

Abstract
Introduction:
Considerable advances in melanoma have been realized through immunotherapy. The principal aim was to determine whether primary tumor characteristics or Next Generation Sequencing (NGS) could serve as markers of immunotherapy response.
Methods and Results:
The study cohort consisted of 67 patients who received immunotherapy for recurrent or metastatic melanoma and for whom primary tumor biopsies and pathology reports were available. A subset of 59 patient tumors were profiled using an NGS panel of 50 cancer-related genes. Objective Response Rate to immunotherapy was assessed using RECIST v1.1 criteria. Progression-free survival (PFS) and overall survival (OS) were used as endpoints. Lymphovascular invasion (LVI) strongly correlated with an increased proportion of immunotherapy responders, (p=0.002). PFS interval (p=0.003) and OS (p=0.0355) were significantly higher in patients with LVI. NRAS mutation was more strongly correlated with an increased proportion of immunotherapy responders (p=0.050). PFS was significantly higher in patients with NRAS mutation (p=0.042); no difference in OS (p=0.111).
Discussion:
This analysis demonstrates an association between lymphovascular invasion and immunotherapy response. Additionally, NGS mutation analysis demonstrated a potential association between NRAS mutations and immunotherapy response.
Keywords: Melanoma, Immunotherapy, Primary Biopsy, Lymphovascular Invasion, Next Generation Sequencing
INTRODUCTION
Treatment of melanoma has grown substantially in recent years to encompass a multitude of novel targeted and immunotherapeutic options. Immune checkpoint inhibitors, which target the cytotoxic T-lymphocyte-associated antigen 4 (CTLA4) and programmed cell death (PD) pathways have significantly enhanced the available therapeutic armamentarium and improved patient outcomes.
Ipilimumab, a CTLA4 inhibitor, is approved in the adjuvant setting and for treatment of advanced, recurrent, and unresectable melanoma, and it was the first treatment in a randomized setting shown to extend survival in advanced melanoma patients (Hodi et al. 2010). Blockade of the PD-1/PD-L1 axis has been shown to induce prominent and durable clinical responses in 18–43% of patients with melanoma malignancies (Topalian et al. 2012). Overall survival (OS) and progression free survival (PFS) data have established PD-1 blockade therapy, such as pembrolizumab and nivolumab, as a standard of care modality for advanced melanoma (Topalian et al. 2014).
While the anti-CTLA4 and PD-1 blockades represent a major step forward in melanoma treatment, a significant subset of patients still fail to respond to immunotherapy treatment. Several host and tumor intrinsic and extrinsic factors such as tumor infiltrating lymphocytes (TILs), (Tumeh et al. 2014), PD-L1 (Weber et al. 2015, Daud et al. 2016), indoleamine 2,3-dioxygenase (IDO), (Hamid et al. 2011), regulatory T cells (Tregs), (Simeone et al. 2014), tumor mutation burden (TMB), (Snyder et al. 2014), gut microbiome (Chaput et al. 2017), and HLA subtype (Inoue et al. 2016) have been explored as potential biomarkers to predict response and resistance to anti-CTLA4 and anti-PD-1 immunotherapies. Unfortunately, the underlying mechanism of tumor progression is still currently unknown (Pitt et al. 2016).
Each patient’s tumor can be interrogated through the use of pathology characteristics, clinical, radiographic and laboratory findings. While BRAF testing is standard-of-care for patients with stage III or IV disease, patients may elect to have their tumor tissue samples undergo additional testing using a validated Next Generation Sequencing (NGS) panel which provides additional information that may include the TMB and other potentially important mutation and fusion information. This robust dataset, which provides unique and patient-specific information, and may be applied to the treatment algorithm in second-line settings (Reiman et al. 2017). Identification of biomarkers is vital to better elucidate why some patients do not respond to therapy and, potentially, to spare patients who are unlikely to respond to immunotherapy from unnecessary risk and treatment associated toxicities, as well as the high incremental cost-effectiveness ratio (ICER) of immunotherapy treatments (Geynisman et al. 2014). Common immunotherapy related adverse events and toxicities (termed immune-related adverse events; irAE) include fatigue, diarrhea, rash, and pruritus, (Hamid et al. 2013), (Robert et al. 2015).
This study aims to identify potential markers of immunotherapy response by evaluating differences in primary biopsy characteristics between responders to immunotherapy and patients with stable disease (SD) or progressive disease (PD). Additionally, by examining tumor mutations using NGS, this study seeks to identify the most common mutations and median number of gene mutations in responders to immunotherapy versus patients with SD or PD.
MATERIALS AND METHODS
Following approval by the Fox Chase Cancer Center’s institutional review board, a melanoma tumor registry was queried for patients diagnosed with American Joint Committee on Cancer (AJCC) pathologic stage I-IV cutaneous melanoma between 2000 and 2018. A retrospective chart review was performed using the prospectively maintained melanoma tumor database. Inclusion criteria included patients who had primary tumor biopsy samples and associated pathology reports. Primary biopsy specimens were reviewed by the Fox Chase pathology department upon patient presentation to the institution. Of the patients with primary biopsy samples and pathology data, database was queried for patients who subsequently received immunotherapy treatment for recurrent or de novo stage IV metastatic melanoma with anti-CTLA4 agent ipilimumab, PD-1 inhibitors pembrolizumab or nivolumab, a combination of ipilimumab and nivolumab, or an anti-CTLA4 or PD-1 inhibitor in combination with another agent as a clinical trial. No adjuvant immunotherapy treated patients were included in this study. Objective response (complete response CR or partial response PR) to initial immunotherapy treatment was assessed using RECIST v1.1 criterion. A subset of patients treated with immunotherapy for advanced melanoma also had tumor tissue samples that underwent Next Generation Sequencing panel testing for 50 cancer-related somatic gene mutations.
Next Generation Sequencing
Next Generation Sequencing was performed at the Fox Chase Cancer Center on primary tumors or metastatic deposits prior to immunotherapy treatment. Hematoxylin and Eosin (H&E) stained tissue sections were used to assess for tumor content. The Ion Torrent Personal Genome Machine was used and analyzed with Torrent Suite Software to determine reportable variants Genomic DNA was extracted from a portion of the patient’s tumor tissue and used for multiplex PCR amplification of targeted regions within a Fox Chase Cancer Center panel of 50 commonly mutated cancer-related gene “hotspots” using Ion AmpliSeq technology. Variants are classified as pathogenic, variant of uncertain significance (VUS), and benign. Tumor mutation burden is not available with this platform. For clinical testing purposes, the lower limit of detection of the assay is approximately 10% mutant allele frequency with variant coverage of at least 250X. All reportable variants were verified using Sanger sequencing.
Statistical Analysis
Wilcoxon rank-sum and Fisher’s exact test were used to evaluate differences in primary biopsy characteristics for continuous and categorical outcomes, respectively. Fisher’s exact test was used to assess differences in the proportion of patients with mutations in specific genes between patients with and without an objective response. In secondary analyses, log-rank tests were used to investigate whether progression free survival (PFS) and/or overall survival (OS) distributions were different between presence of absence lymphovascular invasion, presence or absence of detected mutations in selected genes (i.e. NRAS and BRAF), or long versus short disease free intervals in patients treated for recurrence. Time-to-event end points, including PFS and OS were estimated using the Kaplan-Meier method with pointwise confidence limits. PFS was calculated from first dose of immunotherapy to the last dose of immunotherapy or date of death and censored at the date of last follow up. OS was calculated from the date of first immunotherapy to the date of death and censored at date of last follow up. All tests were two-sided with a 5% type I error. Missing data were not included for analysis. Analyses were performed using SAS version 9.4.
RESULTS
The total eligible study cohort consisted of 67 patients. Median age at diagnosis was 59 years old (range, 27 to 93). Additional patient demographics including gender, sex, site of primary melanoma diagnosis, and initial pathologic AJCC 7th edition TNM tumor staging, are shown in Table 1. Following primary biopsy tumor sample collection and initial melanoma staging and diagnosis, patients with subsequent recurrent (n=58) or de novo stage IV (n=9) melanoma (Figure 1) received immunotherapy of an anti-CTLA4 antibody (n=19), PD-1 inhibitor (n=28), anti-CTLA4/PD-1 inhibitor combination (n=11), or immunotherapy combination in clinical trial (n=9).
Table 1: Patient Demographics.
Study cohort patient demographics includes age at diagnosis, sex, primary melanoma site, pathologic stage, and immunotherapy treatment type.
| Responders | SD or PD | |
|---|---|---|
| N | 27 | 40 |
| Median Age at Diagnosisi (Range) | 60 (30–82) | 59 (27–93) |
| Malei | 20 (74%) | 25 (63%) |
| Primary Site at Diagnosisi | ||
| Extremity | 17 (63%) | 14 (39%) |
| Trunk | 6 (22%) | 15 (38%) |
| Head & Neck | 4 (15%) | 7 (18%) |
| Unknown | 0 (0%) | 2 (5%) |
| TNM Pathologic Stagei | ||
| I | 3 (12%) | 7 (18%) |
| II | 6 (23%) | 12 (32%) |
| III | 13 (50%) | 14 (37%) |
| IV | 4 (15%) | 5 (13%) |
| Treatment | ||
| Anti-CTLA4 | 4 (15%) | 15 (37%) |
| PD-1 Inhibitor | 14 (52%) | 14 (35%) |
| Anti-CTLA4/PD-1 Combination | 5 (18%) | 6 (15%) |
| Clinical Trial Combination | 4 (15%) | 5 (13%) |
Age at diagnosis was missing for 1 patient. Sex and primary site at diagnosis were missing for 2 patients. Pathologic stage data was missing for 3 patients. Missing values were excluded for the analysis.
Figure 1.
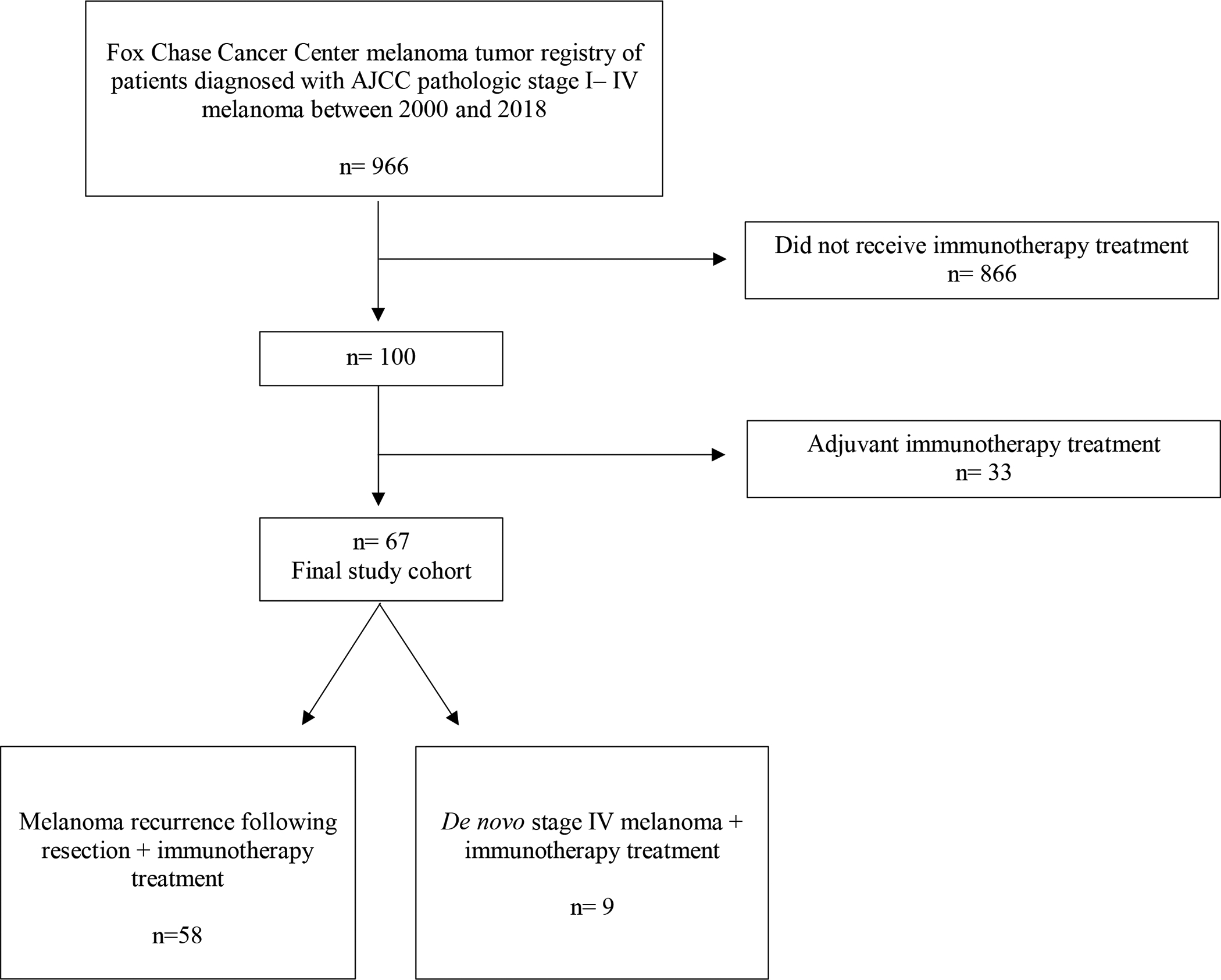
Inclusion diagram.
Primary tumor pathology data included primary site, Breslow thickness, mitotic rate, tumor infiltrating lymphocytes (TILs), lymphovascular invasion, ulceration, radial and vertical growth phases, neurotropism, regression, and satellitosis. Responders had a median Breslow thickness of 2.6 mm compared to 1.9 mm in patients with stable disease (SD) or progressive disease (PD), (p=0.075). There was no statistically significant difference in the proportion of patients with objective response compared to those with SD or PD in the following primary biopsy characteristics, mitotic rate, tumor infiltrating lymphocytes (TILs), ulceration, radial and vertical growth phases, neurotropism, regression, or satellitosis, (Table 2).
Table 2: Primary Tumor Pathology Characteristics.
Characteristics of primary tumors samples from pathology reports. Present (+), absent (−)ii.
| Responders n=27 |
SD or PD n=40 |
p-value | ||
|---|---|---|---|---|
| Median Breslow Thickness (mm) (Range) | 2.6 (0.09–10.0) | 1.9 (0.50–14.0) | 0.075 | |
| Median Mitoses (Range) | 4.0 (0–25) | 4.0 (0–24) | 0.747 | |
| Tumor Infiltrating Lymphocytes (TILs) | Brisk | 2 (10%) | 1 (4%) | 0.449 |
| Non-Brisk | 10 (50%) | 9 (36%) | ||
| − | 8 (40%) | 15 (60%) | ||
| Lymphovascular Invasion | + | 9 (50%) | 1 (5%) | 0.002 |
| − | 9 (50%) | 21 (95%) | ||
| Tumor Ulceration | + | 11 (46%) | 13 (36%) | 0.592 |
| − | 13 (54%) | 23 (64%) | ||
| Radial Growth Phase | + | 4 (40%) | 15 (71%) | 0.127 |
| − | 6 (60%) | 6 (29%) | ||
| Vertical Growth Phase | + | 19 (100%) | 26 (93%) | 0.508 |
| − | 0 (0%) | 2 (7%) | ||
| Neurotropism | + | 2 (13%) | 5 (22%) | 0.681 |
| − | 13 (87%) | 18 (78%) | ||
| Regression | + | 5 (29%) | 6 (29%) | 1.000 |
| − | 12 (71%) | 15 (71%) | ||
| Satellitosis | + | 2 (13%) | 2 (10%) | 1.000 |
| − | 13 (87%) | 19 (90%) | ||
Missing values for each pathology characteristic were excluded for the analysis.
Lymphovascular invasion was seen in 50% (n=9 of 18) of patients with an objective response to immunotherapy compared to 4.6% (n=1 of 21) of SD or PD patients (p=0.002). Kaplan-Meier curves demonstrated PFS was significantly higher in patients with lymphovascular invasion compared to those without (p=0.003), (Figure 2A). Also, a significant difference was found in OS between patients with lymphovascular invasion in primary biopsies compared to those tumors without lymphovascular invasion (p=0.036), (Figure 2B).
Figure 2: PFS and OS Stratified by Lymphovascular Invasion.
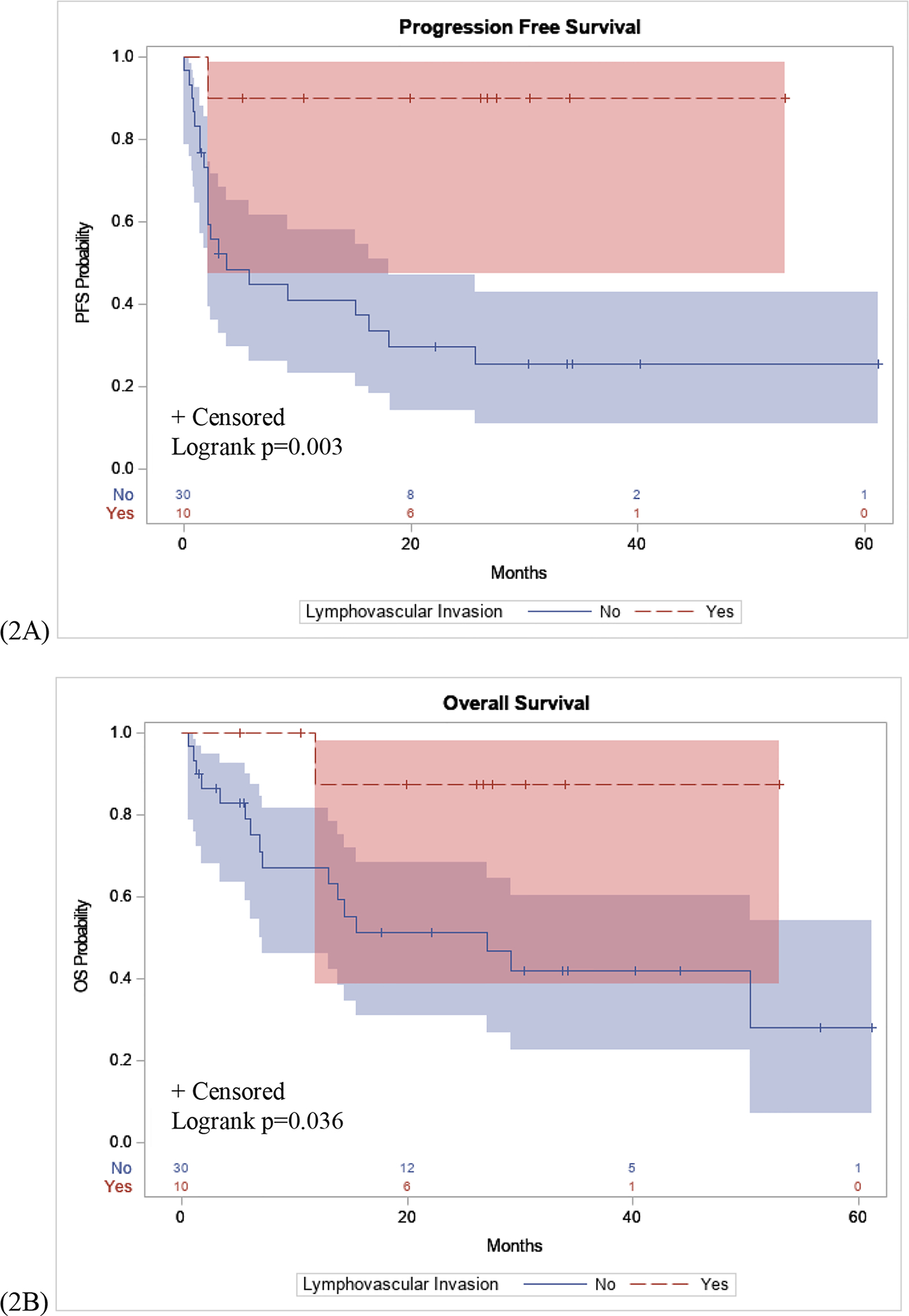
(2A): Patients with lymphovascular invasion seen on primary biopsy samples had a significantly longer PFS interval following immunotherapy compared to patients without lymphovascular invasion (p=0.003). (2B): Significant difference in OS in patients with lymphovascular invasion compared to patients without lymphovascular invasion (p=0.036).
Within the total study cohort, 59 patients also had NGS mutation testing performed on their primary (n=21) or metastatic (n=38) melanoma tumor samples. The median number of mutations in both groups was 1.0, (range 1–3 in responders vs 0–9 in SD or PD patients), (p=0.32). Of responders, 62.5% had 1 mutation, 33.3% had 2 mutations and 4.2% had 3 or more mutations. Among SD or PD patients, 17.1% had no mutations, 54.3% had 1 mutation, 11.4% had 2 mutations, and 17.2% had 3 or more mutations, Table 3).
Table 3: Next Generation Sequencing Profile Characteristics.
Next Generation Sequencing profile, exhibited as the percentage of patients who expressed an individual gene in responders to immunotherapy compared to patients with SD or PDiii.
| Gene | Responders n=27 |
SD or PD n=40 |
p-value |
|---|---|---|---|
| APC | 1 (4.2%) | 2 (6.1%) | 1.000 |
| ATM | 0 (0.0%) | 1 (4.4%) | 1.000 |
| BRAF | 7 (29.2%) | 14 (40.0%) | 0.423 |
| CDKN2A | 1 (4.2%) | 5 (15.6%) | 0.223 |
| CTNNB1 | 0 (0.0%) | 1 (3.1%) | 1.000 |
| EGFR | 2 (8.3%) | 1 (3.1%) | 0.571 |
| FBXW7 | 1 (4.2%) | 1 (3.1%) | 1.000 |
| FGFR2 | 0 (0.0%) | 1 (3.1%) | 1.000 |
| FGFR3 | 0 (0.0%) | 1 (3.1%) | 1.000 |
| FLT3 | 0 (0.0%) | 2 (6.3%) | 0.501 |
| GNAS | 1 (4.2%) | 1 (3.1%) | 1.000 |
| IDH1 | 1 (4.2%) | 1 (3.1%) | 1.000 |
| JAK2 | 0 (0.0%) | 1 (5.3%) | 1.000 |
| KDR | 0 (0.0%) | 1 (3.1%) | 1.000 |
| KIT | 0 (0.0%) | 1 (2.9%) | 1.000 |
| KIT EXON13 | 0 (0.0%) | 1 (3.0%) | 1.000 |
| KRAS | 0 (0.0%) | 1 (3.1%) | 1.000 |
| MET | 0 (0.0%) | 1 (3.1%) | 1.000 |
| NRAS | 13 (54.2%) | 8 (25.0%) | 0.050 |
| PDGFRA | 0 (0.0%) | 1 (3.1%) | 1.000 |
| PTEN | 1 (4.2%) | 2 (6.3%) | 1.000 |
| RB1 | 1 (4.2%) | 1 (3.1%) | 1.000 |
| TP53 | 5 (20.8%) | 7 (21.9%) | 1.000 |
No mutation in this cohort was found in the following genes: ABL1, AKT1, ALK, CDH1, CSF1R, ERBB4, FGFR1, GNA11, GNAQ, HNF1A, HRAS, IDH2, JAK3, MLH1, MPL, NOTCH1, NPM1, PIK3CA, PTPN11, RB1, RET, SMAD4, SMARCB1, SMO, SRC, STK11, VHL. Missing values were excluded for the analysis.
Primary analyses identified associations between NRAS gene mutations from the NGS analysis and patients with an objective response to immunotherapy. There was a greater proportion of patients with an objective response and NRAS positive tumors compared to patients with SD or PD with NRAS positive tumors, (p=0.050), (Figure 3A). Examining the BRAF gene, BRAF mutations in the NGS sequencing panel included BRAF G466V, V600E, V600K, and V600R mutations. Of the 21 BRAF mutated patients, none were found to have a G466V mutation. There was no difference in the proportion of patients with an objective response with BRAF positive tumors compared to patients with SD or PD with BRAF positive tumors, p=0.423 (Figure 3B).
Figure 3: Gene Mutation Profiles.
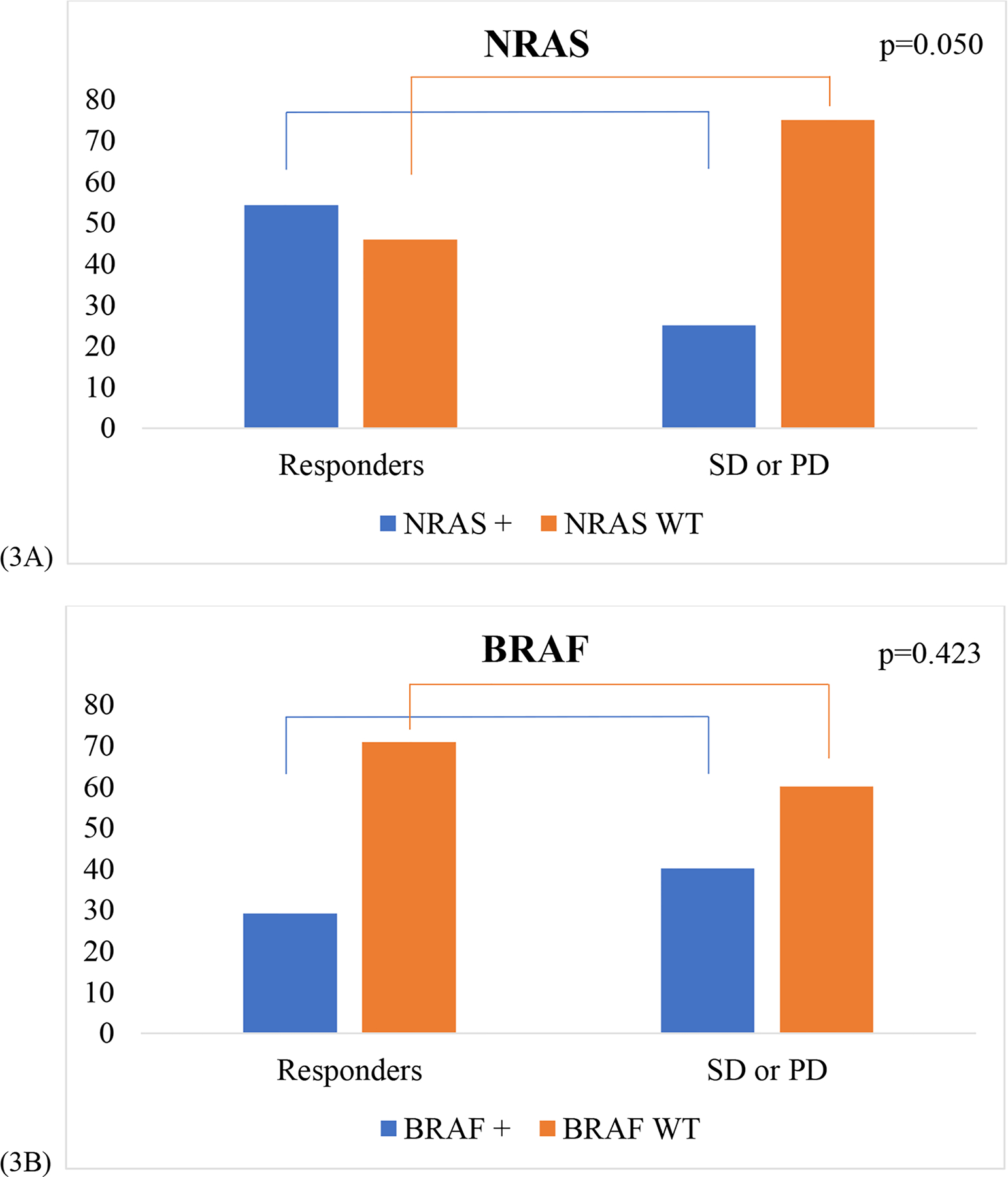
(3A): Proportion of NRAS positive (+) and NRAS WT in responders to immunotherapy compared to patients with SD or PD. A greater proportion of responders to immunotherapy were NRAS positive compared to patients with SD or PD (p=0.050). (3B): Proportion of BRAF positive (+) and BRAF WT in responders to immunotherapy compared to BRAF + and BRAF WT patients with SD or PD (p=0.423).
Post-hoc analysis was performed to assess the relationships between these NGS mutation status with OS and PFS. Kaplan-Meier curves demonstrated that patients who were NRAS positive had a higher PFS probability compared to those who were NRAS WT (p=0.042), (Figure 4A). Conversely, there was no significant difference in OS between patients with an NRAS mutation, compared to NRAS WT (p=0.111), (Figure 4B). Finally, the association between BRAF mutation status and OS and PFS were assessed. There was no significant difference in BRAF mutant positive patients compared to BRAF WT patients in PFS (p=0.661) or OS (p=0.730), in patients who received immunotherapy (Figure 4C, 4D).
Figure 4: PFS and OS Stratified by NRAS and BRAF Mutation.
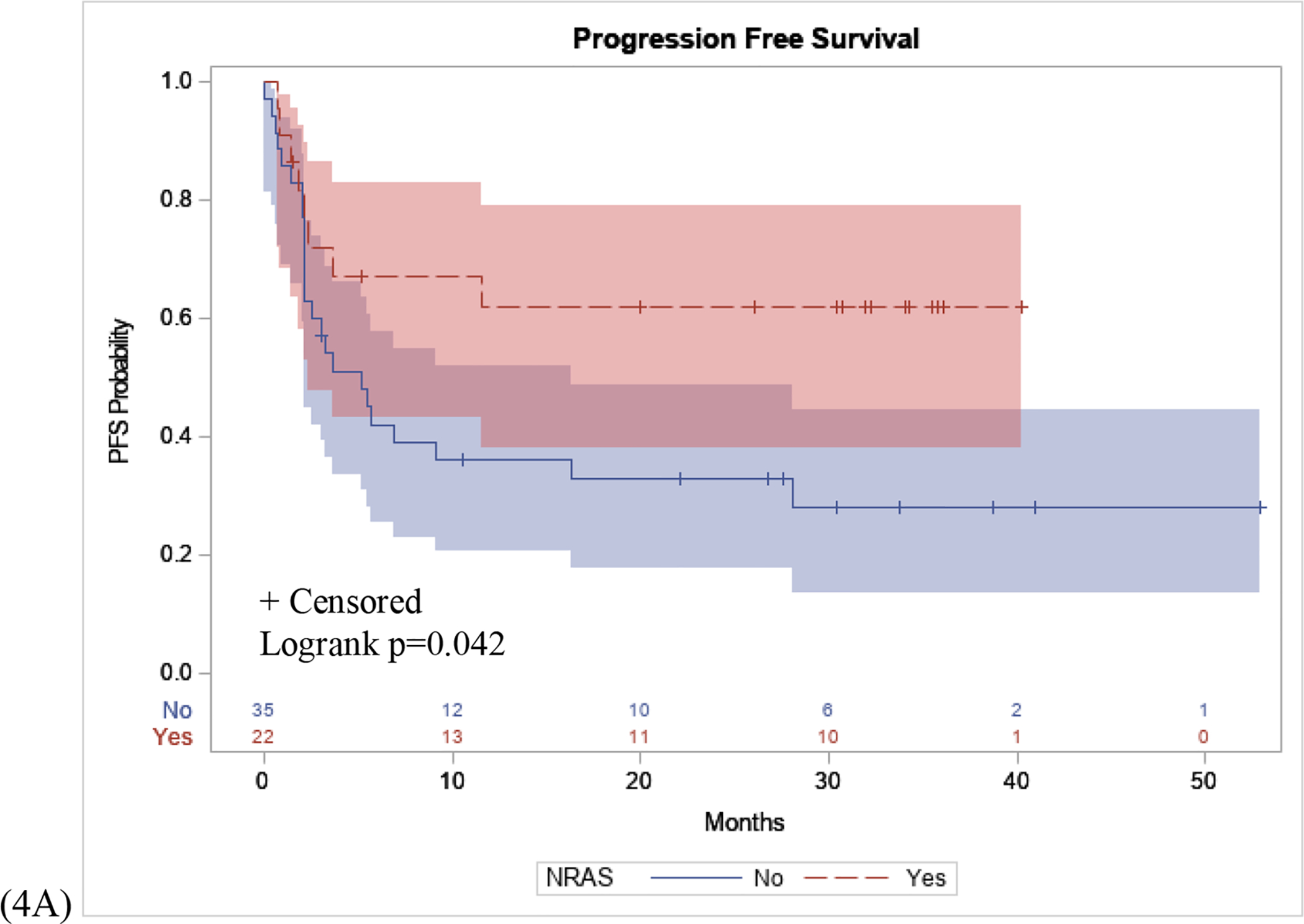
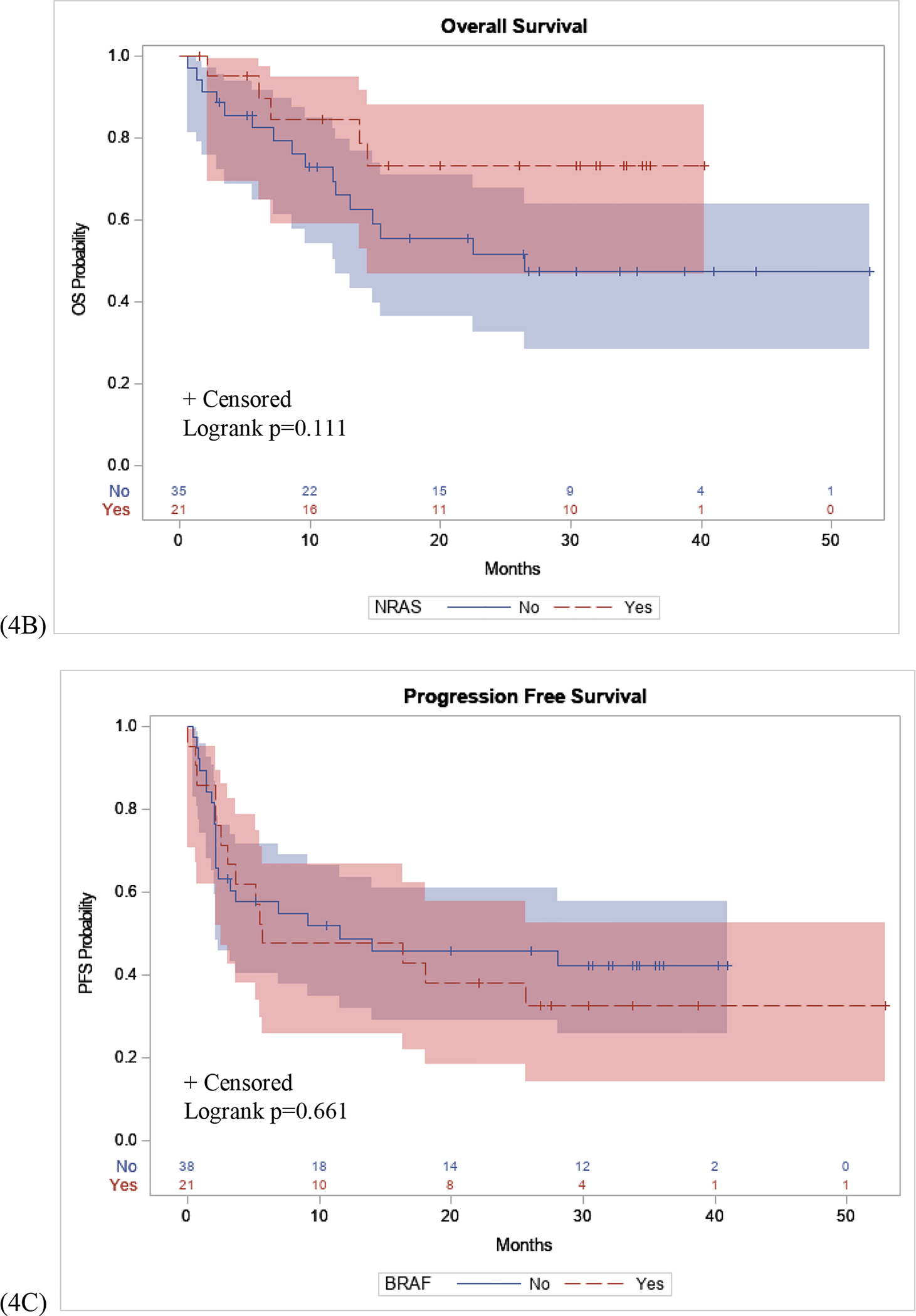
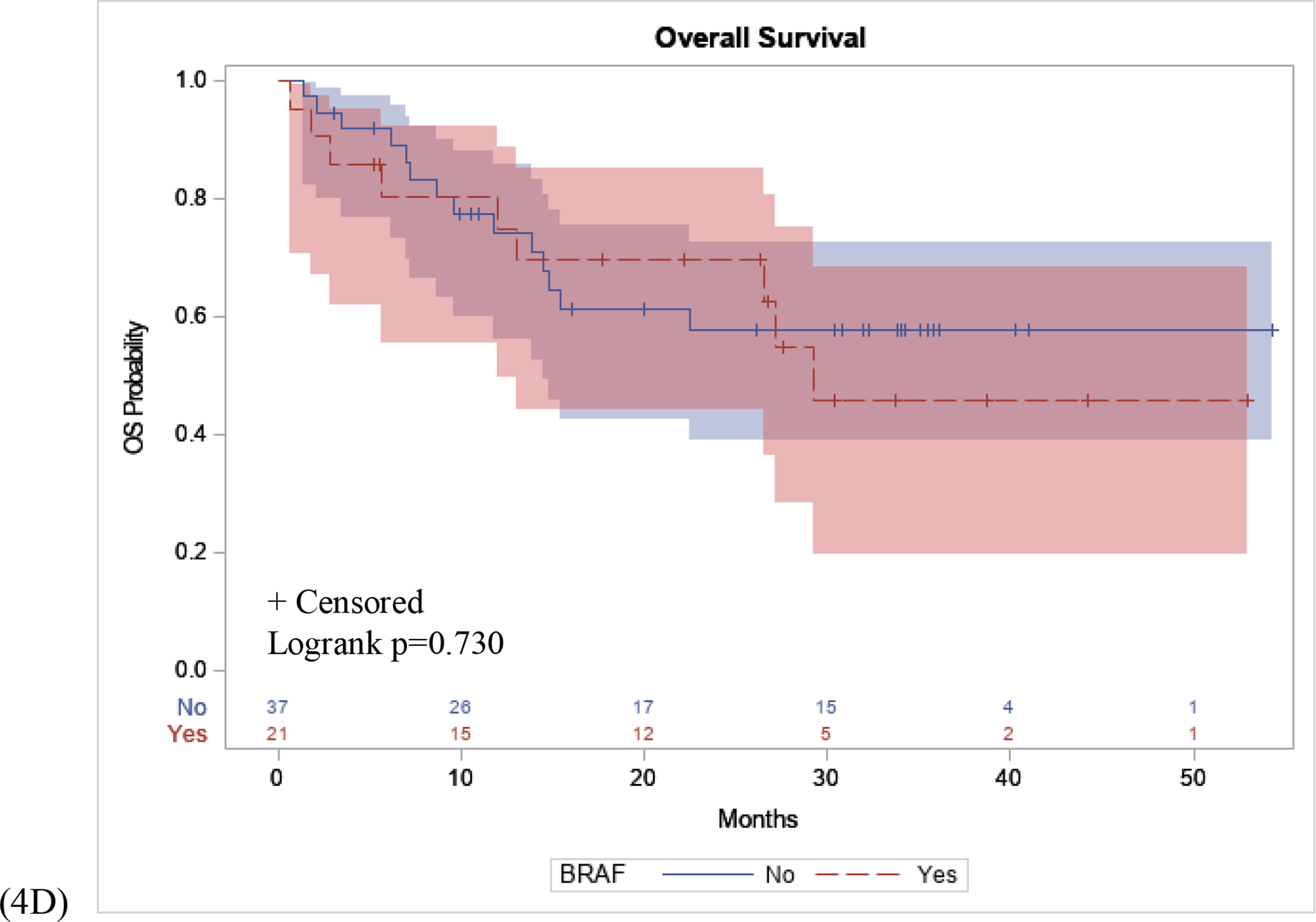
(4A): Patients with an NRAS mutation had a longer PFS interval following initial immunotherapy treatment compared to NRAS WT patients (p=0.042). (4B): No difference in OS in patients with an NRAS mutation compared to NRAS WT patients (p=0.111). (4C) No difference in PFS interval following initial immunotherapy treatment in patients with BRAF mutation compared to BRAF WT patients (p=0.661). (4D) No difference in OS in patients with a BRAF mutation compared to BRAF WT (p=0.730).
For patients treated with immunotherapy for recurrent melanoma after definitive surgical resection, we examined the disease free interval of two cohorts, long disease free interval versus short disease free interval. The greatest risk of recurrence following wide local excision surgery for melanoma has been shown to be within the first 3 years following surgery, with most of that risk within the first 1.5–2 years post-op. If a person recurs within 1 year, it is reasonable to postulate that a patient’s immune system likely is not contributing to stem the recurrence. Therefore, we have defined our disease free cohorts for “short disease free interval” for <1 year, and “long disease free interval” for ≥1 year. Patients with long disease free interval had a significantly longer PFS interval following immunotherapy compared to patients with short disease free interval (p=0.042), (Figure 5A). There was no significant difference in OS in patients with long disease free interval compared to patients with short disease free interval (p=0.280), (Figure 5B).
Figure 5: PFS and OS Stratified by Disease Free Interval.
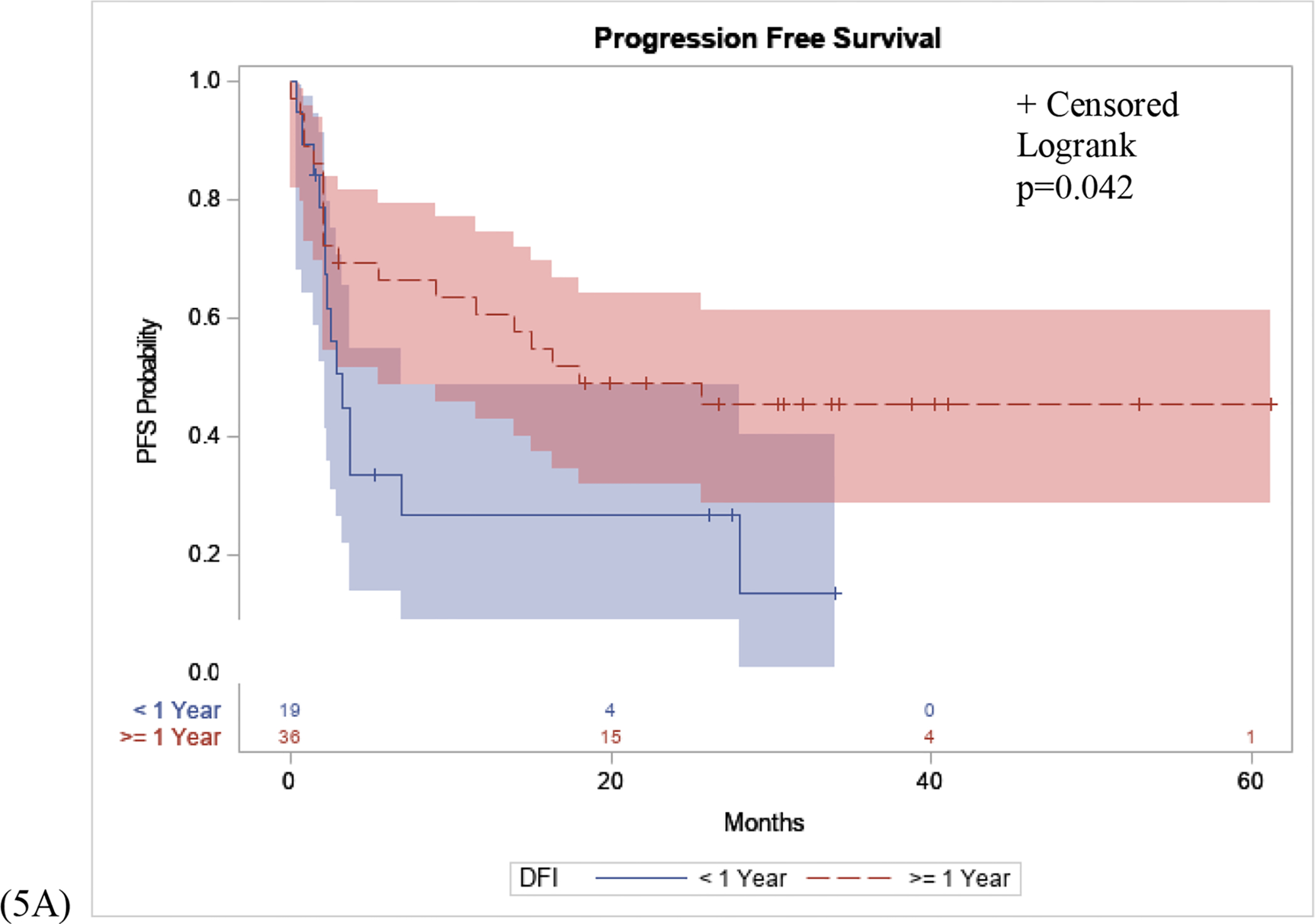
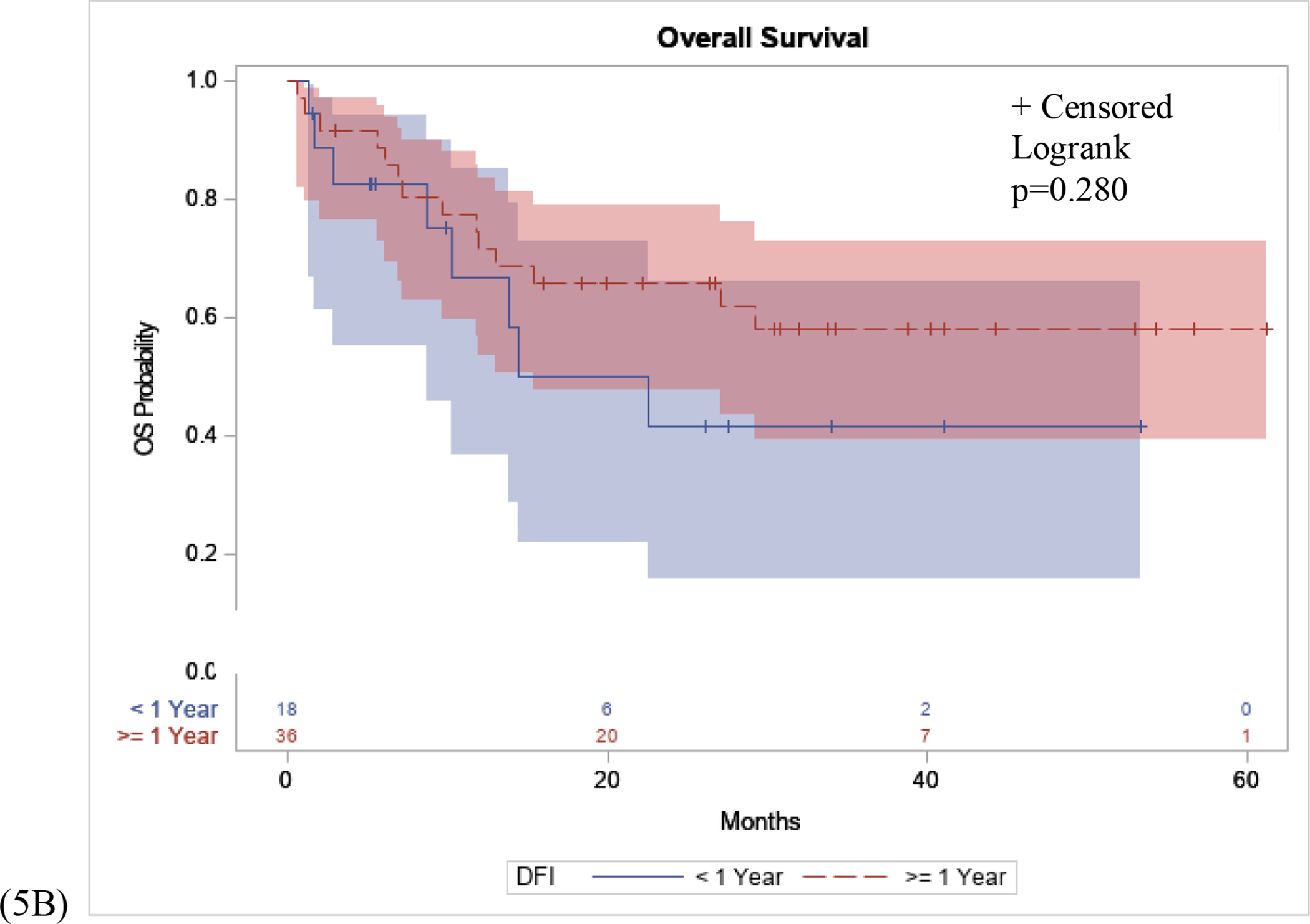
(5A): Patients with long disease free interval (DFI), defined as recurrence ≥1 year following WLE surgery had a significantly longer PFS interval following immunotherapy compared to patients with short disease free interval, defined as recurrence <1 year following WLE surgery (p=0.042). (5B): No significant difference in OS in patients with long disease free interval compared to patients with short disease free interval (p=0.280).
DISCUSSION
Several biomarkers have been proposed as tools to aid in predicting response to immunotherapy in advanced melanoma. The tumor microenvironment, encompassing a greater percentage of tumor infiltrating lymphocytes (TILs), has been shown to correlate with an increased response to anti-PD1 therapy for advanced melanoma (Daud et al. 2016). Additionally, immunohistochemistry (IHC) of PD-L1 staining in metastatic tumors have been shown to be associated with anti-PD1 treatment benefit (Tumeh et al. 2014). However, assays to determine PD-L1 positivity from tumor biopsies remain inconsistent in their clinical protocols. There is currently no standardized detection antibody and variable cutoff determinations for PD-L1 positivity, making the PD-L1 assay a difficult test to be widely used in the clinical setting as an optimal predictive marker of immunotherapy response, (Weber et al. 2013, Patel et al. 2015).
Lymphovascular invasion is a negative prognostic marker in patients diagnosed with melanoma, and is a hallmark of aggressive melanoma (Tas et al. 2017, Scolyer et al, 2013). Sentinel lymph node positivity is associated with decreased 10-year melanoma-specific survival rate (Morton et al. 2014). Yet, sentinel lymph nodes have also been shown to be sites where antitumor immune responses are mounted (Lund et al. 2016). Immune cells are carried within lymphatic vessels and draining lymph nodes are involved in the maintenance of tolerance against self-antigens (Wigg et al. 2012). Additionally, peripherally activated dendritic cells responsible for activating immune responses typically traffic to draining lymph nodes (Platt et al. 2013). Lymphatic endothelial cells further contribute to regional immunity and act as regulators of the immune system to maintain peripheral tolerance by directly inhibiting autoreactive T-cells (Dieterich et al. 2017). Lymphangiogenesis is also involved in a variety of host inflammatory situations such as in melanoma and other cancers (Pasquail et al. 2013). Taken together, lymphatic drainage, lymphatic endothelial cells, and lymphangiogenesis play complex roles in both tumor progression and activation of the immune system in response to active or chronic inflammation.
Given that immunotherapies such as anti-CTLA4 and PD-1 inhibitors have capitalized on harnessing immune-mediated tumor killing and regression, this suggests that endogenous mechanisms regulating immune induction in the tumors may be responsible for resistance to therapy (Crusz et al. 2015, Swartz et al. 2012). Thus, while lymphatic vessels have been explored as conduits for tumor spread, they may also act as regulators of local inflammation and immunity by providing a venue for the regulation of antitumor activity given the importance of endogenous immune response for successful response to immunotherapy. The current paradigm of adaptive immunity induction suggests that tumors release cytokines and chemokines, inducing immune infiltration and an increase in inflammatory mediators, leading to tumor cell killing and the activation of dendritic cells which hone into lymph nodes (Lund et al. 2016). With the transport of antigens, signals, and immune cells through lymphatic vessels, the presence of lymphovascular invasion may bolster the initiation of an immune response against tumors, facilitating an increase between inflammatory cells and immune infiltration in melanoma.
Patients with lymphovascular invasion may therefore exhibit a more robust peripheral T-cell lymphocyte melanoma-specific antigen primed population and may subsequently derive a greater benefit from systemically acting immunotherapies. While a negative prognostic marker of melanoma diagnosis, we hypothesize that patients who exhibit lymphovascular invasion may exhibit increased response to immunotherapy. We postulate that patients with melanoma and associated lymphovascular invasion have an immune system with greater access to and are primed to be exposed to a higher load of tumor antigens. The results of this study suggest that lymphovascular invasion, a reported pathologic feature on the synoptic report, correlates with increased response rates and greater PFS and OS intervals in the setting of immunotherapy for advanced melanoma. It is reasonable then to hypothesize that the presence of lymphovascular invasion, currently related to melanoma tumor progression and nodal involvement, may be associated with greater systemic exposure to peripheral antigen presenting cells and increased response to immunotherapy with an improved PFS and OS.
For patients treated for recurrence, we postulate that a long disease free interval might suggest that such a patient may have active immune-controlled disease. Such patients may feasibly develop an exhausted T-cell phenotype (in which case adding immunotherapy may “waken” the exhausted T-Cells), or their melanoma has mutated and has lost an immune-recognized antigen, allowing it to escape the immune system (in which case immunotherapy might not be beneficial to such patients). Conversely, for patients with a short disease free interval, we postulate that such patients have no immune recognition, thus allowing the melanoma to recur quickly. The addition of immunotherapy to such patients may not be of benefit.
The utilization of Next Generation Sequencing technology has allowed for the testing of millions of DNA segments in parallel, resulting in the sequencing of several cancer-related genes in a single assay. NGS has allowed for improved sensitivity in mutation detection, and has been described as cost effective given its ability to utilize formalin-fixed and paraffin-embedded primary tumor or metastatic lesion specimens with small amounts of DNA (Glenn 2011, Tsongalis et al. 2014). Additionally, studies by Goswami et al. 2015 and Manca et al. 2019 have shown mutational concordance of genetic alterations between melanoma primary tumors and metastatic lesions when pathogenic mutations in driver genes are examined using NGS analysis. The use of the NGS multi-gene screening panel may allow for a more personalized approach to melanoma and cancer therapy given the inclusion of relevant melanoma genes with prognostic, diagnostic, clinical, or treatment value (Bustos et al. 2017). By identifying potentially actionable mutations, the translation of these study results may provide additional insights into the molecular changes that may lead to patients being more or less responsive to subsequent immunotherapy treatment and contribute to the improvement of personalized medicine.
The NGS results in this study validate previous studies examining the impact of NRAS mutations in responders to immunotherapy. Common genetic pathway alterations found in melanoma samples from patient specimens include CDKN2A/B, PTEN, BRAF, TP53, and NRAS (van Herpen et al. 2019). NRAS mutations are found in 15–20% of melanomas and has been associated with a more aggressive phenotype and poorer outcomes compared to patients with NRAS WT melanomas (Jakob et al. 2012). While there are several targeted therapies for patients with BRAF mutant melanomas, limited NRAS-specific targeted therapy is available for those patients who harbor an NRAS mutation (Thumar et al. 2014). The results of this study validate that NRAS positive tumors correlate with increased response to immunotherapy and that NRAS positive patients exhibited a longer PFS interval following first-line immunotherapy treatment.
These results are also in agreement with the work of Dupuis et al. which demonstrated that improved 6-month objective response rates to anti-PD1 immunotherapy were associated with NRAS mutation (Dupuis et al. 2018). Additionally, the results of this study further validate the findings of Muñoz-Couselo et al. and Johnson et al. who demonstrated that patients with NRAS-mutant tumors have enjoyed an improved objective response and median OS to anti-CTLA4, anti-PD1, PD-L1 and IL-2 therapies compared to WT subtypes (Muñoz-Couselo et al. 2017, Johnson DB and Puzanov I 2015, Johnson et al. 2016).
Despite the findings, this study has multiple limitations. This retrospective observational investigation is subject to selection and measurement bias. Additionally, the sample size is small and may under- or over-estimate relationships. Furthermore, with the limited sample size, the feasibility of performing a multivariate analyses to demonstrate pathologic features as independent factors was poor. The inclusion window, which includes a mix of immunotherapy treatment regimens, may be considered too broad. Therefore, future studies to include an additional validation cohort with patients stratified by more distinct immunotherapy treatments and additional patients with primary biopsy samples and pathology reports to include lymphovascular invasion are currently being conducted. Given that the institution is a single regional NCI-designated comprehensive cancer center, the investigative design and institution-specific variability in clinical practice patterns would be optimally controlled by conducting a multicenter study. Additionally, the NGS panel of 50 cancer related genes, used at the time, is limited in relation to other assays able to analyze a wider array of genes. Lastly, given the nature of the institutional melanoma tumor database, we could not account for all variables such as sun exposure, or margin status, for example, that could further impact the interface between putative biomarkers and patient outcomes.
Given the utilization of anti-CTLA4 and PD-1 blockade immunotherapies for recurrent or metastatic melanoma, it is increasingly important to develop robust biomarkers that may optimize treatment of patients. Although it may be that a constellation of variables are needed to accurately predict response to immunotherapy, the current testing of PD-L1 and TMB are of limited utility, thus making it imperative to further identify markers which may be associated with treatment response, or the lack thereof, early in the melanoma disease process. Currently, ongoing efforts at Fox Chase Cancer center include identifying additional patients treated with single agent and combination immunotherapy regimens and stratifying patients based on treatment regimen. Additionally, we are working to further stratify patients based on timing of response to immunotherapy to predict factors that may contribute to exceptional responders versus rapid progression to immunotherapy treatment.
The utilization of primary biopsy pathology reports, with a focus on lymphovascular invasion data, may serve as a rational biomarker data set to help predict response to immunotherapy and, to help predict PFS and OS in patients with advanced melanoma. Additionally, using a validated NGS platform, this analysis validated potential associations between NRAS positive tumors and responders to immunotherapy and prolonged PFS following immunotherapy. The universal practice of obtaining a synoptic primary biopsy pathology report and access to the lymphovascular invasion pathology variable is ideal as a predictive tool regarding response to immunotherapeutics. Looking forward, the drive to identify early predictive factors, tumor mutation profiles, and biomarkers to predict response to immunotherapy will remain crucial in guiding clinical decision making and patient-tailored treatment selection.
SIGNIFICANCE.
This study is unique in its methodology in examining a large data set of melanoma patients with primary biopsy data and conducting a post hoc analysis on a subset of patients who received immunotherapy, or whose tumor samples also underwent NGS molecular testing. The findings in this study demonstrate a potential predictive utility of lymphovascular invasion and response to immunotherapy. To our knowledge, this study represents the first report examining lymphovascular invasion from primary biopsy tissue samples and response to immunotherapy.
ACKNOWLEDGEMENTS
KL, GG, and JMF designed the research studies. ML, SR, AJO, JMF acquired data. KL, IS, MR analyzed data. KL, GG, IS, AJO, JMF wrote the manuscript. MD and ER conducted statistical analysis. BL performed Next Generation Sequencing. HW conducted pathology review. All authors gave individual approval of the submitted version of the manuscript. KL was supported by the Medical Student Rotation grant from the Conquer Cancer ASCO Foundation. This work was supported in part by NCI grant P30 CA006927 for biostatistics support.
Funding
Kimberly Loo was supported by the Medical Student Rotation grant from the Conquer Cancer ASCO Foundation. This work was supported in part by NCI grant P30 CA006927 for biostatistics support.
Footnotes
Conflict of Interest
AJO has received honoraria on advisory boards or providing other scientific content to BMS, Pfizer, Novartis, Merck, Array, Alkermes, EMD Serono, and Iovance and FCCC receives research funding from Alkermes, Amgen, Astellas, BMS, Boston Biomedical, Checkmate Pharmaceuticals, Eli Lilly, EMD Serono, GSK, Immunocore, Incyte, Intensity Therapeutics, Kartos, Nektar, Oncoceutics, Sound Biologics, Takeda, Targovax. All other authors report no commercial or proprietary interest in any product mentioned or concept discussed in this manuscript.
REFERENCES:
- 1.Bd Bustos, Estal RM, Simo GP, et al. (2017). Towards personalized medicine in melanoma: implementation of a clinical next-generation sequencing panel. Scientific Reports, 7:495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chaput N, Lepage P, Coutzac C, et al. (2017) Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Annals of Oncology, 28(6):1368–1379. [DOI] [PubMed] [Google Scholar]
- 3.Crusz SM, Balkwill FR. (2015) Inflammation and cancer: advances and new agents. Nature Reviews Clinical Oncology,12(10):584–96. [DOI] [PubMed] [Google Scholar]
- 4.Daud AI, Loo K, Pauli ML, et al. (2016) Tumor immune profiling predicts response to anti-PD-1 therapy in human melanoma. Journal of Clinical Investigation, 126(9):3447–3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Daud AI, Wolchok JD, Robert C, et al. (2016) Programmed Death-Ligand 1 Expression and Response to the Anti-Programmed Death 1 Antibody Pembrolizumab in Melanoma. Journal of Clinical Oncology, 34(34):4102–4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dupuis F, Lamant L, Gerard E, et al. (2018) Clinical, histological and molecular predictors of metastatic melanoma responses to anti-PD-1 immunotherapy. British Journal of Cancer, 119(2):193–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Geynisman DM, Chen CR, Smieliauskas F, et al. (2014). Economic evaluation of therapeutic cancer vaccines and immunotherapy: A systematic review. Human Vaccines and Immunotherapeutics, 10(11): 3415–3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Glenn TC. (2011) Field guide to next-generation DNA sequencers. Molecular Ecologogy Resources, 11:759–769. [DOI] [PubMed] [Google Scholar]
- 9.Goswami RS, Patel KP, Singh RR et al. (2015) Hotspot mutation panel testing reveals clonal evolution in a study of 265 paired primary and metastatic tumors. Clinical Cancer Research, 21(11): 2644–2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hamid O, Robert C, Daud A, et al. (2013) Safety and tumor responses with lambrolizumab (anti–PD-1) in melanoma. New England Journal of Medicine, 369(2):134–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hamid O, Schmidt H, Nissan A, et al. (2011) A prospective phase II trial exploring the association between tumor microenvironment biomarkers and clinical activity of ipilimumab in advanced melanoma. Journal of Translational Medcine, 9:204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hodi FS, O’Day SJ, McDermott DF, et al. (2010) Improved survival with ipilimumab in patients with metastatic melanoma. New England Journal of Medicine, 363(8):711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Inoue H, Park JH, Kiyotani K, et al. (2016) Intratumoral expression levels of PD-L1, GZMA, and HLA-A along with oligoclonal T cell expansion associate with response to nivolumab in metastatic melanoma. Oncoimmunology, 5(9):e1204507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jakob JA, Bassett RL Jr, Ng CS, et al. (2012) NRAS mutation status is an independent prognostic factor in metastatic melanoma. Cancer, 118(16):4014–4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johnson DB, Lovly CM, Flavin M, et al. (2016) Impact of NRAS mutations for patients with advanced melanoma treated with immune therapies. Cancer Immunology Research, 3(3):288–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johnson DB, Puzanov I. (2015) Treatment of NRAS-mutant melanoma. Current Treatment Options Oncology, 16(4):15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lund AW, Wagner M, Fankhauser M et al. (2016) Lymphatic vessels regulate immune microenvironments in human and murine melanoma. Journal of Clinical Investigation, 126(9): 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manca A, Paliogiannis P, Colombino M et al. (2019) Journal of Translational Medicine, 17:289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morton DL, Thompson JF, Cochran AJ et al. (2014) Final trial report of sentinel-node biopsy versus nodal observation in melanoma. New England Journal of Medicine. 370(7): 599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muñoz-Couselo E, Adelantado EZ, Ortiz C, et al. (2017) NRAS-mutant melanoma: current challenges and future prospect. OncoTargets and Therapy, 10:3941–3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patel SP, Kurzrock R. (2015) PD-L1 expression as a predictive biomarker in cancer immunotherapy. Molecular Cancer Therapies. 14(4): 847–56. [DOI] [PubMed] [Google Scholar]
- 22.Pasquali S, van der Ploeg AP, Mocellin S et al. (2013) Lymphatic biomarkers in primary melanomas as predictors of regional lymph node metastasis and patient outcomes. Pigment Cell & Melanoma Research. 26(3):326–37. [DOI] [PubMed] [Google Scholar]
- 23.Pitt JM, Vetizou M, Daillere R, et al. (2016) Resistance Mechanisms to Immune-Checkpoint Blockade in Cancer: Tumor-Intrinsic and -Extrinsic Factors. Immunity, 44(6):1255–1269. [DOI] [PubMed] [Google Scholar]
- 24.Platt AM, Rutkowski JM, Martel C et al. (2013) Normal dendritic cell mobilization to lymph nodes under conditions of severe lymphatic hypoplasia. Journal of Immunology, 190(9):4608–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reiman A, Kikuchi H, Scocchia D, et al. (2017) Validation of an NGS mutation detection panel for melanoma. BMC Cancer, 17(1):150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robert C, Schachter J, Long GV, et al. (2015) Pembrolizumab versus Ipilimumab in Advanced Melanoma. New England Journal of Medicine, 372(26):2521–2532. [DOI] [PubMed] [Google Scholar]
- 27.Simeone E, Gentilcore G, Giannarelli D, et al. (2014) Immunological and biological changes during ipilimumab treatment and their potential correlation with clinical response and survival in patients with advanced melanoma. Cancer Immunology Immunotherpy, 63(7):675–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Snyder A, Makarov V, Merghoub T, et al. (2014) Genetic basis for clinical response to CTLA4 blockade in melanoma. New England Journal of Medicine, 371(23):2189–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Swartz MA, Lund AW. (2012) Lymphatic and interstitial flow in the tumour microenvironment: linking mechanobiology with immunity. Nature Review Cancer, 12(3):210–9. [DOI] [PubMed] [Google Scholar]
- 30.Tas F, Erturk K. (2017) Histological lymphovascular invasion is associated with nodal involvement, recurrence, and survival in patients with cutaneous malignant melanoma. International Journal of Dermatology, 56(2):166–170. [DOI] [PubMed] [Google Scholar]
- 31.Thumar J, Shahbazian D, Aziz SA, et al. (2014) MEK targeting in N-RAS mutated metastatic melanoma. Molecular Cancer, 13:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Topalian SL, Hodi FS, Brahmer JR, et al. (2012) Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. New England Journal of Medicine, 3366(26):2443–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Topalian SL, Sznol M, McDermott DF, et al. (2014) Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. Journal of Clinical Oncology, 32(10):1020–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tsongalis GJ, Peterson JD, de Abreu FB, et al. (2014) Routine use of the Ion Torrent AmpliSeq™ Cancer Hotspot Panel for identification of clinically actionable somatic mutations. Clin Chem Lab Med. 52(5):707–14. [DOI] [PubMed] [Google Scholar]
- 35.Tumeh PC, Harview CL, Yearley JH, et al. (2014) PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature, 515(7528):568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van Herpen CML, Agarwala SS, Hauschild A, et al. (2019) Biomarkers results from a phase II study of MEK1/2 inhibitor binimetinib (MEK162) in patients with advanced NRAS- or BRAF-mutated melanoma. Oncotarget, 10(19):1850–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weber JS, D’Angelo SP, Minor D, et al. (2015) Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncology, 16(4):375–384. [DOI] [PubMed] [Google Scholar]
- 38.Weber JS, Kudchadkar RR, Yu B, et al. (2013) Safety, efficacy, and biomarkers of Nivolumab with vaccine in Ipilimumab- refractory or -naive melanoma. Journal of Clinical Oncology, 31(34):4311–4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wiig H, Swartz MA. (2012) Interstitial fluid and lymph formation and transport: physiological regulation and roles in inflammation and cancer. Physiology Review, 92(3):1005–60. [DOI] [PubMed] [Google Scholar]