

Abstract
The enhanced permeability and retention (EPR) effect is a unique pathophysiological phenomenon of solid tumors that sees biocompatible macromolecules (>40 kDa) accumulate selectively in the tumor. Various factors have been implicated in this effect. Herein, we report that heme oxygenase‐1 (HO‐1; also known as heat shock protein 32) significantly increases vascular permeability and thus macromolecular drug accumulation in tumors. Intradermal injection of recombinant HO‐1 in mice, followed by i.v. administration of a macromolecular Evans blue–albumin complex, resulted in dose‐dependent extravasation of Evans blue–albumin at the HO‐1 injection site. Almost no extravasation was detected when inactivated HO‐1 or a carbon monoxide (CO) scavenger was injected instead. Because HO‐1 generates CO, these data imply that CO plays a key role in vascular leakage. This is supported by results obtained after intratumoral administration of a CO‐releasing agent (tricarbonyldichlororuthenium(II) dimer) in the same experimental setting, specifically dose‐dependent increases in vascular permeability plus augmented tumor blood flow. In addition, induction of HO‐1 in tumors by the water‐soluble macromolecular HO‐1 inducer pegylated hemin significantly increased tumor blood flow and Evans blue–albumin accumulation in tumors. These findings suggest that HO‐1 and/or CO are important mediators of the EPR effect. Thus, anticancer chemotherapy using macromolecular drugs may be improved by combination with an HO‐1 inducer, such as pegylated hemin, via an enhanced EPR effect. (Cancer Sci 2012; 103: 535–541)
Conventional chemotherapy with small molecule drugs has been used for many types of cancer for decades. However, the therapeutic efficacy remains less than optimal, mostly because of a lack of tumor selectivity, which results in severe adverse side effects and prevents the use of high drug doses.( 1 ) The development of tumor‐targeted chemotherapy is critically important for more successful treatment.
During investigations of targeting drugs to tumors, Matsumura and Maeda( 2 ) found that macromolecular agents larger than 40 kDa selectively accumulate and remain in tumor tissues for long periods. This unique phenomenon in the blood vasculature of solid tumor tissues is quite different from that in normal tissues and was attributed to the unique anatomic and pathophysiologic characteristics of solid tumors. These features include: (i) extensive angiogenesis and hence high vascular density;( 3 , 4 ) (ii) extensive extravasation (vascular permeability) induced by various vascular mediators, including bradykinin,( 5 , 6 , 7 ) nitric oxide (NO),( 7 , 8 ) vascular endothelial growth factor (VEGF),( 9 , 10 ) prostaglandins produced via cyclo‐oxygenases,( 7 ) and matrix metalloproteinases;( 11 ) (iii) defective vascular architecture, such as the lack of a smooth muscle layer and large gaps between vascular endothelial cells;( 12 , 13 ) and (iv) impaired lymphatic clearance from the tumor interstitial space.( 14 , 15 , 16 ) The increased vascular permeability and defective vascular structure allow molecules larger than those subject to renal clearance (i.e. ≥40 kDa) to extravasate gradually, over a long time, into the interstitial space in tumor tissues. Furthermore, the molecules remain in the interstitial space without being cleared because of the impaired lymphatic system in tumors.( 2 , 13 , 14 , 15 , 16 ) This phenomenon was named the enhanced permeability and retention effect (EPR effect) in solid tumors.
In previous studies, we have shown that this EPR effect can be augmented and drug delivery improved two‐ to threefold( 16 , 17 , 18 , 19 , 20 ). One approach used angiotensin (Ang) II‐induced hypertension, during which tumor blood flow was increased selectively.( 16 , 17 ) The AngII‐induced augmentation of the EPR effect was validated not only in animal experiments, but also in the clinical setting with difficult‐to‐treat tumors.( 18 , 19 ) Another approach involved using NO‐generating agents such as nitroglycerin (NTG), which significantly increased the accumulation of macromolecular drugs in tumors.( 20 )
In a completely different series of experiments, we have been working on heme oxygenase‐1 (HO‐1), known as a key factor for supporting rapid tumor growth, as an anticancer target.( 21 , 22 , 23 , 24 ) Heme oxygenase is a key enzyme in heme metabolism, with products including biliverdin, carbon monoxide (CO), and free iron (Fe3+); biliverdin is subsequently converted to bilirubin.( 25 , 26 ) Numerous studies have demonstrated important physiological roles of CO, comparable to those of NO, including vascular dilatation, facilitation of vascular blood flow, and antioxidative and antiapoptotic effects.( 27 , 28 , 29 ) We thus hypothesized that CO‐generating HO‐1 may serve as another factor mediating the EPR effect and may be useful in augmenting chemotherapeutic effects.
In the present study, we used recombinant HO‐1 and a CO‐releasing agent to investigate the effects of HO‐1 and CO on vascular permeability. In addition, by using the water‐soluble macromolecular HO‐1 inducer pegylated hemin (PEG‐hemin), we verified the CO‐ and HO‐1‐induced augmentation of the EPR effect in a murine solid tumor model.
Materials and Methods
Materials. Tricarbonyldichlororuthenium(II) dimer (CORM‐2), zinc protoporphyrin‐IX (ZnPP), and hemin were purchased from Sigma‐Aldrich Chemical (St Louis, MO, USA). Other chemicals of reagent grade were from Wako Pure Chemical Industries (Osaka, Japan) and were used without further purification.
Animals. Male ddY mice, 6 weeks old and weighing 30–35 g, and male SD rat weighing 200‐250 g were obtained from Kyudo (Kumamoto, Japan). Mice were maintained under standard conditions (12‐h dark–light cycle, 23 ± 1°C). All experiments were performed according to the guidelines of the Laboratory Protocol of Animal Handling, Sojo University.
Preparation of recombinant HO‐1 protein. Total mRNA was extracted from rat liver using the Takara RNA PCR Kit (AMV) Ver. 3.0 (Takara Bio, Otsu, Japan), with HO‐1 cDNA being amplified with HO‐1 Cf (gatcagcactagttcatcccagacataacctag) and HO‐1 Cr (gttatgtctgggatgaactagtgct) primers using KOD FX polymerase (Takara). Rat HO‐1 cDNA was then inserted into the pET3c vector through the Nde1 and BamH1 restriction enzyme sites located at the 5′ and 3′ ends, respectively.
Escherichia coli Rosetta‐gami (DE3) bacteria harboring the abovementioned pET3c plasmid encoding rat HO‐1 were cultured in LB medium containing 50 μg/mL ampicillin and 20 μg/mL chloramphenicol. The HO‐1 protein was induced by addition of 10 μM isopropyl β‐d‐thiogalactopyranoside. After 6 h incubation at 37°C with shaking, the bacterial pellet was sonicated (150 W, 20 min) in 50 mM Tris–HCl buffer (pH 8.0) with protease inhibitors (2 mM phenylmethylsulfonyl fluoride and 10 μg/mL leupeptin) at 4°C. Then, rat HO‐1 was partially purified by using ammonium sulfate precipitation (30–60% fraction), after which it was dialyzed against 10 mM potassium phosphate buffer (pH 7.4). It was finally purified by using a DEAE negative ion exchange column; 10 mM phosphate buffer (pH 7.4) containing 250 mM KCl was used for gradient elution. The purity of the rat HO‐1 was demonstrated to be >90% using SDS‐PAGE with Coomassie brilliant blue staining.
Synthesis of PEG‐hemin. The synthesis, purification, and characterization of PEG‐hemin were as described previously.( 30 )
Determination of the effect of HO‐1, HO‐1 inhibitors, and CO on vascular permeability in normal mouse skin. The ddY mice were anesthetized with sodium pentobarbital (83 mg/kg, i.p.). Test samples (50 μL each) were injected intradermally (i.d.) into the dorsal skin of mice, followed immediately by i.v. injection of 10 mg/kg Evans blue dye. Mice were killed 2 h after injection of Evans blue and the amount of extravasated dye in the skin at the site of injection was quantified after extraction with formamide, as described previously.( 2 ) Similar experiments were performed with an in vivo imaging system (NightOWL II; Berthold Technologies, Bad Wildbad, Germany) using a macromolecular fluorescent dye (i.e. rhodamine‐conjugated bovine albumin synthesized in our laboratory; Hideaki Nakamura, Jun Fang, Haibo Qin, Gahininath Y Bharate, Hiroshi Maeda, unpublished data, 2011) instead of Evans blue, with excitation and emission at 540 and 600 nm, respectively.
Induction of HO‐1 activity by PEG‐hemin in sarcoma 180 tumors in mice. Mouse sarcoma S180 cells (2 × 106) were injected s.c. into the dorsal skin of ddY mice. Approximately 8–10 days later, when tumor diameters measured 7–10 mm, PEG‐hemin (10 mg/kg hemin equivalent), which is a water‐soluble macromolecular HO‐1 inducer, was injected i.v. After 24 h, mice were killed; both tumors and normal tissues (liver, muscle) were removed and weighed, with microsomal fractions of each tissue obtained by ultracentrifugation to be used for measurement of HO‐1 activity, as described previously.( 24 , 31 )
Quantification of CO in S180 tumor‐bearing mice after PEG‐hemin treatment. The PEG‐hemin was administered to ddY mice bearing S180 solid tumors as described above. Twenty‐four hours later, mice were killed and blood was collected, with a 0.35‐mL aliquot of the blood sample diluted with 3.65 mL of 0.01 M PBS (pH 7.2) and placed in a 10‐mL glass test tube on ice. The blood was then purged with nitrogen gas, after which the NO donor 3‐(2‐hydroxy‐1‐methyl‐2‐nitrosohydrazino)‐N‐methyl‐1‐propanamine (NOC‐7; Dojin Chemical, Kumamoto, Japan) was added to a final concentration of 1 mM. The test tube was then sealed using paraffin. Under these conditions, excessive NO generated would bind to hemoglobin, so CO would be released instead. After 2 h incubation at room temperature, 1 mL of the gas in the test tubes was used for CO quantification by gas chromatography (TRIlyzer mBA‐3000; TAIYO Instruments, Osaka, Japan).
Effect of HO‐1 on tumor vascular permeability (EPR effect) in S180 tumor‐bearing mice. To induce HO‐1 in tumors, S180 tumor‐bearing ddY mice were injected with 10 mg/kg, i.v., PEG‐hemin. After 24 h, 10 mg/kg, i.v., Evans blue was injected. Then, 24 h after injection of Evans blue, the mice were killed, both the tumors and normal tissues (liver, spleen, and kidney) were removed, and the amount of extravasated dye in the tumors and normal tissues was quantified as described previously.( 2 )
Measurement of tumor blood flow. A laser Doppler flowmeter (ALF21; Advance, Tokyo, Japan) was used to measure blood flow in tumors and normal tissues (liver and muscle) in 10 mg/kg PEG‐hemin‐treated and control mice. In each mouse, the flowmeter probe was inserted into non‐necrotic tumor tissues and blood flow was monitored for 5–10 min until it stabilized.
The effect of CO on tumor blood flow was investigated using the same method, with the exception that mice received intratumoral (i.t.) injections of CORM‐2 (25 nmol in 0.05 mL), and real‐time changes in tumor blood flow before and after CORM‐2 administration were monitored. Measurements were obtained in anesthetized (sodium pentobarbital) mice, as described above.
Statistical analysis. All data are expressed as the mean ± SEM. The significance of differences was evaluated using Student’s t‐test, with significance set at P < 0.05.
Results
Enhanced vascular permeability in normal mouse skin following HO‐1 injection. It was evident that HO‐1 induced dose‐dependent increases in vascular permeability in normal dorsal mouse skin (Fig. 1). However, injection of heat‐inactivated (100°C, 5 min) HO‐1 had a significantly reduced effect (1, 2), which suggests that the enzymatic activity of HO‐1 is necessary for it to enhance vascular permeability. Moreover, the extravasation induced by HO‐1 decreased significantly when HO‐1 was mixed with BSA, a major bilirubin‐binding and probably CO‐binding protein in plasma (Fig. 1). The HO‐1 inhibitor ZnPP completed abolished this effect of HO‐1 on vascular permeability, as assessed using an in vivo imaging system, as described below (Fig. 2). In addition, ZnPP did not significantly decrease the vascular permeability triggered by NO (see Fig. S1), which is considered to occur via the activation of soluble guanylate cyclase (sGC), suggesting the effect of ZnPP on HO‐1 induced vascular permeability, at the concentrations used in the present study, is due mostly to inhibition of HO‐1, although ZnPP has also been reported to inhibit sGC independent of HO‐1.( 32 )
Figure 1.

Effect of heme oxygenase‐1 (HO‐1) on vascular permeability of the dorsal skin in normal mice. Recombinant HO‐1 protein and other agents were administered i.d., followed by i.v. injection of Evans blue (10 mg/kg). The dye was allowed to extravasate for 2 h. (a) Representative image showing the extravasation of blue dye caused by each agent. (b) Quantification of the extravasation of Evans blue from the skin tissues. Alb, albumin. Data are the mean ± SEM (n = 3–4). *P < 0.05, **P < 0.01.
Figure 2.

Vascular permeability induced by heme oxygenase‐1 (HO‐1) and its inhibition in normal mice, as evaluated using an in vivo imaging system. The HO‐1 protein, with or without its inhibitor zinc protoporphyrin‐IX (ZnPP) or the carbon monoxide scavenger hemoglobin (Hb), was administered i.d., followed by i.v. injection of the macromolecular fluorescent dye rhodamine‐conjugated bovine albumin (5 mg/kg rhodamine equivalent). The dye was allowed to extravasate for 1 h. (a) Representative image showing authentic HO‐1‐induced vascular permeability. (b) Quantification of data. Data are the mean ± SEM (n = 3–4). *P < 0.05, **P < 0.01.
More importantly, administration of the CO scavenger hemoglobin completely abolished the enhanced vascular permeability produced by HO‐1 (Fig. 2). These data suggest that CO has an essential role in HO‐1‐induced vascular permeability.
Effect of CO on vascular permeability. To validate the role of CO in vascular permeability, the CO‐releasing agent CORM‐2 was used in the same vascular permeability study. As shown in Figure 3, CORM‐2, at picomolar levels, had significant, dose‐dependent effects on vascular permeability. No such effect was observed when CORM‐2 was decomposed by incubation at room temperature for 24 h to completely liberate CO (Fig. 3). Moreover, ZnPP had no effect on the CORM‐2‐derived CO‐induced increase in vascular permeability (Fig. S1).
Figure 3.

Effect of CORM‐2 on the vascular permeability of the dorsal skin of normal mice. Different concentrations of CORM‐2 were administered i.d., followed by i.v. injection of Evans blue (10 mg/kg). The dye was allowed to extravasate for 2 h. (a) Representative images showing CORM‐2‐induced extravasation of blue dye. (b) Quantification of Evans blue extravasation in the skin. Data are the mean ± SEM (n = 3–4). *P < 0.05, **P < 0.01.
Induction of HO‐1 in tumor tissue by PEG‐hemin. We developed PEG‐hemin, a water‐soluble pegylated HO‐1 inducer, in our laboratory.( 30 ) It behaves as a macromolecular micelle with a molecular mass of 126 kDa, and so may accumulate selectively in solid tumors based on the EPR effect. Accordingly, to investigate the role of HO‐1 in the permeability of the tumor vasculature, we used PEG‐hemin to induce HO‐1 in S180 solid tumors. As expected, HO‐1 activity in tumor tissues was increased significantly after PEG‐hemin treatment (Fig. 4a).
Figure 4.
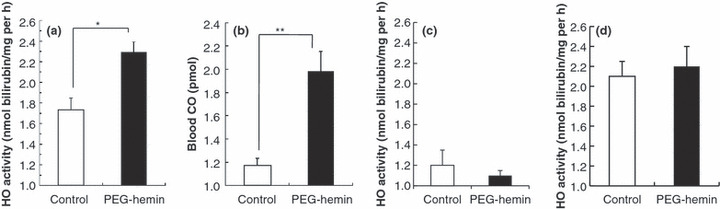
In vivo induction of heme oxygenase‐1 (HO‐1) in tumors and normal tissues (liver, muscle) after pegylated hemin (PEG‐hemin) treatment of tumor‐bearing mice. (a) Twenty‐four hours after i.v. injection of PEG‐hemin (10 mg/kg hemin equivalent), HO‐1 activity, as evidenced by bilirubin formation, was determined in the tumors. (b) The induction of HO‐1 was verified by measuring blood concentrations of carbon monoxide (CO). (c,d) The HO‐1 activity in the liver (c) and muscle (d) was also determined. Data are the mean ± SEM (n = 4). *P < 0.05, **P < 0.01.
We further confirmed this finding by measuring CO concentrations in the circulation, because we have found that circulating levels of CO are positively related to tumor growth and HO‐1 activity in tumors (Jun Fang, Takaaki Akaike, Chiho Taruki, Tomohiro Sawa, Hiroshi Maeda, unpublished observation, 2004). As expected, circulating levels of CO increased significantly after PEG‐hemin treatment, which paralleled the increase in HO‐1 activity (Fig. 4b).
Moreover, increases in HO activity were not found in normal tissue (e.g. muscle) following injection of PEG‐hemin (Fig. 4c). It is known that many macromolecules accumulate in high levels in the liver, primarily through the reticuloendothelial system, and pegylation is widely used to avoid the capturing of macromolecules by the reticuloendothelial clearance system.( 33 ) Thus, in the present study we measured the body distribution of PEG‐hemin in S180 tumor‐bearing mice; high accumulation of PEG‐hemin was observed in tumors and the liver (Fig. S2). However, only non‐significant increases in HO activity were observed in the liver after PEG‐hemin administration (Fig. 4d). This may be probably due to the endogenous presence of a high HO content in the liver and spleen, which are the major organs for heme catabolism.
Involvement of HO‐1 in enhanced vascular permeability in S180 solid tumors. As shown in Figure 5, pretreatment with PEG‐hemin, injected i.v., produced significantly greater extravasation of the Evans blue–albumin complex in tumor tissue, but not in normal tissues. This provides clear evidence that the EPR effect was enhanced in the tumor tissue by PEG‐hemin.
Figure 5.

Accumulation of Evans blue–albumin complex in tumors and normal tissues (liver, kidney, and spleen) after pegylated hemin (PEG‐hemin) treatment of tumor‐bearing mice. Twenty‐four hours after i.v. injection of PEG‐hemin (10 mg/kg hemin equivalent), 10 mg/kg, i.v., Evans blue was injected. After a further 24 h, mice were killed and tissues collected. Control mice were not treated with PEG‐hemin. The blue dye in each tissue was extracted and the amount was quantified. Data are the mean ± SEM (n = 4). *P < 0.05.
Effect of HO‐1 and CO on tumor blood flow. We hypothesized that HO‐1, in addition to augmenting tumor vascular permeability (the EPR effect) as described above, may also improve tumor blood flow, because CO has a vasodilator effect similar to that of NO.( 27 ) Thus, we measured blood flow in S180 solid tumors with and without HO‐1 induction by PEG‐hemin. As anticipated, tumor blood flow increased significantly, by four‐ to fivefold, 24 h after i.v. injection of PEG‐hemin compared with untreated controls (Fig. 6a). This treatment had no effect on the blood flow of normal tissues (i.e. liver and muscle; data not shown). Furthermore, when CORM‐2 was injected directly into tumors, tumor blood flow increased gradually for approximately 2 h, at which time it had increased 1.5–2‐fold of levels seen before CORM‐2 injection (Fig. 6b).
Figure 6.

Changes in tumor blood flow after (a) pegylated hemin (PEG‐hemin) and (b) CORM‐2 treatment of tumor‐bearing mice (control mice were untreated). Mice were injected with either PEG‐hemin (10 mg/kg, i.v.) or the indicated concentrations of CORM‐2 (intratumoral) and tumor blood flow was measured using a laser Doppler flowmeter. Data are the mean ± SEM (n = 4). *P < 0.05.
Discussion
Targeted drug delivery is the key for successful anticancer treatment. In contrast, the use of conventional anticancer drugs results in severe adverse side effects, which prevents the use of higher doses. Consequently, the therapeutic effects of conventional low molecular weight anticancer drugs are limited.
Because of these problems, so‐called molecular target therapy has recently come into focus. This type of therapy is designed to target specific receptors or kinases associated with tumor growth, progression, invasion, and metastasis. However, one problem with molecular target therapy is related to the genetic diversity of human solid tumors, in which target molecules may have mutated.( 34 , 35 ) Another possible problem is that multiple genes may be involved in sophisticated networks that have multiple backup systems for the molecular pathways that are vital for tumor cells. Thus, molecular target therapy, although highly specific for targets, seems to be an approach that would not be able to destroy most, or all, tumor cells.
The discovery of the EPR effect was a considerable breakthrough, leading to a more universal tumor‐targeting mechanism at the tissue or vascular level. That is, EPR effect‐based targeting depends on the unique anatomic and pathophysiologic features of tumor vessels, which are common to most solid tumors. Thus, EPR effect‐based tumor targeting has wider applicability and it is now becoming an increasingly promising paradigm for anticancer drug development.( 16 , 36 , 37 )
We have been seeking to augment the EPR effect even more by using specific features involved in the effect. One example of our success was the application of AngII‐induced hypertension, which induced a two‐ to threefold augmentation of the EPR effect without having any effect on the distribution of drug in normal tissues.(
16
,
18
,
19
,
38
) Another new approach was recently developed with the NO‐releasing agent NTG.(
20
) In hypoxic tumor tissue, NTG is selectively converted to nitrite (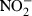 ), which is then converted to NO in the tumor. This event is analogous to the hypoxic conditions in angina pectoris.(
39
) Nitric oxide generated from NTG then increases the delivery of macromolecular drugs to the tumor by two‐ to threefold, resulting in an improved anticancer effect.(
20
)
), which is then converted to NO in the tumor. This event is analogous to the hypoxic conditions in angina pectoris.(
39
) Nitric oxide generated from NTG then increases the delivery of macromolecular drugs to the tumor by two‐ to threefold, resulting in an improved anticancer effect.(
20
)
In addition, CO has been reported as an important endogenous signaling molecule with various biological functions that include regulation of vascular tonus, being involved in antiapoptosis, having anti‐inflammatory effects, and inducing angiogenesis.( 27 , 40 ) Regarding the effect of CO on vasoregulation, most data support a prodilatory role for CO; however, vasoconstrictor effects of CO have also been reported via inhibition of NO synthesis to antagonize NO‐dependent vasodilation( 41 ) or via the induction of a more oxidative stage of the vasculature by CO.( 42 ) These results suggest the complexity of CO‐induced vasoregulation: CO is not necessarily a vasorelaxant and may exhibit an opposite effect depending on the milieu. Notwithstanding, we clearly found that CO increased vascular permeability and blood flow in the present study, suggesting a vasodilatory role for CO in the experimental setting in the present study.
The major source (i.e. >80%) of CO in biological systems is HO‐catalysed heme degradation.( 27 ) In the present study, we found that HO‐1 induced an increase in tumor blood flow (Fig. 6a), which was probably the consequence of the vasorelaxant effect of significantly increased CO levels from HO‐1 induced by PEG‐hemin. This is supported by the finding that direct injection of CORM‐2 into tumors significantly increased tumor blood flow (Fig. 6b). In addition, although circulating levels of CO increased after PEG‐hemin treatment, accompanying the increase in HO activity (CO production) in tumors (Fig. 4b), there was no change in vascular permeability (Fig. 5) and blood flow (data not shown) in normal tissues and organs. This is probably because of the capture of CO by hemoglobin and the autoregulatory function (homeostasis) of normal blood vessels to maintain blood volume.
Furthermore, CO was recently reported to induce VEGF expression via p38 kinase‐dependent activation of specificity protein 1 (SP1) transcriptional factor,( 43 ) which thus induces angiogenesis. It is known that VEGF is an important vascular permeability factor,( 9 , 10 , 44 , 45 ) probably via activation of endothelial NO synthase to generate NO.( 9 ) We thus believe that VEGF may also be involved in the enhanced vascular permeability induced by HO‐1 and CO.
Regarding the vascular pathophysiology of tumors, it should be noted that the role of HO‐1 is not always same as that of CO and will vary depending on the cell context and tumor microenvironment.( 46 , 47 ) For example, although many studies have reported a proangiogenetic effect of HO‐1,( 21 , 48 , 49 ) Ferrando et al. ( 47 ) described an inhibitory effect of HO‐1 on angiogenesis in prostate cancer. The mechanism underlying the proangiogenic effect of HO‐1 under pathological conditions is not clear and, in tumors, may depend on the type of tumor or other undefined factors. However, in the present study we focused our efforts on clarifying the role of the HO‐1 product (CO) in increasing tumor vascular permeability, and not on the effect of HO‐1 per se on angiogenesis, which involves many pathophysiologic effectors.
In the present study, PEG‐hemin induced the expression of HO‐1 in tumor cells by accumulating in tumor tissues, which subsequently induced the generation of CO, resulting in increased tumor vascular permeability and blood flow (Fig. 5). On the basis of these findings, we should be able to improve drug delivery to tumors.
In conclusion, in the present study we found that CO is an important mediator of the EPR effect. The CO‐releasing agent CORM‐2 alone and selective induction of HO‐1 expression in tumor by PEG‐hemin administered i.v. both significantly improved tumor blood flow and extravasation of macromolecules into the tumor by enhancing the EPR effect. Thus, drug delivery based on the EPR effect was augmented. Because of HO induction, and hence CO generation, PEG‐hemin used in combination with chemotherapeutic drugs (especially macromolecular drugs) may hold promise as a new anticancer therapy and warrants further investigation.
Disclosure Statement
The authors declare they have no potential conflicts of interest.
Supporting information
Supporting info item
Supporting info item
Acknowledgments
The authors thank Judith B. Gandy for English language editing of the original manuscript. This work was supported, in part, by Grants‐in‐Aid from the Ministry of Education, Science, Culture, Sports and Technology of Japan (22700927, 17016076, and 20015405) and a Grant‐in‐Aid from the Ministry of Welfare, Health and Labor of Japan (H23‐sanjigann‐ippan‐001).
References
- 1. Hassett MJ, O’Malley AJ, Pakes JR, Newhouse JP, Earle CC. Frequency and cost of chemotherapy‐related serious adverse effects in a population sample of women with breast cancer. J Natl Cancer Inst 2006; 98: 1108–17. [DOI] [PubMed] [Google Scholar]
- 2. Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent SMANCS. Cancer Res 1986; 46: 6387–92. [PubMed] [Google Scholar]
- 3. Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med 1971; 285: 1182–6. [DOI] [PubMed] [Google Scholar]
- 4. Folkman J, Shing Y. Angiogenesis. J Biol Chem 1992; 267: 10931–4. [PubMed] [Google Scholar]
- 5. Maeda H, Matsumura Y, Kato H. Purification and identification of [hydroxyprolyl3]bradykinin in ascitic fluid from a patient with gastric cancer. J Biol Chem 1988; 263: 16051–4. [PubMed] [Google Scholar]
- 6. Matsumura Y, Kimura M, Yamamoto T, Maeda H. Involvement of the kinin‐generating cascade in enhanced vascular permeability in tumor tissue. Jpn J Cancer Res 1988; 79: 1327–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wu J, Akaike T, Maeda H. Modulation of enhanced vascular permeability in tumors by a bradykinin antagonist, a cyclooxygenase inhibitor, and a nitric oxide scavenger. Cancer Res 1998; 58: 159–65. [PubMed] [Google Scholar]
- 8. Maeda H, Noguchi Y, Sato K, Akaike T. Enhanced vascular permeability in solid tumor is mediated by nitric oxide and inhibited by both new nitric oxide scavenger and nitric oxide synthase inhibitor. Jpn J Cancer Res 1994; 85: 331–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Maeda H, Fang J, Inutsuka T, Kitamoto Y. Vascular permeability enhancement in solid tumor: various factors, mechanisms involved and its implications. Int Immunopharmacol 2003; 3: 319–28. [DOI] [PubMed] [Google Scholar]
- 10. Senger DR, Galli SJ, Dvorak AM, Perruzzi CA, Harvey VS, Dvorak HF. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science 1983; 219: 983–5. [DOI] [PubMed] [Google Scholar]
- 11. Wu J, Akaike T, Hayashida K, Okamoto T, Okuyama A, Maeda H. Enhanced vascular permeability in solid tumor involving peroxynitrite and matrix metalloproteinases. Jpn J Cancer Res 2001; 92: 439–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Suzuki M, Takahashi T, Sato T. Medial regression and its functional significance in tumor‐supplying host arteries. A morphometric study of hepatic arteries in human livers with hepatocellular carcinoma. Cancer 1987; 59: 444–50. [DOI] [PubMed] [Google Scholar]
- 13. Skinner SA, Tutton PJ, O’Brien PE. Microvascular architecture of experimental colon tumors in the rat. Cancer Res 1990; 50: 2411–7. [PubMed] [Google Scholar]
- 14. Iwai K, Maeda H, Konno T. Use of oily contrast medium for selective drug targeting to tumor: enhanced therapeutic effect and X‐ray image. Cancer Res 1984; 44: 2115–21. [PubMed] [Google Scholar]
- 15. Maeda H, Sawa T, Konno T. Mechanism of tumor‐targeted delivery of macromolecular drugs, including the EPR effect in solid tumor and clinical overview of the prototype polymeric drug SMANCS. J Control Release 2001; 74: 47–61. [DOI] [PubMed] [Google Scholar]
- 16. Fang J, Nakamura H, Maeda H. The EPR effect: unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev 2010; 63: 136–51. [DOI] [PubMed] [Google Scholar]
- 17. Suzuki M, Hori K, Abe Z, Asito S, Sato H. A new approach to cancer chemotherapy: selective enhancement of tumor blood flow with angiotensin II. J Natl Cancer Inst 1981; 67: 663–9. [PubMed] [Google Scholar]
- 18. Li CJ, Miyamoto Y, Kojima Y, Maeda H. Augmentation of tumor delivery of macromolecular drugs with reduced bone marrow delivery by elevating blood pressure. Br J Cancer 1993; 67: 975–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nagamitsu A, Greish K, Maeda H. Elevating blood pressure as a strategy to increase tumor‐targeted delivery of macromolecular drug SMANCS: cases of advanced solid tumors. Jpn J Clin Oncol 2009; 39: 756–66. [DOI] [PubMed] [Google Scholar]
- 20. Seki T, Fang J, Maeda H. Enhanced delivery of macromolecular antitumor drugs to tumors by nitroglycerin application. Cancer Sci 2009; 100: 2426–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fang J, Akaike T, Maeda H. Antiapoptotic role of heme oxygenase (HO) and the potential of HO as a target in anticancer treatment. Apoptosis 2004; 9: 27–35. [DOI] [PubMed] [Google Scholar]
- 22. Doi K, Akaike T, Fujii S et al. Induction of haem oxygenase‐1 by nitric oxide and ischaemia in experimental solid tumours and implications for tumour growth. Br J Cancer 1999; 80: 1945–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fang J, Sawa T, Akaike T et al. In vivo antitumor activity of pegylated zinc protoporphyrin: targeted inhibition of heme oxygenase in solid tumor. Cancer Res 2003; 63: 3567–74. [PubMed] [Google Scholar]
- 24. Tanaka S, Akaike T, Fang J et al. Antiapoptotic effect of haem oxygenase‐1 induced by nitric oxide in experimental solid tumour. Br J Cancer 2003; 88: 902–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schacter BA. Heme catabolism by heme oxygenase: physiology, regulation, and mechanism of action. Semin Hematol 1988; 25: 349–69. [PubMed] [Google Scholar]
- 26. Maines MD. Heme oxygenase: function, multiplicity, regulatory mechanisms, and clinical applications. FASEB J 1988; 2: 2557–68. [PubMed] [Google Scholar]
- 27. Abraham NG, Kappas A. Pharmacological and clinical aspects of heme oxygenase. Pharmacol Rev 2008; 60: 79–127. [DOI] [PubMed] [Google Scholar]
- 28. Nakao A, Kaczorowski DJ, Wang Y et al. Amelioration of rat cardiac cold ischemia/reperfusion injury with inhaled hydrogen or carbon monoxide, or both. J Heart Lung Transplant 2010; 29: 544–53. [DOI] [PubMed] [Google Scholar]
- 29. Nakao A, Choi AM, Murase N. Protective effect of carbon monoxide in transplantation. J Cell Mol Med 2006; 10: 650–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fang J, Qin H, Seki T et al. Therapeutic potential of pegylated hemin for ROS‐related diseases via induction of heme oxygenase‐1: results from a rat hepatic ischemia/reperfusion injury model. J Pharmacol Exp Ther 2011; 339: 779–89. [DOI] [PubMed] [Google Scholar]
- 31. Drummond GS, Kappas A. Prevention of neonatal hyperbilirubinemia by tin protoporphyrin IX, a potent competitive inhibitor of heme oxygenase. Proc Natl Acad Sci USA 1981; 78: 6466–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Serfass L, Burstyn JN. Effect of heme oxygenase inhibitors on soluble guanylyl cyclase activity. Arch Biochem Biophys 1998; 359: 8–16. [DOI] [PubMed] [Google Scholar]
- 33. Soloman R, Gabizon AA. Clinical pharmacology of liposomal anthracyclines: focus on pegylated liposomal doxorubicin. Clin Lymphoma Myeloma 2008; 8: 21–32. [DOI] [PubMed] [Google Scholar]
- 34. Wood LD, Parsons DW, Jones S et al. The genomic landscapes of human breast and colorectal cancers. Science 2007; 318: 1108–13. [DOI] [PubMed] [Google Scholar]
- 35. Sjöblom T, Jones S, Wood LD et al. The consensus coding sequences of human breast and colorectal cancers. Science 2006; 314: 268–74. [DOI] [PubMed] [Google Scholar]
- 36. Vicent MJ, Ringsdorf H, Duncan R. Polymer therapeutics: clinical applications and challenges for development. Adv Drug Deliv Rev 2009; 61: 1117–20. [DOI] [PubMed] [Google Scholar]
- 37. Matsumura Y, Kataoka K. Preclinical and clinical studies of anticancer agent‐incorporating polymer micelles. Cancer Sci 2009; 100: 572–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Maeda H. Tumor‐selective delivery of macromolecular drugs via the EPR effect: background and future prospects. Bioconjug Chem 2010; 21: 797–802. [DOI] [PubMed] [Google Scholar]
- 39. Maeda H. Nitroglycerin enhances vascular blood flow and drug delivery in hypoxic tumor tissues: analogy between angina pectoris and solid tumors and enhancement of the EPR effect. J Control Release 2010; 142: 296–8. [DOI] [PubMed] [Google Scholar]
- 40. Otterbein LE, Bach FH, Alam J et al. Carbon monoxide has anti‐inflammatory effects involving the mitogen‐activated protein kinase pathway. Nat Med 2000; 6: 422–8. [DOI] [PubMed] [Google Scholar]
- 41. Lin HH, Lai SC, Chau LY. Heme oxygenase‐1/carbon monoxide induces vascular endothelial growth factor expression via p38 kinase‐dependent activation of Sp1. J Biol Chem 2011; 286: 3829–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ferrara N, Henzel WJ. Pituitary follicular cells secrete a novel heparin‐binding growth factor specific for vascular endothelial cells. Biochem Biophys Res Commun 1989; 161: 851–8. [DOI] [PubMed] [Google Scholar]
- 43. Johnson FK, Johnson RA. Carbon monoxide promotes endothelium dependent constriction of isolated gracilis muscle arterioles. Am J Physiol Regul Integr Comp Physiol 2003; 285: R536–41. [DOI] [PubMed] [Google Scholar]
- 44. Lamon BD, Zhang FF, Puri N, Brodsky SV, Goligorsky MS, Nasjletti A. Dual pathways of carbon monoxide‐mediated vasoregulation: modulation by redox mechanisms. Circ Res 2009; 105: 775–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Keck PJ, Hauser SD, Krivi G et al. Vascular permeability factor, an endothelial cell mitogen related to PDGF. Science 1989; 246: 1309–12. [DOI] [PubMed] [Google Scholar]
- 46. Dulak J, Deshane J, Jozkowicz A, Agarwal A. Heme oxygenase‐1 and carbon monoxide in vascular pathobiology: focus on angiogenesis. Circulation 2008; 117: 231–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ferrando M, Gueron G, Elguero B et al. Heme oxygenase 1 (HO‐1) challenges the angiogenic switch in prostate cancer. Angiogenesis 2011; 14: 467–79. [DOI] [PubMed] [Google Scholar]
- 48. Sunamura M, Duda DG, Ghattas MH et al. Heme oxygenase‐1 accelerates tumor angiogenesis of human pancreatic cancer. Angiogenesis 2003; 6: 15–24. [DOI] [PubMed] [Google Scholar]
- 49. Miyake M, Fujimoto K, Anai S et al. Heme oxygenase‐1 promotes angiogenesis in urothelial carcinoma of the urinary bladder. Oncol Rep 2011; 25: 653–60. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting info item
Supporting info item