derived from a multiple-choice survey administered on enrollment in BioMe.1,24 This study was approved by the Icahn School of Medicine at Mount Sinai’s institutional review board.
Generation of Genomic Data and MMR Variant Annotation
Sample preparation and exome sequencing were performed at the Regeneron Genetics Center.25 Post hoc filtering of samples and quality control of sequence data were performed as described previously, resulting in a total of 30,223 samples from participants age 18 years and older.1 Sequence data for the MMR genes (MLH1, MSH2, MSH6, and PMS2) were extracted from exome sequences. Variants were annotated with the Variant Effect Predictor and cross-referenced with the ClinVar database (accessed March 2019).26 Exome sequencing did not capture large genomic rearrangements in the MMR genes or in the 3′ area of EPCAM.
Pathogenic variants in MMR genes were identified based on a pathogenic or likely pathogenic assertion in ClinVar. In addition, we considered predicted loss-of-function (pLOF) variants not classified in ClinVar, including frameshift, stop-gain, start-loss, stop-loss, or canonical splice acceptor or donor. The union of ClinVar pathogenic/likely pathogenic and pLOF variants was termed expected pathogenic (n = 44) and was used to identify variant-positive individuals in BioMe for subsequent analyses. Additional quality control included manual inspection of sequence reads for variant-positive individuals (n = 23) harboring 1 of 7 multiallelic sites annotated as expected pathogenic. Of these, 12 of 15 carriers of MSH6 c.3261dupC and 1 of 2 carriers of MSH6 c.3261del were determined to be false positives and excluded from downstream analyses.
Evaluation of Clinical Characteristics in Variant-Positive Individuals
Individuals harboring expected pathogenic variants in MLH1, MSH2, MSH6, or PMS2 in BioMe, termed variant positive (N = 70), were evaluated for personal and/or family histories of LS-related and other cancers. For this evaluation, we extracted International Classification of Diseases (ICD)-9 and ICD-10 codes related to these cancers from EHRs (Appendix Table A1). Screening or diagnostic testing for CRC and EC was evaluated by extraction of Current Procedural Terminology codes for colonoscopy (45378-45398) and transvaginal or pelvic ultrasound (76830, 76831, 76856, 76857). We carried out medical record review of variant-positive individuals to further evaluate personal and family cancer histories and to evaluate for clinical diagnosis, genetic testing, or tumor screening for LS. These data were supplemented by participant survey data on personal and family cancer histories, which were available for 18 variant-positive individuals. Personal and family cancer histories were used to determine whether variant-positive individuals fulfilled Amsterdam II criteria10 and/or revised Bethesda guidelines11 (Appendix Table A2). Data were summarized using medians for continuous variables and frequencies and percentages for categoric variables.
LS-Related Cancer Case-Control Studies
We evaluated CRC, EC, and breast cancer (BC) risks in LS variant-positive versus variant-negative individuals in BioMe. We excluded individuals within BioMe who were second-degree relatives and closer from these analyses, as previously described.1,27 This exclusion corrects for potential bias in the statistical analysis and is consistent with best practices for genetic association analyses.28 For each cancer type, patient cases were defined as having any ICD-9/10 code for personal history of the cancer, and controls were defined as individuals without any of these codes. For CRC, we tested for association with variant-positive (n = 64) versus variant-negative (n = 26,942) unrelated individuals in BioMe, using the saddlepoint approximation to account for unbalanced case-control ratios29 (implemented via “SPAtest” package, R [version 3.5.3] and adjusting for age, sex, and the first 5 principal components of ancestry). EC and BC were assessed in variant-positive (n = 34) versus variant-negative (n = 15,669) unrelated women, adjusting for age and the first 5 principal components.
RESULTS
We evaluated variants in MLH1, MSH2, MSH6, and PMS2 among 30,223 adult participants of the BioMe Biobank with available exome sequencing data. Demographics of these participants have been described previously: 59% were women, the median age was 59 years, and 74% were non-EA by self-report.1 We identified 44 expected pathogenic variants in MMR genes, including 7 MLH1, 5 MSH2, 14 MSH6, and 18 PMS2 variants (Table 1). There were 70 BioMe participants harboring at least one of these 44 variants. Though the majority (75%) of these variants were observed as singletons, 3 variants were observed in more than 5 individuals each, including MSH2 c.1697del (n = 8), MLH1 c.790+1G>A (n = 6), and PMS2 c.137G>T (n = 6). None of these recurrent variants were observed predominantly in a single ancestry group, and none of the carriers of recurrent variants were related to one another.
TABLE 1.
Expected Pathogenic Variants in MMR Genes Identified Among 30,223 BioMe Participants

Overall, 70 individuals (0.2%) in BioMe harbored expected pathogenic variants in MMR genes (Table 2; Appendix Table A3). PMS2 variants were most frequently identified (41%), followed by MSH6 (23%), MSH2 (19%), and MLH1 (17%). Variant-positive individuals were 56% female, with a median age of 55 years. The overall estimated prevalence of BioMe participants harboring expected pathogenic variants in MMR genes was 1 in 432. Prevalence was unchanged (1 in 428) in an unrelated subset of BioMe that included only 1 individual from each first- or second-degree relationship (n = 27,816). Prevalence varied across self-reported ancestry groups and was highest in individuals of self-reported African American/African (AA) ancestry (1 in 299), with most harboring variants in MSH6 (30%) or PMS2 (39%). Prevalence was lower in EA (1 in 518) and Hispanic/Latinx (H/L; 1 in 654) ancestries.
TABLE 2.
Demographics and Clinical Characteristics of 70 BioMe Participants Harboring Expected Pathogenic Variants in MLH1, MSH2, MSH6, or PMS2

We evaluated for LS-associated clinical characteristics in the 70 variant-positive individuals in BioMe. The overall rate of malignancy was 34%, with the lowest rate in individuals harboring PMS2 (21%) compared with MSH2 (38%), MLH1 (42%), or MSH6 (50%) variants (Table 2). Of the 24 variant-positive individuals with cancer, 13 (54%) had a tumor type associated with LS (Appendix Table A3). LS-related cancers included cancers of the endometrium (n = 7; 18% of women), colorectum (n = 3; 4%), stomach or small bowel (n = 2; 3%), ovary or fallopian tube (n = 1; 3% of women), brain (n = 1, 1%), and cutaneous sebaceous gland (n = 1, 1%). Two individuals had 2 distinct LS-related cancers: an AA woman harboring MSH6 c.892C>T with EC and CRC, and an H/L man harboring MSH2 c.478C>T with small bowel and sebaceous gland cancers. Rates of LS-related cancers varied across ancestry groups, with higher rates in individuals of EA ancestry (27%) and lower rates in those of AA (13%) and H/L (13%) ancestries (Fig 1). Each of the 13 individuals with an LS-related cancer harbored a distinct MMR gene variant, with the exception of 2 individuals with MLH1 c.790+1G>A (one with CRC and the other with gastric cancer) and 2 with MSH2 c.1697del (one with fallopian tube cancer and the other with EC). Other cancer types observed in variant-positive individuals included BC (n = 5); skin cancers, including squamous and basal cell carcinomas and melanoma (n = 4); prostate cancer (n = 3); and lung cancer (n = 2). Two individuals had the same 2 variants, PMS2 c.2444C>T (likely pathogenic) and c.2331dup (pLOF), for which the phase could not be determined. Neither of these individuals had EHR evidence of features consistent with constitutional MMR deficiency (which results from biallelic germline variants in MMR genes), including hematologic, brain, intestinal tract, or other cancers. Only 10 (31%) of 32 women without EC had undergone ultrasonography of the pelvis, and 15 (22%) of 67 individuals without CRC had completed a colonoscopy. Among 34 variant-positive individuals (49%) with a family history of cancer, the majority (n = 19; 56%) had first- and/or second-degree relatives diagnosed with an LS-related cancer (Appendix Table A3). Family members of individuals harboring MLH1 variants had the highest rates of LS-related cancers (50%) or any cancer type (67%) compared with family members of individuals harboring other MMR gene variants (Table 2).
FIG 1.

Rates of Lynch syndrome (LS)–related and overall cancers among self-reported ancestry groups in the BioMe Biobank. Rates of LS-related cancers and any cancer type were evaluated in all 70 BioMe participants harboring LS-associated variants, and across the 3 largest self-reported ancestry groups: African American/African (AA; n = 23), Hispanic/Latinx (H/L; n = 16), and European (EA; n = 15). South Asian (n = 2), East/Southeast Asian (n = 1), multiple selected (n = 8), and other (n = 5) ancestries are not shown.
Review of medical records revealed that 2 variant-positive individuals (3%) had a diagnosis of LS. One was an H/L man harboring MSH2 c.478C>T with duodenal adenocarcinoma and sebaceous gland cancer, consistent with the Muir-Torre variant of LS. IHC for the MMR proteins of both tumors revealed a pattern of absent or reduced MSH2 and absent MSH6. This was the only individual with evidence of IHC or MSI screening in tumor tissue. The second individual with a diagnosis of LS was an EA woman harboring MLH1 c.184C>T with EC and malignant pleural mesothelioma. This was the only individual who fulfilled Amsterdam II criteria. Of the 13 variant-positive individuals with LS-related cancers, 2 fulfilled the revised Bethesda guidelines for tumor testing of MSI: an AA woman harboring MSH6 c.892C>T with CRC, EC, and BC; and a South Asian man harboring MLH1 c.790+1G>A with CRC. Of the 7 ECs and 3 CRCs in our study population, tumor histology was only available for an EA woman harboring PMS2 c.137G>T with endometrioid EC. IHC/MSI screening was not performed.
We tested for association with the most frequently observed cancers—CRC, EC, and BC—in unrelated LS variant-positive versus variant-negative individuals in BioMe (Fig 2). Variant-positive individuals had significantly increased odds of CRC (odds ratio [OR], 5.0; 95% CI, 1.3 to 18.8; P = .02) compared with variant-negative individuals. Variant-positive women had significantly increased odds of EC (OR, 30.8; 95% CI, 9.6 to 99.0; P = 8.5 × 10−9) but not of BC (OR, 2.5; 95% CI, 0.9 to 6.9; P = .08) compared with variant-negative women.
FIG 2.
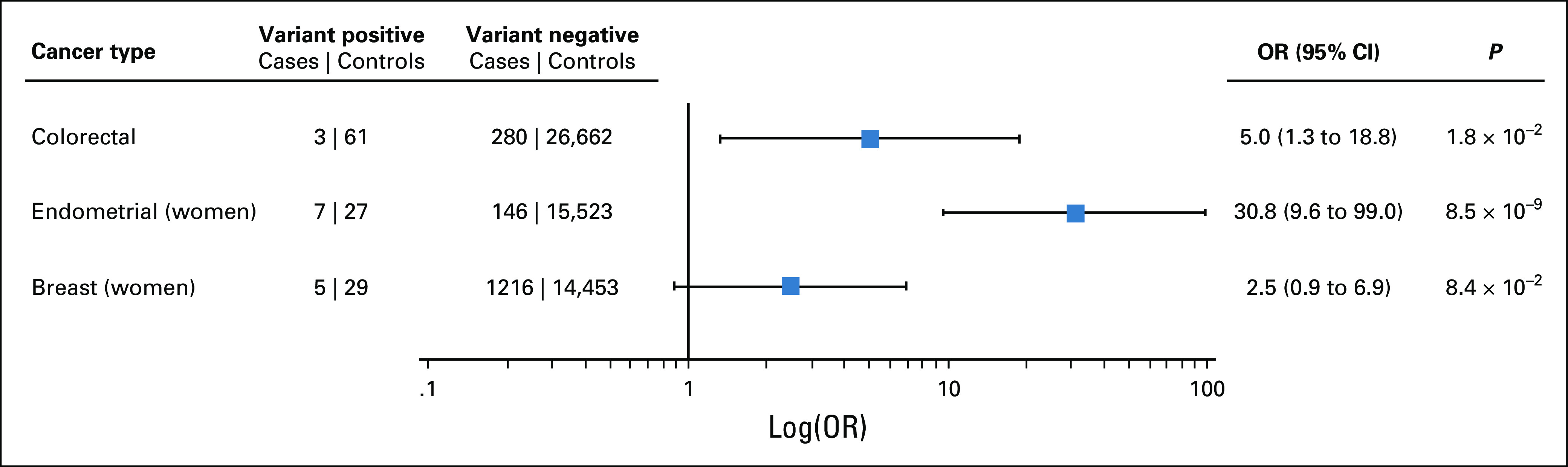
Lynch syndrome (LS)–associated variants are associated with increased risk of colorectal cancer (CRC) and endometrial cancer. CRC, endometrial cancer, and breast cancer cases and controls were defined using International Classification of Diseases (ICD)-9 and ICD-10 codes extracted from electronic health records. We excluded second-degree relatives and closer from these analyses. The odds ratio (OR) for CRC was calculated by saddlepoint approximation, adjusting for age, sex, and the first 5 principal components of ancestry. ORs for endometrial and breast cancers were similarly calculated in women only, adjusting for age and the first 5 principal components.
DISCUSSION
We identified and characterized individuals harboring LS-associated variants in MMR genes within a large, ancestrally diverse biobank in New York City. The overall prevalence of expected pathogenic MMR variants was ∼1 in 430, similar to previous LS prevalence estimates16,23; however, prevalence varied across ancestry groups. Consistent with our previous work identifying genomic risk for BRCA1/2-related cancers,1 we found that the vast majority of variant-positive individuals did not have an awareness of their increased cancer risk.
Expected pathogenic variants in PMS2 and MSH6 were most common, with frequencies of ∼1 in 1,000 and 1 in 1,900. Although up to 90% of LS has been attributed to germline mutations in MLH1 or MSH2, more recent studies have shown PMS2 and MSH6 variants to be more frequent.16,23,30 Prior studies ascertaining patients with CRC and/or meeting clinical diagnostic criteria for LS may have underestimated the prevalence of MSH6 and PMS2 variants, which are thought to be less penetrant than MLH1 and MSH2 variants.22,31 In this study, individuals harboring an expected pathogenic variant in PMS2 had the lowest rates of cancer compared with other MMR genes, suggesting lower penetrance. However, individuals harboring expected pathogenic variants in MSH6 had the highest rates of cancer among variant-positive individuals.
We estimated the prevalence of LS-associated variants in diverse populations, including self-reported AA and H/L ancestry groups, for which estimates did not previously exist. The prevalence in individuals of H/L ancestry was ∼1 in 650, lower than that seen in AA and EA ancestries. The highest prevalence (∼1 in 300) was in individuals of AA ancestry. Few studies have described LS in AA populations to date, despite AA patients having the highest CRC incidence and mortality of all patient populations.32 Guindalini et al33 previously evaluated 51 AA families with pathogenic variants or variants of uncertain significance in an MMR gene and showed that 61% of families had a deleterious variant in MLH1. This is distinct from our findings, in which the majority of AA individuals harbored expected pathogenic variants in MSH6 (30%) or PMS2 (39%).
Expected pathogenic variants in MMR genes were associated with significantly increased risk of EC and CRC in BioMe. EC and CRC were the most frequently observed cancers, affecting 18% of variant-positive women and 4% of variant-positive individuals, respectively. Notably, 4 of 7 women with EC harbored MSH6 variants. Several previous studies have suggested a higher risk for EC in women with pathogenic variants in MSH6 compared with other MMR genes, with an estimated 26% cumulative risk of EC by age 70 years.34-36 Our data did not support an overall association between MMR pathogenic variants and BC risk, which is the subject of ongoing debate.37,38 Some studies have demonstrated increased BC rates in women with MMR pathogenic variants, most recently in MSH6 and PMS2,22,39 while other studies have not.40-42 The highest rate of BC in our study was in women with MSH6 variants (3 of 11). None of the 12 women with a PMS2 variant had evidence of BC.
Formal diagnosis of LS was only documented in EHRs for 2 variant-positive individuals. Lack of awareness of increased cancer risk could be due in part to the diversity of this cohort, as racial and ethnic minorities access genetic counseling and testing considerably less than non-Hispanic whites.43,44 Previous studies have shown that the Amsterdam II criteria identify approximately 13%-67% of families with LS, and the revised Bethesda guidelines detect approximately 78%-91% of patients with LS, both of which are markedly higher than 1% and 3%, respectively, seen in this study.45 Overall, we found that 96% of variant-positive individuals did not meet any current clinical diagnostic criteria for LS testing. This is consistent with the growing concerns that medical history–based screening strategies fail to identify the majority of individuals with high genomic risk for LS and other hereditary cancers.46 Universal tumor screening of all newly diagnosed CRCs and ECs is now recommended to increase the identification of LS.13-15 However, this approach did not appear to enable LS diagnosis in this study: no variant-positive individuals with CRC or EC had EHR documentation of IHC or MSI screening, likely because their cancers were diagnosed before the widespread adoption of tumor screening and/or outside of the health system.
This study had some limitations. First, personal and family cancer histories were obtained retrospectively from EHR data, which may be incomplete and/or inaccurate. This might downwardly bias the estimation of penetrance of LS-associated variants. Additionally, we used exome sequencing to identify expected pathogenic variants in MMR genes; however, this does not capture large genomic rearrangements, such as multiexon deletions or duplications in MMR genes or in EPCAM, which can account for a large portion of LS.30,47 Detection of pathogenic PMS2 variants may have been confounded by numerous homologous pseudogenes, particularly PMS2CL, potentially resulting in a portion of PMS2 variant-positive individuals having false positive results.48 Of note, there were 13 individuals harboring expected pathogenic variants at multiallelic sites who were excluded from this study after manual review of the variants. However, we noted that 1 of these individuals had a personal history of LS-related cancer, and 2 others had first-degree relatives with LS-related cancers. Therefore, the quality control stringency applied may have excluded true variant-positive individuals from the study. Validation by Sanger sequencing or other methods would be needed to confirm the presence of lower-quality variants identified by research exome sequencing. Finally, this study did not include variants of uncertain significance, which can be observed more frequently in non-EA populations1; some of these may eventually be reclassified as pathogenic as knowledge about LS-associated variants continues to grow.
We found that the vast majority of variant-positive individuals did not have EHR evidence of a LS diagnosis, even when they had LS-related cancers, suggesting underrecognition of this syndrome. Genomic screening for LS-associated genomic variants in unselected and diverse populations can be used to identify individuals at elevated risk of CRC, EC, and other cancers. Prospective studies using this genomics-first approach are needed to better understand the penetrance of MMR variants and to inform targeted clinical strategies for the prevention and earlier diagnosis of LS-related cancers.
ACKNOWLEDGMENT
We thank participants of the BioMe Biobank for their permission to use their health and genomic information.
APPENDIX
TABLE A1.
ICD Codes for LS-Related and Other Cancers

TABLE A2.
Amsterdam II Criteria and Revised Bethesda Guidelines for Identification of Individuals at Risk for LS

TABLE A3.
Clinical Characteristics of Individuals Harboring Expected Pathogenic Variants in MMR Genes

PRIOR PRESENTATION
Presented in part at the Annual Meeting of the American Society of Clinical Oncology, Chicago, IL, May 31-June 4, 2019.
SUPPORT
Supported by dedicated funding to the Institute for Genomic Health by the Icahn School of Medicine at Mount Sinai. S.A.S., G.M.B., E.E.K., and N.S.A-H. are supported by the National Institutes of Health, National Human Genome Research Institute (NHGRI) and by National Institute on Minority Health and Health Disparities Grant No. U01 HG009610. E.E.K. and N.S.A.-H. are supported by NHGRI Grant No. U01 HG011176. E.E.K. is supported by NHGRI Grants No. R01 HG010297, U01 HG009080, UM1 HG0089001, and U01 HG007417; National Heart, Lung, Blood Institute Grants No. R01 HL104608, X01 HL1345; and National Institute of Diabetes and Kidney and Digestive Disease Grant No. R01 DK110113. A.L.L. is supported by American Cancer Society Grant No. 129387-MRSG-16-015-01-CPHPS. Exome sequencing of BioMe participants was performed in collaboration with the Regeneron Genetics Center.
AUTHOR CONTRIBUTIONS
Conception and design: Celina Ang, Aimee L. Lucas, Eimear E. Kenny, Noura S. Abul-Husn
Collection and assembly of data: Rachel E. Rosenblum, Meenakshi R. Siggireddi, Sinead Cullina, Gillian M. Belbin, Eimear E. Kenny, Noura S. Abul-Husn
Data analysis and interpretation: Rachel E. Rosenblum, Celina Ang, Sabrina A. Suckiel, Emily R. Soper, Meenakshi R. Sigireddi, Gillian M. Belbin, Aimee L. Lucas, Eimear E. Kenny, Noura S. Abul-Husn
Financial support: Eimear E. Kenny, Noura S. Abul-Husn
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Aimee L. Lucas
Research Funding: Immunovia
Eimear E. Kenny
Honoraria: Illumina, Regeneron
Noura S. Abul-Husn
Honoraria: Genentech
Employment: Regeneron
No other potential conflicts of interest were reported.
REFERENCES
- 1.Abul-Husn NS, Soper ER, Odgis JA, et al. Exome sequencing reveals a high prevalence of BRCA1 and BRCA2 founder variants in a diverse population-based biobank. Genome Med. 2019;12:2. doi: 10.1186/s13073-019-0691-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Manickam K, Buchanan AH, Schwartz MLB, et al. Exome sequencing-based screening for BRCA1/2 expected pathogenic variants among adult biobank participants. JAMA Netw Open. 2018;1:e182140. doi: 10.1001/jamanetworkopen.2018.2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Watson P, Lynch HT. The tumor spectrum in HNPCC. Anticancer Res. 1994;14:1635–1639. [PubMed] [Google Scholar]
- 4.Watson P, Vasen HFA, Mecklin JP, et al. The risk of extra-colonic, extra-endometrial cancer in the Lynch syndrome. Int J Cancer. 2008;123:444–449. doi: 10.1002/ijc.23508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Järvinen HJ, Aarnio M, Mustonen H, et al. Controlled 15-year trial on screening for colorectal cancer in families with hereditary nonpolyposis colorectal cancer. Gastroenterology. 2000;118:829–834. doi: 10.1016/s0016-5085(00)70168-5. [DOI] [PubMed] [Google Scholar]
- 6.de Jong AE, Hendriks YM, Kleibeuker JH, et al. Decrease in mortality in Lynch syndrome families because of surveillance. Gastroenterology. 2006;130:665–671. doi: 10.1053/j.gastro.2005.11.032. [DOI] [PubMed] [Google Scholar]
- 7.Lindor NM, Petersen GM, Hadley DW, et al. Recommendations for the care of individuals with an inherited predisposition to Lynch syndrome: A systematic review. JAMA. 2006;296:1507–1517. doi: 10.1001/jama.296.12.1507. [DOI] [PubMed] [Google Scholar]
- 8.Schmeler KM, Lynch HT, Chen LM, et al. Prophylactic surgery to reduce the risk of gynecologic cancers in the Lynch syndrome. N Engl J Med. 2006;354:261–269. doi: 10.1056/NEJMoa052627. [DOI] [PubMed] [Google Scholar]
- 9.Burn J, Sheth H, Elliott F, et al. Cancer prevention with aspirin in hereditary colorectal cancer (Lynch syndrome), 10-year follow-up and registry-based 20-year data in the CAPP2 study: A double-blind, randomised, placebo-controlled trial. Lancet. 2020;395:1855–1863. doi: 10.1016/S0140-6736(20)30366-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vasen HF, Watson P, Mecklin JP, et al. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology. 1999;116:1453–1456. doi: 10.1016/s0016-5085(99)70510-x. [DOI] [PubMed] [Google Scholar]
- 11.Umar A, Boland CR, Terdiman JP, et al. Revised Bethesda guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst. 2004;96:261–268. doi: 10.1093/jnci/djh034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tranø G, Sjursen W, Wasmuth HH, et al. Performance of clinical guidelines compared with molecular tumour screening methods in identifying possible Lynch syndrome among colorectal cancer patients: A Norwegian population-based study. Br J Cancer. 2010;102:482–488. doi: 10.1038/sj.bjc.6605509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Evaluation of Genomic Applications in Practice and Prevention (EGAPP) Working Group Recommendations from the EGAPP Working Group: Genetic testing strategies in newly diagnosed individuals with colorectal cancer aimed at reducing morbidity and mortality from Lynch syndrome in relatives. Genet Med. 2009;11:35–41. doi: 10.1097/GIM.0b013e31818fa2ff. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Palomaki GE, McClain MR, Melillo S, et al. EGAPP supplementary evidence review: DNA testing strategies aimed at reducing morbidity and mortality from Lynch syndrome. Genet Med. 2009;11:42–65. doi: 10.1097/GIM.0b013e31818fa2db. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Committee on Practice Bulletins ACOG Practice Bulletin No. 147: Lynch syndrome. Obstet Gynecol. 2014;124:1042–1054. doi: 10.1097/01.AOG.0000456325.50739.72. [DOI] [PubMed] [Google Scholar]
- 16.Chen S, Wang W, Lee S, et al. Prediction of germline mutations and cancer risk in the Lynch syndrome. JAMA. 2006;296:1479–1487. doi: 10.1001/jama.296.12.1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peltomäki P, Vasen H. Mutations associated with HNPCC predisposition: Update of ICG-HNPCC/INSiGHT mutation database. Dis Markers. 2004;20:269–276. doi: 10.1155/2004/305058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yurgelun MB, Kulke MH, Fuchs CS, et al. Cancer susceptibility gene mutations in individuals with colorectal cancer. J Clin Oncol. 2017;35:1086–1095. doi: 10.1200/JCO.2016.71.0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hampel H, Frankel WL, Martin E, et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer) N Engl J Med. 2005;352:1851–1860. doi: 10.1056/NEJMoa043146. [DOI] [PubMed] [Google Scholar]
- 20.Moreira L, Balaguer F, Lindor N, et al. Identification of Lynch syndrome among patients with colorectal cancer. JAMA. 2012;308:1555–1565. doi: 10.1001/jama.2012.13088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hampel H, Frankel W, Panescu J, et al. Screening for Lynch syndrome (hereditary nonpolyposis colorectal cancer) among endometrial cancer patients. Cancer Res. 2006;66:7810–7817. doi: 10.1158/0008-5472.CAN-06-1114. [DOI] [PubMed] [Google Scholar]
- 22.Espenschied CR, LaDuca H, Li S, et al. Multigene panel testing provides a new perspective on Lynch syndrome. J Clin Oncol. 2017;35:2568–2575. doi: 10.1200/JCO.2016.71.9260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Win AK, Jenkins MA, Dowty JG, et al. Prevalence and penetrance of major genes and polygenes for colorectal cancer. Cancer Epidemiol Biomarkers Prev. 2017;26:404–412. doi: 10.1158/1055-9965.EPI-16-0693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Belbin GM , Wenric S, Cullina S, et al. 2019 Towards a fine-scale population health monitoring System. bioRxiv:780668.
- 25.Dewey FE, Gusarova V, O’Dushlaine C, et al. Inactivating variants in ANGPTL4 and risk of coronary artery disease. N Engl J Med. 2016;374:1123–1133. doi: 10.1056/NEJMoa1510926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Landrum MJ, Lee JM, Riley GR, et al. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014;42(D1):D980–D985. doi: 10.1093/nar/gkt1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Manichaikul A, Mychaleckyj JC, Rich SS, et al. Robust relationship inference in genome-wide association studies. Bioinformatics. 2010;26:2867–2873. doi: 10.1093/bioinformatics/btq559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wellcome Trust Case Control Consortium Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dey R, Schmidt EM, Abecasis GR, et al. A Fast and accurate algorithm to test for binary phenotypes and its application to PheWAS. Am J Hum Genet. 2017;101:37–49. doi: 10.1016/j.ajhg.2017.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DA Silva FC, Wernhoff P, Dominguez-Barrera C, et al. Update on hereditary colorectal cancer. Anticancer Res. 2016;36:4399–4405. doi: 10.21873/anticanres.10983. [DOI] [PubMed] [Google Scholar]
- 31.Yamano T, Hamanaka M, Babaya A, et al. Management strategies in Lynch syndrome and familial adenomatous polyposis: A national healthcare survey in Japan. Cancer Sci. 2017;108:243–249. doi: 10.1111/cas.13123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Howlader N, Noone AM, Krapcho M, et al (eds): SEER Cancer Statistics Review, 1975-2012. Bethesda, MD, National Cancer Institute, 2014. [Google Scholar]
- 33.Guindalini RS, Win AK, Gulden C, et al. Mutation spectrum and risk of colorectal cancer in African American families with Lynch syndrome. Gastroenterology. 2015;149:1446–1453. doi: 10.1053/j.gastro.2015.07.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramsoekh D, Wagner A, van Leerdam ME, et al. A high incidence of MSH6 mutations in Amsterdam criteria II–negative families tested in a diagnostic setting. Gut. 2008;57:1539–1544. doi: 10.1136/gut.2008.156695. [DOI] [PubMed] [Google Scholar]
- 35.Baglietto L, Lindor NM, Dowty JG, et al. Risks of Lynch syndrome cancers for MSH6 mutation carriers. J Natl Cancer Inst. 2010;102:193–201. doi: 10.1093/jnci/djp473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Talseth-Palmer BA, McPhillips M, Groombridge C, et al. MSH6 and PMS2 mutation positive Australian Lynch syndrome families: Novel mutations, cancer risk and age of diagnosis of colorectal cancer. Hered Cancer Clin Pract. 2010;8:5. doi: 10.1186/1897-4287-8-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Castellsagué E, Foulkes WD. Lynch syndrome: Five unanswered questions. Clin Genet. 2015;87:503–506. doi: 10.1111/cge.12580. [DOI] [PubMed] [Google Scholar]
- 38.Ten Broeke SW, Suerink M, Nielsen M. Response to Roberts et al. 2018: Is breast cancer truly caused by MSH6 and PMS2 variants or is it simply due to a high prevalence of these variants in the population? Genet Med. 2019;21:256–257. doi: 10.1038/s41436-018-0029-1. [DOI] [PubMed] [Google Scholar]
- 39.Roberts ME, Jackson SA, Susswein LR, et al. MSH6 and PMS2 germ-line pathogenic variants implicated in Lynch syndrome are associated with breast cancer. Genet Med. 2018;20:1167–1174. doi: 10.1038/gim.2017.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Couch FJ, Shimelis H, Hu C, et al. Associations between cancer predisposition testing panel genes and breast cancer. JAMA Oncol. 2017;3:1190–1196. doi: 10.1001/jamaoncol.2017.0424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Møller P, Seppälä TT, Bernstein I, et al. Cancer risk and survival in path_MMR carriers by gene and gender up to 75 years of age: A report from the Prospective Lynch Syndrome Database. Gut. 2018;67:1306–1316. doi: 10.1136/gutjnl-2017-314057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stoll J, Rosenthal E, Cummings S, et al. No evidence of increased risk of breast cancer in women with Lynch syndrome identified by multigene panel testing. Precision Oncol. 2020;4:51–60. doi: 10.1200/PO.19.00271. [DOI] [PubMed] [Google Scholar]
- 43.Hall M, Olopade OI. Confronting genetic testing disparities: Knowledge is power. JAMA. 2005;293:1783–1785. doi: 10.1001/jama.293.14.1783. [DOI] [PubMed] [Google Scholar]
- 44.Huo D, Olopade OI. Genetic testing in diverse populations: Are researchers doing enough to get out the correct message? JAMA. 2007;298:2910–2911. doi: 10.1001/jama.298.24.2910. [DOI] [PubMed] [Google Scholar]
- 45.Giardiello FM, Allen JI, Axilbund JE, et al. Guidelines on genetic evaluation and management of Lynch syndrome: A consensus statement by the US Multi-Society Task Force on colorectal cancer. Gastroenterology. 2014;147:502–526. doi: 10.1053/j.gastro.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 46.Murray MF, Evans JP, Khoury MJ. DNA-based population screening: Potential suitability and important knowledge gaps. JAMA. 2019 doi: 10.1001/jama.2019.18640. 323:307-308, [DOI] [PubMed] [Google Scholar]
- 47.Smith MJ, Urquhart JE, Harkness EF, et al. The contribution of whole gene deletions and large rearrangements to the mutation spectrum in inherited tumor predisposing syndromes. Hum Mutat. 2016;37:250–256. doi: 10.1002/humu.22938. [DOI] [PubMed] [Google Scholar]
- 48.De Vos M, Hayward BE, Picton S, et al. Novel PMS2 pseudogenes can conceal recessive mutations causing a distinctive childhood cancer syndrome. Am J Hum Genet. 2004;74:954–964. doi: 10.1086/420796. [DOI] [PMC free article] [PubMed] [Google Scholar]