

INTRODUCTION
Chromosomal instability is one of the characteristics of gastric cancer (GC)1,2 associated with frequent amplifications of receptor tyrosine kinase (RTK)-related genes. Epidermal growth factor receptor (EGFR) is a transmembrane RTK, and its dysregulation is caused by altered EGFR gene that drives cancers.3 Approximately 5%-10% of patients with GC have EGFR amplification, which indicates a poor prognosis.4-6 Several studies suggested the benefit of anti-EGFR therapy for GC with high EGFR copy number (CN).7,8 However, randomized trials failed to demonstrate the survival benefit of anti-EGFR treatments for advanced GC without patient enrichment.9,10
Intratumoral heterogeneity and concurrent genomic alterations in downstream molecules or other signaling pathways have been suggested as possible resistance mechanisms to EGFR-targeted therapies for GC.11 Circulating tumor DNA (ctDNA) analysis is a useful method to detect genomic alterations of tumor cells throughout the body and to identify concurrent heterogeneous resistance mechanisms possibly missed in single-lesion tumor biopsies.12,13 Using serial ctDNA analysis, Maron et al14,15 identified acquired genomic alterations in patients with EGFR-amplified GC, including emergence of EGFR-negative clones; PTEN deletion; KRAS amplification/mutation; NRAS, MYC, and ERBB2 amplification; and GNAS mutations.
We present a patient with EGFR-amplified GC who acquired substantial numbers of EGFR mutations and MET amplification during the cetuximab treatment as detected by serial ctDNA sequencing. Furthermore, genomic characteristics of GC with ctDNA EGFR amplification are summarized. The patient provided written informed consent for the presentation of anonymized clinical information. Our study and reporting of this patient were performed after approval by the institutional review board at the National Cancer Center Japan, our institution.
CASE REPORT
A 42-year-old man underwent distal gastrectomy with lymph node dissection for localized human epidermal growth factor receptor-2–negative GC. Histopathology showed poorly differentiated adenocarcinoma invading into submucosa or deeper with a minor component of moderately to well-differentiated adenocarcinoma within the lamina propria. He received adjuvant chemotherapy with S-1 plus docetaxel followed by S-1 for 1 year. However, he developed multiple lymph node and bone recurrences after 1 month. Irinotecan plus cisplatin and nab-paclitaxel plus ramucirumab were administered; however, the disease progressed within 1 month on each treatment. Nivolumab was initiated; nevertheless, the patient was admitted because of disseminated intravascular coagulation (DIC), with decreased platelet count and fibrinogen level.
ctDNA sequencing was performed using Guardant360 assay (Guardant Health Redwood City, CA), which detects genomic alterations in 74 genes using ctDNA, before first-line chemotherapy. ctDNA sequencing identified EGFR (plasma CN [pCN], 107.9) and BRAF (pCN, 2.9) amplification and RHOA and TP53 point mutations (Table 1). Immunohistochemistry (IHC) analysis showed a strong EGFR expression in 70% of tumor cells (Figs 1A and 1B) in archival surgical samples, whereas mismatch repair proteins were proficient, and chromogenic in situ hybridization for EBV-encoded RNA was negative.
TABLE 1.
Alterations Detected by ctDNA Sequencing at Each Time Point

FIG 1.
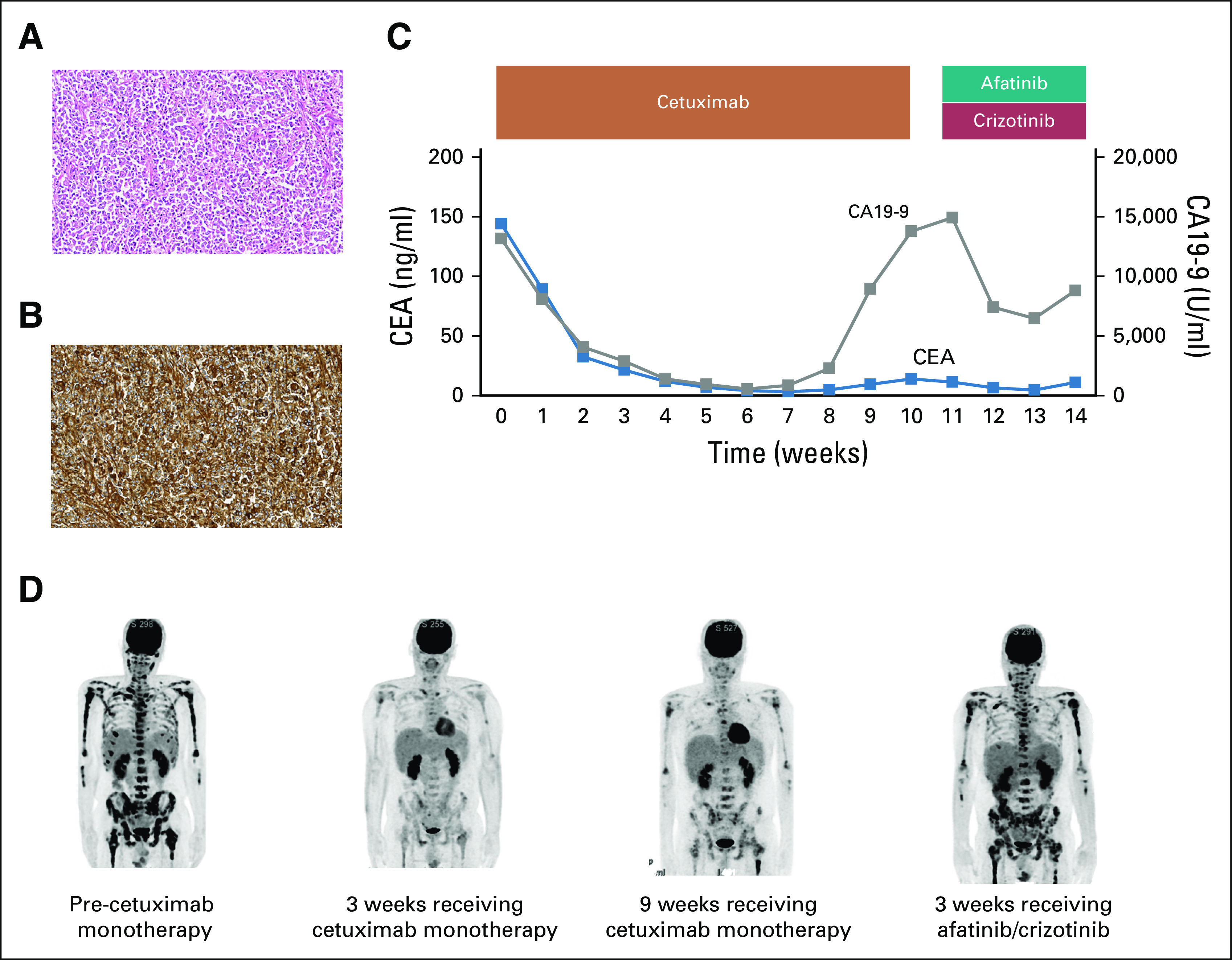
Clinical presentation. (A) Hematoxylin and eosin–stained biopsy specimen of the primary tumor. (B) Immunohistochemistry analysis showing strong epidermal growth factor receptor (EGFR)-positive staining. (C) Course of tumor markers (carcinoembryonic antigen [CEA] and carbohydrate antigen 19-9 [CA 19-9] while receiving treatment with cetuximab and afatinib/crizotinib. (D) Whole-body positron emission tomography–computed tomography scan showing multiple bone metastases pre-cetuximab monotherapy; reduction of [18F]fluorodeoxyglucose (18F-FDG) uptake in bone metastases 3 weeks after the cetuximab monotherapy; reincreased 18F-FDG uptake 9 weeks after the cetuximab monotherapy; and progression 3 weeks after afatinib/crizotinib.
Based on ctDNA sequencing, off-label use of cetuximab, a monoclonal antibody for EGFR, was initiated. Eight days after the treatment initiation, DIC rapidly improved. The positron emission tomography–computed tomography (PET-CT) scan on day 21 showed a significant reduction of [18F]fluorodeoxyglucose uptake in multiple bone metastases, and the serum carcinoembryonic antigen and carbohydrate antigen 19-9 (CA 19-9) levels markedly decreased (Figs 1C and 1D). However, 2 months after the initiation of cetuximab, the patient complained of fatigue, and the serum CA 19-9 increased (Fig 1C). The PET-CT scan on day 63 confirmed bone metastasis progression (Fig 1D).
ctDNA analysis using Guardant360 during disease progression revealed decreased EGFR pCN (to 58.9), emergence of 22 new EGFR mutations, and MET amplification (Figs 2A and 2B; Table 1). EGFR mutations extended from the furin-like domain to beyond the tyrosine kinase domain and included four known pathogenic mutations in the extracellular domain (ECD; Fig 2C).
FIG 2.
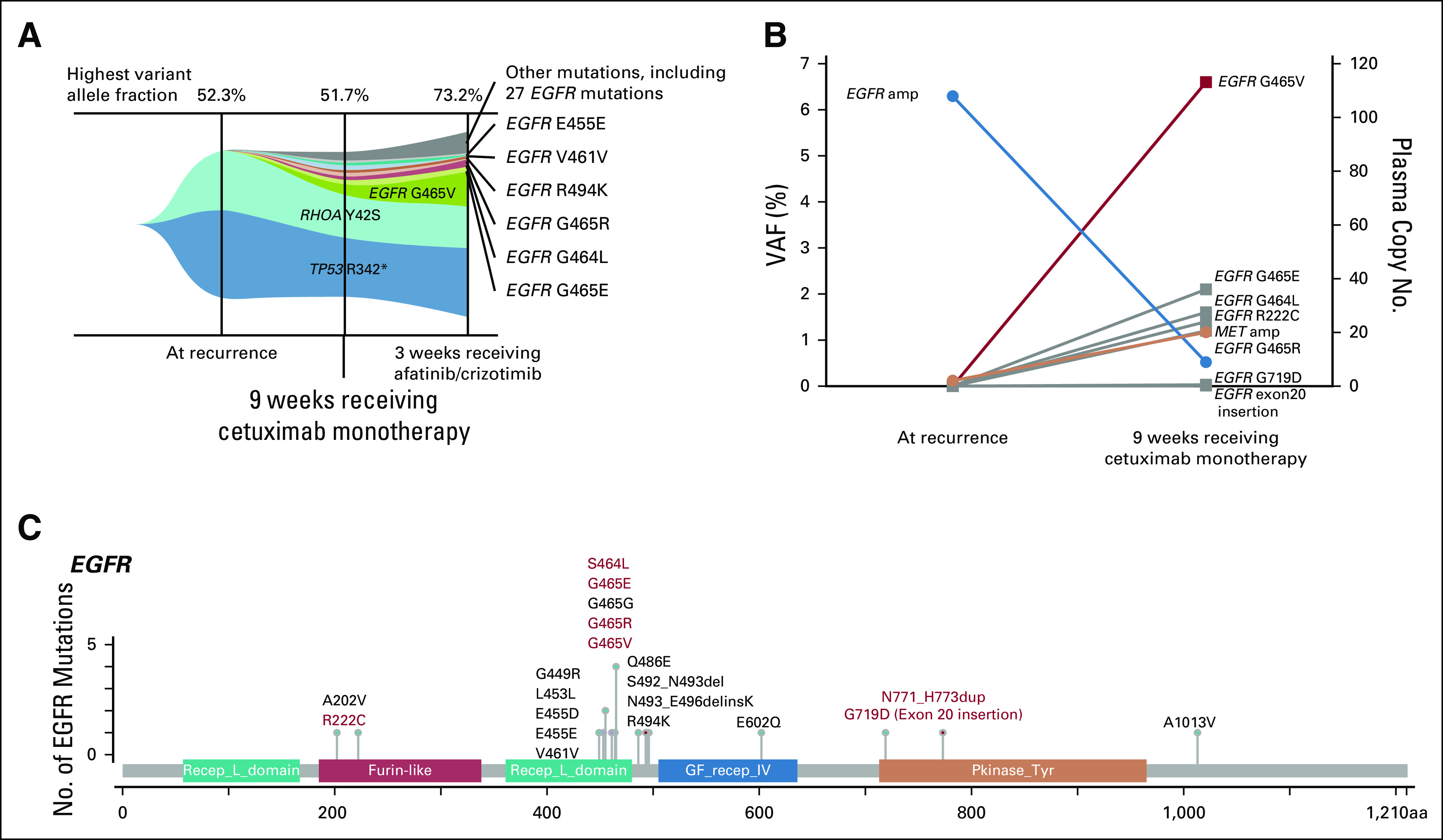
Concurrent emergence of epidermal growth factor receptor (EGFR) mutations and MET amplification. (A) Tumor-response map showing increased genomic diversity through anti-EGFR therapy. (B) Decreased plasma copy number of EGFR and emergence of multiple pathogenic EGFR mutations and MET amplification with cetuximab treatment. (C) Acquired mutations in EGFR domains after cetuximab monotherapy. Actionable variants are highlighted in red. Actionable alterations were annotated using Catalogue of Somatic Mutations in Cancer and genomic visualization tools from cBioPortal were used. VAF, variant allelic frequency.
In an attempt to target acquired EGFR mutations and MET amplification, combination therapy with afatinib and crizotinib was initiated,16 which led to temporary pain relief and decreased serum CA 19-9 levels but was discontinued on day 35 because of progression of bone metastases (Figs 1B and 1C). ctDNA analysis at that time showed the presence of additional EGFR mutations and increased EGFR and MET pCN (Table 1). The patient died of disease progression 2 months after the discontinuation of afatinib and crizotinib.
CTDNA PROFILE OF EGFR-AMPLIFIED GC
To assess the incidence and genomic profiling of GC with EGFR amplification in ctDNA, ctDNA results of GC in our institution were reviewed. EGFR amplification was identified in 26 (18%) of 148 patients with metastatic GC between September 2018 and December 2019. Among them, EGFR pCN was bimodally distributed, with the majority (20; 77%) having low pCN, ranging from 2.2 to 3.4, and the remainder (6; 23%) having pCN of ≥ 3.5, which corresponds to the 90th percentile for EGFR pCN across the Guardant360 database for all tumor types (Fig 3A). Bimodal distribution of EGFR pCN implies that ctDNA sequencing may identify not only homogeneous focal EGFR amplification but also heterogeneity with mixed amplified and nonamplified clones or aneuploidy-associated CN gains, representing low pCN amplifications. Indeed, compared with sample databases tested using tissue-sequencing (GI-SCREEN, our nationwide tissue genotyping study using the Oncomine comprehensive assay [Thermo Fisher Scientific, Waltham, MA], and the Cancer Genome Atlas [TCGA], a publicly available database1), the frequency of all EGFR amplifications was significantly greater in ctDNA, whereas the frequency with high pCN (≥ 3.5) EGFR amplification more closely matched the findings from tissue databases (all-in ctDNA EGFR, 15%; only high pCN EGFR, 4%; GI-SCREEN, 4%; and TCGA, 5%; Fig 3B). We also compared the number of acquired EGFR mutations between our patient with GC and those with 128 metastatic colorectal cancer (mCRC) patients after disease progression with an anti-EGFR therapy. EGFR mutations were detected in 37 patients, including 16 patients with an EGFR amplification. The number of EGFR mutations (range, 1-7; Fig 3C) or known actionable EGFR mutations (range, 1-6; Fig 3D) were lower than those seen in our GC patient with 22 EGFR mutations, including seven known actionable mutations.
FIG 3.

Genomic characteristics of advanced gastric cancer (GC) with epidermal growth factor receptor (EGFR) amplification in circulating tumor DNA (ctDNA). (A) Plasma copy number (pCN) versus ctDNA fraction as the maximum observed variant allelic frequency. (B) Frequency of EGFR-amplified GC in ctDNA versus GI-SCREEN and The Cancer Genome Atlas database. For the ctDNA population, the frequency of all EGFR amplifications and high EGFR pCN in GC is shown, respectively. (C) Distribution of the number of ctDNA EGFR mutations in patients with metastatic colorectal cancer after anti-EGFR therapy. (D) Distribution of the number of ctDNA actionable EGFR mutations in patients with metastatic colorectal cancer after anti-EGFR therapy. (****) P < .0001. NS, not significant; TCGA, The Cancer Genome Atlas.
DISCUSSION
We present a patient with EGFR amplification who acquired EGFR mutations. This patient had markedly high EGFR pCN (107.9) in ctDNA, suggesting EGFR focally amplified disease, confirmed by tissue IHC. Baseline ctDNA sequencing also showed not only concurrent TP53 mutation but also RHOA mutation, reflecting mixed histologic findings.1,17 Our ctDNA genomic profiling study shows that GC with ctDNA EGFR amplification can be divided into two clusters according to pCN. The similar frequency of high EGFR pCN according to ctDNA and EGFR amplification according to tissue analysis suggests that the 90th percentile cutoff for ctDNA most likely enriches for patients with EGFR focally amplified disease, although the cutoff for high EGFR pCN needs to be confirmed in a larger cohort because the pCN can be affected by several factors, including disease burden.13 This interpretation is also supported by a previous report on patients with GC treated with an anti-EGFR antibody-containing regimen, in which responders had a median EGFR pCN of 33.9 compared with 2.5 in nonresponders.15
This patient responded to cetuximab once; however, the disease progressed after only 2 months with numerous acquired mutations throughout EGFR, including the ECD and MET amplification. These heterogeneous resistance alterations might be suggested to be associated with the histopathologic heterogeneity shown in the primary tumor. EGFR ECD mutations are known to indicate anti-EGFR therapy resistance in mCRC due to the interference with binding of anti-EGFR antibodies.18,19 The failure of afatinib-containing treatment despite EGFR tyrosine kinase domain mutation in this patient may be associated with the multiple EGFR ECD mutations, for which the efficacy of afatinib has not been established. Gene amplification has been known to increase the likelihood of new gene mutations and then enhance the growth of subclones harboring a beneficial mutation.20,21 The low variant allelic fractions of acquired EGFR mutations support the hypothesis that subclones with EGFR mutations that occurred in a part of amplified EGFR genes were increased by therapeutic pressure of anti-EGFR therapy. In addition to the heterogeneous and aggressive nature of GC, the remarkably highly amplified EGFR might cause far greater increase of the number of EGFR mutations than seen in mCRC and lead to short duration of response of cetuximab. MET amplification may be associated with resistance to targeted therapies in GC that harbors amplifications of RTK genes.15,22 Of note, the acquired alterations predominantly occurred in chromosome 7. Given the baseline high EGFR pCN, the high instability across chromosome 7 might be associated with the rapid acquisition of resistance in this patient.
The concurrent emergence of the large number of EGFR mutations and MET amplification in this patient and findings of a previous study reporting various types of acquired gene alterations after anti-EGFR therapy,15 indicate that the heterogeneity of EGFR-amplified GC is a great barrier for accurate therapy and warrants a novel strategy to overcome the heterogeneous resistance. Targeting heterogeneous secondary resistance alterations poses a clinical challenge because the majority of emerging mutations are not therapeutically actionable. Several strategies, such as antibody mixture, anti-EGFR combination, and antibody–drug conjugate, have been attempted.23-25
In conclusion, to our knowledge, this is the first report on the occurrence of multiple EGFR ECD mutations as a resistance mechanism to anti-EGFR therapy for EGFR-amplified GC. The use of ctDNA sequencing to identify EGFR-amplified GC and explore the resistance mechanism to anti-EGFR therapy requires additional evaluation to develop effective therapeutic strategies.
SUPPORT
Supported by a grant from the Japan Agency for Medical Research and Development (Grant No. 19ck0106445h0002 to Y.N.) and the National Cancer Center Hospital East.
AUTHOR CONTRIBUTIONS
Conception and design: Yoshiaki Nakamura, Takayuki Yoshino, Kohei Shitara
Administrative support: Hiroya Taniguchi
Collection and assembly of data: Yoshiaki Nakamura, Akinori Sasaki, Hiroki Yukami, Tomoko Jogo, Akihito Kawazoe, Yasutoshi Kuboki, Miho Ozawa, Maho Nakamura, Takayuki Yoshino, Kohei Shitara
Data analysis and interpretation: Yoshiaki Nakamura, Hiroya Taniguchi, Riu Yamashita, Takeshi Kuwata, Takayuki Yoshino, Kohei Shitara
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Yoshiaki Nakamura
Research Funding: Taiho Pharmaceutical (Inst), Guardant Health (Inst), Genomedia (Inst)
Akihito Kawazoe
Honoraria: Ono Pharmaceutical, Taiho Pharmaceutical, Bristol Myers Squibb
Speakers' Bureau: Taiho Pharmaceutical, Ono Pharmaceutical, Bristol Myers Squibb
Research Funding: Ono Pharmaceutical (Inst), Taiho Pharmaceutical (Inst)
Yasutoshi Kuboki
Honoraria: Taiho Pharmaceutical, Bayer, Lilly Japan, Ono Pharmaceutical, Sanofi
Consulting or Advisory Role: Takeda, Boehringer Ingelheim
Research Funding: Taiho Pharmaceutical (Inst), Takeda (Inst), Incyte (Inst), AstraZeneca (Inst), Daiichi Sankyo (Inst), Ono Pharmaceutical (Inst), Boehringer Ingelheim (Inst), Amgen (Inst), Chugai Pharma (Inst), GlaxoSmithKline (Inst)
Hiroya Taniguchi
Honoraria: Bayer, Sanofi, Takeda, Chugai Pharma, Taiho Pharmaceutical, Lilly Japan, Merck Serono, Yakult Honsha, MBL, Bristol Myers Squibb Japan, MSD, Novartis, Daiichi Sankyo, Mitsubishi Tanabe Pharma, Nippon Kayaku
Research Funding: Dainippon Sumitomo Pharma, Array BioPharma, MSD Oncology, Ono Pharmaceutical, Daiichi Sankyo, Sysmex, Novartis, Takeda
Riu Yamashita
Consulting or Advisory Role: Takeda
Takeshi Kuwata
Honoraria: AstraZeneca, MSD, Taiho Pharmaceutical
Research Funding: Daiichi Sankyo, Ono Pharmaceutical
Takayuki Yoshino
Research Funding: Chugai Pharma (Inst), Sumitomo Dainippon (Inst), MSD (Inst), Daiichi Sankyo (Inst), Parexel International (Inst), Ono Pharmaceutical (Inst), Taiho Pharmaceutical (Inst), Amgen (Inst), Sanofi (Inst)
Kohei Shitara
Honoraria: Novartis, AbbVie, Yakult
Consulting or Advisory Role: Astellas Pharma, Lilly, Bristol Myers Squibb, Takeda, Pfizer, Ono Pharmaceutical, MSD, Taiho Pharmaceutical, Novartis, AbbVie, GlaxoSmithKline
Research Funding: Dainippon Sumitomo Pharma (Inst), Lilly (Inst), MSD (Inst), Daiichi Sankyo (Inst), Taiho Pharmaceutical (Inst), Chugai Pharma, Ono Pharmaceutical (Inst), Astellas Pharma (Inst), Medi Science (Inst)
No other potential conflicts of interest were reported.
REFERENCES
- 1.Cancer Genome Atlas Research Network Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202–209. doi: 10.1038/nature13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cristescu R, Lee J, Nebozhyn M, et al. Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat Med. 2015;21:449–456. doi: 10.1038/nm.3850. [DOI] [PubMed] [Google Scholar]
- 3.Normanno N, De Luca A, Bianco C, et al. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene. 2006;366:2–16. doi: 10.1016/j.gene.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 4.Terashima M, Kitada K, Ochiai A, et al. Impact of expression of human epidermal growth factor receptors EGFR and ERBB2 on survival in stage II/III gastric cancer. Clin Cancer Res. 2012;18:5992–6000. doi: 10.1158/1078-0432.CCR-12-1318. [DOI] [PubMed] [Google Scholar]
- 5.Nagatsuma AK, Aizawa M, Kuwata T, et al. Expression profiles of HER2, EGFR, MET and FGFR2 in a large cohort of patients with gastric adenocarcinoma. Gastric Cancer. 2015;18:227–238. doi: 10.1007/s10120-014-0360-4. [DOI] [PubMed] [Google Scholar]
- 6.Liao JB, Lee HP, Fu HT, et al. Assessment of EGFR and ERBB2 (HER2) in gastric and gastroesophageal carcinomas: EGFR amplification is associated with a worse prognosis in early stage and well to moderately differentiated carcinoma. Appl Immunohistochem Mol Morphol. 2018;26:374–382. doi: 10.1097/PAI.0000000000000437. [DOI] [PubMed] [Google Scholar]
- 7.Zhang L, Yang J, Cai J, et al. A subset of gastric cancers with EGFR amplification and overexpression respond to cetuximab therapy. Sci Rep. 2013;3:2992. doi: 10.1038/srep02992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Luber B, Deplazes J, Keller G, et al. Biomarker analysis of cetuximab plus oxaliplatin/leucovorin/5-fluorouracil in first-line metastatic gastric and oesophago-gastric junction cancer: Results from a phase II trial of the Arbeitsgemeinschaft Internistische Onkologie (AIO) BMC Cancer. 2011;11:509. doi: 10.1186/1471-2407-11-509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Waddell T, Chau I, Cunningham D, et al. Epirubicin, oxaliplatin, and capecitabine with or without panitumumab for patients with previously untreated advanced oesophagogastric cancer (REAL3): A randomised, open-label phase 3 trial. Lancet Oncol. 2013;14:481–489. doi: 10.1016/S1470-2045(13)70096-2. [Erratum: Lancet Oncol 14: e254, 2013] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lordick F, Kang YK, Chung HC, et al. Capecitabine and cisplatin with or without cetuximab for patients with previously untreated advanced gastric cancer (EXPAND): A randomised, open-label phase 3 trial. Lancet Oncol. 2013;14:490–499. doi: 10.1016/S1470-2045(13)70102-5. [DOI] [PubMed] [Google Scholar]
- 11.Pectasides E, Stachler MD, Derks S, et al. Genomic heterogeneity as a barrier to precision medicine in gastroesophageal adenocarcinoma. Cancer Discov. 2018;8:37–48. doi: 10.1158/2159-8290.CD-17-0395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Parikh AR, Leshchiner I, Elagina L, et al. Liquid versus tissue biopsy for detecting acquired resistance and tumor heterogeneity in gastrointestinal cancers. Nat Med. 2019;25:1415–1421. doi: 10.1038/s41591-019-0561-9. [Erratum: Nat Med 25:1949, 2019] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakamura Y, Yoshino T. Clinical utility of analyzing circulating tumor DNA in patients with metastatic colorectal cancer. Oncologist. 2018;23:1310–1318. doi: 10.1634/theoncologist.2017-0621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maron SB, Chase LM, Lomnicki S, et al. Circulating tumor DNA sequencing analysis of gastroesophageal adenocarcinoma. Clin Cancer Res. 2019;25:7098–7112. doi: 10.1158/1078-0432.CCR-19-1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maron SB, Alpert L, Kwak HA, et al. Targeted therapies for targeted populations: Anti-EGFR treatment for EGFR-amplified gastroesophageal adenocarcinoma. Cancer Discov. 2018;8:696–713. doi: 10.1158/2159-8290.CD-17-1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kauffmann-Guerrero D, Kahnert K, Kumbrink J, et al. Successful treatment of a patient with NSCLC harboring an EGFR mutation and a concomitant MET exon 14 skipping mutation combining afatinib and crizotinib. Clin Lung Cancer. 2019;20:59–62. doi: 10.1016/j.cllc.2018.09.009. [DOI] [PubMed] [Google Scholar]
- 17.Kakiuchi M, Nishizawa T, Ueda H, et al. Recurrent gain-of-function mutations of RHOA in diffuse-type gastric carcinoma. Nat Genet. 2014;46:583–587. doi: 10.1038/ng.2984. [DOI] [PubMed] [Google Scholar]
- 18.Montagut C, Dalmases A, Bellosillo B, et al. Identification of a mutation in the extracellular domain of the epidermal growth factor receptor conferring cetuximab resistance in colorectal cancer. Nat Med. 2012;18:221–223. doi: 10.1038/nm.2609. [Erratum: Nat Med 18:1445, 2012] [DOI] [PubMed] [Google Scholar]
- 19.Arena S, Bellosillo B, Siravegna G, et al. Emergence of multiple EGFR extracellular mutations during cetuximab treatment in colorectal cancer. Clin Cancer Res. 2015;21:2157–2166. doi: 10.1158/1078-0432.CCR-14-2821. [DOI] [PubMed] [Google Scholar]
- 20.Hendrickson H, Slechta ES, Bergthorsson U, et al. Amplification-mutagenesis: Evidence that “directed” adaptive mutation and general hypermutability result from growth with a selected gene amplification. Proc Natl Acad Sci USA. 2002;99:2164–2169. doi: 10.1073/pnas.032680899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kugelberg E, Kofoid E, Reams AB, et al. Multiple pathways of selected gene amplification during adaptive mutation. Proc Natl Acad Sci USA. 2006;103:17319–17324. doi: 10.1073/pnas.0608309103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sanchez-Vega F, Hechtman JF, Castel P, et al. EGFR and MET amplifications determine response to HER2 inhibition in ERBB2-amplified esophagogastric cancer. Cancer Discov. 2019;9:199–209. doi: 10.1158/2159-8290.CD-18-0598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Montagut C, Argilés G, Ciardiello F, et al. Efficacy of Sym004 in patients with metastatic colorectal cancer with acquired resistance to anti-EGFR therapy and molecularly selected by circulating tumor DNA analyses: A phase 2 randomized clinical trial. JAMA Oncol. 2018;4:e175245. doi: 10.1001/jamaoncol.2017.5245. [Erratum: JAMA Oncol 5:745, 2019] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kato S, Okamura R, Mareboina M, et al. Revisiting epidermal growth factor receptor (EGFR) amplification as a target for anti-EGFR Therapy: Analysis of cell-free circulating tumor DNA in patients with advanced malignancies. JCO Precis Oncol. 2019;3 doi: 10.1200/PO.18.00180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thwaites MJ, Figueredo R, Tremblay G, et al. Abstract 218: AVID100 is an anti-EGFR ADC that promotes DM1-meditated cytotoxicity on cancer cells but not on normal cells. Cancer Res. 2019;79:218. [Google Scholar]