

Abstract
Activation‐induced cytidine deaminase (AID/AICDA) is required for somatic hypermutation and class‐switch recombination of the immunoglobulin gene, and for c‐myc translocation of germinal center‐derived B‐cell lymphoma. In the present study, we attempted to clarify the significance of AID associated with c‐myc in the progression of follicular lymphoma (FL) using RT‐PCR and quantitative real‐time PCR. Tissues from the patients with grade 3 FL expressed relatively higher levels of c‐myc and AID. The samples taken from a patient with FL who died within 2 years after the start of treatment showed either no or low expression of AID, despite expressing high levels of c‐myc. In order to examine the role of AID expression in rapidly progressive FL, the full‐length AID transcript was transfected into AID‐negative cell lines established from different patients with rapidly progressive FL. This led to the establishment of AID‐expressing transfectants with a low proliferation rate and a significantly increased incidence of G0/G1 arrest compared with controls. Our results indicate that AID may act as a negative regulator of cell survival in FL when sufficient c‐myc is expressed. Switch‐off or low expression of AID after c‐myc amplification may correlate with the clinical outcomes of FL. (Cancer Sci 2012; 103: 415–421)
Follicular lymphoma is the most common type of low‐grade lymphoma, accounting for approximately 22% of all non‐Hodgkin’s lymphomas, and shows an indolent clinical course in up to 70% of cases.( 1 , 2 ) Its cell origin is normal GCB lymphocytes with ongoing SHM of the Ig gene in association with AID/AICDA.( 3 , 4 , 5 ) FL cells are typically positive for bcl‐2 protein. Approximately 75–90% of FL cells have a t(14;18)(q32;q21) translocation, which is the recognized hallmark for diagnosis of FL.( 6 ) Although this translocation is thought to be associated with oncogenic change, it is not sufficient to cause FL, because IgH‐bcl‐2 transgenic mice do not develop lymphomas, and IgH‐bcl‐2 translocation can be detected in peripheral blood lymphocytes from healthy individuals.( 7 , 8 , 9 )
Follicular lymphoma can show histological transformation into diffuse aggressive lymphoma during the clinical course in approximately 30% of patients.( 10 , 11 ) This transformation is usually associated with acceleration of the clinical course.( 12 ) Transformed FLs generally retain the t(14;18) translocation,( 13 ) and it is believed that other genetic abnormalities are necessary in order for this transformation to occur. These secondary genetic events associated with histological transformation include c‐myc amplification and translocation,( 14 , 15 ) bcl‐6 translocation,( 16 ) TP53 mutation,( 17 ) P16 gene inactivation,( 18 ) and c‐REL amplification.( 19 )
A series of reports has documented dual‐translocation lymphoma harboring both bcl‐2 and c‐myc translocation, and some of the reported cases have a history of preceding FL.( 20 , 21 ) C‐myc translocation occurs in almost all BLs,( 22 ) 5–15% of DLBCLs, and 2–3% of FLs.( 23 , 24 , 25 ) Although rare cases of histologically typical de novo FL with c‐myc translocation have been reported,( 25 ) c‐myc translocation is thought to occur as an additional genetic abnormality, in order for progression of FL to occur, because bcl‐2 translocation is reportedly an early event in B‐cell development whereas c‐myc translocation is thought to occur in GCB cells, which are AID‐dependent.( 4 , 14 , 26 ) Patients whose FLs show dual translocation of bcl‐2 and c‐myc generally have an extremely poor prognosis. However, few reports have documented other genetic abnormalities of lymphomas carrying c‐myc translocation or amplification during the course of FL.
Activation‐induced cytidine deaminase is required for SHM and CSR of the Ig gene and c‐myc translocation in GCB‐cell lymphoma.( 3 , 26 , 27 ) One report has documented that AID expression was detected in 10 of 15 cases of FL and was associated with ongoing mutation.( 5 ) In a plasmacytoma mouse model, the level of AID expression was correlated with the extent of c‐myc translocation.( 28 ) However, the role of AID in the clinical progression of FL has not been fully clarified. In the present study, we attempted to clarify the significance of AID‐associated c‐myc translocation and amplification during the progression of FL.
Materials and Methods
Patients and clinical samples. Our series included a total of 35 patients with FL treated at Ehime University Hospital (Toon, Japan) and Hiroshima University Hospital (Hiroshima, Japan). Informed consent was obtained from all patients in accordance with the Declaration of Helsinki principles. Chromosomal analyses were carried out by G‐banding and/or FISH. The FLs we examined were divided into three groups: patients with grade 1–2 FL (n = 15); patients with grade 3 FL who survived more than 2 years (n = 14); and patients with RPFL who died within 2 years of the start of treatment (n = 6). As pathologic controls and for genetic comparison, three different types of specimens were also examined, DLBCLs, BLs, and BCLU, and benign reactive hyperplasias. The DLBCLs were subclassified as GCB‐cell‐like lymphoma or ABC‐like lymphoma according to the classification of Hans et al. ( 29 ) This study was approved by the Ethics Committee for Clinical Studies at Ehime University Graduate School of Medicine (study # 1105004).
Cell lines and culture conditions. A human BL cell line (Raji), one BCLU cell line (Oki1), and three RPFL cell lines (established from patients at Ehime University Hospital) were incubated at 37°C under 5% CO2 in RPMI‐1640 medium supplemented with 10% heat‐inactivated FBS, 2 mmol/L glutamine, 1 mmol/L sodium pyruvate, 10 mmol/L HEPES buffer, 100 μg/mL streptomycin, and 100 U/mL penicillin.
Reverse transcription‐PCR and qRT‐PCR. Total RNAs were extracted from lymph node tissues or peripheral blood using an RNeasy Mini Kit (Qiagen, Tokyo, Japan). One microgram of total RNA was used for cDNA synthesis. Primer sequences for RT‐PCR are shown in Table S1. qRT‐PCR assay was carried out using TaqMan technology (Applied Biosystems, Foster City, CA, USA). The specific numbers for the TaqMan Gene Expression Assay primers were Hs00757808_m1 for AID and Hs99999003_m1 for c‐myc. The internal control was GAPDH (Hs99999905_m1). The expression levels of AID and c‐myc were corrected by reference to that of GAPDH, and the relative expression levels for each sample were calculated using ΔΔC t values with a control sample.
Western blot analysis. Cells were lysed with cell lysis buffer. After sonication and centrifugation, the supernatant was collected as the lysate. After addition of SDS buffer, each lysate was separated by 10% SDS‐PAGE, and transferred to nitrocellulose membranes. The blots were incubated with rabbit anti‐human AID polyclonal antibody (Abcam, Cambridge, UK), and rabbit anti‐human GAPDH polyclonal antibody (Abcam) was used as a control. These antibodies were detected with a donkey polyclonal secondary antibody against rabbit IgG (Abcam).
Immunohistochemistry for AID. Cells were fixed in 4% paraformaldehyde and paraffin‐embedded. After deparaffinization and blocking of endogenous peroxidase activity, the sections were incubated overnight at 4°C with a primary anti‐AID rabbit polyclonal antibody (Abcam) at a dilution of 1:250. After washing and incubation with peroxidase‐conjugated goat polyclonal secondary antibody against rabbit IgG (Dako, Kyoto, Japan), the sections were reacted with diaminobenzidine and counterstained with hematoxylin.
Transfection. Human full‐length AID was amplified from cDNA of a BL cell line (Raji) using the primers for transfection (Table S1). The PCR products were cloned into an expression vector. An HIV‐based self‐inactivating lentiviral expression vector, CSII‐CMV‐MCS‐IRES‐Bsd, and two packaging vectors (pCAG‐HIVgp and pCMV‐VSV‐G‐RSV‐Rev) were kindly provided by Dr. Hiroyuki Miyoshi (Riken BioResource Center, Wako, Japan). Lentiviral particles were generated using standard transfection procedures. To evaluate the expression of AID, transduced cells were examined by qRT‐PCR, Western blotting analysis, and immunohistochemical staining. Vector‐only transfectants and parental cells were used as controls.
Estimation of cell proliferation using [3H]thymidine incorporation. Each of the cell lines was seeded in a 96‐well plate in triplicate at 1 × 104 cells per well. Cells cultured for 12 h were pulsed with 0.5 mCi [3H]thymidine, then harvested onto glass filters with a cell harvester (Labo Science, Tokyo, Japan) after 24, 48, and 72 h. The uptake of [3H]thymidine was monitored using a liquid scintillation counter (LSC‐5100; Aloka, Tokyo, Japan). Experimental data were expressed as counts per minute (mean ± SD).
Cell cycle assay. Cell cycle analysis was carried out using a Cycletest Plus DNA Reagent kit (BD Biosciences, Franklin Lakes, NJ, USA) in accordance with the manufacturer’s instructions. Each of the cell lines was analyzed for cell cycle distribution in comparison with the control.
Statistical analysis. Statistical significance of differences in the expression of AID and c‐myc was determined by non‐parametric Mann–Whitney U‐test at a significance level of P < 0.05.
Results
If a B‐cell lymphoma has t(14;18) and c‐myc amplification and/or translocation, it would be diagnosed as “double hit” or “dual translocation”; also, some B‐cell lymphomas are unclassifiable, as they have features intermediate between DLBCL and BL (BCLU). It is also known that some of these aggressive lymphomas are derived from FL. However, it is not appropriate to designate this type of lymphoma as tFL, because some FLs with grade IIIB morphology, which are included in the tFL category, are potentially curable with current chemotherapies. Within the past 10 years, our institution has treated 11 patients with FL that showed an aggressive and refractory clinical course. A consistent clinical feature was that the FL was rapidly progressive and chemotherapy‐resistant, and expressed a high degree of Ki‐67 immunostaining. Out of these 11 patients, six clinical samples were available for genetic analyses in this study. The clinical characteristics of the six patients (FL30–FL35) with RPFL who died within 2 years (five patients within 5 months) after chemotherapy are shown in Table 1. We had isolated mRNAs from tissue biopsy samples from these six patients with RPFL before treatment, and also mRNA from additional clinical samples obtained from two of the patients (FL30 and FL31) shortly before their death. Moreover, we had established three RPFL cell lines out of the six patients with RPFL (FL30, FL31, and FL32). Using the above clinical samples, we tried to identify differences between the RPFLs designated here and other aggressive B‐cell lymphomas, together with the genes involved in their pathogenesis. For this purpose, we designed and carried out the following experiments.
Table 1.
Clinical characteristics of six patients with rapidly progressive follicular lymphoma (FL)
| Case | Age (years)/gender | FLIPI | Extranodal sites | Immunohistochemistry | Karyotype | Therapy | Survival (months) |
|---|---|---|---|---|---|---|---|
| FL30 | 73/F | 4 | BM, CNS | CD5−, CD10+, CD19+, CD20+, CD79a+, bcl‐2+ | t(14;18)(q32;q21), t(8;22)(q24; q11.2) | R‐CHOP | 5 |
| FL31 | 54/M | 4 | Gingiva | CD5−, CD10+, CD19+, CD20+, CD79a+, bcl‐2+ | t(14;18)(q32;q21), c‐myc rearrangement (FISH) | R‐CHOP, EPOCH | 4 |
| FL32 | 47/M | 2 | BM | CD5−, CD10+, CD19+, CD20+, CD79a+, bcl‐2+ | t(14;18)(q32;q21), t(8;22)(q24; q11.2) | R‐CHOP, Flu | 4 |
| FL33 | 48/F | 3 | None | CD5−, CD10+, CD19+, CD20+, CD79a+, bcl‐2+ | t(14;18)(q32;q21) | R‐CHOP, R‐DEVIC, Allo BMT | 20 |
| FL34 | 43/M | 3 | Ascites | CD5−, CD10+, CD19+, CD20+, CD79a+, bcl‐2+ | t(14;18)(q32;q21) | R‐CHOP, R‐MECP | 5 |
| FL35 | 55/M | 4 | BM | CD5−, CD10+, CD19+, CD20+, CD79a+, bcl‐2+ | nd | R‐CHOP | 3 |
Allo BMT, allogenic bone marrow transplant; BM, bone marrow; CNS, central nervous system; EPOCH, etoposide, prednisolone, vincristine, cyclophosphamide and doxorubicin; F, female; FLIPI, Follicular Lymphoma International Prognostic Index; Flu, fludarabine; M, male; nd; not done; R‐CHOP, rituximab, cyclophosphamide, doxorubicin, vincristine and prednisolone; R‐DEVIC, rituximab, dexamethasone, etoposide, ifosfamide and carboplatin; R‐MECP, rituximab, mitoxantrone, etoposide, carbonplatin and prednisolone.
Gene expression in RPFLs and negative expression of AID. Our initial focus was to place RPFL in its appropriate context within the cytologic and genetic spectrum of cell classification, that is, to determine whether RPFL, although an advanced and rare disease, is nevertheless grouped within the FL category, or represents a transformed DLBCL. To clarify this issue, we examined the gene expression of RPFL compared with other types of lymphoma. We selected bcl‐6 as a GCB cell marker, IRF4 (MUM‐1) and NF‐κB as ABC markers, and bcl‐2 as a hallmark of FL, and carried out RT‐PCR analysis. As shown in Figure 1, the RPFLs still had enough expression of bcl‐2 and GCB‐associated genes, and strong expression of c‐myc, suggesting that the RPFLs we sampled were classifiable as GC‐derived FL expressing c‐myc. As a marker of GCB cells, we also examined the AID transcript, which is required for c‐myc translocation. We had expected significant amounts of AID expression in samples of c‐myc‐associated RPFL. However, two clinical tissue samples from the patients (FL30 and FL31) with RPFL at the progressive stage (just before death) showed complete shutdown of the AID transcript, even though the same cases had expressed AID and c‐myc before the start of treatment (Fig. 2). This finding was contrary to expectation. Moreover, interferon regulatory factor 8, which regulates AID expression in GCs,( 30 ) was also shut down in tissue samples from patients with RPFL at the progressive stage (data not shown), suggesting that transcriptional signaling upstream of AID is downregulated.
Figure 1.
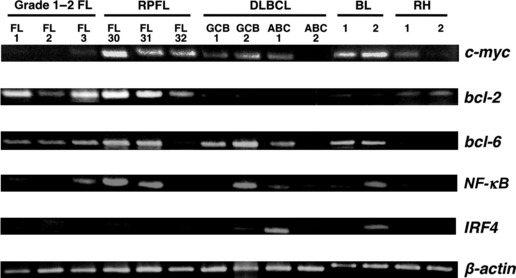
Relative expression of genes in c‐myc‐associated rapidly progressive follicular lymphoma (RPFL) determined by RT‐PCR. Expression of c‐myc, bcl‐2, bcl‐6, NF‐κB, and IRF‐4 in c‐myc‐associated RPFL (n = 3) was compared to that in grade 1–2 follicular lymphoma (FL) (n = 3), germinal center B (GCB)‐like type diffuse large B‐cell lymphoma (DLBCL) (n = 2), activated B cell (ABC)‐like type DLBCL (n = 2), Burkitt’s lymphoma (BL) (n = 2), and reactive hyperplasia (RH) (n = 2). β‐actin was used as an endogenous control.
Figure 2.
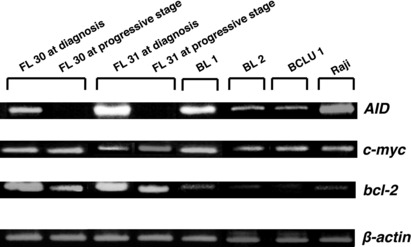
Expression of activation‐induced cytidine deaminase (AID), c‐myc, and bcl‐2 in c‐myc‐associated rapidly progressive follicular lymphoma (RPFL), Burkitt’s lymphoma (BL), and B‐cell lymphoma, unclassifiable with features intermediate between DLBCL and BL (BCLU), determined by RT‐PCR. Expression of c‐myc, AID, and bcl‐2 was examined in tissue biopsy samples from patients with c‐myc‐associated RPFL (n = 2), BL (n = 2), and BCLU (n = 1) before treatment, and two additional second‐biopsy specimens from RPFL patients at the progressive stage (shortly before death). A human BL cell line (Raji) was used as a positive control. β‐actin was used as an endogenous control.
Expression of AID and c‐myc in FLs determined using qRT‐PCR. We next examined the relationship between AID and c‐myc expression in an additional series of 35 clinical samples from patients with FL (Fig. 3). Samples from the patients with grade 3 FL expressed relatively higher levels of the c‐myc transcript than samples from the patients with grade 1–2 FL (P = 0.02; Fig. 3A). Additionally, non‐significantly high levels of AID expression were observed in tissues from the patients with grade 3 FL (Fig. 3A). Interestingly, all four samples from the patients (FL32–FL35) with RPFL expressed no, or only low levels of, AID transcript, even though they expressed high levels of c‐myc (Fig. 3B). These results suggested that overexpression of c‐myc might be correlated with the biological transformation and clinical progression of FL, and also that AID might play a partial role in disease progression. In contrast, low expression of AID may be associated with the clinical features and outcome of patients with RPFL.
Figure 3.
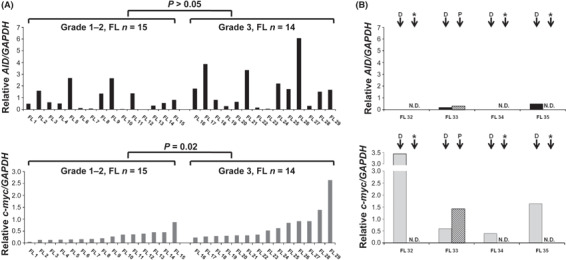
Expression of activation‐induced cytidine deaminase (AID) and c‐myc in follicular lymphomas (FLs) and rapidly progressive FLs determined by quantitative real‐time PCR. Expression levels of AID and c‐myc normalized to that of GAPDH in grade 1–2 FL (n = 15) and grade 3 FL (n = 14) (A), and rapidly progressive FL (n = 4) (B) were analyzed. Top panel: a series of experiments showing AID expression. Bottom panel: a series of experiments showing c‐myc expression. *N.D., not done. D, samples obtained at diagnosis; P, samples obtained at progressive stage (shortly before death).
Biological changes resulting from AID transfection in AID‐negative cell lines. We established three different cell lines from patients with RPFL (FL30, FL31, and FL32) (Fig. 4), and none of them expressed the AID transcript (Fig. 5A). In order to examine the role of AID expression in RPFL, the full‐length AID gene was transfected into two of these AID‐negative cell lines (FL30 and FL32), and three clones from the FL30 patient and two clones from the FL32 patient were established. Differences in the expression levels of AID among the transfectants were determined using qRT‐PCR (Fig. 5A). Western blot analysis showed that the AID‐transfectants established from FL30 expressed the AID protein (Fig. 5B). Immunohistochemical analysis showed that the transfectants expressed significant amounts of AID protein, predominantly in the cytoplasm (Fig. 5C,D).
Figure 4.
Morphological features of rapidly progressive follicular lymphoma (FL) cell lines. Three cell lines established from different patients with rapidly progressive FL (FL30, FL31, and FL32) indicate medium‐sized morphology compared to other cell lines derived from Burkitt’s lymphoma (Raji) and B‐cell lymphoma, unclassified (Oki1).
Figure 5.
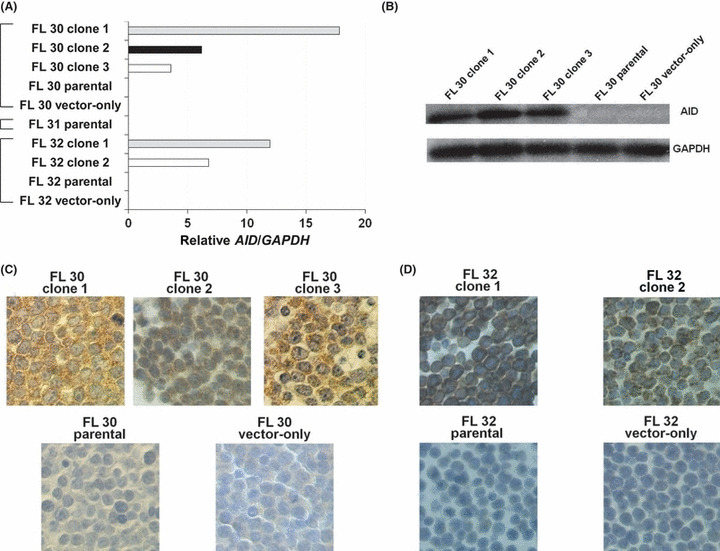
Establishment of activation‐induced cytidine deaminase (AID)‐positive transfectants. The full‐length AID transcript was transfected into three AID‐negative cell lines established from different patients with rapidly progressive follicular lymphoma (FL; designated FL30 and FL32). Three clones from the FL30 patient and two clones from the FL32 patient were established. (A) Differences in the expression levels of AID among the transfectants were determined by quantitative real‐time PCR. (B) Western blot analysis showed that the AID‐transfectants expressed AID protein. (C) Immunohistochemical analysis showed that the transfectants expressed significant amounts of AID protein, predominantly in the cytoplasm. (D) Similar results were obtained for the transfectants established from FL32.
Interestingly, three AID‐positive clones from FL30 and two AID‐positive transfectants from FL32 with different levels of AID expression showed damping of cell proliferation proportional to the genetic level of AID expression, relative to the controls (Fig. 6). Cell cycle analysis showed that the AID‐positive transfectants established from FL30 had an increased cell population in G0/G1 phase and a reduced population in S phase, suggesting that G0/G1 arrest was responsible for the reduction in cell proliferation relative to the parental cells and the vector‐only transfectant used as controls (Fig. 7A). Similar results were obtained for the AID‐positive transfectants from FL32 (Fig. 7B). These results indicated that AID acts as a negative regulator of FL progression in c‐myc‐associated RPFL.
Figure 6.
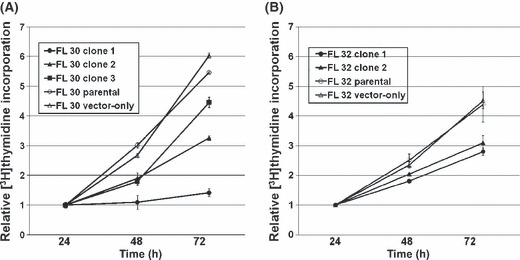
Cell growth of activation‐induced cytidine deaminase (AID)‐positive transfectants. The AID‐positive transfectants showed low proliferation rates compared with the parental cells and vector‐only transfectant used as controls. Cells cultured for 12 h were pulsed with 0.5 mCi [3H]thymidine, then harvested onto glass filters after 24, 48, and 72 h. (A) Three AID‐positive clones from a patient with follicular lymphoma (FL), designated FL30, expressing different levels of AID showed damping of cell proliferation that was proportional to the expression level of the AID gene, compared with controls. (B) Similar results were obtained for AID‐positive clones from patient FL32.
Figure 7.
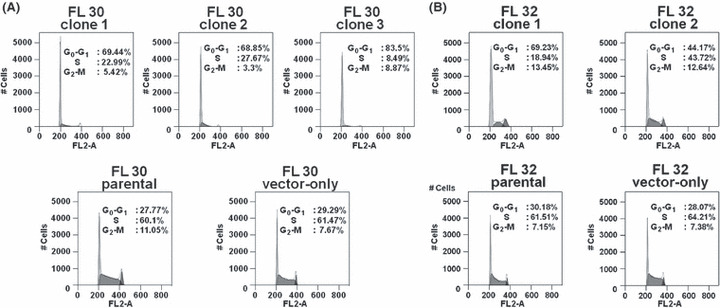
Cell cycle characteristics of activation‐induced cytidine deaminase (AID)‐positive transfectants. (A) AID‐positive transfectants from a patient with follicular lymphoma (FL), designated FL30, showed an increased population of cells in G0/G1 phase and a reduced population in S phase relative to the parental cells and vector‐only transfectant used as controls. (B) Similar results were obtained for AID‐positive clones from patient FL32.
Discussion
During clinical observation of patients with FL, clinicians sometimes encounter cases that show accelerated tumor progression, for which the term “RPFL” in this article has been adopted. The majority of RPFLs are refractory to any antitumor agents, and their unresponsiveness to myeloablative therapies renders them incurable. These tumor cells express a high level of Ki‐67,( 31 ) a cell proliferation marker, suggesting that molecules involved in the cell cycle are involved in the pathogenesis.
The initial focus of our present study was to clarify whether RPFL is pathologically classifiable as FL, or as BCLU, that is, so‐called Burkitt‐like DLBCL. Our present series of RPFLs showed strong expression of c‐myc, retained expression of bcl‐2, and negativity for IRF4 (MUM‐1).( 32 ) Morphologically, they were medium‐sized cleaved‐cell lymphomas, suggesting that RPFL is a lymphoma with a pathological and genetic background of FL, harboring not only t(14;18) but also secondary genetic events. Recent clinical studies have indicated that c‐myc translocation, known to be one of the secondary genetic events occurring after bcl‐2 translocation, is accompanied by an aggressive clinical course. Many hematopathologists classify these lymphomas as a distinct subtype of non‐Hodgkin’s lymphoma, designated double (dual) hit lymphoma,( 33 ) in which c‐myc is intrinsically involved in the lymphomagenesis. Among our present cases, a split signal and/or translocation of c‐myc was detectable in three samples out of six RPFLs that were testable using FISH and/or G‐banding analysis, indicated that some of the RPFLs were such double hit lymphomas. Unlike previous studies of double hit lymphomas, however, we chose to focus directly on the expression of c‐myc, because overexpression rather than translocation of c‐myc would be more directly associated with the clinical features and aggressiveness of RPFL. Our results indicated that tumors with a high level of c‐myc expression had a more aggressive morphology and clinical course, similar to the cases reported by Lossos et al. ( 15 ) C‐myc thus appears to be a powerful predictor of the clinical course of FL, especially RPFL.
Our next objective was to clarify how c‐myc‐associated genes were involved in the pathogenesis of RPFL. The t(14;18) translocation is thought to occur early in B‐cell lymphoma through expression of RAG1/2.( 34 ) In contrast, most chromosomal breakpoints that involve c‐myc and the IgH locus are mediated by AID and not by RAG1/2.( 35 ) Therefore, the AID transcript would be a likely candidate gene for involvement in c‐myc‐associated pathogenesis. We expected that AID would be overexpressed in the advanced stages of FL. However, we found that RPFLs showed no levels, or only low levels, of AID expression, even though they expressed high levels of c‐myc. This tendency was especially evident in the more advanced stages of RPFL (in the cases of FL30 and FL31). In other words, overexpression of both c‐myc and AID was not sufficient to trigger an aggressive clinical course of FL. In fact, patient FL29 in the present series, whose histologically typical grade 3 FL showed both t(14;18) and c‐myc translocation to the IgH locus, t(8;14)(q24;q32), with high expression of both c‐myc and AID, achieved complete remission after chemotherapy. Five years later, this patient suffered relapse as FL with t(14;18) as the sole genetic abnormality (an additional genetic abnormality of t(8;14)(q24;q32) having disappeared), and expressed low levels of c‐myc and AID (data not shown). After rituximab monotherapy, this patient maintained a second complete remission for 6 years. The clinical course of this patient suggested that clonal evolution may have been induced by AID as a second‐hit genetic event, and that overexpression of c‐myc after induction of AID expression may be insufficient for malignant transformation of FL.
Activation‐induced cytidine deaminase is known to deaminate cytidine residues in DNA by converting them to uridine residues, resulting in DNA cleavage for CSR or c‐myc translocation.( 3 , 26 , 27 , 36 ) Hardianti et al. ( 5 ) reported that the SHM rate of the Ig gene in AID‐negative FL is generally higher than that of AID‐positive FL, suggesting that B lymphocytes might require AID to maintain ongoing mutation after DNA cleavage, resulting in SHM. However, cells may downregulate AID in GCs. This phenomenon indicates that AID may be necessary for c‐myc translocation, although the effect of DNA cleavage and subsequent c‐myc translocation by AID may be insufficient for maintaining the gene activation necessary for RPFL progression. A recent basic analysis has indicated increased proliferation and decreased apoptosis of AID−/− B lymphocytes in comparison with AID‐expressing B lymphocytes derived from the GC. In addition, expression of AID was shown to lead to the generation of point mutations for SHM and CSR, resulting in DNA damage and apoptosis,( 37 ) probably in the deregulated expression of anti‐apoptotic factors such as bcl‐2 and bcl‐XL.( 38 ) These findings suggest that cell‐cycle arrest or apoptosis can be ultimately induced when sufficient AID is subsequently expressed in B lymphocytes after CSR. Interestingly, among our sources of lymphoma samples, two BLs and one de novo Burkitt‐like DLBCL (BCLU1) did express c‐myc and AID, but not bcl‐2, and they did not undergo G0/G1 arrest (data not shown). These in vivo and in vitro findings suggest that the pathogenesis of c‐myc‐associated FL may be different from the pathogenesis of BL and de novo Burkitt‐like DLBCL, and that downregulation of AID in cooperation with the bcl‐2 family may be one of the clinically observed mechanisms operating in RPFL. These phenomena might drive the rapid progression of c‐myc‐associated FLs.
In conclusion, our findings suggest a possible role of AID expression and downregulation in the progression of FL. AID might not only induce CSR of the Ig and c‐myc translocation, but could also be a factor involved in histological regulation and progression of FL. Switch‐off or low expression of AID after c‐myc amplification could be a potentially useful marker for prognostication of FL.
Disclosure Statement
The authors have no conflict of interest.
Abbreviations
- ABC
activated B cell
- AID/AICDA
activation‐induced cytidine deaminase
- BCLU
B‐cell lymphoma, unclassifiable with features intermediate between diffuse large B‐cell lymphoma and Burkitt’s lymphoma
- BL
Burkitt’s lymphoma
- CSR
class‐switch recombination
- DLBCL
diffuse large B‐cell lymphoma
- FL
follicular lymphoma
- GC
germinal center
- GCB
germinal center B
- Ig
immunoglobulin gene
- qRT‐PCR
quantitative real‐time PCR
- RPFL
rapidly progressive follicular lymphoma
- SHM
somatic hypermutation
- tFL
transformed follicular lymphoma
Supporting information
Supporting info item
Acknowledgments
We are grateful for the skilled technical assistance of Dr Kozo Nagai, Dr Akiyoshi Nagatoshi, and Dr Kenji Kameda, Ehime University (Toon, Japan).
References
- 1. Armitage JO, Weisenburger DD. New approach to classifying non‐Hodgkin’s lymphomas: clinical features of the major histologic subtypes. Non‐Hodgkin’s Lymphoma Classification Project. J Clin Oncol 1998; 16: 2780–95. [DOI] [PubMed] [Google Scholar]
- 2. Horning SJ. Natural history of and therapy for the indolent non‐Hodgkin’s lymphomas. Semin Oncol 1993; 20: 75–88. [PubMed] [Google Scholar]
- 3. Martin A, Bardwell PD, Woo CJ, Fan M, Shulman MJ, Scharff MD. Activation‐induced cytidine deaminase turns on somatic hypermutation in hybridomas. Nature 2002; 415: 802–6. [DOI] [PubMed] [Google Scholar]
- 4. Smit LA, Bende RJ, Aten J, Guikema JE, Aarts WM, van Noesel CJ. Expression of activation‐induced cytidine deaminase is confined to B‐cell non‐Hodgkin’s lymphomas of germinal‐center phenotype. Cancer Res 2003; 63: 3894–8. [PubMed] [Google Scholar]
- 5. Hardianti MS, Tatsumi E, Syampurnawati M et al. Activation‐induced cytidine deaminase expression in follicular lymphoma: association between AID expression and ongoing mutation in FL. Leukemia 2004; 18: 826–31. [DOI] [PubMed] [Google Scholar]
- 6. Rowley JD. Chromosome studies in the non‐Hodgkin’s lymphomas: the role of the 14;18 translocation. J Clin Oncol 1988; 6: 919–25. [DOI] [PubMed] [Google Scholar]
- 7. McDonnell TJ, Deane N, Platt FM et al. bcl‐2‐immunoglobulin transgenic mice demonstrate extended B cell survival and follicular lymphoproliferation. Cell 1989; 57: 79–88. [DOI] [PubMed] [Google Scholar]
- 8. Dolken G, Illerhaus G, Hirt C, Mertelsmann R. BCL‐2/JH rearrangements in circulating B cells of healthy blood donors and patients with nonmalignant diseases. J Clin Oncol 1996; 14: 1333–44. [DOI] [PubMed] [Google Scholar]
- 9. Yasukawa M, Bando S, Dolken G et al. Low frequency of BCL‐2/J(H) translocation in peripheral blood lymphocytes of healthy Japanese individuals. Blood 2001; 98: 486–8. [DOI] [PubMed] [Google Scholar]
- 10. Cullen MH, Lister TA, Brearley RI, Shand WS, Stansfeld AG. Histological transformation of non‐Hodgkin’s lymphoma: a prospective study. Cancer 1979; 44: 645–51. [DOI] [PubMed] [Google Scholar]
- 11. Bastion Y, Sebban C, Berger F et al. Incidence, predictive factors, and outcome of lymphoma transformation in follicular lymphoma patients. J Clin Oncol 1997; 15: 1587–94. [DOI] [PubMed] [Google Scholar]
- 12. Montoto S, Davies AJ, Matthews J et al. Risk and clinical implications of transformation of follicular lymphoma to diffuse large B‐cell lymphoma. J Clin Oncol 2007; 25: 2426–33. [DOI] [PubMed] [Google Scholar]
- 13. Maeshima AM, Omatsu M, Nomoto J et al. Diffuse large B‐cell lymphoma after transformation from low‐grade follicular lymphoma: morphological, immunohistochemical, and FISH analyses. Cancer Sci 2008; 99: 1760–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. De Jong D, Voetdijk BM, Beverstock GC, van Ommen GJ, Willemze R, Kluin PM. Activation of the c‐myc oncogene in a precursor‐B‐cell blast crisis of follicular lymphoma, presenting as composite lymphoma. N Engl J Med 1988; 318: 1373–8. [DOI] [PubMed] [Google Scholar]
- 15. Lossos IS, Alizadeh AA, Diehn M et al. Transformation of follicular lymphoma to diffuse large‐cell lymphoma: alternative patterns with increased or decreased expression of c‐myc and its regulated genes. Proc Natl Acad Sci USA 2002; 99: 8886–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Akasaka T, Lossos IS, Levy R. BCL6 gene translocation in follicular lymphoma: a harbinger of eventual transformation to diffuse aggressive lymphoma. Blood 2003; 102: 1443–8. [DOI] [PubMed] [Google Scholar]
- 17. Lo Coco F, Gaidano G, Louie DC, Offit K, Chaganti RS, Dalla‐Favera R. p53 mutations are associated with histologic transformation of follicular lymphoma. Blood 1993; 82: 2289–95. [PubMed] [Google Scholar]
- 18. Pinyol M, Cobo F, Bea S et al. p16(INK4a) gene inactivation by deletions, mutations, and hypermethylation is associated with transformed and aggressive variants of non‐Hodgkin’s lymphomas. Blood 1998; 91: 2977–84. [PubMed] [Google Scholar]
- 19. Davies AJ, Rosenwald A, Wright G et al. Transformation of follicular lymphoma to diffuse large B‐cell lymphoma proceeds by distinct oncogenic mechanisms. Br J Haematol 2007; 136: 286–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Johnson NA, Savage KJ, Ludkovski O et al. Lymphomas with concurrent BCL2 and MYC translocations: the critical factors associated with survival. Blood 2009; 114: 2273–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tomita N, Tokunaka M, Nakamura N et al. Clinicopathological features of lymphoma/leukemia patients carrying both BCL2 and MYC translocations. Haematologica 2009; 94: 935–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hecht JL, Aster JC. Molecular biology of Burkitt’s lymphoma. J Clin Oncol 2000; 18: 3707–21. [DOI] [PubMed] [Google Scholar]
- 23. Kramer MH, Hermans J, Wijburg E et al. Clinical relevance of BCL2, BCL6, and MYC rearrangements in diffuse large B‐cell lymphoma. Blood 1998; 92: 3152–62. [PubMed] [Google Scholar]
- 24. Niitsu N, Okamoto M, Miura I, Hirano M. Clinical features and prognosis of de novo diffuse large B‐cell lymphoma with t(14;18) and 8q24/c‐MYC translocations. Leukemia 2009; 23: 777–83. [DOI] [PubMed] [Google Scholar]
- 25. Christie L, Kernohan N, Levison D et al. C‐MYC translocation in t(14;18) positive follicular lymphoma at presentation: an adverse prognostic indicator? Leuk Lymphoma 2008; 49: 470–6. [DOI] [PubMed] [Google Scholar]
- 26. Ramiro AR, Jankovic M, Eisenreich T et al. AID is required for c‐myc/IgH chromosome translocations in vivo. Cell 2004; 118: 431–8. [DOI] [PubMed] [Google Scholar]
- 27. Revy P, Muto T, Levy Y et al. Activation‐induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper‐IgM syndrome (HIGM2). Cell 2000; 102: 565–75. [DOI] [PubMed] [Google Scholar]
- 28. Takizawa M, Tolarova H, Li Z et al. AID expression levels determine the extent of cMyc oncogenic translocations and the incidence of B cell tumor development. J Exp Med 2008; 205: 1949–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hans CP, Weisenburger DD, Greiner TC et al. Confirmation of the molecular classification of diffuse large B‐cell lymphoma by immunohistochemistry using a tissue microarray. Blood 2004; 103: 275–82. [DOI] [PubMed] [Google Scholar]
- 30. Lu R. Interferon regulatory factor 4 and 8 in B‐cell development. Trends Immunol 2008; 29: 487–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang SA, Wang L, Hochberg EP, Muzikansky A, Harris NL, Hasserjian RP. Low histologic grade follicular lymphoma with high proliferation index: morphologic and clinical features. Am J Surg Pathol 2005; 29: 1490–6. [DOI] [PubMed] [Google Scholar]
- 32. Naresh KN. MUM1 expression dichotomises follicular lymphoma into predominantly, MUM1‐negative low‐grade and MUM1‐positive high‐grade subtypes. Haematologica 2007; 92: 267–8. [DOI] [PubMed] [Google Scholar]
- 33. Aukema SM, Siebert R, Schuuring E et al. Double‐hit B‐cell lymphomas. Blood 2011; 117: 2319–31. [DOI] [PubMed] [Google Scholar]
- 34. Tsai AG, Lu H, Raghavan SC, Muschen M, Hsieh CL, Lieber MR. Human chromosomal translocations at CpG sites and a theoretical basis for their lineage and stage specificity. Cell 2008; 135: 1130–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dorsett Y, Robbiani DF, Jankovic M, Reina‐San‐Martin B, Eisenreich TR, Nussenzweig MC. A role for AID in chromosome translocations between c‐myc and the IgH variable region. J Exp Med 2007; 204: 2225–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chaudhuri J, Tian M, Khuong C, Chua K, Pinaud E, Alt FW. Transcription‐targeted DNA deamination by the AID antibody diversification enzyme. Nature 2003; 422: 726–30. [DOI] [PubMed] [Google Scholar]
- 37. Zaheen A, Boulianne B, Parsa JY, Ramachandran S, Gommerman JL, Martin A. AID constrains germinal center size by rendering B cells susceptible to apoptosis. Blood 2009; 114: 547–54. [DOI] [PubMed] [Google Scholar]
- 38. Jankovic M, Robbiani DF, Dorsett Y et al. Role of the translocation partner in protection against AID‐dependent chromosomal translocations. Proc Natl Acad Sci USA 2010; 107: 187–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting info item