

Abstract
Olaparib (AZD2281) is an orally active Poly(ADP‐ribose) polymerase (PARP) inhibitor with favorable antitumor activity in advanced ovarian and breast cancers with BRCA1/2 mutations in Western (USA and European) studies. This Phase I dose‐finding study evaluated the tolerability, pharmacokinetics, PARP inhibitory activity, and antitumor activity of olaparib in Japanese patients with solid tumors. Olaparib was administered as a single‐dose on day 1, followed by twice‐daily dosing for 28 days from 48 h after a single dose. Doses were escalated from 100 mg b.i.d. in successive cohorts, up to a maximum of 400 mg b.i.d. The present study enrolled 12 patients (n = 3, 3, and 6 in 100, 200 and 400‐mg b.i.d. levels, respectively). The most common adverse events were nausea, increased blood creatinine, decreased hematocrit, leukopenia and lymphopenia; dose‐limiting toxicities were not observed up to and including the 400‐mg b.i.d. dose level. Following twice‐daily dosing, olaparib showed no marked increase in exposure at steady state over that expected from the single‐dose pharmacokinetics. PARP‐1 inhibition was observed from the 100‐mg b.i.d. dose level in peripheral blood mononuclear cells from 6 h post‐dose on day 1 during the multiple‐dosing period. A patient with metastatic breast cancer (100 mg b.i.d.) had a partial response for 13 months and four patients (two each in the 200 and 400‐mg b.i.d. levels) had stable disease >8 weeks. Olaparib was well tolerated up to the 400‐mg b.i.d. dose in Japanese patients with solid tumors. Preliminary evidence of antitumor activity was observed. (Cancer Sci 2012; 103: 504–509)
Poly(ADP‐ribose) polymerase (PARP) has an established role in DNA repair via two pathways, base excision repair (BER) and homologous recombination (HR), which compensate for each other when one is compromised.( 1 ) DNA single‐strand breaks are repaired by the BER pathway, of which PARP is a key component. If the BER pathway is compromised, for example by inhibition of PARP, it becomes non‐functional for DNA repair, which can lead to DNA double‐strand breaks. DNA double‐strand breaks are usually repaired by the HR repair pathway, key components of which are the tumor‐suppressor genes BRCA1 and BRCA2.( 2 ) However, in tumors that lack BRCA1 and BRCA2 proteins, the HR repair pathway is non‐functional, leaving only the BER pathway to repair any DNA breaks. This represents an opportunity for PARP inhibition, as blockade of BER will render tumor cells unable to repair DNA damage, a concept known as synthetic lethality.( 3 ) PARP inhibitors might also have potential in other non‐BRCA‐dependent HR‐deficient tumors that involve dysfunction of other proteins in the HR repair pathway. A number of preclinical studies in cells and xenograft tumor models support the proposal that PARP inhibitors might be effective targeted therapies for tumors defective in the HR pathway.( 4 , 5 , 6 , 7 , 8 , 9 )
Olaparib (AZD2281) is an orally active PARP inhibitor that induces synthetic lethality in HR‐deficient cells such as those with homozygous BRCA mutations.( 7 , 8 , 9 ) In a Phase I study of Western patients with solid tumors, olaparib was shown to be well tolerated.( 10 , 11 ) The dosing regimen increased from 10 mg once daily for two of every 3 weeks, to 60 mg and above twice daily continuously. Evaluable objective tumor responses were observed in patients treated with olaparib at doses from 100 mg b.i.d. The most commonly reported toxicities were grade 1 and 2 nausea and fatigue; dose‐limiting toxicities (DLT) were observed in one of eight patients receiving olaparib 400 mg b.i.d. and two of five patients receiving olaparib 600 mg b.i.d. The incidence of toxicities observed at the olaparib 600‐mg b.i.d. dose level was considered unacceptable and the maximum tolerated dose (MTD) was determined as 400 mg b.i.d.( 10 , 11 ) More recently, olaparib 400 mg b.i.d. was shown to be well tolerated with evidence of antitumor activity in two Phase II studies in BRCA1 or BRCA2 mutation carriers with advanced ovarian or breast cancer.( 12 , 13 ) Furthermore, in a Phase II study of patients with ovarian and breast cancer, evidence of antitumor activity of olaparib was reported in patients with high‐grade serous ovarian cancer with and without BRCA mutations.( 14 )
Based on the findings of the Phase I study in Western patients with solid tumors,( 10 , 11 ) we conducted a Phase I dose‐finding study in Japanese patients with advanced solid tumors to assess the safety and tolerability, the pharmacokinetic (PK) profile including potential inter‐ethnic differences as compared with the previous Phase I study, the PARP inhibitory activity in peripheral blood mononuclear cells (PBMC), and the preliminary antitumor activity of olaparib at doses up to and including 400 mg b.i.d.
Materials and Methods
Patients. Eligible patients aged 20–74 years with histologically or cytologically confirmed solid tumors for whom no standard therapy existed or no standard therapies were likely to result in durable remission were included in the study. All patients were also required to have: World Health Organization (WHO) performance status ≤2; adequate bone marrow, hepatic and renal functions (absolute neutrophil count ≥1500/mm3, platelet count ≥100 000/mm3, hemoglobin ≥9.0 g/dl, serum total bilirubin ≤1.5 times the normal upper limit, aspartate aminotransferase [AST] ≤80 IU/l, alanine aminotransferase [ALT] ≤80 IU/l, serum creatinine level ≤1.2 mg/dl); life expectancy ≥3 months; and a minimum washout period of 4 weeks after any previous anticancer therapy. BRCA1 or BRCA2 mutation carrier status was not required for eligibility, and was not evaluated in this study. Exclusion criteria included: serious pre‐existing medical conditions, such as significant cardiovascular disease and psychogenic disorders; family history of long QT syndrome or QTc ≥ 450 ms; persistent CTCAE ≥ grade 2 toxicities (excluding alopecia) caused by prior medication; symptomatic brain metastases; pregnancy or lactation; and hepatitis B or C or human immunodeficiency virus infections.
Written informed consent was obtained from all patients. The study was approved by the institutional review boards and ethics committees at the National Cancer Center and conducted in accordance with the Good Clinical Practice guidelines and the Declaration of Helsinki.
Study design. Olaparib was supplied by AstraZeneca KK (Osaka, Japan) as 50‐mg capsules, which were administered orally, morning and evening; study patients were required to fast for at least 1 h before and 2 h after administration. This study involved two phases: a dose‐escalation phase and a dose‐expansion phase. Each phase was divided into two periods: a single‐dose period (dosing on the morning of day 1) and a multiple‐dose period of continuous twice‐daily dosing for 28 days (one cycle) starting on the morning of multiple‐dose period day 1 (48 h after the initial single dose).
Based on the previously reported Phase I study in a Western patient population,( 10 ) the starting dose of olaparib was determined as 100 mg b.i.d., escalating to 200 mg b.i.d. and up to 400 mg b.i.d.( 10 ) Dose escalation above 400 mg b.i.d. was not planned because in the previous Phase I study in Western patients, the olaparib 600‐mg b.i.d. dose level was considered unacceptable and the MTD was determined as 400 mg b.i.d.( 10 ) A minimum of three patients were entered into each cohort. Additional cohorts of patients received escalating doses of olaparib (200 and 400 mg b.i.d.) until DLT were observed in more than two of three patients in a cohort, or the previously determined MTD of 400 mg b.i.d. was reached. If the MTD was reached, that dose level was expanded to a maximum of six patients. If the MTD was not reached, olaparib 400 mg b.i.d. was administered to a maximum of six patients as an expanded cohort. Any patient in whom a DLT occurred had their olaparib dose withheld until the adverse event (AE) had resolved to baseline or grade 1. A maximum of 2 weeks’ dosing delay was allowed, and if the AE had resolved, olaparib was restarted at the lower dose level, except the initial dose level. If the AE did not resolve within 2 weeks, the patient was withdrawn from the study. A DLT was defined as any of the following events that were considered by the investigator to be related to olaparib treatment and which occurred during the first cycle of treatment, irrespective of whether the observed AE were eliminated or reduced: grade 4 neutropenia lasting for >5 days or grade 4 neutropenia with neutropenic fever/sepsis, grade 4 thrombocytopenia or persistent non‐hematological grade 3 or 4 toxicity despite implementation of supporting therapy, or any other toxicity that was judged by the investigators to be a DLT.
Assessments.
Evaluations and follow‐up studies. Complete clinical assessments, including physical examination, WHO performance status, blood pressure, weight, electrocardiogram (ECG), chest X‐ray and routine laboratory tests, were evaluated in all patients before study entry and prior to each subsequent treatment cycle. Routine laboratory tests included a complete blood count and differential testing of electrolytes, total serum protein, albumin, total serum bilirubin, AST, ALT, alkaline phosphatase, lactic dehydrogenase, gamma‐glutamyl transferase, serum creatinine, serum blood urea nitrogen, glucose, adequate tumor markers and urinalysis. With the exception of tumor markers, these laboratory tests were repeated weekly. Tumor markers and ECG were assessed before each treatment cycle; however, the ECG was assessed weekly only in the initial treatment cycle. Safety and tolerability were evaluated by recording the incidence of AE according to Common Terminology Criteria for Adverse Events (CTCAE version 3.0, http://cstep.cancer.gov/protocolDevelopment/electronic_applications/ctc.htm#ctc_30). Tumor radiological assessments by computed tomography or magnetic resonance imaging scans were carried out every two cycles and evaluated according to Response Evaluation Criteria In Solid Tumors (RECIST) version 1.0.( 15 )
Pharmacokinetics. Pharmacokinetic evaluation was performed in all patients. Blood samples were collected to determine plasma concentrations of olaparib following single‐dose and multiple‐dose capsule administration. For the single‐dose period, samples were collected pre‐dose, and then at 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, 24 and 48 h following dosing. For the multiple‐dosing period, blood samples were collected pre‐dose on the morning of days 3 and 15, and then at 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 8 and 10 h following the morning dose on day 15 (all samples were collected prior to the evening dose).
All of the obtained plasma samples were analyzed by an independent bioanalytical company (Covance Laboratories, Harrogate, UK) using manual solid phase extraction followed by liquid chromatography with tandem mass spectrometric detection. PK parameters evaluated following single dosing included maximal plasma concentration (C max), time to maximal plasma concentration (t max), half‐life (t ½), area under the plasma concentration–time curve (AUC) from 0 to 10 h and from 0 to infinity (AUC 0–10h and AUC 0–∞, respectively), plasma clearance (CL/F), volume of distribution at steady state (V ss /F) and mean residence time (MRT). PK parameters evaluated following multiple dosing included C max, t max and AUC 0–10h. Partial area (AUC 0–10h) was assessed for multiple‐dose administration in preference to AUC at dosing interval (AUC 0–12h) due to practical reasons (late‐night sampling was avoided to maximize compliance to the study design). To enable comparison between multiple and single olaparib doses, it was decided that AUC 0–10h should also be the preferred measurement assessed for single‐dose administration. PK parameters were calculated using non‐compartmental analyses with WinNonlin (version 3.1; Pharsight, Cary, NC, USA).
Poly(ADP‐ribose) polymerase 1 inhibitory activity. Blood samples were also collected for evaluation of the PARP‐1 inhibitory dose range in PBMC during screening (two samples at least 24 h apart), at 6 h following the first dose of olaparib on day 1 of the olaparib multiple‐dosing period, and pre‐dose on days 8 and 15. From the blood samples, whole cell extracts were produced, aliquoted and stored at −80 °C. The total protein content of each sample was determined by a Bradford Coomassie assay and samples were diluted to a fixed protein concentration before analysis. PARP‐1 inhibition was assessed by means of functional electro‐chemiluminescence based immunoassays utilizing the MesoScale Discovery technology platform.( 16 ) For each patient sample, two assays were performed in parallel, one to measure total PARP‐1 protein content and the other to measure PARP activity (PAR forming ability). The ratio of PARP activity/total PARP‐1 content was then used as the final measure of normalized PARP activity in each sample.
Statistical methods. Descriptive statistics were used to define the trial population, safety, tolerability, PK and pharmacodynamic data, and the observed tumor response.
Results
Patients. From November 2007 to June 2009, 13 patients were enrolled in the study and 12 patients received olaparib (n = 3 100 mg b.i.d., n = 3 200 mg b.i.d. and n = 6 400 mg b.i.d.). A patient with breast cancer did not receive treatment because she did not fulfill the eligibility criteria. Table 1 shows the baseline demographics and characteristics. The study included six male and six female patients, and the median age was 57 years. The primary tumor type was breast cancer and all 12 patients discontinued study treatment due to disease progression. The median duration of study treatment was 30 days (range 9–436).
Table 1.
Demographics and baseline characteristics
| 100 mg b.i.d. (n = 3) | 200 mg b.i.d. (n = 3) | 400 mg b.i.d. (n = 6) | |
|---|---|---|---|
| Age years, median (range) | 62.0 (42–62) | 67.0 (54–67) | 51.5 (39–69) |
| Sex, n | |||
| Male | 2 | 2 | 2 |
| Female | 1 | 1 | 4 |
| WHO performance status, n | |||
| 0 | 2 | 2 | 2 |
| 1 | 1 | 1 | 4 |
| Primary tumor, n | |||
| Breast | 1 | 0 | 3 |
| NSCLC | 0 | 1 | 1 |
| Thymus | 0 | 0 | 1 |
| Esophagus | 1 | 0 | 0 |
| Liver | 0 | 0 | 1 |
| Colon | 1 | 0 | 0 |
| Rectal | 0 | 1 | 0 |
| Unknown primary | 0 | 1 | 0 |
NSCLC, non‐small‐cell lung cancer; WHO, World Health Organization.
Safety. No DLT were observed with olaparib capsule treatment at the doses up to and including 400 mg b.i.d.; the dose escalation was then halted as 400 mg b.i.d. had been previously determined as the MTD in Western patients.( 10 ) The 400‐mg b.i.d. cohort was expanded to six patients and the acceptable tolerability of olaparib of 400 mg b.i.d. was confirmed. Overall, the most commonly reported AE were nausea (50%), blood creatinine increase (33%), hematocrit decrease (33%), leukopenia (33%) and lymphopenia (33%) (Table 2). All AE were graded 1 or 2, except for grade 3 ALT increase and anemia, described below. A total of three patients had dose interruptions and/or dose reductions due to AE after the second cycle of treatment; one patient with metastatic breast cancer in the 100‐mg b.i.d. cohort required a dose interruption due to grade 3 ALT increase that was resolved within 1 week and treatment was restarted at the same dose level; one patient with metastatic breast cancer in the 400‐mg b.i.d. cohort required a dose reduction to 200 mg b.i.d. due to grade 3 anemia in the second cycle of treatment; and one patient with NSCLC in the 400‐mg b.i.d. cohort had a dose interruption due to anemia, and discontinued the study treatment during the dose interruption due to progressive disease.
Table 2.
Adverse events reported in ≥2 patients overall
| Patients with adverse events, n | 100 mg b.i.d. (n = 3) | 200 mg b.i.d. (n = 3) | 400 mg b.i.d. (n = 6) | Total n (%) |
|---|---|---|---|---|
| Any adverse events | 3 | 3 | 6 | 12 (100) |
| Nausea | 1 | 1 | 4 | 6 (50.0) |
| Blood creatinine increased | 0 | 1 | 3 | 4 (33.3) |
| Leukopenia | 1 | 0 | 3 | 4 (33.3) |
| Lymphopenia | 1 | 1 | 2 | 4 (33.3) |
| Hematocrit decreased | 2 | 0 | 2 | 4 (33.3) |
| Anemia | 0 | 0 | 3 | 3 (25.0) |
| Anorexia | 0 | 1 | 2 | 3 (25.0) |
| Red blood cell count decreased | 1 | 0 | 2 | 3 (25.0) |
| Hyperglycemia | 1 | 1 | 1 | 3 (25.0) |
| Dysgeusia | 0 | 0 | 2 | 2 (16.7) |
| Pruritus | 0 | 0 | 2 | 2 (16.7) |
| Thrombocytopenia | 0 | 0 | 2 | 2 (16.7) |
| Hemoglobin decreased | 0 | 0 | 2 | 2 (16.7) |
| Vomiting | 0 | 1 | 1 | 2 (16.7) |
| Fatigue | 0 | 1 | 1 | 2 (16.7) |
| Rash | 0 | 1 | 1 | 2 (16.7) |
| Alanine aminotransferase increased | 1 | 0 | 1 | 2 (16.7) |
| Aspartate aminotransferase increased | 1 | 0 | 1 | 2 (16.7) |
| Gamma‐glutamyltransferase increased | 1 | 0 | 1 | 2 (16.7) |
| Constipation | 1 | 1 | 0 | 2 (16.7) |
Pharmacokinetics. Pharmacokinetic (PK) profiles were assessed after single dosing (day 1) in all 12 patients and after multiple twice‐daily dosing (day 15) in 11 patients (Table 3). The plasma‐concentration–time profiles following both single and multiple dosing are shown in Figure 1. Following single dosing, olaparib at all dose levels was rapidly absorbed with median t max values of 1–2.4 h across the dose groups, with an individual patient range of 0.5–4.2 h. Thereafter, plasma concentrations appeared to decline biphasically, with a mean terminal half‐life of approximately 7–11 h and an individual patient range of 3.4–19 h. Following multiple dosing, absorption of olaparib was still rapid, with median t max values of 1.5–2.1 h across the dose groups, with an individual patient range of 1.5–6.2 h. Due to limited sampling points (up to 10 h post‐morning dose), determination of the terminal half‐lives from these multiple‐dose profiles was not possible.
Table 3.
Pharmacokinetic (PK) parameters of olaparib following single dosing and multiple dosing for 15 days
| PK parameters† | Olaparib dose | ||
|---|---|---|---|
| Single dose | |||
| 100 mg b.i.d (n = 3) | 200 mg b.i.d (n = 3) | 400 mg b.i.d (n = 6) | |
| C max (μg/mL) | 2.2 (43.2) | 3.4 (19.8) | 4.8 (15.6) |
| t max (h), median (range) | 1.0 (0.5–1.4) | 2.1 (1.5–3.0) | 2.4 (1.6–4.2) |
| AUC 0–10h (μg.h/mL) | 9.32 (59.4) | 14.5 (21.1) | 24.9 (29.0) |
| AUC 0–∞ (μg.h/mL) | 13.7 (101.4) | 20.2 (33.5) | 37.0 (37.8) |
| t ½ (h), mean (range) | 7.8 (3.4–15.3) | 6.9 (4.6–11.2) | 10.7 (3.8–18.9) |
| CL/F (l/h) | 7.3 (101.3) | 9.9 (33.5) | 10.8 (37.7) |
| V ss /F (l) | 57.6 (30.7) | 74.6 (13.8) | 107 (37.6) |
| Multiple doses | |||
|---|---|---|---|
| 100 mg b.i.d. (n = 3) | 200 mg b.i.d. (n = 3) | 400 mg b.i.d. (n = 5) | |
| C max (μg/mL) | 3.7 (88.1) | 4.8 (31.6) | 5.9 (19.7) |
| t max (h), median (range) | 1.6 (1.5–6.2) | 1.5 (1.5–1.9) | 2.1 (1.5–4.0) |
| AUC 0–10h (μg.h/mL) | 21.9 (160.9) | 23.8 (36.6) | 33.3 (22.3) |
AUC 0–10h, area under the plasma concentration–time curve from 0 to 10 h; AUC 0–∞, area under the plasma concentration–time curve from 0 to infinity; CL/F, apparent oral clearance; C max, maximum plasma concentration; t ½, terminal half‐life; t max, time to maximal plasma concentration; V ss /F, apparent volume of distribution at steady state. †All values are geometric mean (CV%) unless otherwise stated.
Figure 1.
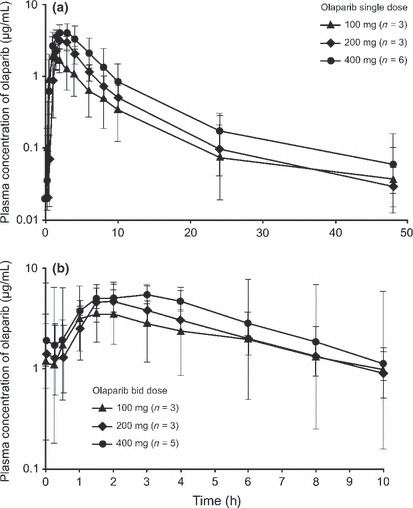
Geometric mean plasma‐concentration profiles of olaparib following: (a) single dosing and (b) twice‐daily dosing for 15 days.
Olaparib exposure (C max and AUC) increased less than dose proportionally following single and multiple dosing, with a fourfold increase in dose from 100 to 400 mg b.i.d. resulting in 1.6‐fold and 1.5‐fold increases in geometric mean C max and AUC 0–10h, respectively. Olaparib showed no marked increase in exposure at steady state over the expectation from a single dose. However, one patient in the 100‐mg cohort had a marked increase in C max and AUC following multiple dosing, resulting in considerably higher plasma concentrations of olaparib than the remaining two patients in the same cohort (C max was three to fourfold higher and AUC 0–10h was five to ninefold higher), although there was no marked difference observed in the plasma‐concentration profile.
These PK profiles determined across the dose ranges investigated here were similar to those reported in the previous Phase I study.( 10 )
Poly(ADP‐ribose) polymerase 1 inhibitory activity in peripheral blood mononuclear cells. Poly(ADP‐ribose) polymerase 1 inhibitory activity in PBMC was evaluated in all 12 patients. The two baseline measurements (taken 24 h apart during screening) collected for each patient had, on average, an intra‐patient variability of 11.5% (range, 0.6–32.3) of the mean. The inhibitory activity on days 8 and 15 in the multiple‐dose period could not be assessed in one patient in the 400‐mg b.i.d. group due to progressive disease on day 7. PARP‐1 activity was inhibited by more than 50% at 6 h post‐dose on day 1 and continued until pre‐dose on day 15 of the multiple‐dosing period in all patients (Table 4). The extent of PARP‐1 inhibitory activity did not grow with increased olaparib doses.
Table 4.
PAR:PARP‐1 ratio at baseline and following multiple dosing of olaparib
| Olaparib dose | |||||||
|---|---|---|---|---|---|---|---|
| 100 mg b.i.d. (n = 3) | 200 mg b.i.d. (n = 3) | 400 mg b.i.d. (n = 6)† | |||||
| PAR:PARP‐1 ratio | % change from baseline | PAR:PARP‐1 ratio | % change from baseline | PAR:PARP‐1 ratio | % change from baseline | ||
| Single dose | Baseline | 3.67 (0.87) | 2.51 (0.67) | 2.75 (1.0) | |||
| Multiple dose | Day 1, 6 h | 1.63 (0.52) | –55.9 (6.24) | 0.97 (0.29) | –61.1 (7.45) | 1.34 (0.65) | –52.4 (8.79) |
| Day 8, pre‐dose | 1.50 (0.35) | –59.1 (3.18) | 0.907 (0.34) | –64.2 (8.52) | 1.05 (0.58) | –60.7 (12.2) | |
| Day 15, pre‐dose | 1.61 (0.54) | –56.5 (6.66) | 0.619 (0.42) | –76.4 (14.1) | 1.21 (0.51) | –51.0 (25.4) | |
All values are presented as mean (SD). †n = 5 for days 8 and 15 of the multiple‐dose period.
Efficacy. All 12 patients were evaluable for antitumor response, and the best overall responses are shown in Table 5. A patient with metastatic breast cancer who had a family history of breast cancer in the 100‐mg b.i.d. cohort had a durable partial response for approximately 13 months.
Table 5.
Best overall responses
| Response | Olaparib dose | Total (n = 12) | ||
|---|---|---|---|---|
| 100 mg, b.i.d (n = 3) | 200 mg, b.i.d (n = 3) | 400 mg, b.i.d (n = 6) | ||
| CR | 0 | 0 | 0 | 0 |
| PR | 1 | 0 | 0 | 1 |
| SD >8 weeks | 0 | 2 | 2 | 4 |
| PD | 2 | 1 | 4 | 7 |
CR, complete response; PD, progressive disease; PR, partial response; SD, stable disease.
Discussion
We conducted a Phase I dose‐finding study of the oral PARP inhibitor olaparib (capsule formulation) in Japanese patients with solid tumors, and confirmed that it is well tolerated with a manageable toxicity profile at doses up to and including 400 mg b.i.d., the previously defined MTD in Western patients.( 10 )
Overall, the AE profile of olaparib in this study was similar to that observed previously in larger studies in Western patients,( 10 , 12 , 13 ) and no new safety concerns were identified. Consistent with the Western studies, nausea was the most common AE in this Japanese patient population, occurring in 50% of patients. Other common AE included blood creatinine increase, hematocrit decrease, leukopenia and lymphopenia. All AE were generally mild to moderate in intensity (grade 1 or 2); the only two exceptions were grade 3 AST elevation and anemia, which were managed effectively with dose pause and dose reduction, respectively. No DLT were reported in this study.
Absorption of olaparib was moderately rapid at all dose levels following both single and multiple dosing, with maximal plasma concentrations achieved by 2.0–2.4 h. Elimination was assessable only for the single‐dose period, with plasma concentrations declining biphasically with a half‐life of approximately 7–11 h. Biphasic elimination of olaparib has been previously described in Western patients with solid tumors following olaparib single dosing.( 10 ) Apparent plasma clearance of olaparib was low, and the apparent volume of distribution at steady‐state values indicated that olaparib was distributed outside the central compartment, although not extensively. As reported in the Western study,( 10 ) olaparib exposure does not increase proportionally with doses escalated above 100 mg following either single or multiple twice‐daily dosing. Although one patient in the 100‐mg b.i.d. cohort showed a marked increase in the plasma concentration and exposure to olaparib following multiple dosing, there was no marked difference following single dosing. Background demographic characteristic and laboratory test data for this patient were generally similar to the other 11 patients, so it was not possible to clarify the underlying cause of this observation.
There was preliminary evidence of antitumor activity in Japanese patients with solid tumors, with a durable partial response for approximately 13 months in one patient with metastatic breast cancer in the 100‐mg b.i.d. group. An additional four patients had stable disease >8 weeks. Further, more than 50% inhibition of PARP‐1 activity in PBMC was observed in all patients by 6 h and continued up until day 15 (last evaluated day) at olaparib doses of 100–400 mg b.i.d. These results are consistent with those reported for the Western Phase I dose‐escalation study, in which maximal PARP‐1 inhibition was reached with a 60‐mg dose.( 10 ) In consideration of these previous findings, our data indicate that maximal inhibition was achieved at the lowest dose administered (100 mg). These findings show that pharmacological and biological activity is evident at olaparib doses of ≥100 mg b.i.d., although the extent of PARP‐1 inhibitory activity did not increase proportionally with increased doses of olaparib.
In this Phase I study, the dose escalation of olaparib was planned up to 400 mg b.i.d., according to the following reasons: (i) in the previously reported Phase I study on Western patients, the olaparib 600‐mg b.i.d. dosing regimen was considered unacceptable and the MTD was determined as 400 mg b.i.d.;( 10 ) (ii) the fundamental and efficient global strategy for new anti‐cancer drug development; and (iii) the formulation of olaparib available for use in this study was a 50‐mg capsule (one capsule is 21.8 mm in length), requiring patients to swallow 16 capsules per day to achieve the Western MTD of 400 mg b.i.d. This “pill burden” has previously been acknowledged as having the potential to compromise patient convenience and compliance,( 17 ) thereby introducing further issues into the assessment of higher dose levels in this study. Although an MTD was not achieved in the present study, we have demonstrated that the tolerability and PK profiles, and PARP‐1 inhibitory activity, were similar to those reported in the previous Phase I study.( 10 ) No inter‐ethnic differences were observed between our findings and those previously reported.( 10 ) Due to the predominance of our use of the previous Western Phase I study as a guide to design this dose‐finding Phase I study, the dose escalation planned in this current study is limiting; indeed, this design would be considered unacceptable if it was a first‐in‐human Phase I study.
Although patients in this study were not tested for BRCA mutations, in future studies, it might be interesting to evaluate the antitumor activity of olaparib in Japanese patients with and without BRCA mutations. Studies in Western patients have demonstrated the potential for olaparib in a broader patient population, including Phase II studies in patients with high‐grade serous ovarian cancer and triple‐negative breast cancer.( 14 , 18 ) Although no Japanese patients with ovarian cancer were included in the current study, it would be interesting to undertake future studies in ovarian cancer and broader patient populations.
In conclusion, olaparib was well tolerated with a manageable toxicity profile at doses up to and including 400 mg b.i.d. in Japanese patients with solid tumors. Preliminary evidence of antitumor activity was also observed, as well as PARP‐1 inhibition in PBMC within the doses investigated. No inter‐ethnic differences were observed when compared with the previous Phase I study.( 10 ) The dose range between 100 and 400 mg b.i.d. could be considered appropriate for further investigation in this population. An international (including Japan) Phase II study of olaparib 400 mg b.i.d. in patients with ovarian cancer has commenced (ClinicalTrials.gov identifier NCT01081951).
Disclosure Statement
This study was sponsored by AstraZeneca. T Kawata and X Shi are employees of, and own shares in, AstraZeneca KK. Other authors have no potential conflict of interest information to disclose.
Acknowledgments
We thank Dr Juliet Fawcett from Mudskipper who provided editorial assistance funded by AstraZeneca.
An abstract of preliminary results from this study was presented at ESMO 2010: Yamamoto N, Nokihara H, Yamada Y, Goto Y, Tanioka M, Shibata T, Yamada K, Asahina H, Tamura T. Encouraging data from a Phase I study of the first oral PARP inhibitor olaparib in Japanese patients with solid tumors. Ann Oncol 2010; 21 (Suppl 8): viii 176 (abst 537P).
ClinicalTrials.gov identifier NCT00572364.
References
- 1. Amé J‐C, Spenlehauer C, de Murcia G. The PARP superfamily. Bioessays 2004; 26: 882–93. [DOI] [PubMed] [Google Scholar]
- 2. Gudmundsdottir K, Ashworth A. The roles of BRCA1 and BRCA2 and associated proteins in the maintenance of genomic stability. Oncogene 2006; 25: 5864–74. [DOI] [PubMed] [Google Scholar]
- 3. Ashworth A. A synthetic lethal therapeutic approach: Poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double‐strand break repair. J Clin Oncol 2008; 26: 3785–90. [DOI] [PubMed] [Google Scholar]
- 4. Bryant HE, Schultz N, Thomas HD et al. Specific killing of BRCA2‐deficient tumours with inhibitors of poly(ADP‐ribose) polymerase. Nature 2005; 434: 913–7. [DOI] [PubMed] [Google Scholar]
- 5. Farmer H, McCabe N, Lord CJ et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005; 434: 917–21. [DOI] [PubMed] [Google Scholar]
- 6. McCabe N, Lord CJ, Tutt AN, Martin NM, Smith GC, Ashworth A. BRCA2‐deficient CAPAN‐1 cells are extremely sensitive to the inhibition of Poly (ADP‐Ribose) polymerase: an issue of potency. Cancer Biol Ther 2005; 4: 934–6. [DOI] [PubMed] [Google Scholar]
- 7. Hay T, Jenkins H, Sansom OJ, Martin NM, Smith GC, Clarke AR. Efficient deletion of normal Brca2‐deficient intestinal epithelium by poly(ADP‐ribose) polymerase inhibition models potential prophylactic therapy. Cancer Res 2005; 65: 10145–8. [DOI] [PubMed] [Google Scholar]
- 8. Evers B, Drost R, Schut E et al. Selective inhibition of BRCA2‐deficient mammary tumor cell growth by AZD2281 and cisplatin. Clin Cancer Res 2008; 14: 3916–25. [DOI] [PubMed] [Google Scholar]
- 9. Rottenberg S, Jaspers JE, Kersbergen A et al. High sensitivity of BRCA1‐deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci USA 2008; 105: 17079–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fong PC, Boss DS, Yap TA et al. Inhibition of poly(ADP‐ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med 2009; 361: 123–34. [DOI] [PubMed] [Google Scholar]
- 11. Fong PC, Yap TA, Boss DS et al. Poly(ADP)‐ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum‐free interval. J Clin Oncol 2010; 28: 2512–9. [DOI] [PubMed] [Google Scholar]
- 12. Tutt A, Robson M, Garber JE et al. Oral poly(ADP‐ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof‐of‐concept trial. Lancet 2010; 376: 235–44. [DOI] [PubMed] [Google Scholar]
- 13. Audeh MW, Carmichael J, Penson RT et al. Oral poly(ADP‐ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof‐of‐concept trial. Lancet 2010; 376: 245–51. [DOI] [PubMed] [Google Scholar]
- 14. Gelmon KA, Hirte HW, Robidoux A et al. Can we define tumors that will respond to PARP inhibitors? A phase II correlative study of olaparib in advanced serous ovarian cancer and triple negative breast cancer. J Clin Oncol 2010; 28: abst 3002. [Google Scholar]
- 15. Therasse P, Arbuck SG, Eisenhauer EA et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 2000; 92: 205–16. [DOI] [PubMed] [Google Scholar]
- 16. Menear KA, Adcock C, Boulter R et al. 4‐[3‐(4‐cyclopropanecarbonylpiperazine‐1‐carbonyl)‐4‐fluorobenzyl]‐2H‐phthalazin‐ 1‐one: a novel bioavailable inhibitor of poly(ADP‐ribose) polymerase‐1. J Med Chem 2008; 51: 6581–91. [DOI] [PubMed] [Google Scholar]
- 17. Molife R, Kaye S, Forster M et al. A phase I study to determine the comparative bioavailability of two different oral formulations of the PARP inhibitor, olaparib (AZD2281), in patients with advanced solid tumors. J Clin Oncol 2010; 28: abst 2599. [Google Scholar]
- 18. Ledermann J, Harter P, Gourley C et al. Phase II randomized placebo‐controlled study of olaparib (AZD2281) in patients with platinum‐sensitive relapsed serous ovarian cancer (PSR SOC). J Clin Oncol 2011; 29: abst 5003. [Google Scholar]